

Abstract
Constitutively active KRAS mutations are among the major drivers of lung cancer, yet the identity of molecular co‐operators of oncogenic KRAS in the lung remains ill‐defined. The innate immune cytosolic DNA sensor and pattern recognition receptor (PRR) Absent‐in‐melanoma 2 (AIM2) is best known for its assembly of multiprotein inflammasome complexes and promoting an inflammatory response. Here, we define a role for AIM2, independent of inflammasomes, in KRAS‐addicted lung adenocarcinoma (LAC). In genetically defined and experimentally induced (nicotine‐derived nitrosamine ketone; NNK) LAC mouse models harboring the Kras G12D driver mutation, AIM2 was highly upregulated compared with other cytosolic DNA sensors and inflammasome‐associated PRRs. Genetic ablation of AIM2 in Kras G12D and NNK‐induced LAC mouse models significantly reduced tumor growth, coincident with reduced cellular proliferation in the lung. Bone marrow chimeras suggest a requirement for AIM2 in Kras G12D‐driven LAC in both hematopoietic (immune) and non‐hematopoietic (epithelial) cellular compartments, which is supported by upregulated AIM2 expression in immune and epithelial cells of mutant KRAS lung tissues. Notably, protection against LAC in AIM2‐deficient mice is associated with unaltered protein levels of mature Caspase‐1 and IL‐1β inflammasome effectors. Moreover, genetic ablation of the key inflammasome adapter, ASC, did not suppress Kras G12D‐driven LAC. In support of these in vivo findings, AIM2, but not mature Caspase‐1, was upregulated in human LAC patient tumor biopsies. Collectively, our findings reveal that endogenous AIM2 plays a tumor‐promoting role, independent of inflammasomes, in mutant KRAS‐addicted LAC, and suggest innate immune DNA sensing may provide an avenue to explore new therapeutic strategies in lung cancer.
Keywords: cell proliferation, inflammasome, innate immunity, lung cancer, pattern recognition receptors
Our findings define an inflammasome‐independent role for the cytosolic DNA sensor AIM2 in mutant KRAS lung cancer, and suggest that AIM2 represents an attractive new target of innate immunity for the development of therapies for lung cancer and potentially other mutant KRAS‐addicted cancers.
1. INTRODUCTION
Non–small‐cell lung cancer (NSCLC) comprises ~85% of all lung cancers, and is the leading cause of cancer‐related mortality worldwide, with the majority of NSCLC patients presenting with the histological subtype lung adenocarcinoma (LAC). 1 , 2 Lung cancer is strongly associated with tobacco smoking, and a common disease‐associated consequence of the genotoxicity of tobacco smoke carcinogens such as nitrosamine 4‐(methylnitrosamino)‐1‐(3‐pyridyl)‐1‐butanone (nicotine‐derived nitrosamine ketone; NNK) is activating mutations (e.g., G12C, G12D) in the KRAS proto‐oncogene, present in 25%–30% of NSCLC patients and linked to poor survival. 3 , 4 , 5 , 6 , 7 Notably, mouse strains that express human KRAS G12C or mouse Kras G12D mutant alleles in the lung epithelium spontaneously develop LAC. 8 , 9 , 10 Prolonged tobacco smoke exposure in the lung also triggers overactivation of the innate immune response, leading to chronic inflammation that is associated with ~80% of NSCLC cases. 11 , 12 Furthermore, a low‐grade inflammatory response (predominantly comprising innate immune macrophages and neutrophils) occurs in the lungs of the Kras G12D LAC mouse model. 13 , 14 , 15 Despite the association between chronic lung inflammation and NSCLC, the identity of disease‐associated molecular regulators of innate immunity in NSCLC remains obscure.
A recent clinical trial (CANTOS) involving anti‐IL‐1β therapy on atherosclerotic patients unexpectedly suggested that targeting the pro‐inflammatory cytokine IL‐1β may be beneficial in preventing NSCLC. 16 The production of mature IL‐1β, and the related IL‐18 cytokine, is governed by multiprotein “inflammasome” complexes of innate immunity containing specific pattern recognition receptors (PRRs), the best documented being NOD‐like receptors NLRC4, NLRP1, NLRP3, NLRP6 and NLRP12, along with the cytosolic DNA sensor, Absent‐in‐melanoma 2 (AIM2). 17 , 18 Upon ligand sensing, these PRRs associate with the adapter protein Apoptosis‐associated Speck‐like protein containing a CARD (ASC) to form distinct oligomeric inflammasome complexes that recruit Caspase‐1 to catalyze maturation of inactive pro‐IL‐18 and pro‐IL‐1β into bioactive, secreted IL‐18 and IL‐1β cytokines. 17 , 18 Among these PRRs, AIM2 has recently attracted considerable interest for its diverse and often opposing tumor‐promoting or tumor‐inhibiting activities in numerous cancers (e.g., colorectal, liver, gastric), which can be either inflammasome‐dependent or inflammasome‐independent. 19 , 20 , 21 , 22 , 23 In NSCLC, studies on the role of AIM2 have been limited to wild‐type or mutant KRAS immortalized human NSCLC cell lines, with contrasting inflammasome‐dependent or inflammasome‐independent tumor‐promoting functions assigned to AIM2 irrespective of KRAS mutation status. 24 , 25
To define the in vivo role of endogenous AIM2 in NSCLC, here we coupled the genetic ablation of AIM2 with bona fide preclinical genetically engineered Kras G12D and NNK tobacco carcinogen‐induced LAC mouse models. We revealed that upregulated expression of AIM2 in both the immune and epithelial cell compartments of the lung promotes mutant KRAS lung carcinogenesis, independent of inflammasome activity. We also demonstrate that genetic ablation of the ASC inflammasome adapter has no effect on the LAC phenotype of Kras G12D mice. Collectively, these in vivo findings define an inflammasome‐independent role for AIM2 in mutant KRAS LAC, and suggest that AIM2 represents an attractive target of innate immunity to develop therapies for NSCLC and potentially other KRAS‐addicted cancers.
2. MATERIALS AND METHODS
2.1. Human biopsies
Human lung tissue biopsies (Table S1) were either fixed with 4% paraformaldehyde and paraffin embedded, or snap‐frozen in liquid nitrogen. Studies were approved by the Monash Health Human Research Ethics Committee, and formal written informed patient consent was obtained prior to blood and tissue collection.
2.2. Animal studies
All mice were housed under specific pathogen‐free conditions. Experiments were approved by the Hudson Animal Ethics Monash Medical Centre “B” Committee, and were conducted in accordance with the ARRIVE guidelines. 26
2.2.1. Kras G12D model
Male and female 6‐week‐old Kras LSL‐G12D/+ mice on a mixed 129Sv × C57BL/6 background were subjected to intranasal inhalation of 5 × 106 plaque‐forming units of Adenovirus Cre recombinase (Ad‐Cre; University of Iowa), generating “Kras G12D mice.” 9 By 6 weeks post inhalation, Kras G12D mice develop diffuse atypical adenomatous hyperplasia (AAH) and adenomas in the lung, which further progress to sporadic adenocarcinoma in situ (AIS) by 12 weeks post inhalation. 9 , 14 , 15 As a lesion‐free control, Kras LSL‐G12D/+ mice received an equal volume of phosphate‐buffered saline (PBS) to generate “Kras WT mice.” 14 Kras LSL‐G12D/+ mice 9 were mated with Aim2 −/− 27 or Pycard −/− 28 mice on a 129Sv × C57BL/6 background, and all Kras G12D mouse strains were housed under specific pathogen‐free conditions.
For inhibitor studies, Kras G12D mice were injected (intraperitoneal) with a single dose of either the p38 MAPK inhibitor (SB203580; 10 mg/kg, Sigma), the ERK1/2 inhibitor (U0126; 10 mg/kg, Cell Signaling Technology), or dimethyl sulfoxide (DMSO) vehicle (control) as described previously. 15
2.2.2. NNK model
Wild‐type and Aim2 −/− male and female mice aged 6–8 weeks on a “pseudo‐A/J” background were intraperitoneal injected three times on alternate days with NNK (100 mg/kg, Toronto Research Chemicals) dissolved in PBS, or the equivalent volume of PBS as a vehicle control. 29 , 30 Mice were culled at 20 weeks post injections.
2.2.3. Bone marrow chimeras
Kras LSL‐G12D/+ male and female mice aged 6 weeks were lethally irradiated (single 9.5 Gy dose) and reconstituted with 5 × 106 unfractionated donor bone marrow cells from Kras G12D mice or Kras G12D:Aim2 −/− mice. Recipient mice were inhaled with Ad‐Cre 8 weeks thereafter, and were culled 6 weeks following inhalation.
