

Abstract
Background
As the diagnosis of Parkinson's disease (PD) is fundamentally clinical, the usefulness of ioflupane (123I) single‐photon emission computed tomography (SPECT) or DaTSCAN as a diagnostic tool has been a matter of debate for years. The performance of DaTSCAN is generally recommended in the follow‐up of patients with a clinically uncertain diagnosis, especially in those with a suspected essential tremor, drug‐induced parkinsonism, or vascular parkinsonism. However, there is a dearth of DaTSCAN findings regarding neurodegenerative parkinsonisms besides PD and atypical parkinsonisms. To date, a specific nigrostriatal dopamine uptake pattern that would help differentiate PD from the most frequent atypical parkinsonisms is yet to be described. This fact is further complicated by the possible visualization of abnormalities in the uptake pattern in patients with rarer neurodegenerative parkinsonisms.
Objectives
We aimed to summarize the current literature regarding DaTSCAN findings in patients with rare neurodegenerative parkinsonisms.
Methods
The PubMed database was systematically screened for studies in English or Spanish up to October 15, 2023, using search terms “DaTSCAN”, “ioflupane”, “DaT‐SPECT”, “123I‐FP‐CIT SPECT”, “dopamine transporter imaging”, and “[123I] FP‐CIT SPECT”. Duplicated publications and studies regarding PD, atypical parkinsonisms, dystonia‐parkinsonism, essential tremor, and parkinsonism due to non‐degenerative causes were excluded.
Results
The obtained results were reviewed and summarized, including DaTSCAN findings in fragile X‐associated tremor/ataxia syndrome, prion diseases, Huntington's disease, spinocerebellar ataxia, hereditary spastic paraparesis, metabolic disorders, and other diseases (anti‐IgLON5 disease, ring chromosome 20 syndrome, chorea‐acanthocytosis, and neuronal ceroid lipofuscinosis).
Conclusions
This review highlights the need to determine in the future the utility and cost‐effectiveness of DaTSCAN, both as a diagnostic and a prognostic tool, in patients with parkinsonian symptoms in rare neurodegenerative diseases.
Keywords: SPECT, DaTSCAN, Parkinsonism, nigrostriatal pathway
Much has been achieved from a physiopathological, anatomopathological, clinical, and therapeutical point of view since Parkinson's disease (PD) was first described. However, to date, a diagnostic marker is missing, and the diagnosis of PD remains a fundamentally clinical—and, thus, a complex and potentially error‐bound—process. The current clinical diagnosis of PD relies on “red flags” and supporting criteria, some of them implying the longitudinal follow‐up of the patient. Therefore, it is not surprising that autopsy diagnostic confirmation in patients clinically diagnosed with PD ranges from 75 to 95%, according to different data. 1 , 2 As a result, physicians sometimes use complementary tests, such as pharmacological tests, neurosonography, and/or structural and functional neuroimaging to facilitate the differential diagnosis of the disease, particularly at its onset.
Dopamine transporters, localized at the presynaptic terminal of the nigrostriatal pathway in the putamen and caudate nucleus, are responsible for dopamine reuptake at the synaptic cleft to regulate the levels of the neurotransmitter, and the number of these transporters is associated with neuron loss in the mesencephalic substantia nigra. Alterations in the presynaptic dopaminergic pathway can be evaluated via single‐photon emission computerized tomography (SPECT) imaging using [123I]2β‐carbomethoxy‐3β‐(4‐iodophenyl)‐N‐(3‐fluoropropyl) nortropane, also known as 123I‐FP‐CIT, ioflupane, or DaTSCAN, a cocaine analog that reversibly binds to presynaptic dopamine transporters with high affinity, or via positron emission tomography (PET) using 3,4‐dihydroxy‐6‐[18F]fluoro‐L‐phenylalanine (F18‐DOPA), which allows the visualization of the dopamine precursor L‐DOPA. 3
The use of DaTSCAN has an A recommendation level in the European Federation of the Neurological Societies guidelines for PD diagnosis, 4 and is considered a useful diagnostic tool in patients presenting clinically inconclusive parkinsonism or tremor, particularly at the onset of the symptoms, as it allows the detection of dopaminergic neuron loss even before the start of clinical manifestations in individuals carrying PD‐related genetic mutations. This technique differentiates PD patients from healthy individuals with approximately 90% sensitivity 5 and offers a low false‐positive rate, frequently related to medications that can alter image analysis (antidepressants, anticholinergics, opioids, anesthetics, etc.). 6 , 7 Likewise, up to 15% of the patients meeting PD clinical criteria but showing predominant resting and postural tremor in the upper limbs might have normal results in this test, known as scans without evidence of dopaminergic deficit (SWEDD). 8
Approximately 1–2% of the global population is thought to be affected by PD. In some studies, the accuracy of the diagnosis would be mathematically identical to that of DaTSCAN. 9 Consequently, the utility of DaTSCAN versus clinical diagnosis has been a matter of debate since it was first approved by the United States Food and Drug Administration “to assist in the evaluation of adult patients with suspected parkinsonian syndromes (PS)”. As established by the European Medicines Agency, DaTSCAN is indicated for patients presenting clinically uncertain PS and to assist in the differentiation between Lewy body dementia (LBD) and Alzheimer's disease (AD). Thus, an abnormal DaTSCAN result would support the presence of nigrostriatal degeneration (PD, multiple system atrophy (MSA), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), LBD), while a normal result would suggest a diagnosis of pharmacological, psychogenic, or vascular parkinsonism (except when a striatal structure is directly affected by an infarct 10 in the latter case), dystonic tremor, essential tremor, or levodopa‐responsive dystonia. 11
However, the capacity of DaTSCAN to reflect the loss of integrity of the terminal fields of the nigrostriatal neurons regardless of the etiology limits its utility to differentiate among PD and other causes of dopaminergic deficiency at this level, such as atypical parkinsonisms. Moreover, its lack of specificity to differentiate among such diseases might reflect its inability to assist with decisions regarding the treatment of the patient, as it cannot predict which patients could respond to dopaminergic therapy at an early stage. 12 Nonetheless, the application of advanced image analysis techniques to differentiate between PD and atypical parkinsonisms has offered encouraging results, 13 and a recent meta‐analysis reported that DaTSCAN results modified the management of approximately half the patients tested for suspected PS and led to a change in the diagnosis of one‐third of them. 14
Over the years, DaTSCAN imaging has been performed in an increasing number of patients with suspicion of atypical parkinsonism or tremor. In this sense, DaTSCAN abnormalities have also been described in some patients who have received radiotherapy, 15 with structural lesions such as meningiomas, 16 and, more recently, patients with acute hypokinetic‐rigid syndrome following SARS‐CoV‐2 infection. 17 However, there is a scarcity of data regarding potential DaTSCAN abnormalities in patients presenting tremor and/or parkinsonism due to neurodegenerative causes beyond PD or parkinsonism plus syndromes.
In this review, we aim to summarize the published literature regarding potential DaTSCAN abnormal results observed in patients with fragile X‐associated tremor/ataxia syndrome, prion diseases, Huntington's disease, spinocerebellar ataxias, hereditary spastic paraparesis, some metabolic disorders, and other illnesses such as anti‐IgLON5 disease, ring chromosome 20 syndrome, chorea‐acanthocytosis, and neuronal ceroid lipofuscinosis. We also highlight the need to evaluate in the future the cost‐effectiveness of DaTSCAN, not only as a diagnostic aid tool but also in terms of its utility to predict clinical evolution and therapeutic response in these diseases.
Materials and Methods
A systematic review of the literature was performed using the PubMed database and search terms “DaTSCAN”, “ioflupane”, “DaT‐SPECT”, “123I‐FP‐CIT SPECT”, “dopamine transporter imaging”, and “[123I] FP‐CIT SPECT”, up to October 15, 2023. All results referencing DaTSCAN results in neurodegenerative diseases presenting tremor and/or parkinsonism were selected. Exclusion criteria were as follows: PD and atypical parkinsonisms (PSP, CBD, LBD, MSA); dystonia‐parkinsonism; essential tremor; and parkinsonism due to non‐degenerative causes (structural lesions, infections, pharmacological parkinsonism, vascular parkinsonism, normal pressure hydrocephalus, cranioencephalic trauma). Duplicated clinical studies were also excluded. Only publications written in English or Spanish were included.
