

Key Points
-
•
CD19-CD28 provides effective CD28 costimulation to glofitamab-activated T cells without superagonistic properties.
-
•
Triple combination with CD19–4-1BBL further prolongs antitumor responses.
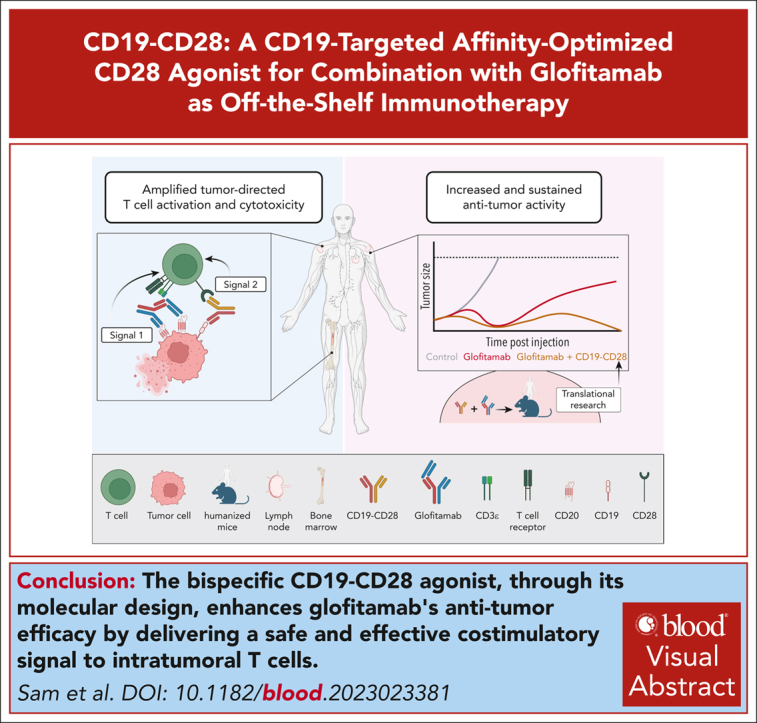
Visual Abstract
Abstract
Effective T-cell responses not only require the engagement of T-cell receptors (TCRs; “signal 1”), but also the availability of costimulatory signals (“signal 2”). T-cell bispecific antibodies (TCBs) deliver a robust signal 1 by engaging the TCR signaling component CD3ε, while simultaneously binding to tumor antigens. The CD20-TCB glofitamab redirects T cells to CD20-expressing malignant B cells. Although glofitamab exhibits strong single-agent efficacy, adding costimulatory signaling may enhance the depth and durability of T-cell–mediated tumor cell killing. We developed a bispecific CD19-targeted CD28 agonist (CD19-CD28), RG6333, to enhance the efficacy of glofitamab and similar TCBs by delivering signal 2 to tumor-infiltrating T cells. CD19-CD28 distinguishes itself from the superagonistic antibody TGN1412, because its activity requires the simultaneous presence of a TCR signal and CD19 target binding. This is achieved through its engineered format incorporating a mutated Fc region with abolished FcγR and C1q binding, CD28 monovalency, and a moderate CD28 binding affinity. In combination with glofitamab, CD19-CD28 strongly increased T-cell effector functions in ex vivo assays using peripheral blood mononuclear cells and spleen samples derived from patients with lymphoma and enhanced glofitamab-mediated regression of aggressive lymphomas in humanized mice. Notably, the triple combination of glofitamab with CD19-CD28 with the costimulatory 4-1BB agonist, CD19–4-1BBL, offered substantially improved long-term tumor control over glofitamab monotherapy and respective duplet combinations. Our findings highlight CD19-CD28 as a safe and highly efficacious off-the-shelf combination partner for glofitamab, similar TCBs, and other costimulatory agonists. CD19-CD28 is currently in a phase 1 clinical trial in combination with glofitamab. This trial was registered at www.clinicaltrials.gov as #NCT05219513.
Sam and colleagues present preclinical data regarding a novel bispecific CD19-targeted CD28 agonist to enhance the efficacy of glofitamab, a CD20 T-cell bispecific antibody (TSB) that directs T cells to CD20-expressing malignant B cells. This CD19-CD28 TSB is an “off-the-shelf” product that in studies in humanized mice enhances the antitumor efficacy of glofitamab and now awaits clinical evaluation.
Introduction
T cells are crucial for antitumor immune responses due to their ability to recognize and eliminate cancer cells.1,2 The T-cell receptor (TCR)–CD3 complex is key in this process, binding to specific antigens on target cells and transmitting activation signals through its associated CD3γ, δ, ε, and ζ domains. This antigen-dependent signal, termed “signal 1,” establishes the foundation of T-cell activation and expansion. However, full T-cell functionality requires a second signal, “signal 2,” provided by costimulatory receptors such as CD28, which is abundantly expressed on naïve and antigen-experienced T cells, including tumor-infiltrating lymphocytes.3, 4, 5, 6 Ligation of CD28 is strictly required for T-cell priming, clonal expansion, cytokine production, target cell lysis, and the establishment of durable T-cell memory.3 In natural T-cell responses, TCR stimulation without costimulation eventually leads to T-cell unresponsiveness.3
Synthetic redirection of T cells to destroy tumor cells has been used in cancer immunotherapy,2,7 with promising approaches including tumor-targeted T-cell bispecific antibodies (TCBs) and chimeric antigen receptor–modified T cells (CAR-Ts). TCBs bind to tumor antigens and simultaneously activate T cells via CD3 engagement. CAR-Ts are patient-derived T cells genetically engineered to express an extracellular tumor antigen–binding domain coupled to an intracellular signaling domain that activates TCR and costimulatory signaling pathways.8 In cases of B-cell non-Hodgkin lymphoma (B NHL), TCBs have shown strong antitumor efficacy.9, 10, 11, 12 Glofitamab, a CD20-targeted TCB engaging T cells via CD3ε, was recently approved by both the US Food and Drug Administration and the European Medicines Agency for treating relapsed/refractory diffuse large B-cell lymphoma (R/R DLBCL), after 39% of patients achieved a complete response in an open-label, multicenter, single-arm study.13 These observations with TCB antibodies show some similarities to the outcomes seen with second-generation CAR-Ts, which incorporate both signal 1 and signal 2 within their CAR designs. However, the production of CAR-Ts involves a complex 3-week manufacturing process, requiring specific infrastructure, which limits availability and delays treatment onset, thus presenting a significant challenge for patients battling a highly aggressive disease.