2.3. Histology and immunohistochemistry
Dissected human and mouse lungs were formalin‐fixed and paraffin‐embedded (FFPE) for sectioning (4–6 μm). Human lung tissues were stained with antibodies against AIM2 (HPA031365, Atlas Antibodies) and p20 Caspase‐1 (Cleaved Asp210; PA5‐38099, Invitrogen). 20 Mouse tissues were stained with hematoxylin and eosin (H&E) and subjected to blinded histological evaluation. Immunohistochemistry was performed with antibodies against AIM2 (63660, Cell Signaling Technology) thyroid transcription factor‐1 (TTF‐1; ab76013, Abcam), Ki67 (ab16667, Abcam), Proliferating Cell Nuclear Antigen (PCNA; ab18197, AbCam), CD45 (550539, BD Biosciences), B220 (550286, BD Biosciences), CD3 (559974, BD Biosciences), cleaved Caspase‐3 (9661, Cell Signaling Technology), pThr202/pTyr204 ERK1/2 MAPK (9101S, Cell Signaling Technology), pThr180/pTyr182 p38 MAPK (4511S, Cell Signaling Technology), phospho‐NF‐κB p65 (3031S, Cell Signaling Technology), phospho‐Akt (4060S, Cell Signaling Technology) and cleaved Caspase‐1 (PA5‐38099, Invitrogen), followed by counterstaining with hematoxylin. 15 , 20 Digital photomicrographs (60× high‐power fields) were viewed using ImageJ software (National Institutes of Health, USA), and positive cellular staining was quantified manually per high‐power field (n = 20). 20
2.4. Immunofluorescence
Human FFPE lung sections were stained with antibodies against AIM2 (HPA031365, Atlas Antibodies), CC10 (sc‐365992, Santa Cruz Biotechnology), SP‐C (sc‐518029, Santa Cruz Biotechnology), and CD45 (555480, BD Biosciences), using Alexa Fluor‐conjugated secondary antibodies (Invitrogen). 15 , 20 Nuclear staining was detected using 4′,6‐diamidino‐2‐phenylindole (DAPI), and sections subjected to the above staining protocol in the absence of primary antibodies served as negative controls to indicate the level of background autofluorescence.
2.5. ELISA and immunoblotting
ELISAs for IL‐1β (R&D Systems) were performed on mouse serum. Total protein lysates were prepared from snap‐frozen mouse lung tissues and subjected to immunoblotting with antibodies against Caspase‐1 (p45/p20) (AG‐20B‐0042‐C100, AdipoGen), Gasdermin D (ab209845, Abcam), IL‐1β (BAF401, R&D Systems) and β‐tubulin (ab6046, Abcam). 20 Protein bands were visualized using enhanced chemiluminescence with the Chemidoc Imaging System (BioRad), and quantified by densitometry using ImageJ software.
2.6. RNA isolation and gene expression
Total RNA was isolated from snap‐frozen human and mouse lung tissues using TRIzol (Sigma), and quantitative RT‐PCR (qPCR) was performed on cDNA with SYBR Green (Life Technologies) using the 7900HT Fast RT‐PCR System (Applied Biosystems). Gene expression data acquisition and analyses were performed using the Sequence Detection System Version 2.4 software (Applied Biosystems), and are normalized to 18S rRNA (human, RNA18S1; mouse, Rn18s). Fold change of gene expression was determined using the delta–delta cycle threshold (C t) method. Primer sequences are available upon request.
2.7. The Cancer Genome Atlas data analysis
RNA‐Seq HTSeq gene counts and clinical data from the TCGA‐LUAD cohort were obtained using the TCGAbiolinks package. 31 KRAS‐mutant status was obtained from the GDC Data Portal website. 32 Counts were processed and differential expression analysis was performed using the edgeR package 33 to create a DGEList object, and gene annotation information was obtained using the Homo.sapiens package. Recurrent solid tumor samples were excluded and samples were grouped by normal (i.e., non‐tumor) tissue, KRAS wild‐type primary tumor or KRAS‐mutant primary tumor. Lowly expressed genes were removed using the “filterByExpr” function with a minimum count of 5. Normalization factors were calculated using the TMM method and robustified dispersions were estimated. 34 , 35 A quasi‐likelihood model was fit using the “glmQLFit” function, 36 and the “glmTreat” function was used to compare the wild‐type KRAS and mutant KRAS primary tumors to normal controls, with a treat fold change of 1.5. 37 Differentially expressed genes were selected with a false discovery rate of <0.05. Expression levels, expressed as log2 counts per million, were plotted for genes of interest. BRAF‐, EGFR‐, and ERBB2‐mutant statuses were also obtained from the GDC Data Portal, and EML4–ALK‐fusion status was obtained from an analysis of TCGA gene fusions. 38 For each of these, differential expression analyses comparing normal tissue, wild‐type primary tumor and mutant primary tumor were performed as for KRAS.
2.8. Statistics
Statistics were performed using GraphPad Prism software (version 9.3.1), with D'Agostino and Pearson omnibus K2 normality tests. Significance between two groups of normal distribution was determined using Student's t‐test and Mann–Whitney test was performed for non‐normally distributed data or smaller data sets. One‐way analysis of variance (ANOVA) was used to determine differences among three or more groups for normally distributed data, and Kruskal–Wallis test for non‐normally distributed data or smaller data sets. A p‐value < 0.05 was considered statistically significant, as indicated in figure legends along with experimental sample sizes, when relevant. All data were expressed as the mean ± standard error of the mean (SEM).
3. RESULTS
3.1. Elevated expression of AIM2 among inflammasome‐associated PRRs and cytosolic DNA sensors in human mutant KRAS LAC
In human NSCLC, AIM2 gene expression has previously been reported to be upregulated and prognostic for poor survival outcomes of patients. 25 However, the expression profile of AIM2 compared with other inflammasome‐associated PRRs or cytosolic DNA sensors in NSCLC, including the major NSCLC subtype, mutant KRAS‐addicted LAC, is unknown. Interrogation of TCGA RNA‐Seq datasets from LAC patients revealed that AIM2 was the only inflammasome‐associated PRR significantly upregulated in KRAS‐mutant LAC, and also KRAS wild‐type (WT) LAC, tumor tissues compared with non‐tumor control tissues (Figure 1A). Compared with other well‐known cytosolic DNA sensors, AIM2 mRNA levels were also the highest and most significantly upregulated (Figure S1A). Interestingly, AIM2 mRNA levels were also significantly higher in other molecular subtypes of NSCLC, such as those that are mutant for EGFR, BRAF, EML4‐ALK, or ERBB2 (Figure S1B).
FIGURE 1.
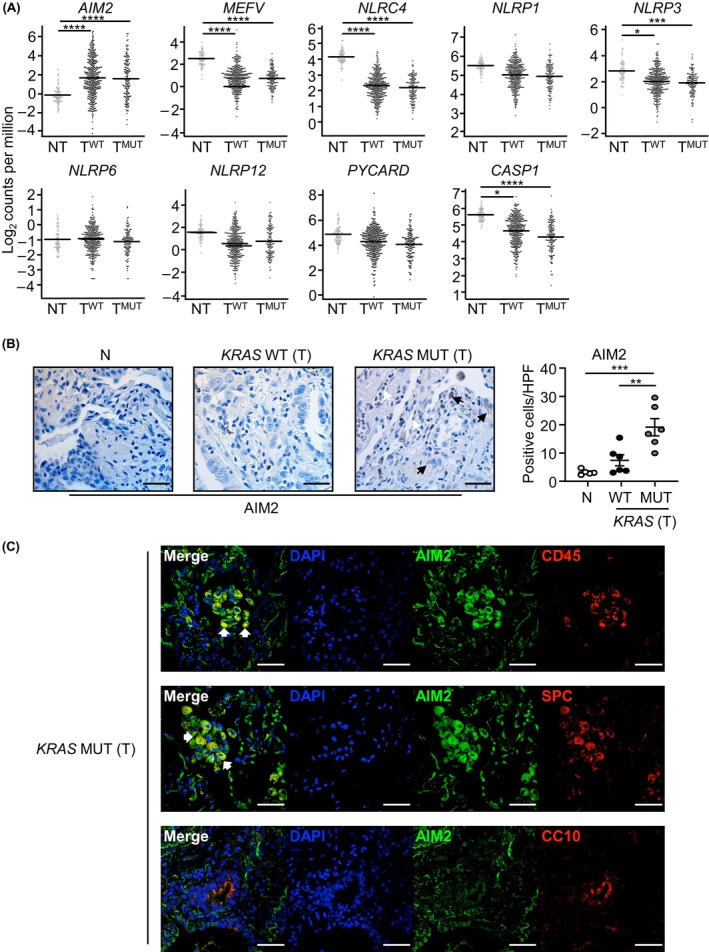
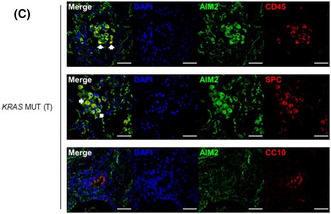
Elevated AIM2 expression in immune and epithelial cells of human KRAS‐mutant LAC lung biopsies. (A) Gene expression of inflammasome‐associated components in KRAS‐mutant (MUT) tumor (T; n = 138) and KRAS wild‐type (WT) tumor (n = 375) versus non‐tumor (NT; n = 59) tissues from TCGA LAC patients. False discovery rate adjusted p‐values: *p < 0.05, ***p < 0.001, ****p < 0.0001. (B) Representative images of AIM2‐stained lung sections from non‐cancer (N) and tumor (T) tissues from an Australian LAC patient cohort stratified into KRAS wild‐type or mutant. Scale bars: 100 μm. The graph depicts quantification of AIM2‐positive cells/high‐power field (HPF) in human lung biopsies (n = 5–6/group). (C) Representative immunofluorescence images of lung tumor sections from a KRAS‐mutant LAC patient co‐stained for AIM2 (green) and total immune cells (CD45, red, top panel), alveolar type‐II cells (surfactant protein‐C (SPC), red, middle panel), and club cells (CC10, red, bottom panel). DAPI nuclear staining is blue. Scale bars: 50 μm. White arrowheads, representative dual‐positive AIM2‐expressing immune (top panel) and alveolar type‐II (middle panel) cells.