Due to the scarce number of publications meeting the inclusion criteria, the results were divided into the following sub‐categories, according to the pathology: fragile X‐associated tremor/ataxia syndrome, prion diseases, Huntington's disease, spinocerebellar ataxia, hereditary spastic paraparesis, metabolic disorders, and other disorders.
Results
Fragile X‐Associated Tremor/Ataxia Syndrome
The fragile X‐associated tremor/ataxia syndrome (FXTAS) is a late‐onset neurodegenerative disorder that commonly affects men carrying an expansion in the premutation range (50–200 repetitions) of the CGG triplet in the FMR1 gene. Clinically, FXTAS is characterized by cerebellar ataxia or intention tremor, variably associated with other symptoms such as peripheral neuropathy, autonomic dysfunction, dementia, and parkinsonism.
The pathophysiological mechanism underlying parkinsonism in these patients is unknown. The currently available evidence supports the participation of both the cerebellum and the basal ganglia, particularly in those cases in which parkinsonism is a predominant clinical feature. T2‐weighted magnetic resonance imaging (MRI) studies have shown changes in the subcortical periventricular white matter, primarily affecting the middle cerebellar peduncles. Conversely, DaTSCAN imaging studies have shown contradictory results, displaying normal images in some patients 18 , 19 and abnormal results in others, 20 , 21 , 22 even with similar clinical phenotypes (Table 1). In a recent series that included 22 FXTAS patients, 47% of them showed abnormal results in DaTSCAN images, 23 suggesting that not all patients presenting expansions of the FMR1 gene will have a normal DaTSCAN or that some patients might present comorbid PD. The potential participation of the basal ganglia, and, particularly, that of the dopaminergic system projected towards the putamen, suggests the presence of a degenerative process affecting the nigrostriatal dopaminergic terminals from the very onset of the disease. 22
TABLE 1.
Summary of the available literature regarding potential DaTSCAN abnormal results associated with fragile X‐associated tremor/ataxia syndrome
| Research | Patients with DaTSCAN results | Clinical findings | DaTSCAN findings | Research conclusions |
|---|---|---|---|---|
| Ceravolo et al, 2005 18 | 4 | The patients presented mild to moderate parkinsonism and a poor or absent response to dopaminergic therapy. | Striatal DaTSCAN uptake was normal. | Parkinsonism in these patients could potentially be caused by postsynaptic malfunction of the nigrostriatal pathway or dysfunction of other neurotransmitter systems. |
| Hall et al, 2010 19 | 3 | The patients presented mild to moderate parkinsonism and a positive response to dopaminergic therapy. | Striatal DaTSCAN uptake was normal. | Parkinsonism in these patients could be due to the dysfunction of systems other than the presynaptic dopaminergic pathway and may potentially explain some parkinsonian SWEDD cases. |
| Healy et al, 2009 20 | 1 | The patient presented severe parkinsonism. Response to dopaminergic therapy was poor. | DaTSCAN uptake was reduced. | FXTAS‐associated parkinsonism is caused by mixed pre and postsynaptic degeneration; alternatively, the patient might present both FXTAS and idiopathic PD. |
| Scaglione et al, 2008 21 | 3 | One patient presented prominent parkinsonism. Response to dopaminergic therapy was poor in all patients. | DaTSCAN uptake was reduced, with an asymmetrical pattern. | The presence of the FXTAS premutation and leukoencephalopathy points at subcortical gray matter damage with midbrain atrophy and striatal degeneration. |
| Madeo et al, 2013 22 | 1 | The patient presented subtle clinical signs of tremor and typical MRI features. | DaTSCAN uptake was reduced, with an asymmetrical pattern. | The FXTAS premutation might cause a dysfunction of the bidirectional circuit connecting the cerebellum and the basal ganglia, providing a mechanism through which abnormal signals from the basal ganglia could affect the cerebellar function, and vice versa. |
| Apartis et al., 2012 23 | 22 | 43% of the patients had no family history of FXTAS, with 60% of them presenting parkinsonism. | 47% of the patients showed abnormal DaTSCAN results. | The results of this study highlight the need to extend the criteria evaluated during the diagnosis of FXTAS, including tremor, peripheral neuropathy, and MRI abnormalities. |
Abbreviations: SWEDD, Scan Without Evidence of Dopaminergic Deficit; FXTAS, Fragile X‐Associated Tremor/Ataxia Syndrome; PD, Parkinson's Disease; MRI, Magnetic Resonance Imaging.
Nevertheless, DaTSCAN results do not allow us to predict which FXTAS patients will respond to dopaminergic treatment, as some patients with normal DaTSCAN results have shown some response to this therapy. This might be related to the cutoff considered normal for the test, or to a potential placebo effect. 19 Thus, some patients with expansions of the FMR1 gene in the premutation range and showing dopaminergic treatment‐responsive parkinsonism would have a normal DaTSCAN according to the cutoff established for PD and would also explain some SWEDD cases.
Prion Diseases
Prion diseases in general, and Creutzfeldt‐Jakob disease (CJD) in particular, can sometimes include parkinsonian symptoms as part of their heterogeneous phenotype (Table 2). These symptoms might be a consequence of the involvement of subcortical structures such as the caudate and putamen and would be associated with the presence of hyperintensities in the basal ganglia in MRI diffusion‐weighted sequences in approximately 70% of the patients. 28
TABLE 2.
Summary of the available literature regarding potential DaTSCAN abnormal results associated with prion diseases
| Research | Patients with DaTSCAN results | Clinical findings | DaTSCAN findings | Research conclusions |
|---|---|---|---|---|
| Creutzfeldt‐Jakob disease | ||||
| Hinnell et al, 2011 24 | 1 | The patient presented parkinsonism signs. He later developed startle myoclonus and died shortly after that. Sporadic CJD was diagnosed during autopsy. | DaTSCAN performed a year into the illness showed reduced caudate and putamen uptake. | Although abnormal DaTSCAN results are typically associated with PD or Parkinson plus syndromes, CJD should also be considered during the differential diagnosis process. |
| Tomizawa et al, 2020 25 | 1 | The patient initially presented cognitive dysfunction and hand tremor that rapidly progressed to dementia and parkinsonism. MRI findings were normal. Response to dopaminergic therapy was poor. Almost 3 years after the disease onset, MRI showed cortical hyperintensities. | DaTSCAN showed bilateral decreases in radiotracer striatal binding. | The initial diagnosis of LBD turned out to be wrong, with the later MRI and DaTSCAN findings pointing to CJD, ultimately confirmed by a genetic analysis. This led the authors to suggest the performing of periodical DWI‐MRI examinations for patients presenting rapidly progressive dementia of unidentified origin. |
| Renard et al, 2010 26 | 1 | The patient presented cognitive deficit and a symmetric extrapyramidal syndrome without tremor. | DaTSCAN results showed a presynaptic bilateral decrease of the dopaminergic function, predominant in the head of the caudate nucleus. | DaTSCAN studies, so far used mostly to differentiate between LBD and AD, should be interpreted cautiously as they may be misleading. |
| Fatal familial insomnia | ||||
| Fukuoka et al, 2018 27 | 1 | The patient presented with dementia, visual hallucinations, and parkinsonism. Response to dopaminergic therapy was poor. FFI was diagnosed during autopsy. | DaTSCAN results showed a decrease in the presynaptic striatal dopaminergic function. | This is the first study reporting an FFI case in which parkinsonism was the initial symptom of the illness, with DaTSCAN confirming the damage in both presynaptic and postsynaptic dopaminergic systems. |
Abbreviations: CJD, Creutzfeldt‐Jakob Disease; PD, Parkinson's Disease; MRI, Magnetic Resonance Imaging; LBD, Lewy Body Dementia; DWI, Diffusion‐Weighted Imaging; AD, Alzheimer's Disease; FFI, Fatal Familial Insomnia.