We hypothesized that a tumor-targeted CD28 agonistic antibody, designed to provide signal 2 as off-the-shelf combination partner to TCBs, including glofitamab, could further enhance glofitamab’s efficacy. However, efforts to harness CD28 signaling require caution, because the first-in-human trial with the CD28 agonistic, antibody TGN1412 resulted in a life-threatening cytokine release syndrome in 6 healthy volunteers.14 In this trial, TGN1412 caused systemic T-cell activation in consequence of its signal-1–independent activity, a phenomenon termed superagonism.
Here, we report the generation and evaluation of a CD19-targeted CD28 agonist (CD19-CD28), that offers costimulation exclusively in the presence of signal 1 and amplifies glofitamab’s efficacy. Furthermore, humanized mouse models emerged as an adequate approach for exploratory CD28 safety assessment and human dose prediction. These models guided dosing and scheduling in an ongoing phase 1 clinical trial, which assesses the safety, tolerability, pharmacokinetics (PK), and efficacy of CD19-CD28 in combination with glofitamab in patients with R/R NHL (NCT05219513).
Methods
Detailed methods are available in the supplemental Data, available on the Blood website.
Design and production of monovalent CD28 antibody derivatives and CD19-CD28 bispecific antibodies
Key amino acids of the TGN1412 antibody (murine binder 5.11A) were replaced to prevent degradation and reduce affinity. Several variable heavy and variable light variants were designed. Mutations were introduced to the constant region of the human immunoglobulin G1 (IgG1) heavy chains to prevent binding to Fcγ receptors and C1q. Monovalent CD28 antibodies and bispecific CD19-CD28 antibodies were generated with specific mutations to prevent mispairing of heavy and light chains and produced by transient transfection of Expi293F cells. More details can be found in the supplemental Methods.
In vitro binding of CD19-CD28 to human CD19 and CD28
Binding of CD19-CD28 to human CD28 was evaluated using Chinese hamster ovary (CHO-kl) cells genetically modified to overexpress human CD28. To monitor nonspecific binding interactions, a DP47 IgG, germ line antibody without specificity, was included as negative control. Binding was measured by flow cytometry. A detailed protocol can be found in the supplemental Methods.
In vitro activity of CD19-CD28 in Jurkat IL-2 promoter reporter cells
Jurkat interleukin-2 (IL-2) promoter reporter cells were incubated with NALM-6 target cells alongside increasing concentrations of CD19-CD28 and a suboptimal dose of glofitamab. IL-2 promoter activation was assessed by measuring luciferase activity in a Tecan Spark 10M, as described in the supplemental Methods.
Ability of CD19-CD28 to activate human PBMCs
Peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats and stimulated with the indicated compounds in round-bottom 96-well plates at 37°C. After 48 hours, cytokine release was measured using the Bio-Plex Pro Human Cytokine 17-plex or 8-plex kit (Bio-Rad). After 72 hours, T-cell activation markers were assessed via flow cytometry, as described in the supplemental Methods.
Ex vivo characterization of CD19-CD28 using samples from patients with lymphoma
DLBCL PBMC samples were purchased from Discovery Life Sciences. A spleen sample from a patient with B-cell lymphoma (BCL) was purchased from iSpecimen. PBMCs were depleted of B cells using CD20 microbeads (Miltenyi Biotec) and stimulated with the indicated. Splenocytes were stimulated with glofitamab alone or in combination with CD19-CD28 and/or CD19–4-1BBL. Supernatants were harvested and cytokine levels were measured using a cytokine bead array (BD Biosciences), according to the manufacturer’s instructions. Sample and assay preparation are described in detail in the supplemental Methods.
In vivo mouse models
Three- to 4-week-old female NOD.Cg-PrkdcscidIL2rgtm1Wjl/SzJ (NSG) mice were humanized via human stem cell engraftment as described in the supplemental Methods. In some cases, humanized BRGS-CD47 mice were directly purchased from Jackson laboratory.
For xenograft models in humanized mice, NALM-6, WSU-DLCL2, or OCI-Ly18 cells were injected subcutaneously into the right flank of mice, and the tumor volume was assessed by caliper measurement. For in vivo efficacy assessment in disseminated WSU-DLCL2-Fluc model, mice received cell injection IV and tumor burden was measured via bioluminescence imaging as described in the supplemental Methods.
Mice were randomized and treatments were injected IV in histidine buffer.
Single dose PK study in human FcRn Tg32 mice
PK properties of the CD19-CD28 agonist were assessed at indicated time points in human FcRn Tg32 upon a single IV injection. Serum samples were analyzed for quantification of CD19-CD28 using a CD19–binding competent enzyme-linked immunosorbent assay (ELISA) as described in more detail in the supplemental Methods.
RNA sequencing and analysis of tumor tissues
Azenta Life Sciences performed RNA sequencing of tumor tissue. RNA extraction, sequencing protocols, and differential gene expression analysis were performed as described in the supplemental Methods.
Confocal imaging and 3D image processing
Sections of 70 μm of fixed tumor samples were stained with the indicated antibodies (see supplemental Methods). Images were captured using a Leica SP8 inverted confocal microscope and a 40× lens, with a resolution of 512 × 512 pixels and a z-spacing of 1.5 μm. Image quantification was performed in Imaris 9.6 (Bitplane). Neighborhood analysis for cluster identification is described in the supplemental Methods.
In silico modeling and translation of nonhuman PK for dose range projection
A MATLAB Simbiology 2020b in silico model was developed to simulate receptor occupancy and trimeric complex formation (ie, synapse formation between cancer target cells and immune cells) across various CD19-CD28 concentrations using the respective target binding affinities.
The generated efficacy data in humanized mice were compared with the in silico–predicted bell shape. With that, 2 pharmacodynamic (PD) thresholds were derived from the study and scaled to human: the minimal pharmacologically active dose (mPAD) and a maximal effective concentration (ECmax), which correspond to the lowest effective dose and the dose leading to maximum tumor growth inhibition, respectively.
Human equivalent doses were defined using allometric scaling of PK parameters from hFcRn tg32 mice. The predicted human PK was used to define the doses expected to lead to mPAD and ECmax and to predict the expected human PK after various doses. The clinical PK observed in patients was compared with preclinical predictions to evaluate its accuracy.
Plasma cytokine assessment and PK measurement in patients enrolled in the phase 1 clinical trial of glofitamab in combination with CD19-CD28 (NCT05219513)
Patient plasma samples were analysis at protocol-specified time points using validated multiplex immunoassays on a ProteinSimple Ella platform. Concentrations of binding-competent CD19-CD28 in human serum samples were determined using a fully validated ELISA method as described in supplemental Methods.