We further investigated the expression status of AIM2 in the prevalent NSCLC molecular subtype, KRAS‐mutant LAC. Semi‐quantitative immunohistochemical analyses of an independent LAC patient cohort also confirmed significantly increased numbers of AIM2‐positive cells in tumors from KRAS‐mutant LAC patients versus KRAS WT LAC patients, and tumor‐free lung tissues (Figure 1B). The morphology of AIM2‐positive cells was consistent with epithelial and immune cell types, with predominant cytoplasmic AIM2 positivity (Figure 1B). Dual immunofluorescence staining with an anti‐AIM2 antibody together with antibodies against either pan‐immune cells (CD45) or distinct epithelial cell subtypes associated with cells of origin in LAC, namely alveolar type‐II (surfactant protein‐C, SPC) or Club (CC10) cells, verified that AIM2 was predominantly expressed in the cytoplasm of SP‐C‐positive alveolar type‐II epithelial cells, as well as CD45‐positive immune cells, of KRAS‐mutant LAC patient tumors (Figure 1C).
In contrast with AIM2, gene expression levels for key inflammasome components ASC (encoded by PYCARD) and Caspase‐1 (CASP1) were not elevated in tumors of KRAS‐mutant LAC patients (Figure 1A). Furthermore, immunohistochemical cellular staining levels for cleaved (i.e., active) Caspase‐1 (indicative of inflammasome activation) were comparable between tumor‐free lung tissues and tumors of KRAS‐mutant and WT LAC patients, and no significant positive correlation was observed between AIM2 and cleaved Caspase‐1 cellular positivity in human lung tissues (Figure S1C–E). Collectively, these findings suggest that elevated AIM2 expression in human KRAS‐mutant LAC may be independent of inflammasome activation.
3.2. Genetic ablation of upregulated AIM2 expression in KrasG12D mice abrogates LAC
To date, the in vivo expression of endogenous AIM2 in bona fide preclinical models of NSCLC, including oncogenic KRAS‐driven LAC, remains unknown. In the lungs of lesion‐bearing Kras G12D mice at 6 weeks post Ad‐Cre inhalation (to induce expression of the oncogenic Kras G12D allele), among inflammasome‐associated components and cytosolic DNA sensors, only Aim2 mRNA levels were significantly upregulated compared with lesion‐free lungs from control Kras WT (PBS‐inhaled) mice (Figure 2A,B). These observations are consistent with our clinical data indicating that AIM2 is upregulated in KRAS‐mutant LAC. In this regard, as both p38 and ERK1/2 MAPKs are downstream effectors of KRAS, we assessed whether these pathways were involved in the regulation of AIM2 expression by performing immunohistochemical staining analyses (consistent with our clinical data) in Kras G12D mice that were treated with an ERK (U0126) or p38 (SB203580) MAPK inhibitor. As shown in Figure S2A,B, AIM2 protein expression was significantly reduced in lung lesions of Kras G12D mice treated with the p38, but not ERK1/2, MAPK inhibitor, suggesting that the p38 ERK MAPK pathway contributes to the induction of AIM2 in KRAS‐mutant LAC.
FIGURE 2.
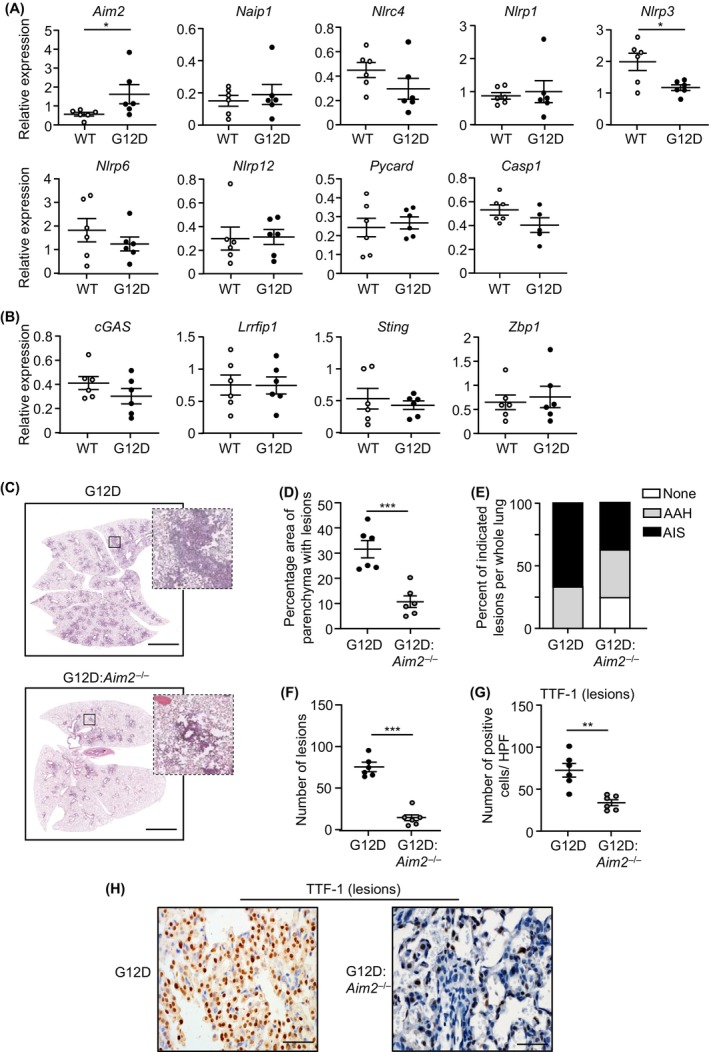
AIM2 deficiency suppresses oncogenic Kras‐induced LAC. (A, B) qPCR of (A) inflammasome‐associated pattern recognition receptors and effectors, and (B) cytosolic DNA sensors, in mouse lungs (n = 6/genotype). *p < 0.05, Student's t‐test. (C) Representative low‐power images of H&E‐stained lung sections from Kras G12D and Kras G12D:Aim2 −/− mice at 6 weeks post Ad‐Cre inhalation. Insets depict magnified areas comprising lesions in the low‐power images (open squares). Scale bars: 3 mm. (D–F) Quantification of (D) lung parenchyma area containing tumor lesions, (E) lesion histological classification, and (F) tumor incidence, per whole mouse lung (n = 6/genotype). ***p < 0.001, Student's t‐test. AAH, atypical adenomatous hyperplasia; AIS, adenocarcinoma in situ. (G) Quantification of TTF‐1‐positive cells/high‐power field (HPF) in lung lesions (n = 6/genotype). **p < 0.01, Student's t‐test. (H) Representative images of TTF‐1‐stained lung sections from Kras G12D and Kras G12D:Aim2 −/− mice at 6 weeks post Ad‐Cre. Scale bars: 100 μm.
We next assessed whether AIM2 contributed to the pathogenesis of oncogenic KRAS‐driven LAC by generating Kras G12D mice homozygous null for AIM2 (Kras G12D:Aim2 −/−). At 6 weeks post Ad‐Cre inhalation, the area of lung parenchyma affected by diffuse AAH and AIS lesions in Kras G12D:Aim2 −/− mice was significantly reduced by 64% compared with Kras G12D littermates (Figure 2C–E; Figure S2C). The lungs of Kras G12D:Aim2 −/− mice also contained markedly lower numbers (by 80%) of lesions versus Kras G12D mice (Figure 2F). Furthermore, the numbers of morphologically distinguishable alveolar epithelial type‐II cells positive for the LAC marker, TTF‐1, were significantly reduced in lesions and histologically normal lung parenchyma of Kras G12D:Aim2 −/− versus Kras G12D mice (Figure 2G,H; Figure S2D,E). At 12 weeks following Ad‐Cre inhalation, the lungs of Kras G12D:Aim2 −/− mice also developed fewer advanced AIS lesions and TTF‐1‐positive cells versus age‐matched Kras G12D mice (Figure S2F–K). Collectively, these findings suggest a key role for AIM2 in promoting Kras G12D‐induced LAC.
3.3. AIM2 contributes to augmented tumor cell proliferation and inflammation in Kras G12D ‐induced LAC
Next, we assessed the cellular processes by which AIM2 promotes KRAS‐mutant LAC. In the lungs of Kras G12D:Aim2 −/− mice, the abrogated LAC phenotype associated with significantly reduced cellular proliferation throughout lesions and unaffected parenchyma, measured by cellular reactivity to proliferation markers Ki67 and PCNA, compared with Kras G12D mice (Figure 3A–D; Figure S3A,B). In contrast, levels of cellular apoptosis (i.e., cleaved Caspase‐3 immunostaining) throughout the lungs of Kras G12D and Kras G12D:Aim2 −/− mice were comparable (Figure 3E,F; Figure S3C,D). The suppressed LAC phenotype in Kras G12D:Aim2 −/− mice did not appear to associate with alterations in angiogenesis, as qPCR gene expression analyses showed a comparable profile of an angiogenic gene signature 14 , 15 in the lungs of Kras G12D and Kras G12D:Aim2 −/− mice (Figure S3E).