However, the potential secondary damage to the dopaminergic nigrostriatal pathway has not been studied in depth. DaTSCAN could be a useful tool to confirm the presence of dopaminergic striatal degeneration in vivo. Hinnell et al verified the utility of this technique to show the abnormal uptake by both the caudate and the putamen, showing abnormal DaTSCAN results even before the onset of parkinsonian symptoms. 24
Based on neuroimaging findings, it has been suggested that these patients might present a double cause of parkinsonism, with both neurodegenerative and structural parkinsonism, although the findings in diffusion‐weighted sequences could also be explained by the spongiform changes typically observed in CJD patients. This would be consistent with the knowledge provided by necropsy studies (loss of dopaminergic neurons in the substantia nigra, caudate nucleus, and putamen), supporting the hypothesis of a pre‐ and postsynaptic degeneration of the nigrostriatal pathway in CJD. 29
Tomizawa et al presented a case with clinical and radiological findings initially compatible with those of LBD. However, as the disease progressed, MRI evaluation of the patient started to show cortical hyperintensities on diffuse‐weighted sequences. Furthermore, a DaTSCAN examination at 30 months post‐disease onset showed bilateral decreases in radiotracer striatal binding. Ultimately, a genomic DNA analysis confirmed the diagnosis of genetic CJD. This led the authors to suggest the performing of periodical DWI‐MRI examinations for patients presenting rapidly progressive dementia of unidentified origin. 25
Fatal familial insomnia (FFI) typically presents motor symptoms during its clinical evolution, including myoclonic jerks, ataxia, spasticity, dysarthria, dysphagia, and parkinsonism. The most recognizable histological change in FFI is the atrophy of the anterior and mediodorsal nuclei of the thalamus, although different degrees of atrophy and reactive astrogliosis of other thalamic nuclei, cerebral and cerebellar cortex and olives, and apoptotic neurons in the neocortex and striatum have also been described. DaTSCAN might allow the direct evaluation of presynaptic neuron degeneration and reduced uptake in the striatum, offering high sensitivity for parkinsonism in FFI, but at the expense of a low specificity. This might be of particular interest, as some FFI patients do not present insomnia and other typical manifestations at the onset of the disease, but do show clinical symptoms of parkinsonism similar to those of LBD as well as cognitive decline and visual hallucinations. 27 In these cases, the abnormal DaTSCAN result could lead to a misdiagnosis, especially if the response to dopaminergic therapy and/or chronology of the patient's symptoms are not taken into account.
In summary, parkinsonism in prion diseases could be explained by lesions in the basal ganglia affecting the lenticular nucleus and/or by degeneration of the nigrostriatal pathway. However, the utility of DaTSCAN in patients presenting cognitive decline has only been studied with the aim of differentiating between AD and LBD; thus, in patients showing both cognitive decline and an abnormal DaTSCAN result, assessing the semiology and chronology of cognitive symptomatology remains essential to reach an appropriate diagnosis. 26 Therefore, it would be the combination of DaTSCAN with other techniques, such as MRI or electroencephalogram that would facilitate an early diagnosis of CJD.
Huntington's Disease
Huntington's disease (HD) is an autosomal dominant hereditary disease caused by an abnormally unstable expansion of the CAG triplet in the IT15 gene in chromosome 4p16.3. Although HD is typically characterized by chorea, other types of abnormal movements such as bradykinesia and rigidity are often present as well (Table 3). Mulroy et al documented a case of HD‐like 2 exhibiting the chorea and cognitive decline typically associated with HD, but also parkinsonism. 30 This form of HD, resulting from a ≥40 repetition of the CTG/CAG triplet within the JPH3 gene in chromosome 16q24.3, 33 has only been described in patients of African descent. 34 Both qualitative and quantitative DaTSCAN imaging analysis showed a bilateral asymmetric reduction of radiotracer uptake in the striatum. This case, the first describing DaTSCAN findings for this disease, highlighted the potential heterogeneity of HD symptoms. 30
TABLE 3.
Summary of the available literature regarding potential DaTSCAN abnormal results associated with Huntington's disease
| Research | Patients with DaTSCAN results | Clinical findings | DaTSCAN findings | Research conclusions |
|---|---|---|---|---|
| Mulroy et al, 2020 30 | 1 | The patient presented cognitive impairment, chorea, and parkinsonism. Dopaminergic therapy was rejected by the patient. | DaTSCAN analysis revealed a bilateral asymmetric reduction of radiotracer uptake in the striatum. | This is the first case describing DaTSCAN findings for HD‐like 2. The case further confirms the potential variability of HD symptoms. |
| Gamez et al, 2010 31 | 12 | The patients presented varying degrees of clinical severity as per UHDRS scores. | DaTSCAN striatal uptake was reduced in the putamen in seven patients and the caudate and putamen in one patient. Reduction of specific striatal binding at the putamen level as confirmed in four patients. | This is the first study reporting the dysfunction of the pre‐synaptic dopaminergic system in HD, with DaTSCAN confirming the reduction of radiotracer uptake at the presynaptic level. |
| Gamez et al, 2014 32 | 4 | The patients presented varying degrees of clinical severity as per UHDRS scores. | DaTSCAN uptake showed a mean annual decline of 5.8% in the caudate and 9.6% in the putamen after a 2‐year follow‐up. | The authors propose the use of DaTSCAN as a progression biomarker for presynaptic dopaminergic degeneration in HD and neuroprotective therapy effectiveness. |
Abbreviations: HD, Huntington's Disease; UHDRS, Unified Huntington's Disease Rating Scale.
The most striking neuropathological change of HD is the gradual loss of striatal medium spiny neurons, although degenerative alterations have also been described in other regions of the brain. Dopaminergic neurotransmission seems to play an important role in the physiopathology of HD symptoms, as postmortem studies have shown a remarkable reduction in striatal postsynaptic dopaminergic receptors. However, the possibility of a presynaptic dopaminergic neuron malfunction is a matter of controversy.
DaTSCAN qualitative and quantitative analyses have shown a reduced uptake in the putamen of these patients, although this appears to not be related to the clinical severity of the disease. 31 While radiotracers [11C]raclopride, 35 [18F]fluorodeoxyglucose, 36 and [18F]MNI‐659 37 have already been employed to successfully document HD progression, DaTSCAN could also be a useful tool to evaluate the progression of the presynaptic dopaminergic degeneration associated to the disease, acting as a biomarker and offering the possibility to objectively evaluate the effectiveness of future neuroprotective treatments. 32 From a practical point of view, a patient without a known familial history of HD could be erroneously diagnosed with PD in the initial stages of the disease. In this context, an abnormal DaTSCAN result could further add to the clinical ambiguity.
Spinocerebellar Ataxia
Except for spinocerebellar ataxia type 6 (SCA6), which is a typically cerebellar syndrome, the rest of the SCAs are multisystem disorders characterized by a wide range of non‐cerebellar symptoms, including movement disorders (Table 4). Indeed, patients presenting two of the most frequent types of SCAs, SCA2 and SCA3, can show degeneration of the substantia nigra. The parkinsonian symptoms in these patients only evolve rarely, and when this happens, symptoms typically show a positive response to levodopa. Only some patients with different types of SCA present parkinsonian symptoms, independently of an involvement of the nigrostriatal pathway. It has been suggested that lesions in the motor area of the subthalamic nucleus could prevent the manifestation of parkinsonism in SCA2 and SCA3 patients, in which the cerebellar dysfunction would further mask these other symptoms.
TABLE 4.
Summary of the available literature regarding potential DaTSCAN abnormal results associated with spinocerebellar ataxia
| Research | Patients with DaTSCAN results | Clinical findings | DaTSCAN findings | Research conclusions |
|---|---|---|---|---|
| Spinocerebellar ataxia type 3 | ||||
| Pulido‐Valdeolivas et al, 2016 38 | 17 | The patients presented varying degrees of clinical severity as per SARA scores. | 76.5% of patients showed abnormal DaTSCAN results. | Classical symptoms of presynaptic damage in the dopaminergic system are present in SCA3 patients only when major damage appears. SCA3 patients might show abnormal DaTSCAN results even in the absence of clinical extrapyramidal signs. |
| Bonnet et al, 2012 39 | 2 | The patients presented varying degrees of clinical severity as per SARA scores. Patient 2 showed a complete response to dopaminergic therapy, while patient 4 showed no response. | DaTSCAN results showed a severe presynaptic bilateral decrease of radiotracer uptake, especially in the putamen. | The lack of response to dopaminergic therapy in patient 4 could be due to the patient's notable alteration of the dopaminergic pathway and mild‐to‐moderate cerebellar dysfunction. |
| Spinocerebellar ataxia type 2 | ||||
| Miyaue et al, 2017 40 | 4 | SCA2 patients presented parkinsonism with varying degrees of clinical severity and a good response to dopaminergic therapy in most cases. | DaTSCAN results showed reduced striatal uptake, more prominent in the caudate. | The observation of reduced striatal DaTSCAN uptake even in SCA2 early stages could be used as a marker for early dopaminergic therapy administration, thus improving the quality of life of these patients. |
| Spinocerebellar ataxia type 6 | ||||
| Xie et al, 2016 41 | 12 | None of the patients presented evident symptoms of parkinsonism. | DaTSCAN results showed no striatal DAT loss in the putamen and caudate. | Neither parkinsonism nor dopaminergic dysfunction are part of the pathology of SCA6. |
| Spinocerebellar ataxia type 12 | ||||
| Latorre et al, 2019 42 | 1 | The patient presented parkinsonism signs and a poor response to dopaminergic therapy. | DaTSCAN results showed reduced uptake in the putamen. |
This is the first study describing a case of genetically confirmed SCA12 showing both atypical parkinsonism signs and abnormal DaTSCAN results. |
Abbreviations: SCA, Spinocerebellar Ataxia; SARA, Scale for Assessment and Rating of Ataxia; DAT, Dopamine Transporter.