Experiments involving animal models, as presented in this manuscript, were conducted following strict ethical guidelines in the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC)-accredited animal facility of the Roche Innovation Center Zurich. The experimental design and procedures were thoroughly reviewed and approved by the internal Institutional Animal Care and Use Committee. This approval process ensured that all efforts were made to minimize animal suffering. The number of animals used was kept to the minimum necessary to achieve valid results. All animal housing, care, and experimental procedures were in compliance with the guidelines set by GV-Solas, Felasa, and TierschG. As part of a multicenter international clinical trial (NCT05219513), patients have signed informed consent, and ethics committees and health authorities of respective sites/countries have approved the study.
Results
Design and functional evaluation of an affinity-optimized CD19-targeted CD28 agonist
We aimed to create CD19-CD28 bispecific antibodies with optimized properties (Figure 1A). Published evidence indicates that high affinities for CD3 can restrict the tumor distribution of TCB antibodies, due to their sequestration in T-cell–rich tissues.15 CD28 is abundantly expressed on T cells in blood and lymphoid organs, which could represent a considerable sink for CD28-targeted antibodies. To address this concern, we generated 31 CD28 antigen–binding domains starting from TeGenero’s TGN1412 binding moiety by introducing point mutations into the complementarity determining regions, aiming to reduce CD28 affinity. Furthermore, we removed potential sources of protein instability, including unpaired cysteines, asparagine deamidation sites, and tryptophane residues from the original sequence, because these might compromise structural in vivo integrity. In vitro binding assays led to the selection of 6 candidates for functional in vitro characterization in a tumor-targeted format (supplemental Figure 1A-D).
Figure 1.

Design and functional evaluation of an affinity-optimized CD19-targeted CD28 agonist. (A) CD19-CD28 is composed of one CD19 binder and one CD28 binder. The Fc part is devoid of FcγR binding (huIgG1 PGLALA). Heterodimerization and correct assembly are achieved via knob into hole (kih) mutation and CrossMab technology. (B) Binding of CD19-CD28 affinity variants to human CD28 on CHO cells, genetically modified to overexpress CD28. To monitor unspecific binding interactions, a DP47 huIgG1 was included as negative control. Binding was assessed via flow cytometry. Dots show individual values of technical duplicates. (C) Luciferase activity in an IL-2 reporter assay with Jurkat IL-2 promoter cells after 6 hours of stimulation with increasing concentrations (0.5 pM to 200 nM) of CD19-CD28 and 10 nM glofitamab. NALM-6 cells served as target cells (E:T ratio 5:1). Dots show individual values of technical duplicates. (D) Experimental design of an in vivo biodistribution study. Non–tumor bearing humanized NSG mice (3 mice per group) were treated with vehicle (histidine buffer) or 5 mg/kg of untargeted, Alexa-Fluor-647-(AF647)-labeled CD28 affinity variants. (E-F) Blood and splenic T cells were analyzed for drug binding via flow cytometry. Bars show mean + standard error of the mean (SEM) of 3 animals per group and time point. Dots show values of individual mice. (G) Experimental design of in vivo efficacy study. Humanized NSG mice (9-10 mice per group) were subcutaneously (s.c.) injected with 1 × 106 NALM-6 lymphoma cells in 1 flank. After 18 days, mice were treated IV with vehicle (histidine buffer), 0.15 mg/kg glofitamab, and 1 mg/kg CD19-CD28 variants according to the depicted timeline. (H) Tumor volumes shown as mean + SEM of 9 to 10 mice per group. Significance was calculated using an unpaired, 2-tailed Student t test. ∗∗P < .01. (I) Immunohistochemical analysis of CD8+ T-cell infiltration in tumors on study day 56. Upper row, ×2 original magnification. Lower row, ×20 original magnification. Images were captured with a VS120 virtual slide microscope (Olympus) and analyzed with Tissue Studio software (Definiens) for cell quantification. E:T, effector to target; glofit., glofitamab.
From these experiments, we identified 3 candidates: a low-affinity variant (CD28LOW; with a dissociation constant [Kd] of 36 nM), an intermediate affinity variant (CD28MED; Kd, 10 nM), and the original high-affinity TGN1412 (CD28HIGH; Kd, 1 nM), which was optimized for stability by removal of an unpaired cysteine (Figure 1A). These were converted into bispecific monovalent 1 + 1 heterodimeric CD19-CD28 bispecific antibodies. Both, CD28LOW and CD28MED showed reduced CD28 (Figure 1B) but comparable CD19 binding (supplemental Figure 1E). All variants activated Jurkat IL-2 promoter reporter cells in a dose-dependent manner when combined with glofitamab in the presence of CD19-expressing NALM-6 lymphoma cells (Figure 1C).
Next, we examined the biodistribution of the 3 untargeted CD28 variants in humanized NSG mice, which display functional human T cells, to see whether reduced CD28 affinity would lessen drug sequestration in T-cell–rich areas (Figure 1D). Indeed, drug binding to T cells in both blood and spleen was most pronounced with the CD28HIGH binder, followed by CD28MED and CD28LOW (Figure 1E-F), indicating that lower CD28 affinity reduces peripheral sequestration. Consequently, we excluded CD28HIGH from further analyses.
We evaluated whether the remaining CD28 variants could enhance glofitamab-mediated antitumor response in humanized NSG mice bearing subcutaneous NALM-6 lymphomas (Figure 1G). Although both variants showed a combination effect, the CD28LOW proved significantly more effective (Figure 1H) and led to superior intratumoral CD8+ T-cell infiltration than CD19-CD28MED (Figure 1I).
In conclusion, CD19-CD28LOW, in combination with glofitamab, showed comparable in vitro potency with higher affinity variants, superior in vivo efficacy, and minimal peripheral T-cell binding. Thus, CD19-CD28LOW, now referred to as CD19-CD28, was selected for further evaluation.