FIGURE 3.
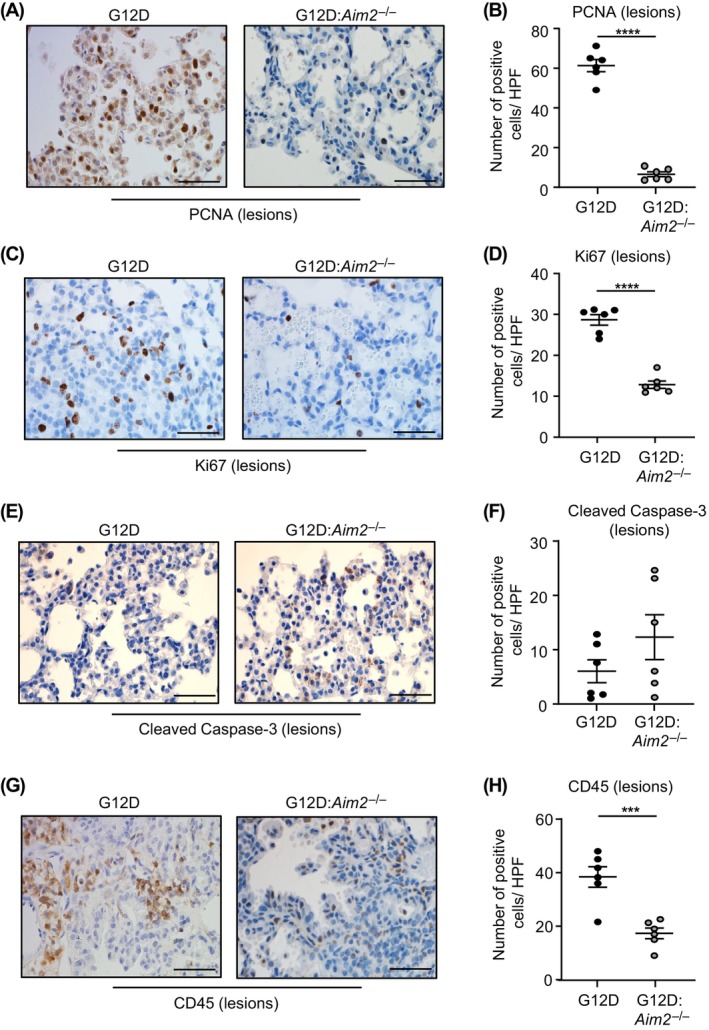
Reduced tumor cell proliferation and inflammation in Kras G12D:Aim2 −/− mouse lungs. (A, C, E, G) Representative images of lung sections containing lesions from Kras G12D and Kras G12D:Aim2 −/− mice at 6 weeks post Ad‐Cre immunostained with antibodies against (A) PCNA, (C) Ki67, (E) cleaved Caspase‐3, and (G) CD45. Scale bars: 100 μm. (B, D, F, H) Quantification of positive cells/high‐power field (HPF) in lesion‐bearing mouse lungs immunostained with antibodies against (B) PCNA, (D) Ki67, (F) cleaved Caspase‐3, and (H) CD45 (n = 6/genotype). ***p < 0.001, ****p < 0.0001, Student's t‐test.
As oncogenic KRAS can trigger an inflammatory tumor‐promoting microenvironment in the lung coincident with immune cell infiltrates, 13 , 15 we investigated whether suppressed lung tumorigenesis in Kras G12D:Aim2 −/− mice is associated with reduced infiltration of immune cells. Indeed, immunohistochemistry revealed a significant reduction in the numbers of CD45‐positive total immune cells in Kras G12D:Aim2 −/− lung lesions and unaffected parenchyma compared with their Kras G12D counterparts (Figure 3G,H; Figure S3F,G). Furthermore, the lower numbers of total immune cell infiltrates in Kras G12D:Aim2 −/− lung lesions largely corresponded to F4/80‐positive macrophages, as well as CD3‐positive T and B220‐positive B cells (Figure 4A–F).
FIGURE 4.
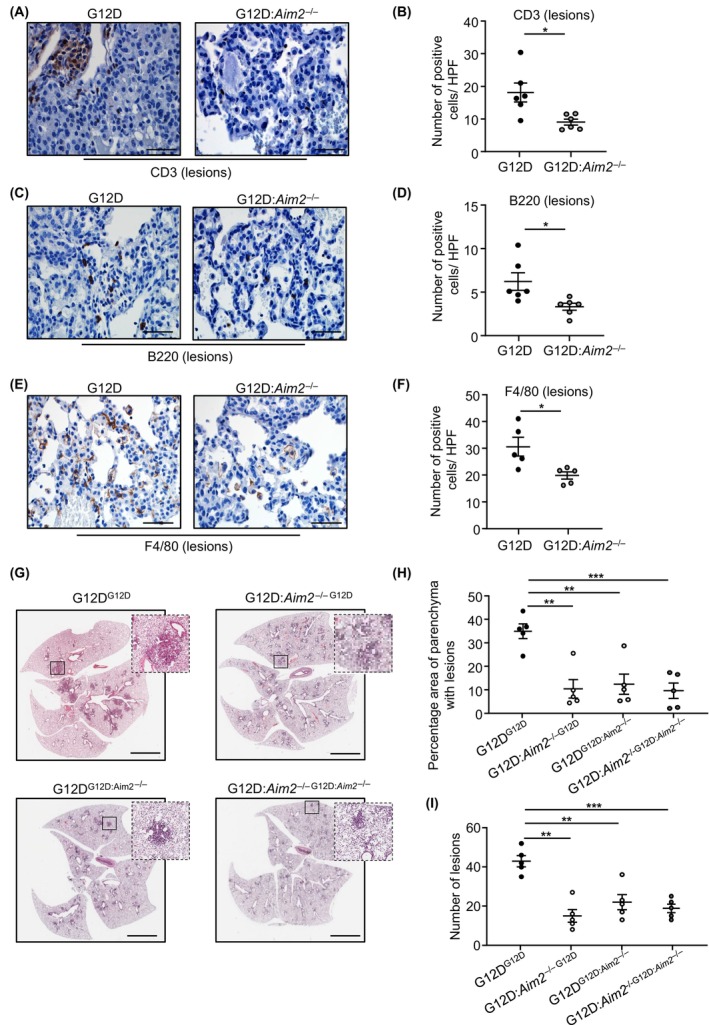
AIM2 promotion of KRAS‐driven LAC augments immune cell infiltrates, and requires hematopoietic and non‐hematopoietic cellular compartments. (A, C, E) Representative images of lung sections containing lesions from Kras G12D and Kras G12D:Aim2 −/− mice at 6 weeks post Ad‐Cre inhalation immunostained with antibodies against (A) CD3, (C) B220 and (E) F4/80. Scale bars: 100 μm. (B, D, F) Quantification of positive cells/high‐power field (HPF) in lesion‐bearing mouse lungs immunostained with antibodies against (B) CD3, (D) B220 and (F) F4/80 (n = 6/genotype). *p < 0.05, Student's t‐test. (G) Representative images of H&E‐stained lung sections from Kras G12D (G12D) or Kras G12D:Aim2 −/− (G12D:Aim2 −/−) recipient mice reconstituted with Kras G12D or Kras G12D:Aim2 −/− donor bone marrow (indicated in superscript font). Insets depict magnified areas comprising lesions in the low‐power images (open squares). Scale bars: 3 mm. (H, I) Quantification of (H) lung parenchyma area containing tumor lesions, and (I) tumor incidence, per whole mouse lung in chimeras (n = 5/group). **p < 0.01, ***p < 0.001; One‐way ANOVA.
Considering AIM2 was predominantly expressed in both alveolar type‐II epithelial and CD45‐positive immune cells in KRAS‐mutant LAC (Figure 1C), we investigated whether AIM2 expression in myeloid cells contributed to Kras G12D‐driven LAC by generating reciprocal bone marrow chimeras between Kras G12D and Kras G12D:Aim2 −/− mice. The absence of AIM2 in either bone marrow‐derived hematopoietic (e.g., myeloid) cells (Kras G12D recipients reconstituted with Kras G12D:Aim2 −/− donor bone marrow; G12DG12D:Aim2−/−) or non‐hematopoietic (e.g., epithelial) cells (Kras G12D:Aim2 −/− recipients reconstituted with Kras G12D donor bone marrow; G12D:Aim2 −/−G12D) led to similar marked protection against the LAC phenotype of the recipient mice, comparable with that upon AIM2 deficiency in both hematopoietic and non‐hematopoietic compartments (G12D:Aim2 −/−G12D:Aim2−/−) (Figure 4G–I). Therefore, these findings further suggest that AIM2 expression in lung epithelial and immune cells promotes mutant KRAS‐driven LAC.