SCA3, also known as Machado‐Joseph disease, is one of the most frequent forms of SCA. SCA3 is caused by an expansion of the CAG triplet in the ATXN3 gene, inherited in an autosomal dominant manner. The disorder shows a highly variable age at onset and clinical manifestations, possibly related to the number of repetitions of the triplet, and includes a progressive cerebellar dysfunction associated with parkinsonism, pyramidal signs, neuropathy, and external ophthalmoplegia in a variable manner. In a study by Pulido‐Valdeolivas et al including 17 SCA3 patients, it was reported that 76.5% of them presented anomalies in the DaTSCAN visual analysis, with higher rates in those patients showing anomalous alternating movements and nose‐finger tests on examination. 38 This contrasts with the low percentage of patients showing extrapyramidal symptoms on examination, which might be explained by the presence of sub‐clinical damage in the presynaptic pathway; thus, the typical symptomatology of the disease (bradykinesia, rigidity, hypomimia, and resting tremor) would only appear in those patients with higher damage. 38 The opposite can also occur, with some patients with tremor showing normal DaTSCAN results. For this reason, it has been suggested that there is little correlation between striatal dopamine uptake and clinical phenotype. 39
SCA2 is caused by the abnormal repetition of the CAG triplet in the ATXN2 gene, typically manifesting in the third or fourth decade of life. Although clinical manifestations are generally cerebellar in origin, some patients present parkinsonian or motor neuron symptoms through the course of the disease, sometimes even prior to manifestations of ataxia. Many SCA2 patients (even the ones not presenting parkinsonism, as it occurs in SCA3) have shown a reduced DaTSCAN striatal uptake that is less pronounced in the caudate nucleus and putamen. 40 This might allow their differentiation from patients with PD. As the reported DaTSCAN findings would take place in the early stages of the disease, Miyaue et al have proposed that the quality of life of patients with SCA2 could be improved with levodopa treatment. 40
The next most frequent form of SCA is SCA6, in which the presence of parkinsonism seems to be less prevalent than in other subtypes. Indeed, no objective DaTSCAN abnormalities have been uniformly reported in these patients, which would support a normal dopaminergic function in this SCA subtype. 41
The scarce prevalence of the other SCA subtypes explains the lack of DaTSCAN findings in these patients. Nonetheless, recent research has described abnormal results for this test in patients with SCA12, which is typically characterized by an action tremor in the upper limbs similar to that observed for essential tremor and cerebellar ataxia often manifested in the evolution of the disease. 42
Hereditary Spastic Paraparesis
Hereditary spastic parapareses are characterized by progressive spasticity and paresis of the lower limbs, secondary to a degenerative axonopathy of the corticospinal tract. Other myelopathy symptoms, such as urinary urgency and a mild decline of the deep sensory modalities, can also be present sometimes, which might make it necessary to establish a differential diagnosis with hereditary ataxias (Table 5).
TABLE 5.
Summary of the available literature regarding potential DaTSCAN abnormal results associated with hereditary spastic paraparesis
| Research | patients with DaTSCAN results | Clinical findings | DaTSCAN findings | Research conclusions |
|---|---|---|---|---|
| Hereditary spastic paraparesis | ||||
| Onder et al, 2023 43 | 1 | The patient presented lower extremity spasticity, gradually deteriorating gait, constipation, sleep alterations, bilateral bradykinesia, rigidity, and apparent postural instability. MRI showed severe spinal atrophy and mild cerebellar atrophy. Levodopa treatment alternated between on and off periods. | DaTSCAN results showed a bilateral asymmetric reduction of radiotracer uptake in the striatum. | The dopaminergic neuron vulnerability in this HSP patient might point to the contribution of central dopaminergic neuron degeneration to the phenotype of the condition. |
| Spastic paraplegia type 7 | ||||
| Bellini et al, 2021 44 | 1 | The patient presented ataxia concomitant with mild parkinsonism and pyramidal signs, and urinary urge incontinence. | DaTSCAN results showed bilateral striatal degeneration. | The joint evaluation of DaTSCAN imaging and clinical findings can help to differentiate MSA from other causes of adult‐onset ataxia, such as in the case described in this study. |
| Spastic paraplegia type 11 | ||||
| Anheim et al, 2009 45 | 2 | Both patients presented early‐onset parkinsonism. Response to dopaminergic therapy was moderate at first, then progressively weaker in one patient, while the second patient showed no response. | DaTSCAN results showed reduced striatal uptake. | The authors present the first case of hereditary SPG11 linked to abnormal DaTSCAN results and an atypical juvenile parkinsonism phenotype. |
Abbreviations: MRI, Magnetic Resonance Imaging; HSP, Hereditary Spastic Paraparesis; MSA, Multiple System Atrophy; SPG, Spastic Paraplegia.
Onder and Comoglu reported an unusual case of suspected HSP in which the diagnosis could not be confirmed by genetic analysis. The patient, whose family history was unremarkable, presented some common HSP symptoms, but also atypical pyramidal signs and parkinsonism. Nonetheless, MRI examination showed mild atrophy of the cerebellum and severe cervical and thoracic spinal cord atrophy, and the bilateral reduction of radiotracer uptake in the striatum observed in the DaTSCAN analysis further supported the HSP diagnosis. 43
Spastic paraplegia type 7 (SPG7) is one of the most frequent forms of hereditary autosomal recessive spastic paraparesis, with up to 57% of the patients presenting clinical ataxia and approximately a third of them showing objective signs of cerebellar atrophy in neuroimaging studies. As a result of this symptomatology that includes ataxia and dysautonomia, some SPG7 cases have been misdiagnosed in the literature as MSA. 46 Differential diagnosis in these cases can be complex, as some SPG7 patients can also show abnormal DaTSCAN results. 44
Autosomal recessive hereditary spastic paraparesis with thinning of the corpus callosum is an uncommon neurodegenerative disorder frequently caused by mutations in the SPG11 gene, which encodes the spatacsin protein, in the 15q chromosome. These patients present progressive spastic paraparesis and mental retardation frequently associated with peripheral neuropathy, typically occurring in the first two decades of life. The disorder is named after the distinctive thinning of the corpus callosum observed in brain MRI images that is commonly accompanied by changes in the periventricular white matter. Anheim et al reported the possibility of early disease onset in the form of juvenile parkinsonism with resting tremor, akinesia, and rigidity, which might even show a moderate initial response to levodopa and pathological DaTSCAN results. 45
Metabolic Disorders
Fahr Disease
Fahr disease is characterized by a degeneration of the basal ganglia accompanied by neuropsychiatric clinical manifestations (Table S1). The presence of parkinsonism in dystonia syndromes, including Fahr disease, suggests the potential existence of an anatomical and functional link between this symptom and the dopaminergic system; however, if this link takes place at the presynaptic level is currently unknown. Interestingly, Paghera et al described a case of Fahr disease with asymmetric DaTSCAN uptake in the basal ganglia, possibly reflecting presynaptic failure, but the lack of a specific uptake pattern complicates the differential diagnosis between these patients and those presenting other types of parkinsonism and/or dystonia. 47 Conversely, Kalampokini et al presented a case of Fahr disease due to hypoparathyroidism in which the patient presented parkinsonism with severe rigidity in the limbs, bilateral calcifications in the brain confirmed by both CT and MRI examination, and a normal DaTSCAN result. 48 However, a previous report of Fahr disease due to hypoparathyroidism had previously shown asymmetrical radiotracer uptake in the putamen, although the presence of calcifications was symmetric. 49 These contradictory results point to currently unknown factors, besides the presence of calcifications, affecting the dopaminergic activity in this rare disorder.