CD19-CD28 is not superagonistic and relies on signal 1
Previous studies linked TGN1412’s superagonistic activity to CD28 binding bivalency and FcγRIIb crosslinking, presumably allowing TGN1412 to create linear CD28 arrays, resulting in aggregated signaling components sufficient to surpass the threshold for T-cell activation.16, 17, 18 Therefore, we compared the monotherapeutic activity of CD19-CD28 and TGN1412 to induce cytokine secretion and immune cell activation (Figure 2). In vitro, only TGN1412 triggered a dose-dependent cytokine release in healthy donor–derived PBMCs (Figure 2A). In humanized NSG mice, TGN1412 elicited a robust cytokine response, peaking at 4 hours (Figure 2B-C), but no cytokine release was detected with CD19-CD28. Because humanized NSG mice maintain substantial levels of murine Fc receptors in nonhematopoietic cells, we also included a superagonistic anti-human CD28 mouse IgG1 antibody capable of interacting with murine Fc receptors. Nonetheless, we observed no discernable difference in cytokine responses compared with the native TGN1412 IgG4 (Figure 2C). By day 6, animals receiving TGN1412 (huIgG4 or muIgG1) displayed marked activation of T cells, including regulatory T cells, in both spleens and thymuses (supplemental Figure 2). In contrast, no cytokine release was detected in humanized NSG mice when treated with CD19-CD28 (Figure 2C). CD19-CD28 only induced a dose-dependent upregulation of T-cell activation markers and proinflammatory cytokines in human PBMCs when combined with glofitamab (Figure 2D; supplemental Figure 3). From these results, we inferred that CD19-CD28 is not superagonistic. Instead, it offers genuine costimulation dependent on binding to CD19 and on the presence of signal 1. Furthermore, we addressed the impact of CD19 expression on our bispecific antibody's activity. Our in vitro studies (supplemental Figure 10) show that T-cell activation by our CD19-CD28 bispecific agonist correlates with CD19 expression levels, yet effective T-cell activation is achieved even at reduced CD19 levels. The concentration threshold at which CD19-CD28 begins to activate T cells remains consistent. This underscores the robustness of the agonist's potency, affirming that the initial concentration required for activity is stable across different levels of target expression.
Figure 2.

CD19-CD28 is not superagonistic and relies on signal 1. (A) Activity of human PBMCs in response to TGN1412 and CD19-CD28. PBMCs isolated from buffy coats of 3 donors were stimulated with TGN1412 or CD19-CD28 (dose titration, 0-500 nM) and cytokine release was analyzed after 48 hours via multiplex analysis. Bars show mean + SEM of technical triplicates from 3 donors. (B) Experimental design of in vivo cytokine release evaluation. Non–tumor bearing humanized NSG mice (3 mice per group) were treated IV with vehicle (histidine buffer), CD19-CD28 (10 or 1 mg/kg), TGN1412 huIgG4 (10 mg/kg), or TGN1412 msIgG1 (10 mg/kg). (C) Multiplex analysis of serum cytokines at indicated time points after treatment. The absolute cytokine values are shown in supplemental Table 1. (D) Human PBMCs from 4 healthy donors were stimulated with glofitamab (0, 1, 10, or 100 pM) and increasing concentrations of CD19-CD28 (0-200 nM). Upper row, the percentage of CD25 expression on CD8+ T cells was analyzed by flow cytometry after 72 hours. Bars show mean + SEM of technical triplicates for each donor. Dots show individual values. Lower row, multiplex analysis of IL-6 in culture supernatants after 48 hours. Bars show mean + SEM of technical triplicates. Dots show individual values. The shades of gray correspond to different donors, with each donor consistently represented by the same shade across all treatment conditions.
CD19-CD28 boosts the efficacy of glofitamab in a dose-dependent manner in vivo and enhances its activity on patient-derived T cells
We next explored the potency of CD19-CD28 in more challenging scenarios, including assays with patient-derived T cells and aggressive in vivo lymphoma mouse models. In a disseminated DLBCL WSU-DLCL2-Fluc tumor model in humanized mice (Figure 3A), the combination of glofitamab and CD19-CD28 achieved complete tumor regression (Figure 3B), with significantly superior efficacy compared with glofitamab monotherapy at 17 and 20 days after tumor cell injection (Figure 3C). By study day 10, animals receiving either vehicle or only CD19-CD28 began to exhibit weight loss attributed to tumor progression (Figure 3D). Mice treated with glofitamab, either alone or in combination with CD19-CD28, initially lost weight, which is associated with the mode of action of TCB and their induced cytokine release.10 Mice in these groups subsequently recovered, with no additional weight loss indicative of tumor progression, suggesting robust antitumor activity and supporting the bioluminescence data (Figure 3D). Distinguishing early weight loss attributable to cytokine release from later weight decline associated with tumor advancement is critical. In the same in vivo model, using different doses of CD19-CD28, we observed a bell-shaped dose response for CD19-CD28 and an optimal efficacious dose range of 0.1 to 1 mg/kg in mice. The bioluminescence data, generated 3 days after the first CD19-CD28 injection, from this dose escalation experiment have been successfully overlaid with an in silico model for trimeric complex formation. The good agreement of model and observed data further support the proposed optimal dose range window (Figure 3E). The preclinical PK was translated to patients based on allometric scaling from human FcRn Tg32 mice. The projected PK was overlaid with clinically observed PK data and shows a good agreement between the projected and observed clinical PK overall. This overlay also provides guidance in terms of how well the PK is matching the predicted efficacy thresholds derived based on the preclinical prediction for trimeric complex formation (maximum bell shape) and observed PD readouts (mPAD and ECmax) from the humanized mouse study (Figure 3F).
Figure 3.

CD19-CD28 boosts the efficacy of glofitamab in a dose-dependent manner in vivo and enhances its activity on patient-derived T cells. (A) Experimental design of in vivo efficacy study. Humanized NSG mice bearing orthotopic WSU-DLCL2-Fluc tumors (8 mice per group) were treated with vehicle (histidine buffer), glofitamab (0.15 mg/kg), and CD19-CD28 (1 mg/kg) IV according to the displayed timeline. (B) Visualization of tumor growth in treated mice after luciferin injection. (C) Tumor burden evaluated by bioluminescence signal (total flux, photons/second) calculated as the mean radiance integrated over the region of interest. Dots represent individual mice. Bars show the median signal + IQR for each treatment group. Statistical analysis was performed using an ordinary 1-way analysis of variance (ANOVA) with Fisher least significant test. ∗∗∗P < .0001. (D) Body weight kinetics. Dots represent means -SEM of 8 mice per group. (E) Overlay of in vivo dose finding experiment with in silico modeling for trimeric complex formation. (F) Overlay of predicted human PK from on hFcRn tg32 mice and preclinical efficacy thresholds based on in vivo studies in NSG mice with PK measurements in participants of the ongoing CD19-CD28 phase 1 trial (NCT05219513). (G) PBMCs from 2 patients with DLBCL (patient 1 and 2) were depleted for internal B cells and were stimulated with the indicated treatments (glofitamab, untargeted TCB, and CD19-CD28 were used at a concentration of 1 nM) in the presence of NALM-6 target cells (E:T ratio: 3:1). Graphs show IFNγ and granzyme B release after 72 hours, assessed via cytokine bead array. Bars show means and dots show individual values from technical triplicates (patient 1) and duplicates (patient 2). BLI, bioluminescence imaging; IQR, interquartile range; max, maximum.