3.4. AIM2 contributes to Kras G12D ‐induced LAC, independent of inflammasome activation, via STAT3, p38 and ERK MAPK signaling cascades
AIM2 can modulate tumorigenesis both dependent and independent of inflammasome activation, 20 , 21 , 22 , 23 , 24 , 25 , 39 yet the mechanism of action of AIM2 in a bona fide in vivo lung cancer model is ill‐defined. Immunoblotting for the cleaved p20 subunit of Caspase‐1 revealed that despite marginally elevated levels of pro‐Caspase‐1 (p45) in the lungs of Kras G12D versus control Kras WT mice, cleaved Caspase‐1 protein levels were not significantly altered irrespective of the activation or expression status of KRAS or AIM2, respectively (Figure 5A,B). Furthermore, in Kras G12D and Kras G12D:Aim2 −/− mouse lungs, immunoblotting for Gasdermin D, an important mediator of Caspase‐1 (i.e., inflammasome)‐mediated cellular death, indicated that expression levels of full‐length Gasdermin D were also comparable, and cleaved (mature N‐terminal p31 fragment) Gasdermin D was barely detectable (Figure S4A). Similarly, immunoblotting and ELISA indicated unchanged levels of pro (p31) and/or mature (p17) IL‐1β in the lungs and serum, respectively, of Kras G12D versus control Kras WT mice (Figure 5A–C). Immunohistochemistry also indicated comparable cellular staining levels for cleaved Caspase‐1 in lung lesions of Kras G12D and Kras G12D:Aim2 −/− mice (Figure 5D,E). The lungs, but not serum, of Kras G12D:Aim2 −/− mice displayed elevated levels of pro and mature forms of IL‐1β, which remains unexplained and could not be accounted for by any corresponding increase in Il1b mRNA levels in Kras G12D:Aim2 −/− mouse lungs (Figure S4B). These observations suggest that AIM2 promotes oncogenic KRAS‐induced LAC independent of inflammasome activity.
FIGURE 5.
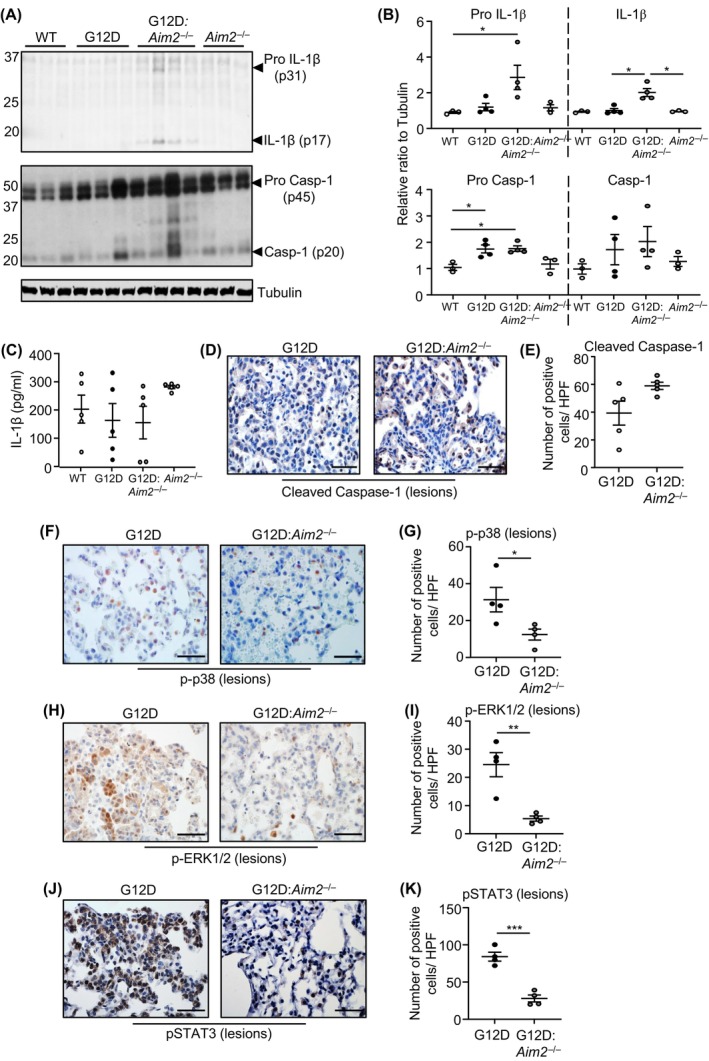
AIM2‐driven LAC in Kras G12D mice is independent of inflammasome activation, yet aligns with STAT3, ERK1/2 and p38 MAPK signaling. (A) Immunoblots of individual lung lysates from Kras WT, Kras G12D, Kras G12D:Aim2 −/− and Aim2 −/− mice at 6 weeks post Ad‐Cre (Kras G12D, Kras G12D:Aim2 −/−) or PBS vehicle (Kras WT, Aim2 −/−) inhalation with indicated antibodies. (B) Densitometry of blots from (A), with expression levels relative to Tubulin loading. *p < 0.05, one‐way ANOVA. (C) ELISA for total IL‐1β protein levels in the serum of mice at 6 weeks post inhalations (n = 5/genotype). (D, F, H, J) Representative images of (D) cleaved Caspase‐1, (F) phosphorylated (p) p38 MAPK, (H) pERK1/2, and (J) pSTAT3 immunostaining of lung sections containing lesions from Kras G12D and Kras G12D:Aim2 −/− mice at 6 weeks post Ad‐Cre. Scale bars: 100 μm. (E, G, I, K) Quantification of (E) cleaved Caspase‐1, (G) pp38 MAPK, (I) pERK1/2, and (K) pSTAT3 positive cells/high‐power field (HPF) in mouse lung lesions (n = 4/genotype). *p < 0.05, **p < 0.01, ***p < 0.001, Student's t‐test.
The requirement for AIM2 in Kras G12D‐dependent LAC is associated with activation of signaling cascades implicated in cellular proliferation and lung tumorigenesis. Specifically, immunohistochemistry revealed that cellular staining levels of phosphorylated (activated) p38 and ERK1/2 MAPKs, and the latent oncogenic transcription factor STAT3, were significantly upregulated in lesion‐bearing Kras G12D mouse lungs versus their Kras G12D:Aim2 −/− counterparts (Figure 5F–K). By contrast, numbers of cells positively stained for phosphorylated p65 NF‐κB or Akt were not reduced in lung lesions of Kras G12D:Aim2 −/− mice, although phosphorylated Akt immunostaining levels were slightly elevated in Kras G12D:Aim2 −/− mouse lungs (Figure S4C–F).
3.5. Genetic ablation of AIM2 abrogates NNK‐induced lung carcinogenesis
To further verify an in vivo inflammasome‐independent role for AIM2 in LAC, we used the NNK tobacco carcinogen‐induced LAC mouse model, a molecular hallmark of which is activating mutations in codon 12 of Kras. 7 Indeed, we observed comparable levels of pro and mature Caspase‐1 and IL‐1β in lung tissues, and serum levels of secreted IL‐1β, in pseudo‐A/J WT mice administered with PBS or NNK over 20 weeks (Figure 6A; Figure S5A), at which time NNK reproducibly induces LAC. 7 , 29 , 30 qPCR expression profiling again revealed that among inflammasome‐associated PRRs (plus ASC and Caspase‐1) and cytosolic DNA sensors, only Aim2 mRNA levels were significantly upregulated by NNK in mouse lungs (Figure 6B; Figure S5B). Moreover, tumor incidence in NNK‐treated pseudo‐A/J Aim2 −/− mice was significantly reduced to ~50% (11.42 ± 2.21 lesions) of their WT counterparts (22.08 ± 2.18 lesions) (Figure 6C,D), and immunohistochemistry indicated significantly reduced numbers of TFF‐1‐positive alveolar type‐II cells in pseudo‐A/J Aim2 −/− mouse lung lesions (Figure 6E,F).
FIGURE 6.