Succinic Semialdehyde Dehydrogenase Deficiency Disorder
Frantellizzi et al described a significant global reduction of DaTSCAN uptake in a patient presenting the uncommon autosomal recessive disorder succinic semialdehyde dehydrogenase deficiency, thus raising new physiopathological concerns regarding this disease (Table S1). 50
GM1 Gangliosidosis
Adult GM1 gangliosidosis is often manifested as generalized dystonia followed by rigid‐akinetic parkinsonism (Table S1). The selective involvement of the basal ganglia and the clinical phenomenology typical of the disease are thought to be caused by the high turnover of the GM1 ganglioside in this region. Marangi et al described a reduced DaTSCAN uptake in the basal ganglia in a patient affected by this disorder, suggesting a presynaptic dopaminergic deficiency possibly related to lesion topography. 51 The reduced DaTSCAN uptake in both the putamen and the striatum reported by Koya Kutti et al further confirmed the presence of presynaptic dopaminergic dysfunction associated with this disorder. 52
Neurodegeneration with Brain Iron Accumulation Disorders (NBIA)
This group of hereditary, neurodegenerative disorders is characterized by the formation of abnormal iron deposits in the brain, typically present in the globus pallidus and substantia nigra and resulting in miscellaneous clinical manifestations, including different degrees of dystonia, chorea, spasticity, and cognitive decline (Table S1). Kang and Minoshima described a case of pantothenate kinase‐associated neurodegeneration showing late‐onset parkinsonism and a potential presynaptic nigrostriatal degeneration of the dopaminergic pathway, as evidenced by the DaTSCAN results, albeit with a non‐specific and variable pattern. 53 Other rare NBIA disorders that present with parkinsonism have shown heterogeneous results in DaTSCAN studies. This includes Kufor‐Rakeb disease (caused by a mutation in the ATP13A2 gene and showing marked asymmetrical reduced DaTSCAN uptake in both the caudate and putamen) 54 and fatty acid hydroxylase‐associated neurodegeneration (also known as SPG35, caused by a mutation in the FA2H gene, which reportedly presented normal DaTSCAN results). 55 CoA synthase protein‐associated neurodegeneration, caused by mutations in the CoASY gene, also belongs to this category of NBIA with clinical parkinsonism, 56 although there are no DaTSCAN data available related to this condition so far.
Other Disorders
Anti‐IgLON5 Disease
The uncommon disorder anti‐IgLON5 disease can present a myriad of symptoms that include both neurodegenerative and autoimmune processes; however, the physiopathology of the disease remains unknown (Table S2). Approximately 65% of these patients present movement disorders that may include chorea, hand tremor, myoclonic movements of the limbs, and symmetrical parkinsonism without tremor.
To date, DaTSCAN results have been reported for only four patients with anti‐IgLON5 disease, 57 , 58 , 59 , 60 with three of them showing a reduced striatal uptake (Fig. 1). 57 , 59 , 60 These DaTSCAN abnormalities support a neurodegenerative origin of the disease, also reflected by the presence of three‐ (3R) and four‐repeat (4R) tau aggregates in the hypothalamus and the tegmentum of the brainstem with a rostro‐caudal gradient.
Figure 1.
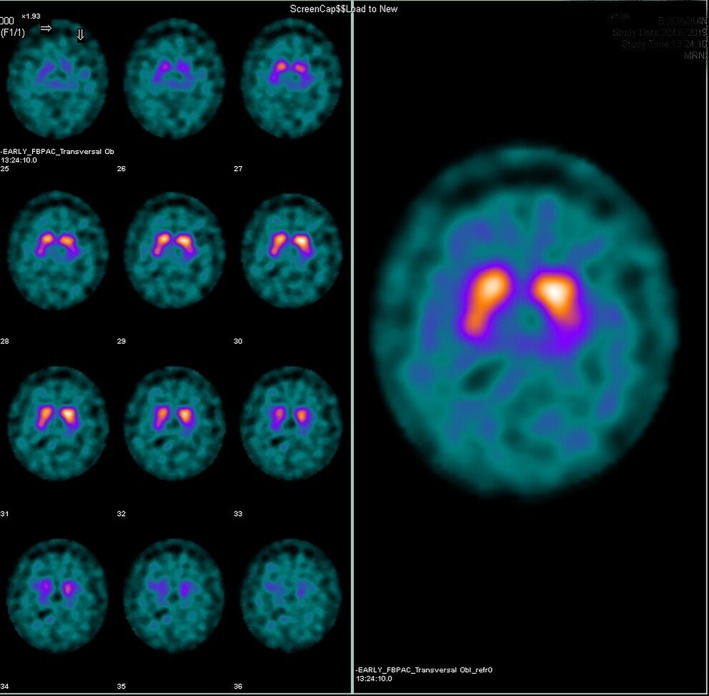
Ioflupane single‐photon emission computed tomography scan from a patient with anti‐IgLON5 disease. The image shows an alteration of the left presynaptic dopaminergic pathway with a reduced uptake in the left putamen region.
Nonetheless, these reports emphasize the need to include anti‐IgLON5 disease in the differential diagnosis of movement disorders, particularly that of atypical parkinsonism associated with sleep disorders, supranuclear gaze palsy, gait instability, and orofacial dyskinesia. However, it is yet unknown whether abnormal DaTSCAN results corroborate the neurodegenerative etiology of this condition or whether they correlate with the various clinical symptoms. 57
Ring Chromosome 20 Syndrome
The potential participation of the dopaminergic system in the propagation of epileptic seizures has gained attention in recent years, and dopamine metabolism is also being investigated in relation to genetic epilepsies, such as that presented by ring chromosome 20 syndrome patients. In a study conducted by Del Sole et al, some of these patients showed a nigrostriatal dopaminergic deficiency in their DaTSCAN results, with a negative correlation between ring chromosome 20 mosaicism and ioflupane uptake in the putamen and caudate nucleus (Table S2). 61
Chorea‐Acanthocytosis
Chorea‐acanthocytosis and McLeod syndrome are the main manifestations of neuroacanthocytosis, a group of rare disorders characterized by neuromuscular abnormalities, a shorter life expectancy, progressive and irreversible hyperkinesia, epilepsy (in the case of chorea‐acanthocytosis), and myocardiopathy (in the case of McLeod syndrome). Both disorders present a progressive atrophy of the caudate nucleus that has been related to the presence of clinical bradykinesia and a reduced DaTSCAN binding to the dopamine transporter in the striatum (Table S2). 62
Neuronal Ceroid Lipofuscinosis
Neuronal ceroid lipofuscinoses (NCL), a group of lysosomal disorders characterized by the accumulation of abnormal storage material in neurons and other cell types, are the most common cause of dementia in children and young adults. The different NCL subtypes are associated with 14 different gene mutations, commonly inherited in an autosomal recessive manner. 63 , 64 These neurodegenerative disorders present a broad range of symptoms, including vision loss, movement disorders, dementia, and epilepsy. Lange et al reported a case of adult‐onset NCL type 5, confirmed by genetic analysis, in which MRI examination of the patient showed progressive brain atrophy and leukoencephalopathy over the course of six years. DaTSCAN analysis further demonstrated an asymmetrical decrease of the presynaptic dopamine transporter density, a finding consistent with the severe parkinsonism observed in the patient (Table S2). 65
DaTSCAN as a Diagnostic and Prognostic Tool
The identification of PD and other atypical parkinsonisms is traditionally based on semiology. Due to the progressive course of these disorders, an early diagnosis of degenerative parkinsonisms is oftentimes complex and subjected to under‐ and overdiagnosis in variable degrees. The use of different laboratory tests and neuroimaging as supporting diagnostic tools mainly aims to exclude other pathologies, as at present there are no biomarkers available that allow a positive diagnosis of these disorders in their early stages.