Furthermore, we evaluated the responsiveness of patient-derived T cells to CD19-CD28 ex vivo and found that CD19-CD28 in combination with glofitamab, but not in its absence, enhanced granzyme B and interferon-γ (IFNγ) secretion in PBMC-derived T cells of 2 patients with DLBCL (Figure 3G). Collectively, these findings highlight the potential of CD19-CD28 to enhance the therapeutic efficacy of T-cell engagers including glofitamab.
CD19-CD28 increases proinflammatory T-cell signatures in tumors and facilitates transendothelial migration
Glofitamab monotherapy and its combination with CD19-CD28 promote tumor T-cell infiltration (supplemental Figures 4C and 5B). However, combination treatment increased intratumoral CD8+ and CD4+ T-cell counts compared with glofitamab alone by day 8 after therapy onset (study day 25) in WSU-DLCL2–bearing humanized NSG mice (supplemental Figure 4). Intriguingly, the combination treatment shifted the intratumoral CD8+/regulatory T-cell ratio in favor of CD8+ T cells, suggesting the induction of a highly inflammatory tumor milieu (supplemental Figure 4D). In subcutaneous OCI-Ly18 tumors, we confirmed that both glofitamab and the combination with CD19-CD28 increased tumor infiltration of CD28-expressing T cells (supplemental Figure 5A-D). In this model, the addition of CD19-CD28 to glofitamab led to a marked increase in IFNγ pathway genes, followed by significantly increased cytotoxic T-cell and decreased B-cell signatures, compared with glofitamab monotherapy (Figure 4B; supplemental Figure 5E). Furthermore, although glofitamab increased the overall ICAM1 expression in tumors, the addition of CD19-CD28 skewed ICAM1 expression toward endothelial cells, suggesting that CD19-CD28 might promote transendothelial T-cell migration (Figure 4C-D). A neighborhood analysis in 20 μm radius regions of the tumor tissue indicated a trend in spatial association between endothelial and CD8+ T cells, which is observed more often in the combination treatment (supplemental Figure 5F-H).
Figure 4.

CD19-CD28 increases proinflammatory T-cell signatures in tumors and facilitates transendothelial migration. (A) Experimental design of in vivo efficacy study. OCI-Ly18 tumor-bearing humanized NSG mice were treated with vehicle (histidine buffer, 10 mice per group), glofitamab (0.15 mg/kg or 1 mg/kg, 25 mice per group), and CD19-CD28 (1 mg/kg, 25 mice per group). On day 19 and 21, five scouts per group were euthanized for analysis. Gpt: Gazyva (obinutuzumab) pretreatment, 30 mg/kg. (B) Gene signature analysis via RNA sequencing from frozen tumor tissue at different time points. Color code shows change between glofitamab monotherapy vs combination with CD19-CD28 (red denotes higher and blue lower expression in the combination). Statistical significance was calculated using ∗P < .05, ∗∗P < .01 (adjusted for multiple testing using FDR correction). Individual genes are shown in supplemental Figure 5E (C) Immunofluorescence microscopy of tumors on study day 29, showing ICAM1 (red), CD31 (green), and CD8 (cyan). Images were captured on a Leica SP8 inverted confocal microscope using a 40× lens, with a resolution of 512 × 512 pixels and a z-spacing of 1.5 μm. (D) Image quantification was performed with Imaris 9.6. Dot plots show spots created on ICAM1 signal per μm3 (left) and spots created on ICAM1-expressing endothelial (CD31+) cells of total ICAM1-expressing cells in tumors (right) on study day 29, quantified by fluorescence microscopy. Dots represent intratumoral regions from 1 or more mice. One-way ANOVA with Tukey multiple comparison test: ∗P < .05; ∗∗P < .01. FDR, false discovery rate; ICAM1, intercellular adhesion molecule 1.
These findings suggests that CD19-CD28 enhances the antitumor efficacy of glofitamab by amplifying tumor T-cell infiltration and proinflammatory cytokine profiles at early stages and promoting cytotoxic T-cell signatures at subsequent stages of antitumor T-cell responses.
Triple combination of glofitamab with CD19-CD28 and CD19–4-1BBL deepens and prolongs antitumor responses in vivo
Research in third generation CAR-T therapies suggests that combining CD28 and 4-1BB signaling achieves more robust and durable antitumor responses than each costimulatory signals on its own.19 To explore whether the same principle can be transferred to bispecific antibody approaches, we evaluated the ability of glofitamab in double and triple combinations with CD19-CD28 and the previously described costimulatory molecule CD19–4-1BBL20 to activate patient-derived T cells and prolong antitumor responses in established tumors in humanized mice (Figure 5). The combination of glofitamab and CD19-CD28 induced the expression of 4-1BB in human PBMC-derived T cells (Figure 5A), indicating that CD19-CD28 may amplify T-cell responsiveness to CD19–4-1BBL stimulation. In line with this, the triple combination revealed highest levels of granzyme B, IFNγ, IL-8, and IL-2 in splenic T cells from a patient with DLBCL (Figure 5B). In vivo, combining glofitamab with either CD19-CD28 or CD19–4-1BBL showed a prolongation of the antitumor activity over glofitamab monotherapy in OCI-Ly18 tumors. Yet, these dual combinations eventually only resulted in a delay of tumor growth for most animals (Figure 5D). However, as seen in 2 independent in vivo experiments, the triple combination of glofitamab with CD19-CD28 and CD19–4-1BBL consistently led to long-term tumor control (Figure 5E; supplemental Figure 6). This was especially evident in animals treated first with glofitamab and CD19-CD28, followed by glofitamab and CD19–4-1BBL. This outcome underscores the significance of determining the most effective sequence at which costimulatory signals are introduced.
Figure 5.

Triple combination of glofitamab with CD19-CD28 and CD19–4-1BBL deepens and prolongs antitumor responses in vivo. (A) PBMC-derived T cells from a healthy donor were stimulated with glofitamab and a dose titration of CD19-CD28. Graphs show 4-1BB expression of CD4+ and CD8+ T cells after 48 hours, assessed via flow cytometry. Shown are mean + SEM of technical triplicates. (B) Cytokine release of splenocytes derived from a patient with BCL after 72 hours of stimulation with 25 pM glofitamab and/or CD19-CD28 or CD19–4-1BBL at 0.5 or 1 nM. Cytokines were assessed via cytokine bead array. Bars show means and dots indicate individual values of technical duplicates. (C) Experimental design of in vivo efficacy study in OCI-Ly18 tumor-bearing humanized NSG mice. Twelve mice per group received vehicle (histidine buffer) or obinutuzumab pretreatment (Gpt, 30 mg/kg), followed by weekly injections of glofitamab (5 mg/kg) alone or in combination with CD19-CD28 (1 mg/kg), CD19–4-1BBL (1 mg/kg) according to the depicted scheme. Costimulatory antibodies were injected simultaneously with glofitamab. (D-E) Tumor volumes assessed via caliper. Each line represents tumor volume over time in 1 mouse.