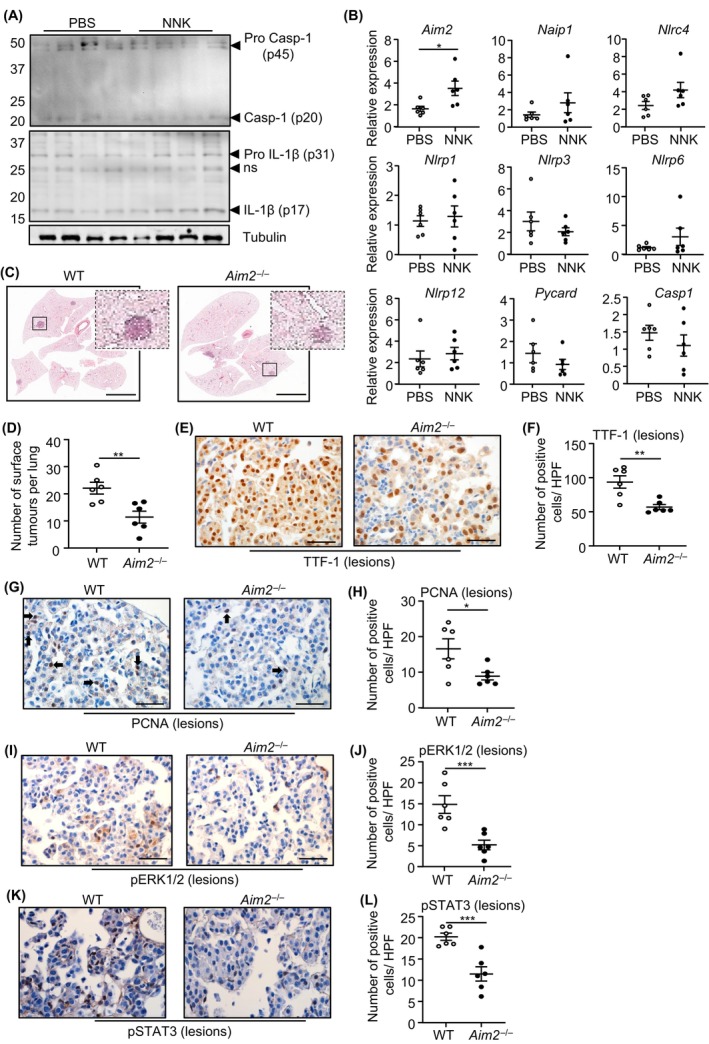
AIM2 promotes NNK tobacco carcinogen‐induced LAC independent of inflammasome activation. (A) Immunoblots with antibodies of individual lung lysates from pseudo‐A/J wild‐type (WT) mice at 20 weeks post NNK or PBS administration. ns, non‐specific. (B) qPCR of inflammasome‐associated genes in lungs from pseudo‐A/J WT mice at 20 weeks post NNK or PBS (n = 6/genotype). *p < 0.05, Student's t‐test. (C) Representative low‐power images of H&E‐stained lung sections from WT and Aim2 −/− pseudo‐A/J mice at 20 weeks post NNK or PBS. Insets depict magnified areas comprising lesions in the low‐power images (open squares). Scale bars: 3 mm. (D) Quantification of surface tumor lesions/whole mouse lung in WT and Aim2 −/− pseudo‐A/J mice at 20 weeks post NNK (n = 6/genotype). **p < 0.01, Student's t‐test. (E, G, I, K) Representative images of (E) TTF‐1, (G) PCNA, (I) pERK1/2 MAPK, and (K) pSTAT3 immunostaining of lung lesions from WT and Aim2 −/− pseudo‐A/J mice at 20 weeks post NNK. Scale bars: 100 μm. (F, H, J, L) Quantification of (F) TTF‐1, (H) PCNA, (J) pERK1/2 MAPK, and (L) pSTAT3 positive cells/high‐power field (HPF) in mouse lung lesions (n = 6/genotype). *p < 0.05, ***p < 0.001, Student's t‐test.
We next investigated whether elevated AIM2 expression also augmented proliferation within the lung epithelium in response to NNK. Indeed, the suppressed NNK‐induced tumor phenotype of Aim2 −/− mouse lungs was accompanied by reduced PCNA‐positive cellular staining compared with WT controls. Also, the lower proliferative potential within the lungs of NNK‐administered Aim2 −/− mice was associated with reduced phosphorylation of ERK1/2 and p38 MAPK, and STAT3 compared with WT counterparts (Figure 6I–L; Figure S5C,D). Collectively, these data revealed that AIM2 independent of inflammasomes cooperates with NNK to promote LAC.
3.6. ASC inflammasome adapter does not contribute to mutant KRAS LAC
We next assessed the LAC phenotype in Kras G12D:Pycard −/− mice in which the inflammasome adapter ASC had been ablated. At 6 and 12 weeks post Ad‐Cre inhalation, the area of lung parenchyma comprising lesions, and the number of lung lesions, in Kras G12D:Pycard −/− mice were comparable with Kras G12D littermates (Figure 7A–C; Figure S6A–E). Similarly, in lung lesions from Kras G12D:Pycard −/− and Kras G12D mice at 6 weeks and/or 12 weeks post Ad‐Cre inhalation, immunohistochemistry revealed comparable numbers of TTF‐1‐positive and PCNA‐positive cells (Figure 7D–G; Figure S6F,G). By contrast, numbers of CD45‐positive pan‐immune cell infiltrates were significantly reduced (by 35%) in lung lesions from Kras G12D:Pycard −/− versus Kras G12D mice (Figure 7H,I). In lung lysates from Ad‐Cre inhaled Kras G12D:Pycard −/− and Kras G12D mice, immunoblotting indicated similar levels of pro and mature Caspase‐1 and IL‐1β, and serum IL‐1β levels were unchanged in both Ad‐Cre‐inhaled genotypes (Figure 7J–L). Collectively, these findings suggested that ASC‐containing inflammasomes do not contribute to Caspase‐1 activity during LAC, nor play a major role in promoting Kras G12D‐induced LAC.
FIGURE 7.
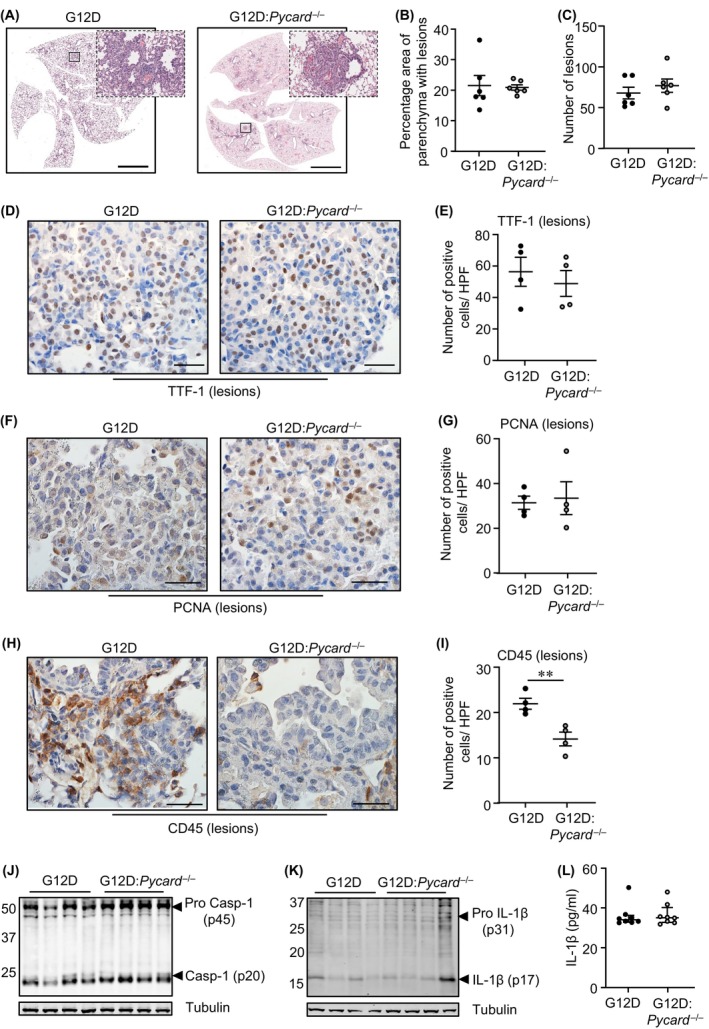
Genetic ablation of ASC in Kras G12D:Pycard −/− mice does not suppress LAC. (A) Representative low‐power images of H&E‐stained lung sections from Kras G12D and Kras G12D:Pycard −/− mice at 6 weeks post Ad‐Cre inhalation. Insets depict magnified areas comprising lesions in the low‐power images (open squares). Scale bars: 5 mm. (B, C) Quantification of (B) lung parenchyma containing tumor lesions, and (C) tumor incidence, per whole mouse lung (n = 6/genotype). (D, F, H) Representative images of lung sections containing lesions from Kras G12D and Kras G12D:Pycard −/− mice at 6 weeks post Ad‐Cre inhalation immunostained with antibodies against (D) TTF‐1, (F) PCNA, and (H) CD45. Scale bars: 100 μm. (E, G, I) Quantification of positive cells/high‐power field (HPF) in lesion‐bearing mouse lungs immunostained with antibodies against (E) TTF‐1, (G) PCNA, and (I) CD45 (n = 4/genotype). **p < 0.01, Student's t‐test. (J, K) Immunoblots of lung lysates from Kras G12D and Kras G12D:Pycard −/− mice at 6 weeks post Ad‐Cre with antibodies against pro and mature (J) Caspase‐1 (p45, p20) and (K) IL‐1β (p31, p17). (L) ELISA for total IL‐1β protein levels in the serum of mice at 6 weeks post inhalations (n = 8/genotype).