DaTSCAN can help to document the degeneration of the nigrostriatal pathway at the presynaptic level in degenerative parkinsonisms, making it a useful tool for the differentiation between these and other types of tremors and/or parkinsonisms, such as essential tremor, pharmacological parkinsonism, or vascular parkinsonism. However, its limited capacity to distinguish between the different forms of degenerative parkinsonism dismisses it as a potential biomarker, and its recommended use is currently limited to patients with an uncertain diagnosis after several evaluations and clinical tests.
Nonetheless, the utilization of DaTSCAN is becoming more frequent and extended, and the number of patients presenting rare degenerative parkinsonisms subjected to this test is on the rise. Parkinsonism is not a constant finding in these degenerative disorders, frequently of a genetic nature, and in most cases an adequate correlation between phenotype and genotype could not be established.
It is important to acknowledge the advantages and limitations of DaTSCAN as a cost‐effective diagnostic tool, as it permits to avoid the expenses associated with incorrect treatments in patients misdiagnosed with PS and reduces the gap in undiagnosed patients that do present PS. 66 However, the diagnosis of PD and other types of parkinsonism must still be based on clinical criteria. The utilization of supplementary tests such as DaTSCAN in patients presenting a clinically defined PS lacks diagnostic cost‐effectiveness and can overwhelm the health system, with an estimated 50% of DaTSCAN tests requested by general neurologists offering no clinical utility. 67 Different committees of experts in movement disorders recommend that these kinds of tests be requested only by specialists in these syndromes, particularly in patients with uncertain diagnoses after several evaluations. 67 , 68 This includes patients showing subtle early symptoms, sub‐optimal response to levodopa, a prominent action tremor, pharmacological parkinsonism, and other patients presenting parkinsonism with lower limb predominance or other less frequent clinical manifestations. 69
Conclusions
This review summarizes DaTSCAN findings in some of these pathologies, including the fragile X‐associated tremor/ataxia syndrome, prion diseases, Huntington's disease, spinocerebellar ataxia, hereditary spastic paraparesis, some metabolic diseases, and other uncommon disorders. It is important to address the potential bias associated with the selection of literature regarding abnormal DaTSCAN results exclusively. As most studies are published specifically because of their abnormal results, there is a lack of information regarding the potential contributions of studies in which DaTSCAN results follow a conventional pattern. Research has shown a reduced DaTSCAN uptake at the basal ganglia level in some patients with these diseases, suggesting that the presence of parkinsonism in these patients is indeed showing a degeneration of the nigrostriatal pathway, thus potentially benefiting from dopaminergic treatment similar to that seen in the more frequent degenerative parkinsonisms such as PD, LBD, PSP, MSA, or CBD. However, although this reduced DaTSCAN uptake would be present with a higher probability in patients with parkinsonian symptoms, it has also been observed in asymptomatic patients. Thus, it is unknown if DaTSCAN could predict the clinical evolution in those patients showing abnormal results in the test but not showing parkinsonism yet. Another potential explanation for these abnormal findings is the co‐existence with one of the most frequent and better‐known forms of neurodegenerative parkinsonism.
In this regard, the potential role of DaTSCAN uptake as a biomarker of clinical evolution and treatment response in patients with rare forms of parkinsonism still needs to be determined. However, it is unlikely that the results of this test in patients with an uncertain diagnosis and uncommon clinical phenomenology will establish a definitive diagnosis due to the lack of differential findings among the diverse etiologies of parkinsonism. For these reasons, further studies are required to determine the real benefit of performing this test in this group of patients.
Author Roles
(1) Research project: A. Conception, B. Organization, C. Execution, D. Supervision; (2) Manuscript: A. Writing of the first draft, B. Editing, C. Review and Critique.
S.Q.: 1A, 1B, 1C, 2A
R.S.F.: 2B, 2C
M.A.B.: 1D, 2C
Disclosures
Ethical Compliance Statement: The authors confirm that neither the approval of an institutional review board nor patient consent was required for this work. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflict of Interest: This research received no specific grant from any funding agency in the public, commercial, or not‐for‐profit sectors. The authors have no conflicts of interest to declare regarding this work.
Financial Disclosures for the Previous 12 Months: MÁB is CEO and Board Member of Cells IA Technologies. The remainder of the authors have no financial disclosures to declare.
Supporting information
Table S1. Summary of the available literature regarding potential DaTSCAN abnormal results associated with metabolic disorders.
Table S2. Summary of the available literature regarding potential DaTSCAN abnormal results associated with other neurodegenerative pathologies.
References
- 1. Tolosa E, Wenning G, Poewe W. The diagnosis of Parkinson's disease. Lancet Neurol 2006;5(1):75–86. 10.1016/S1474-4422(05)70285-4. [DOI] [PubMed] [Google Scholar]
- 2. Postuma RB, Berg D, Stern M, et al. MDS clinical diagnostic criteria for Parkinson's disease. Mov Disord 2015;30(12):1591–1601. 10.1002/mds.26424. [DOI] [PubMed] [Google Scholar]
- 3. Verger A, Grimaldi S, Ribeiro M, Frismand S, Guedj E. Single photon emission computed tomography/positron emission tomography molecular imaging for parkinsonism: a fast‐developing field. Ann Neurol 2021;90(5):711–719. 10.1002/ana.26187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berardelli A, Wenning GK, Antonini A, et al. EFNS/MDS‐ES recommendations for the diagnosis of Parkinson's disease. Eur J Neurol 2013;20(1):16–34. 10.1111/ene.12022. [DOI] [PubMed] [Google Scholar]
- 5. Marshall VL, Reininger CB, Marquardt M, et al. Parkinson's disease is overdiagnosed clinically at baseline in diagnostically uncertain cases: a 3‐year European multicenter study with repeat [ 123 I]FP‐CIT SPECT. Mov Disord 2009;24(4):500–508. 10.1002/mds.22108. [DOI] [PubMed] [Google Scholar]
- 6. Darcourt J, Booij J, Tatsch K, et al. EANM procedure guidelines for brain neurotransmission SPECT using 123I‐labelled dopamine transporter ligands, version 2. Eur J Nucl Med Mol Imaging 2010;37(2):443–450. 10.1007/s00259-009-1267-x. [DOI] [PubMed] [Google Scholar]
- 7. Chahid Y, Sheikh ZH, Mitropoulos M, Booij J. A systematic review of the potential effects of medications and drugs of abuse on dopamine transporter imaging using [123I]I‐FP‐CIT SPECT in routine practice. Eur J Nucl Med Mol Imaging 2023;50(7):1974–1987. 10.1007/s00259-023-06171-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Marek K, Seibyl J, Eberly S, et al. Longitudinal follow‐up of SWEDD subjects in the PRECEPT study. Neurology 2014;82(20):1791–1797. 10.1212/WNL.0000000000000424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. de la Fuente‐Fernandez R. Role of DaTSCAN and clinical diagnosis in Parkinson disease. Neurology 2012;78(10):696–701. 10.1212/WNL.0b013e318248e520. [DOI] [PubMed] [Google Scholar]