In summary, our data highlight the potential of bispecific antibody therapies to evoke more robust and durable antitumor responses when leveraging multiple costimulatory signals.
Humanized mouse models are adequate for exploratory safety evaluation and dose finding
The life-threatening cytokine release syndrome in the first-in-human trial with TGN1412 occurred, although the initial drug dosage was 500 times below the presumed safe dose established in cynomolgus monkey studies.21,22 Subsequent research highlighted the differences in CD28 expression on cynomolgus monkey vs human T cells as a potential explanation why no toxicity had been observed in prior toxicology studies.23, 24, 25, 26
In light of these findings, we evaluated the predictivity of cynomolgus monkey studies for safety assessment of CD19-CD28. In vitro, we directly compared the responsiveness of isolated T cells from 4 cynomolgus monkeys with those from 4 humans to CD19-CD28. Unlike their human counterparts, T cells from cynomolgus monkeys remained unresponsive to the combined stimulation with CD19-CD28 and glofitamab (supplemental Figure 7). Our data further confirm that CD28 expression in cynomolgus T cells is lower than that of human T cells (supplemental Figure 8A-B). This difference is particularly pronounced in specific T-cell subsets, including CD4+ effector T cells, effector memory T cells, and terminally differentiated effector T cells (supplemental Figure 8C-D). Notably, these subsets have been identified as the primary contributors to the TGN1412-mediated cytokine release.23, 24, 25, 26
Conversely, T cells isolated from spleens of humanized NSG mice exhibited responses to CD19-CD28 and glofitamab that were comparable with their human counterparts in vitro (supplemental Figure 9). Specifically, the minimal concentration of CD19-CD28 required to produce an additive effect to glofitamab was consistent between the different human T-cell sources, suggesting humanized mice as a suitable model for human dose predictions. Importantly, the control bispecific antibody DP47-CD28 showed no detectable activity in T cells from either species, indicating that the observed effects are dependent on targeted crosslinking.
Pretreatment with Gpt followed by sequential administration of glofitamab and CD19-CD28 mitigates cytokine release
When the combination treatment was administered simultaneously to tumor-free mice, we observed a robust peripheral cytokine response accompanied by rapid weight loss in all animals, which lead to premature termination of this group (Figure 6B-C). In contrast, although the sequential combination with a 3-day interval between glofitamab and CD19-CD28 did reduce the severity of peripheral cytokine release, a pronounced body weight loss was still evident in these mice. However, pretreatment with obinutuzumab (Gpt) before the sequential administration effectively mitigated severe weight loss (Figure 6C), cytokine release (Figure 6B), and peripheral T-cell activation (Figure 6D-I).
Figure 6.

Pretreatment with Gpt, followed by sequential administration of glofitamab and CD19-CD28 mitigates cytokine release. (A) Non–tumor bearing humanized NSG mice (6 mice per group) were treated according to the depicted scheme, using the following doses: Gpt, 30 mg/kg; glofitamab, 0.15 mg/kg; CD19-CD28, 1 mg/kg. Histidine buffer was used as vehicle. Glofitamab and CD19-CD28 were either administered simultaneously (sim.) or sequentially (seq.), with a 3-day interval between treatments. Seq. and sim. combinations were either administered with or without Gpt 7 days before therapy. (B) Multiplex analysis of cytokines in serum at 4, 24, and 72 hours after last therapy injection. The absolute cytokine values are reported in supplemental Table 2. (C) Body weight kinetics. Dots show mean + SEM of body weight change of 6 mice per group. (D-I) Comparative analysis of T-cell activation and B-cell depletion in spleens, assessed via flow cytometry on study day 6. Bars show means, and dots indicate values of individual remaining mice at study termination. (J) Line plot shows mean fold change of plasma cytokine concentrations at indicated time points in patients with R/R NHL enrolled in the phase 1 clinical trial of glofitamab in combination with CD19-CD28 (NCT05219513). The error bars indicate SEM, and the dashed horizontal line indicates the baseline. The timing of each drug administration is shown by the dashed arrows. CxDx, cycle x day x.
Furthermore, we assessed the influence of Gpt on tumor growth inhibition and PD effects within the tumor in a separate in vivo study. Although Gpt did not demonstrate a potent antitumor response, it did lead to an increase of intratumoral T-cell frequency (supplemental Figure 11). These findings are in line with previously published data.10
These findings further guided scheduling in the ongoing phase 1 clinical trial of glofitamab in combination with CD19-CD28 in patients with R/R NHL (NCT05219513). In line with the preclinical findings, Gpt followed by the stepup dosing of glofitamab and the sequential administration of CD19-CD28 a week later mitigated systemic cytokine release as measured by changes in the concentration of proinflammatory cytokines in plasma at the end of first CD19-CD28 infusion (Figure 6J). These observations were in line with the clinically observed safety profile of the combination (data not shown) and demonstrates the translatability of the preclinical models.
Discussion
This study introduces CD19-CD28, a novel CD19-targeted CD28 agonistic bispecific antibody, delivering safe and strictly signal 1–dependent costimulation to T cells for immunotherapy of hematological malignancies. In humanized NSG mouse models with aggressive lymphomas, CD19-CD28 amplified the efficacy of glofitamab, a CD20-targeted CD3 T-cell engager recently approved by both the US Food and Drug Administration and the European Medicines Agency for the treatment of R/R DLBCL.
Therapeutic exploration of CD28 agonism has been approached with caution due to severe toxicity observed in a first-in-human trial with the CD28 superagonist TGN1412.14 In CD19-CD28, monovalent CD28 binding and a PGLALA mutated Fc region prevent superagonism. In this study, monotherapeutic CD19-CD28 remained inactive in both in vivo and in vitro scenarios in which TGN1412 induced substantial cytokine release and T-cell activation.