4. DISCUSSION
Chronic pulmonary inflammation triggered by dysregulated activation of innate (and adaptive) immune responses plays a key role in promoting lung cancers, including oncogenic KRAS‐addicted NSCLC. 11 , 12 , 13 , 40 However, the identity of innate immune molecular regulators that coordinate these tumor‐promoting cellular processes in the lung remains ill‐defined. Here, we revealed that the innate immune PRR and cytosolic DNA sensor AIM2, expressed in hematopoietic (immune) and non‐hematopoietic (epithelial) cells, promoted mutant KRAS‐driven lung tumorigenesis, independent of inflammasomes, by augmenting cellular proliferation and infiltration of immune/inflammatory cells. An independent role for inflammasomes in KRAS‐mutant LAC was also supported by the genetic ablation of the ASC inflammasome adapter having no effect on suppressing LAC in Kras G12D mice. ASC deficiency in Kras G12D mice did alleviate immune cell infiltration, albeit without any reduction in inflammasome activation (mature Caspase‐1 and IL‐1β levels). This suggests that ASC can induce inflammatory responses in the lung independently of inflammasomes, which may align with the notion that ASC and its formation of oligomeric specks are not always required for Caspase‐1 (inflammasome) activation. 41
Indirect evidence that inflammasomes and associated PRRs may contribute to NSCLC was first suggested by the serendipitous finding that CANTOS trial participants receiving anti‐IL‐1β therapy compared with placebo had a significantly lower occurrence of lung cancer. 16 In support of the notion that therapeutic targeting of the IL‐1β inflammasome effector cytokine may be efficacious in NSCLC, treatment of Kras G12D mice with an anti‐IL‐1β monoclonal antibody suppressed lung tumor burden. 42 Although upregulation of IL‐1β is a feature of NSCLC patients, 43 the mechanistic basis for its overexpression during lung carcinogenesis is unknown. Furthermore, a link between inflammasome activation and expression levels of mature IL‐1β protein in NSCLC has not been reported. Based on our current findings, we propose that inflammasome activation does not contribute to high IL‐1β levels in NSCLC patients. Alternatively, other mechanisms may include transcriptional upregulation of IL1B associated with the inflammatory response in the lung (e.g., driven by NF‐κB activation), which is supported by correlations between IL‐1β production and neutrophil infiltrates in KRAS‐mutant LAC. 42 , 44 , 45 Furthermore, in NSCLC, the processing of mature IL‐1β can be mediated by serine proteases (e.g., cathepsin G) in the lung. 45
The role of AIM2 in cancer is complex, with reports using Aim2 −/− mice coupled to in vivo cancer models indicating that AIM2 possesses opposing pro‐tumor and anti‐tumor properties either dependent or independent of inflammasomes. 21 In experimentally induced hepatocellular carcinoma, AIM2 inflammasome activation in liver‐resident Kupffer cells promotes inflammation‐associated tumorigenesis, whereas in a gastric cancer mouse model (gp130 F/F) AIM2 promotes tumorigenesis independent of inflammasomes by directing cancer cell‐intrinsic migration. 19 , 20 Conversely, AIM2 can protect against intestinal tumorigenesis in a cancer cell‐autonomous manner by impairing survival and proliferation of intestinal epithelial and stem cells, independent of inflammasomes. 22 , 23 Although the mechanisms underlying the pleiotropy of AIM2 in cancer remain ill‐defined, they are most likely to be influenced by differential dependency on numerous factors including the type of inflammasome, cell and tissue, microbial versus host origins of the activating ligand (DNA), and stage of disease. 21 , 22 , 23 In the lung, here we provide evidence from immunofluorescence co‐localization and bone marrow chimeras that AIM2 expression in both alveolar type‐II epithelial and infiltrating immune cells promotes a hyper‐proliferative and chronic inflammatory environment to support tumorigenesis. Interestingly, a recent study demonstrated that AIM2 expression also in alveolar type‐II cells promoted pulmonary emphysema, 46 which is a prominent risk factor for NSCLC. However, unlike in NSCLC, AIM2 disease activity in emphysema is associated with inflammasomes, and independent of its immune cell expression and effect on infiltrating immune cells. 46 It is likely that quantitative and qualitative differences in activating DNA agonists, for instance damaged double‐stranded DNA that is a hallmark of tobacco smoke exposure, 47 as well as the inflammatory environment which can be influenced by oncogenic KRAS in NSCLC, 13 influence AIM2‐mediated disease outcome in the lung. In addition, we propose that upstream mechanisms governing the expression of AIM2 will also influence its disease activity, with our findings here suggesting that p38 MAPK, potentially downstream of activated (mutant) KRAS, contributes to the upregulated expression of AIM2 in lung cancer. In this regard, we observed that AIM2 protein, but not mRNA, expression levels were upregulated in human KRAS‐mutant versus KRAS‐WT NSCLC, which highlights the likely complex mechanism(s) for transcriptional versus translational regulation of AIM2 in human lung cancer, the latter being dependent (at least in part) on KRAS.
Limited numbers of previous studies investigating the role of AIM2 in lung cancer using human immortalized cell lines have yielded contradictory results. In two overlapping studies using KRAS‐mutant NSCLC cell lines A549 and H460, genetic modulation of AIM2 by artificial overexpression or siRNA‐mediated knockdown increased or reduced, cell proliferation and migration, respectively. 24 , 48 The authors reported that these AIM2‐mediated cellular changes coincided with corresponding alterations in levels of mature Caspase‐1 and IL‐1β, which is consistent with AIM2 inflammasome activation. By contrast, another similar study to genetically modify AIM2 expression in human NSCLC cell lines H358 (KRAS‐mutant) and H1975 (KRAS‐WT) reported that AIM2 also promoted cell proliferation and migration, yet independent of inflammasomes. 25 Although an explanation for the contrasting inflammasome‐dependent versus inflammasome‐independent roles of AIM2 was not provided, a cautious interpretation is warranted using immortalized human cell lines in artificial cell culture systems and subcutaneous xenografts in immunocompromised mice. This is especially pertinent when investigating the complex role of immune system regulators such as AIM2 in disease models, whereby the pathological activity of endogenous AIM2 in its native (lung) environment is strongly influenced by its temporal and spatial expression in multiple cell types (epithelial, immune), as well as exposure to endogenous DNA ligands. Importantly, the bona fide preclinical Kras G12D and NNK tobacco carcinogen‐induced NSCLC mouse models in our current study recapitulate key molecular and cellular features, including an inflammatory tumor microenvironment, of human lung cancer, thus enabling elucidation of the AIM2‐driven tumor‐promoting molecular and cellular processes in the native lung. In this respect, our preclinical in vivo models, revealing that AIM2‐driven cellular proliferation of the transformed lung epithelium is associated with activation of the ERK MAPK pathway, validate the cell line‐based report of an inflammasome‐independent role for AIM2 in NSCLC, in which AIM2 knockdown in human NSCLC cell lines suppressed cellular proliferation and strongly inhibited ERK MAPK activation. 25 Interestingly, AIM2 augmentation of ERK MAPK signaling in NSCLC cell lines was linked to dysregulated dynamics of mitochondrial fission and fusion, which in turn upregulated reactive oxygen species production and subsequent ERK MAPK activity, an observation that warrants further interrogation in an in vivo setting.
In summary, our study addresses a significant knowledge gap, and provides much needed clarity, on the in vivo molecular and cellular mechanisms by which AIM2 contributes to the pathogenesis of lung cancer. While our study focused on KRAS‐mutant NSCLC, we note that AIM2 gene expression was also upregulated in other molecular subtypes of NSCLC (e.g., EGFR‐mutant, BRAF‐mutant), suggesting a potentially broader role for AIM2 in potentiating human lung carcinogenesis. Emerging clinical data suggest that KRAS‐mutant NSCLC patients can benefit from immune‐based therapy using adaptive immune checkpoint inhibitors (anti‐PD‐1/PD‐L1), either as monotherapy or in concert with standard‐of‐care chemotherapy. 49 While this sets a precedent for targeting adaptive immunity as a promising treatment modality in KRAS‐mutant lung cancers, the utility of therapeutically targeting components of innate immunity in these cancers is unknown. Therefore, our study raises the enticing prospect that regulators of innate immunity, such as AIM2, that promote the pathogenesis of lung cancers present as new immune‐based therapeutic targets for the future clinical management of KRAS‐mutant NSCLC.
AUTHOR CONTRIBUTIONS
Mohammad Alanazi: Data curation; investigation; methodology. Teresa Weng: Data curation; investigation; methodology. Louise McLeod: Data curation; methodology. Linden J. Gearing: Data curation; formal analysis; software. Julian A. Smith: Resources. Beena Kumar: Formal analysis; resources. Mohamed I. Saad: Data curation; formal analysis; supervision. Brendan J. Jenkins: Conceptualization; formal analysis; funding acquisition; project administration; resources; supervision; writing – original draft; writing – review and editing.
ACKNOWLEDGEMENTS
We thank staff at the Monash Histology Platform – MHTP Node, for their expertise and assistance.
FUNDING INFORMATION
This work was supported in part by the Operational Infrastructure Support Program of the Victorian Government of Australia. M.A. was supported by a University of Tabuk postgraduate student scholarship (Ministry of Education, Kingdom of Saudi Arabia), T.W. by an Australian Government Research Training Program Scholarship, and B.J.J. by a National Health and Medical Research Council of Australia Senior Medical Research Fellowship.
CONFLICT OF INTEREST STATEMENT
The author declares no conflict of interest.
ETHICS STATEMENTS
Approval of the research protocol by an Institutional Reviewer Board: The study was approved by the Monash Health Human Research Ethics Committee (ID 13058A).
Informed Consent: Written informed patient consent was obtained prior to blood and tissue collection from patients.
Registry and the Registration No. of the study/trial: N/A.
Animal Studies: The study was approved by the Hudson Animal Ethics Monash Medical Centre “B” Committee (ID MMCB/2021/29).
Supporting information
Appendix S1.
Alanazi M, Weng T, McLeod L, et al. Cytosolic DNA sensor AIM2 promotes KRAS‐driven lung cancer independent of inflammasomes. Cancer Sci. 2024;115:1834‐1850. doi: 10.1111/cas.16171
Mohammad Alanazi and Teresa Weng contributed equally to this work.