- 10. Morbelli S, Esposito G, Arbizu J, et al. EANM practice guideline/SNMMI procedure standard for dopaminergic imaging in parkinsonian syndromes 1.0. Eur J Nucl Med Mol Imaging 2020;47(8):1885–1912. 10.1007/s00259-020-04817-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kagi G, Bhatia KP, Tolosa E. The role of DAT‐SPECT in movement disorders. J Neurol Neurosurg Psychiatry 2010;81(1):5–12. 10.1136/jnnp.2008.157370. [DOI] [PubMed] [Google Scholar]
- 12. Perlmutter JS, Eidelberg D. To scan or not to scan: DaT is the question. Neurology 2012;78(10):688–689. 10.1212/WNL.0b013e3182494c72. [DOI] [PubMed] [Google Scholar]
- 13. Badoud S, Van De Ville D, Nicastro N, Garibotto V, Burkhard PR, Haller S. Discriminating among degenerative parkinsonisms using advanced 123 I‐ioflupane SPECT analyses. Neuroimage Clin 2016;12:234–240. 10.1016/j.nicl.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bega D, Kuo PH, Chalkidou A, et al. Clinical utility of DaTscan in patients with suspected parkinsonian syndrome: a systematic review and meta‐analysis. NPJ Parkinsons Dis 2021;7(1):43. 10.1038/s41531-021-00185-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mehanna R, Jimenez‐Shahed J, Itin I. Three cases of levodopa‐resistant parkinsonism after radiation therapy. Am J Case Rep 2016;17:916–920. 10.12659/AJCR.900537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Erro R, Pappata S, Picillo M, et al. Teaching NeuroImages: pseudo‐abnormal DaTscan findings in meningioma‐induced parkinsonism. Neurology 2013;80(13):e147. 10.1212/WNL.0b013e318289709d. [DOI] [PubMed] [Google Scholar]
- 17. Méndez‐Guerrero A, Laespada‐García MI, Gómez‐Grande A, et al. Acute hypokinetic‐rigid syndrome following SARS‐CoV‐2 infection. Neurology 2020;95(15):e2109–e2118. 10.1212/WNL.0000000000010282. [DOI] [PubMed] [Google Scholar]
- 18. Ceravolo R, Antonini A, Volterrani D, et al. Dopamine transporter imaging study in parkinsonism occurring in fragile X premutation carriers. Neurology 2005;65(12):1971–1973. 10.1212/01.wnl.0000188821.51055.52. [DOI] [PubMed] [Google Scholar]
- 19. Hall DA, Jennings D, Seibyl J, Tassone F, Marek K. FMR1 gene expansion and scans without evidence of dopaminergic deficits in parkinsonism patients. Parkinsonism Relat Disord 2010;16(9):608–611. 10.1016/j.parkreldis.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Healy DG, Bressman S, Dickson J, et al. Evidence for pre and postsynaptic nigrostriatal dysfunction in the fragile X tremor‐ataxia syndrome. Mov Disord 2009;24(8):1245–1247. 10.1002/mds.22267. [DOI] [PubMed] [Google Scholar]
- 21. Scaglione C, Ginestroni A, Vella A, et al. MRI and SPECT of midbrain and striatal degeneration in fragile X‐associated tremor/ataxia syndrome. J Neurol 2008;255(1):144–146. 10.1007/s00415-007-0711-8. [DOI] [PubMed] [Google Scholar]
- 22. Madeo G, Alemseged F, di Pietro B, Schillaci O, Pisani A. Early abnormalities in 123I‐ioflupane (DaTSCAN) imaging in the fragile X‐associated tremor ataxia syndrome (FXTAS): a case report. Neurol Sci 2013;34(8):1475–1477. 10.1007/s10072-012-1223-6. [DOI] [PubMed] [Google Scholar]
- 23. Apartis E, Blancher A, Meissner WG, et al. FXTAS: new insights and the need for revised diagnostic criteria. Neurology 2012;79(18):1898–1907. 10.1212/WNL.0b013e318271f7ff. [DOI] [PubMed] [Google Scholar]
- 24. Hinnell C, Buxton‐Thomas M, Sibtain N, Samuel M. Striatal 123I‐Ioflupane SPECT abnormality in sporadic Creutzfeldt–Jakob disease. J Neurol 2011;258(5):948–950. 10.1007/s00415-010-5860-5. [DOI] [PubMed] [Google Scholar]
- 25. Tomizawa Y, Taniguchi D, Furukawa Y. Genetic Creutzfeldt‐Jakob disease mimicking dementia with Lewy bodies: clinical and radiological findings. J Neurol Sci 2020;409:116604. 10.1016/j.jns.2019.116604. [DOI] [PubMed] [Google Scholar]
- 26. Renard D, Collombier L, Ayrignac X, Bouttier F, Kotzki PO, Labauge P. [123I] FP‐CIT SPECT in Creutzfeldt–Jakob disease. J Neurol 2010;257(10):1754–1755. 10.1007/s00415-010-5594-4. [DOI] [PubMed] [Google Scholar]
- 27. Fukuoka T, Nakazato Y, Yamamoto M, Miyake A, Mitsufuji T, Yamamoto T. Fatal familial insomnia initially developing parkinsonism mimicking dementia with Lewy bodies. Intern Med 2018;57(18):2719–2722. 10.2169/internalmedicine.0573-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. García C, de León S, Cabello JP, Ortiz R, Vaamonde J. Parkinsonism associated with pathological 123I‐FP‐CIT SPECT (DaTSCAN) results as the initial manifestation of sporadic Creutzfeldt‐Jakob disease. Case Rep Neurol Med 2018;2018:1–3. 10.1155/2018/5157275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vital A, Fernagut PO, Canron MH, et al. The Nigrostriatal pathway in Creutzfeldt‐Jakob disease. J Neuropathol Exp Neurol 2009;68(7):809–815. 10.1097/NEN.0b013e3181abdae8. [DOI] [PubMed] [Google Scholar]
- 30. Mulroy E, Latorre A, Menozzi E, Teh PC, Magrinelli F, Bhatia KP. Huntington disease like 2 (HDL‐2) with parkinsonism and abnormal DAT‐SPECT – a novel observation. Parkinsonism Relat Disord 2020;71:46–48. 10.1016/j.parkreldis.2020.01.008. [DOI] [PubMed] [Google Scholar]
- 31. Gamez J, Lorenzo‐Bosquet C, Cuberas‐Borrós G, Carmona F, Hernández‐Vara J, Castilló J, Castell‐Conesa J. Does reduced [123I]‐FP‐CIT binding in Huntington's disease suggest pre‐synaptic dopaminergic involvement? Clin Neurol Neurosurg 2010;112(10):870–875. 10.1016/j.clineuro.2010.07.014. [DOI] [PubMed] [Google Scholar]
- 32. Gamez J, Lorenzo‐Bosquet C, Cuberas‐Borrós G, et al. Progressive presynaptic dopaminergic deterioration in Huntington disease. Clin Nucl Med 2014;39(3):e227–e228. 10.1097/RLU.0b013e31828162cd. [DOI] [PubMed] [Google Scholar]
- 33. Holmes SE, O'Hearn E, Rosenblatt A, et al. A repeat expansion in the gene encoding junctophilin‐3 is associated with Huntington disease–like 2. Nat Genet 2001;29(4):377–378. 10.1038/ng760. [DOI] [PubMed] [Google Scholar]
- 34. Anderson DG, Walker RH, Connor M, Carr J, Margolis RL, Krause A. A systematic review of the Huntington disease‐like 2 phenotype. J Huntingtons Dis 2017;6(1):37–46. 10.3233/JHD-160232. [DOI] [PubMed] [Google Scholar]
- 35. Tang C, Feigin A. Monitoring Huntington's disease progression through preclinical and early stages. Neurodegener Dis Manag 2012;2(4):421–435. 10.2217/nmt.12.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pagano G, Niccolini F, Politis M. Current status of PET imaging in Huntington's disease. Eur J Nucl Med Mol Imaging 2016;43(6):1171–1182. 10.1007/s00259-016-3324-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Russell DS, Jennings DL, Barret O, et al. Change in PDE10 across early Huntington disease assessed by [ 18 F]MNI‐659 and PET imaging. Neurology 2016;86(8):748–754. 10.1212/WNL.0000000000002391. [DOI] [PubMed] [Google Scholar]
- 38. Pulido‐Valdeolivas I, Gómez‐Andrés D, Sanz‐Gallego I, Rausell E, Arpa J. Patterns of motor signs in spinocerebellar ataxia type 3 at the start of follow‐up in a reference unit. Cerebellum Ataxias 2016;3(1):4. 10.1186/s40673-016-0042-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bonnet C, Apartis E, Anheim M, et al. Tremor‐spectrum in spinocerebellar ataxia type 3. J Neurol 2012;259(11):2460–2470. 10.1007/s00415-012-6531-5. [DOI] [PubMed] [Google Scholar]
- 40. Miyaue N, Tada S, Ando R, et al. DAT SPECT may have diagnostic value in prodromal SCA2 patients with parkinsonism. Parkinsonism Relat Disord 2017;44:137–141. 10.1016/j.parkreldis.2017.08.012. [DOI] [PubMed] [Google Scholar]