Follow-up studies of the TGN1412 trial have shown that the calculations of a safe starting dose for humans based on preclinical testing in cynomolgus monkeys was flawed by discrepancies in CD28 expression levels in cynomolgus monkey and human T cells.26 We found that, unlike their human counterparts, cynomolgus monkey PBMCs did not respond to CD19-CD28 in vitro. Conversely, humanized NSG mice exhibited profound cytokine responses to TGN1412 and responded to CD19-CD28 only when combined with glofitamab. Although not considered feasible for long-term toxicological studies, humanized NSG mice can be highly informative for assessing early PD and acute safety events related to human lymphocytes, such as T cells, as witnessed with TGN1412. Corroborating this notion, human dose predictions and scheduling based on humanized mouse models and human FcRn Tg32 mice have proven accurate for CD19-CD28, and no serum cytokine release has occurred upon first infusion of CD19-CD28 in the ongoing phase 1 clinical trial in patients with R/R NHL (NCT05219513). Interestingly, another study describing a MUC16-CD28 bispecific antibody found a dose-dependent response in cynomolgus monkey T cells in combination with a MUC16-specific T-cell engager.27 Furthermore, a CD22-CD28 bispecific antibody described by Wei et al in 2022 showed limited activity and no toxicity in vivo but augmented T-cell activation when combined with odronextamab, a CD20-CD3 bispecific TCB in primate studies.28 These findings indicate that additional factors, such as distinct affinities to CD28 or the nature of the signal 1 provider, may influence the predictivity of a preclinical model to anticipate the clinical safety of CD28 agonistic antibodies.
The evident success of next-generation CAR-T technology underscores the importance of integrating costimulatory signaling in therapeutic approaches based on T-cell redirection.29, 30, 31 Prior studies support the idea that this principle applies to bispecific antibody approaches as well; adding a tumor-specific antigen xCD28 antibody to tumor-specific antigen xCD3 antibodies enhanced the (artificial) immune synapse and significantly improved antitumor activity of T cells.27 Furthermore, tumor-targeted 4-1BBL fusion proteins effectively activated T cells and supported TCB-mediated tumor regression in mouse models.20 Consistent with these findings, our study reaffirms the advantages of introducing costimulatory signaling to bispecific antibody approaches.
Research on third generation CAR-Ts has indicated an added advantage of merging costimulatory signals, particularly CD28 and 4-1BB.19 Importantly, our findings suggest that the sequence in which costimulatory antibodies are administered may influence their antitumor efficacy. Administering both costimulatory molecules simultaneously can enhance tumor control, but this is dependent on the timing of the combination treatment, as evidenced by 2 separate studies discussed here. Specifically, it was observed that administering CD19-CD28 before CD19–4-1BBL resulted in superior tumor control compared with the reverse sequence or concomitant treatment. These findings may be in line with the generalized notion that although CD28 costimulation provides an early and hard hit T-cell response, subsequent 4-1BB costimulation is responsible for a durable and persistent T-cell response. In addition, these preclinical data hint at a potential advantage of bispecific antibody approaches. Unlike CAR-Ts, which deliver both stimulatory signals at once, bispecific antibodies allow for the timing of each signal to be adjusted. This flexibility in scheduling could potentially leverage the unique benefits of each costimulatory pathway more effectively.
It is important to note that target antigen expression is a critical factor for the success of targeted therapies. Although CD19– B NHLs are uncommon at initial diagnosis, CD19 downregulation can emerge in a subset of patients with DLBCL after CD19–CAR-T therapy, with incidences up to 30%.32 Despite the majority of patients with R/R NHL retaining CD19 expression even after CAR-T therapy, the increasing utilization of CAR-T as a second-line treatment and the risk of disease escape via CD19 antigen loss underscore the importance of evaluating this combination therapy in earlier treatment lines, ideally before CAR-T administration.
In conclusion, this study contributes to the mounting evidence supporting the advantages of incorporating costimulatory signaling into T-cell–engaging antibody strategies. Notably, we showcase that CD28 agonism can be tailored to offer genuine costimulation in its physiological role, strictly dependent on signal 1. CD19-CD28 could serve as a readily available, off-the-shelf combination partner to glofitamab and other costimulatory antibodies, bypassing the need for genetic T-cell modification and special infrastructure required for CAR-T approaches.
Conflict-of-interest disclosure: J.S., T.H., C. Kuettel, C.C., S.H., G.G., K.K., M.L., M.J.E., D.M., C.J., K.L., S.M.K., B.G., J.M., S.K., L.K., S.W., S.B., S. Gebhardt, A.V., B.A., V.N., D.S., M. Bez, E.B., J.E., C.H., F.L., S.J., A.S., M.L.C., S.C., M. Bacac, S. Gasser, C. Klein, and P.U. are employees of Roche and hold ownership of Roche stocks and patents. The remaining authors declare no competing financial interests.
Acknowledgments
Authorship
Contribution: C. Klein and P.U. contributed to the concepts and idea; J.S., C. Klein, P.U., M. Bacac, S. Gasser, and S.C. contributed to the design of all in vitro and in vivo experiments; T.H. and G.G. contributed to and coordinated antibody design and engineering; S.K. designed and coordinated in vivo and in vitro safety assessment; M.L. and M.J.E. designed and coordinated preclinical in vivo pharmacodynamic analyses and in silico prediction analysis; C.J. and S.M.K. designed and conducted the clinical pharmacodynamics analysis; J.T., S.H., C.C., C. Kuettel, J.-B.P.V., J.M., and B.G. designed, coordinated, and performed in vitro assays and analyzed, discussed, and interpreted the results; D.M. analyzed preclinical RNA sequencing data; J.S., E.B., B.A., A.S., M.L.C., V.N., F.L., L.K., D.S., S.B., S. Gebhardt, A.V., M. Bez, J.E., C.H., and S.J. designed, coordinated, and performed in vivo and ex vivo experiments and analyzed, discussed, and interpreted the results; K.K. and S.W. designed and analyzed the clinical biomarker data; K.L. designed and coordinated clinical trial; J.S. and J.T. wrote the manuscript; and all authors reviewed and edited the manuscript.
Footnotes
RNAsequencing raw data is currently uploaded to Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo/; accession number GSE260674).
Original data and more detailed information on methodologies and protocols are available upon reasonable request from the corresponding author, Johannes Sam (johannes.sam@roche.com).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Supplementary Material
References
- 1.Ahmed H, Mahmud AR, Siddiquee MFR, et al. Role of T cells in cancer immunotherapy: opportunities and challenges. Cancer Pathog Ther. 2023;1(2):116–126. doi: 10.1016/j.cpt.2022.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20(11):651–668. doi: 10.1038/s41577-020-0306-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Esensten JH, Helou YA, Chopra G, Weiss A, Bluestone JA. CD28 costimulation: from mechanism to therapy. Immunity. 2016;44(5):973–988. doi: 10.1016/j.immuni.2016.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lavin Y, Kobayashi S, Leader A, et al. Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell. 2017;169(4):750–765.e17. doi: 10.1016/j.cell.2017.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tirosh I, Izar B, Prakadan SM, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352(6282):189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zheng C, Zheng L, Yoo JK, et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell. 2017;169(7):1342–1356.e16. doi: 10.1016/j.cell.2017.05.035. [DOI] [PubMed] [Google Scholar]
- 7.Zhukovsky EA, Morse RJ, Maus MV. Bispecific antibodies and CARs: generalized immunotherapeutics harnessing T cell redirection. Curr Opin Immunol. 2016;40:24–35. doi: 10.1016/j.coi.2016.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao Y, Wang Y, Luo F, Chu Y. Optimization of T cell redirecting strategies: obtaining inspirations from natural process of T cell activation. Front Immunol. 2021;12 doi: 10.3389/fimmu.2021.664329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun LL, Ellerman D, Mathieu M, et al. Anti-CD20/CD3 T cell–dependent bispecific antibody for the treatment of B cell malignancies. Sci Transl Med. 2015;7(287) doi: 10.1126/scitranslmed.aaa4802. [DOI] [PubMed] [Google Scholar]
- 10.Bacac M, Colombetti S, Herter S, et al. CD20-TCB with obinutuzumab pretreatment as next-generation treatment of hematologic malignancies. Clin Cancer Res. 2018;24(19):4785–4797. doi: 10.1158/1078-0432.CCR-18-0455. [DOI] [PubMed] [Google Scholar]
- 11.Bannerji R, Arnason JE, Advani R, et al. Emerging clinical activity of REGN1979, an anti-CD20 x anti-CD3 bispecific antibody, in patients with relapsed/refractory follicular lymphoma (FL), diffuse large B-cell lymphoma (DLBCL), and other B-cell non-Hodgkin lymphoma (B-NHL) subtypes [abstract] Blood. 2018;132(suppl 1):1690. [Google Scholar]
- 12.Budde LE, Sehn LH, Assouline S, et al. Mosunetuzumab, a full-length bispecific CD20/CD3 antibody, displays clinical activity in relapsed/refractory B-cell non-Hodgkin lymphoma (NHL): interim safety and efficacy results from a phase 1 study [abstract] Blood. 2018;132(suppl 1):399. [Google Scholar]
- 13.Dickinson MJ, Carlo-Stella C, Morschhauser F, et al. Glofitamab for relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2022;387(24):2220–2231. doi: 10.1056/NEJMoa2206913. [DOI] [PubMed] [Google Scholar]
- 14.Suntharalingam G, Perry MR, Ward S, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355(10):1018–1028. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]
- 15.Mandikian D, Takahashi N, Lo AA, et al. Relative target affinities of T-cell–dependent bispecific antibodies determine biodistribution in a solid tumor mouse model. Mol Cancer Ther. 2018;17(4):776–785. doi: 10.1158/1535-7163.MCT-17-0657. [DOI] [PubMed] [Google Scholar]
- 16.Hussain K, Hargreaves CE, Roghanian A, et al. Upregulation of FcγRIIb on monocytes is necessary to promote the superagonist activity of TGN1412. Blood. 2015;125(1):102–110. doi: 10.1182/blood-2014-08-593061. [DOI] [PubMed] [Google Scholar]
- 17.Poirier N, Blancho G, Vanhove B. CD28-specific immunomodulating antibodies: what can be learned from experimental models? Am J Transplant. 2012;12(7):1682–1690. doi: 10.1111/j.1600-6143.2012.04032.x. [DOI] [PubMed] [Google Scholar]
- 18.Beyersdorf N, Hanke T, Kerkau T, Hünig T. Superagonistic anti-CD28 antibodies: potent activators of regulatory T cells for the therapy of autoimmune diseases. Ann Rheum Dis. 2005;64(suppl 4):iv91–iv95. doi: 10.1136/ard.2005.042564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramos CA, Rouce R, Robertson CS, et al. In vivo fate and activity of second-versus third-generation CD19-specific CAR-T cells in B cell non-Hodgkin’s lymphomas. Mol Ther. 2018;26(12):2727–2737. doi: 10.1016/j.ymthe.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Claus C, Ferrara C, Xu W, et al. Tumor-targeted 4-1BB agonists for combination with T cell bispecific antibodies as off-the-shelf therapy. Sci Transl Med. 2019;11(496) doi: 10.1126/scitranslmed.aav5989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Attarwala H. TGN1412: from discovery to disaster. J Young Pharm. 2010;2(3):332–336. doi: 10.4103/0975-1483.66810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eastwood D, Bird C, Dilger P, et al. Severity of the TGN 1412 trial disaster cytokine storm correlated with IL-2 release. Br J Clin Pharmacol. 2013;76(2):299–315. doi: 10.1111/bcp.12165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eastwood D, Findlay L, Poole S, et al. Monoclonal antibody TGN1412 trial failure explained by species differences in CD28 expression on CD4+ effector memory T-cells. Br J Pharmacol. 2010;161(3):512–526. doi: 10.1111/j.1476-5381.2010.00922.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Findlay L, Eastwood D, Stebbings R, et al. Improved in vitro methods to predict the in vivo toxicity in man of therapeutic monoclonal antibodies including TGN1412. J Immunol Methods. 2010;352(1-2):1–12. doi: 10.1016/j.jim.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 25.Pallardy M, Hünig T. Primate testing of TGN1412: right target, wrong cell. Br J Pharmacol. 2010;161(3):509–511. doi: 10.1111/j.1476-5381.2010.00925.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stebbings R, Eastwood D, Poole S, Thorpe R. After TGN1412: recent developments in cytokine release assays. J Immunotoxicol. 2013;10(1):75–82. doi: 10.3109/1547691X.2012.711783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Skokos D, Waite JC, Haber L, et al. A class of costimulatory CD28-bispecific antibodies that enhance the antitumor activity of CD3-bispecific antibodies. Sci Transl Med. 2020;12(525) doi: 10.1126/scitranslmed.aaw7888. [DOI] [PubMed] [Google Scholar]
- 28.Wei J, Montalvo-Ortiz W, Yu L, et al. CD22-targeted CD28 bispecific antibody enhances antitumor efficacy of odronextamab in refractory diffuse large B cell lymphoma models. Sci Transl Med. 2022;14(670) doi: 10.1126/scitranslmed.abn1082. [DOI] [PubMed] [Google Scholar]
- 29.Schuster SJ, Svoboda J, Chong EA, et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N Engl J Med. 2017;377(26):2545–2554. doi: 10.1056/NEJMoa1708566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531–2544. doi: 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schuster SJ, Bishop MR, Tam CS, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019;380(1):45–56. doi: 10.1056/NEJMoa1804980. [DOI] [PubMed] [Google Scholar]
- 32.Plaks V, Rossi JM, Chou J, et al. CD19 target evasion as a mechanism of relapse in large B-cell lymphoma treated with axicabtagene ciloleucel. Blood. 2021;138(12):1081–1085. doi: 10.1182/blood.2021010930. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.