- 41. Xie T, Appelbaum D, Bernard J, Padmanaban M, Pu Y, Gomez C. Evaluation of parkinsonism and striatal dopamine transporter loss in patients with spinocerebellar ataxia type 6. J Neurol 2016;263(11):2302–2307. 10.1007/s00415-016-8261-6. [DOI] [PubMed] [Google Scholar]
- 42. Latorre A, del Gamba C, Menozzi E, Balint B, Brugger F, Bhatia KP. Abnormal DaTSCAN and atypical parkinsonism in SCA12. Mov Disord Clin Pract. 2019;6(5):400–402. 10.1002/mdc3.12751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Onder H, Comoglu S. A rare patient with hereditary spastic Paraparesis with parkinsonism. Asian J Neurosurg 2023;18(1):216–218. 10.1055/s-0043-1764117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bellini G, del Prete E, Unti E, Frosini D, Siciliano G, Ceravolo R. Positive DAT‐SCAN in SPG7: a case report mimicking possible MSA‐C. BMC Neurol 2021;21(1):328. 10.1186/s12883-021-02345-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Anheim M, Lagier‐Tourenne C, Stevanin G, et al. SPG11 spastic paraplegia. J Neurol 2009;256(1):104–108. 10.1007/s00415-009-0083-3. [DOI] [PubMed] [Google Scholar]
- 46. Salgado P, Latorre A, del Gamba C, Menozzi E, Balint B, Bhatia KP. SPG7: the great imitator of MSA‐C within the ILOCAs. Mov Disord Clin Pract. 2019;6(2):174–175. 10.1002/mdc3.12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Paghera B, Caobelli F, Giubbini R. 123 I‐Ioflupane SPECT in Fahr disease. J Neuroimaging 2013;23(1):157–158. 10.1111/j.1552-6569.2011.00581.x. [DOI] [PubMed] [Google Scholar]
- 48. Kalampokini S, Georgouli D, Dadouli K, et al. Fahr's syndrome due to hypoparathyroidism revisited: a case of parkinsonism and a review of all published cases. Clin Neurol Neurosurg 2021;202:106514. 10.1016/j.clineuro.2021.106514. [DOI] [PubMed] [Google Scholar]
- 49. Caranci G, Grillea G, Barbato F, et al. Scintigraphic, neuroradiological and clinical comparison in two patients with primary sporadic and two with secondary Fahr's disease. Neurol Sci 2011;32(2):337–341. 10.1007/s10072-010-0444-9. [DOI] [PubMed] [Google Scholar]
- 50. Frantellizzi V, Pontico M, Pani A, de Feo MS, de Vincentis G. 123I‐FP‐CIT brain SPECT findings in succinic Semialdehyde dehydrogenase (SSADH) deficiency. Curr Radiopharm 2021;14(1):78–83. 10.2174/1874471013666200325101302. [DOI] [PubMed] [Google Scholar]
- 51. Marangi A, Tagliapietra M, Vicenzi V, Pasquin I, Salviati A. Teaching NeuroImages: brain MRI and DaT‐SPECT imaging in adult GM1 gangliosidosis. Neurology 2018;91(2):e187–e188. 10.1212/WNL.0000000000005775. [DOI] [PubMed] [Google Scholar]
- 52. Koya Kutty S, Magrinelli F, Milner AV, Bhatia KP. Abnormal <scp>DaTscan</scp> in <scp>GM1</scp> ‐Gangliosidosis type <scp>III</scp> manifesting with dystonia‐parkinsonism. Mov Disord Clin Pract 2022;9(6):825–828. 10.1002/mdc3.13512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kang A, Minoshima S. 123I‐Ioflupane SPECT findings of Pantothenate kinase–associated neurodegeneration. Clin Nucl Med 2014;39(9):849–851. 10.1097/RLU.0000000000000409. [DOI] [PubMed] [Google Scholar]
- 54. Santoro L, Breedveld GJ, Manganelli F, et al. Novel ATP13A2 (PARK9) homozygous mutation in a family with marked phenotype variability. Neurogenetics 2011;12(1):33–39. 10.1007/s10048-010-0259-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Benger M, Mankad K, Proukakis C, Mazarakis ND, Kinali M. The interaction of genetic mutations in PARK2 and FA2H causes a novel phenotype in a case of childhood‐onset movement disorder. Front Neurol 2019;10:555. 10.3389/fneur.2019.00555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dusi S, Valletta L, Haack TB, et al. Exome sequence reveals mutations in CoA synthase as a cause of neurodegeneration with brain iron accumulation. Am J Hum Genet 2014;94(1):11–22. 10.1016/j.ajhg.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. González‐Ávila C, Casado L, Muro García I, Villacieros‐Álvarez J, Vivancos J, Quintas S. Altered ioflupane single‐photon emission computed tomography in anti‐IgLON5 disease: a new case mimicking probable progressive supranuclear palsy and review of the literature. Eur J Neurol 2021;28(4):1392–1395. 10.1111/ene.14634. [DOI] [PubMed] [Google Scholar]
- 58. Brüggemann N, Wandinger KP, Gaig C, Sprenger A, Junghanns K, Helmchen C, Münchau A. Dystonia, lower limb stiffness, and upward gaze palsy in a patient with IgLON5 antibodies. Mov Disord 2016;31(5):762–764. 10.1002/mds.26608. [DOI] [PubMed] [Google Scholar]
- 59. Fuseya K, Kimura A, Yoshikura N, Yamada M, Hayashi Y, Shimohata T. Corticobasal syndrome in a patient with <scp>anti‐IgLON5</scp> antibodies. Mov Disord Clin Pract 2020;7(5):557–559. 10.1002/mdc3.12957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Montojo T, Piren V, Benkhadra F, Codreanu A, Diederich NJ. Gaze palsy, sleep and gait disorder, as well as Tako‐Tsubo syndrome in a patient with IgLON5 antibodies. Mov Disord Clin Pract 2017;4(3):441–443. 10.1002/mdc3.12407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. del Sole A, Chiesa V, Lucignani G, et al. Exploring dopaminergic activity in ring chromosome 20 syndrome: a SPECT study. Quart J Nucl Med Mol Imaging 2010;54(5):564–569. [PubMed] [Google Scholar]
- 62. Niemelä V, Salih A, Solea D, et al. Phenotypic variability in chorea‐acanthocytosis associated with novel VPS13A mutations. Neurol Genet 2020;6(3):e426. 10.1212/NXG.0000000000000426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schulz A, Kohlschütter A, Mink J, Simonati A, Williams R. NCL diseases — clinical perspectives. Biochim Biophys Acta (BBA) – Mol Basis Dis 2013;1832(11):1801–1806. 10.1016/j.bbadis.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jalanko A, Braulke T. Neuronal ceroid lipofuscinoses. Biochim Biophys Acta (BBA) – Mol Cell Res 2009;1793(4):697–709. 10.1016/j.bbamcr.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 65. Lange LM, Schell N, Tunc S, Shoukier M, Weißbach A, Hellenbroich Y, Brüggemann N. Atypical parkinsonism with pathological dopamine transporter imaging in neuronal ceroid Lipofuscinosis type 5. Mov Disord Clin Pract 2022;9(8):1116–1119. 10.1002/mdc3.13562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bajaj N, Hauser RA, Grachev ID. Clinical utility of dopamine transporter single photon emission CT (DaT‐SPECT) with (123I) ioflupane in diagnosis of parkinsonian syndromes. J Neurol Neurosurg Psychiatry 2013;84(11):1288–1295. 10.1136/jnnp-2012-304436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gajos A, Dąbrowski J, Bieńkiewicz M, Płachcińska A, Kuśmierek J, Bogucki A. Should non‐movement specialists refer patients for SPECT‐DaTSCAN? Neurol Neurochir Pol 2019;53(2):138–143. 10.5603/PJNNS.a2019.0011. [DOI] [PubMed] [Google Scholar]
- 68. Graebner AK, Tarsy D, Shih LC, et al. Clinical Impact of 123I‐Ioflupane SPECT (DaTscan) in a Movement Disorder Center. Neurodegener Dis 2017;17(1):38–43. 10.1159/000447561. [DOI] [PubMed] [Google Scholar]
- 69. Isaacson JR, Brillman S, Chhabria N, Isaacson SH. Impact of DaTscan imaging on clinical decision making in clinically uncertain Parkinson's disease. J Parkinsons Dis 2021;11(2):885–889. 10.3233/JPD-202506. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary of the available literature regarding potential DaTSCAN abnormal results associated with metabolic disorders.
Table S2. Summary of the available literature regarding potential DaTSCAN abnormal results associated with other neurodegenerative pathologies.