REFERENCES
- 1. Wong MCS, Lao XQ, Ho KF, Goggins WB, Tse SLA. Incidence and mortality of lung cancer: global trends and association with socioeconomic status. Sci Rep. 2017;7(1):14300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Inamura K. Lung cancer: understanding its molecular pathology and the 2015 WHO Classification. Front Oncol. 2017;7:193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ahrendt SA, Decker PA, Alawi EA, et al. Cigarette smoking is strongly associated with mutation of the K‐ras gene in patients with primary adenocarcinoma of the lung. Cancer. 2001;92(6):1525‐1530. [DOI] [PubMed] [Google Scholar]
- 4. Arbour KC, Jordan E, Kim HR, et al. Effects of Co‐occurring genomic alterations on outcomes in patients with KRAS‐mutant non‐small cell lung cancer. Clin Cancer Res. 2018;24(2):334‐340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Meng D, Yuan M, Li X, et al. Prognostic value of K‐RAS mutations in patients with non‐small cell lung cancer: a systematic review with meta‐analysis. Lung Cancer. 2013;81(1):1‐10. [DOI] [PubMed] [Google Scholar]
- 6. Román M, Baraibar I, López I, et al. KRAS oncogene in non‐small cell lung cancer: clinical perspectives on the treatment of an old target. Mol Cancer. 2018;17(1):33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Akopyan G, Bonavida B. Understanding tobacco smoke carcinogen NNK and lung tumorigenesis. Int J Oncol. 2006;29(4):745‐752. [PubMed] [Google Scholar]
- 8. Jackson EL, Willis N, Mercer K, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K‐ras. Genes Dev. 2001;15(24):3243‐3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. DuPage M, Dooley AL, Jacks T. Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc. 2009;4(7):1064‐1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li S, Liu S, Deng J, et al. Assessing therapeutic efficacy of MEK inhibition in a KRAS(G12C)‐driven mouse model of lung cancer. Clin Cancer Res. 2018;24(19):4854‐4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Walser T, Cui X, Yanagawa J, et al. Smoking and lung cancer: the role of inflammation. Proc Am Thorac Soc. 2008;5(8):811‐815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Milette S, Fiset PO, Walsh LA, Spicer JD, Quail DF. The innate immune architecture of lung tumors and its implication in disease progression. J Pathol. 2019;247(5):589‐605. [DOI] [PubMed] [Google Scholar]
- 13. Ji H, Houghton AM, Mariani TJ, et al. K‐ras activation generates an inflammatory response in lung tumors. Oncogene. 2006;25(14):2105‐2112. [DOI] [PubMed] [Google Scholar]
- 14. Brooks GD, McLeod L, Alhayyani S, et al. IL6 trans‐signaling promotes KRAS‐driven lung carcinogenesis. Cancer Res. 2016;76(4):866‐876. [DOI] [PubMed] [Google Scholar]
- 15. Saad MI, Alhayyani S, McLeod L, et al. ADAM17 selectively activates the IL‐6 trans‐signaling/ERK MAPK axis in KRAS‐addicted lung cancer. EMBO Mol Med. 2019;11(4):e9976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wong CC, Baum J, Silvestro A, et al. Inhibition of IL1beta by Canakinumab may Be effective against diverse molecular subtypes of lung cancer: An exploratory analysis of the CANTOS trial. Cancer Res. 2020;80(24):5597‐5605. [DOI] [PubMed] [Google Scholar]
- 17. Malik A, Kanneganti TD. Inflammasome activation and assembly at a glance. J Cell Sci. 2017;130(23):3955‐3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Karki R, Kanneganti TD. Diverging inflammasome signals in tumorigenesis and potential targeting. Nat Rev Cancer. 2019;19(4):197‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Martínez‐Cardona C, Lozano‐Ruiz B, Bachiller V, et al. AIM2 deficiency reduces the development of hepatocellular carcinoma in mice. Int J Cancer. 2018;143(11):2997‐3007. [DOI] [PubMed] [Google Scholar]
- 20. Dawson RE, Deswaerte V, West AC, et al. STAT3‐mediated upregulation of the AIM2 DNA sensor links innate immunity with cell migration to promote epithelial tumourigenesis. Gut. 2022;71(8):1515‐1531. [DOI] [PubMed] [Google Scholar]
- 21. Man SM, Jenkins BJ. Context‐dependent functions of pattern recognition receptors in cancer. Nat Rev Cancer. 2022;22(7):397‐413. [DOI] [PubMed] [Google Scholar]
- 22. Man SM, Zhu Q, Zhu L, et al. Critical role for the DNA sensor AIM2 in stem cell proliferation and cancer. Cell. 2015;162(1):45‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wilson JE, Petrucelli AS, Chen L, et al. Inflammasome‐independent role of AIM2 in suppressing colon tumorigenesis via DNA‐PK and Akt. Nat Med. 2015;21(8):906‐913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang M, Jin C, Yang Y, et al. AIM2 promotes non‐small‐cell lung cancer cell growth through inflammasome‐dependent pathway. J Cell Physiol. 2019;234(11):20161‐20173. [DOI] [PubMed] [Google Scholar]
- 25. Qi M, Dai D, Liu J, et al. AIM2 promotes the development of non‐small cell lung cancer by modulating mitochondrial dynamics. Oncogene. 2020;39(13):2707‐2723. [DOI] [PubMed] [Google Scholar]
- 26. Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8(6):e1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jakobs C, Perner S, Hornung V. AIM2 drives joint inflammation in a self‐DNA triggered model of chronic polyarthritis. PloS One. 2015;10(6):e0131702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ozoren N, Masumoto J, Franchi L, et al. Distinct roles of TLR2 and the adaptor ASC in IL‐1beta/IL‐18 secretion in response to Listeria monocytogenes . J Immunol. 2006;176(7):4337‐4342. [DOI] [PubMed] [Google Scholar]
- 29. Saad MI, McLeod L, Yu L, et al. The ADAM17 protease promotes tobacco smoke carcinogen‐induced lung tumorigenesis. Carcinogenesis. 2020;41(4):527‐538. [DOI] [PubMed] [Google Scholar]
- 30. Miller A, Brooks GD, McLeod L, Ruwanpura S, Jenkins BJ. Differential involvement of gp130 signalling pathways in modulating tobacco carcinogen‐induced lung tumourigenesis. Oncogene. 2015;34(12):1510‐1519. [DOI] [PubMed] [Google Scholar]
- 31. Mounir M, Lucchetta M, Silva TC, et al. New functionalities in the TCGAbiolinks package for the study and integration of cancer data from GDC and GTEx. PLoS Comput Biol. 2019;15(3):e1006701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Grossman RL, Heath AP, Ferretti V, et al. Toward a shared vision for cancer genomic data. N Engl J Med. 2016;375(12):1109‐1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA‐seq data. Genome Biol. 2010;11(3):R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Phipson B, Lee S, Majewski IJ, Alexander WS, Smyth GK. Robust Hyperparameter estimation protects against hypervariable genes and improves power to detect differential expression. Ann Appl Stat. 2016;10(2):946‐963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lun AT, Chen Y, Smyth GK. It's DE‐licious: a recipe for differential expression analyses of RNA‐seq experiments using quasi‐likelihood methods in edgeR. Methods Mol Biol. 2016;1418:391‐416. [DOI] [PubMed] [Google Scholar]
- 37. McCarthy DJ, Smyth GK. Testing significance relative to a fold‐change threshold is a TREAT. Bioinformatics. 2009;25(6):765‐771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gao Q, Liang WW, Foltz SM, et al. Driver fusions and their implications in the development and treatment of human cancers. Cell Rep. 2018;23(1):227‐238.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ma X, Guo P, Qiu Y, et al. Loss of AIM2 expression promotes hepatocarcinoma progression through activation of mTOR‐S6K1 pathway. Oncotarget. 2016;7(24):36185‐36197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lavin Y, Kobayashi S, Leader A, et al. Innate immune landscape in early lung adenocarcinoma by paired single‐cell analyses. Cell. 2017;169(4):750‐765.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nagar A, Rahman T, Harton JA. The ASC speck and NLRP3 inflammasome function are spatially and temporally distinct. Front Immunol. 2021;12:752482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yuan B, Clowers MJ, Velasco WV, et al. Targeting IL‐1beta as an immunopreventive and therapeutic modality for K‐ras‐mutant lung cancer. JCI Insight. 2022;7(11):e157788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Millares L, Barreiro E, Cortes R, et al. Tumor‐associated metabolic and inflammatory responses in early stage non‐small cell lung cancer: local patterns and prognostic significance. Lung Cancer. 2018;122:124‐130. [DOI] [PubMed] [Google Scholar]
- 44. Dimitrakopoulos FD, Kottorou AE, Kalofonou M, Kalofonos HP. The fire within: NF‐κB involvement in non‐small cell lung cancer. Cancer Res. 2020;80(19):4025‐4036. [DOI] [PubMed] [Google Scholar]
- 45. McLoed AG, Sherrill TP, Cheng DS, et al. Neutrophil‐derived IL‐1beta impairs the efficacy of NF‐κB inhibitors against lung cancer. Cell Rep. 2016;16(1):120‐132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ruwanpura SM, McLeod L, Dousha LF, et al. Cross‐talk between IL‐6 trans‐signaling and AIM2 inflammasome/IL‐1beta axes bridge innate immunity and epithelial apoptosis to promote emphysema. Proc Natl Acad Sci USA. 2022;119(36):e2201494119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hecht SS. Tobacco carcinogens, their biomarkers and tobacco‐induced cancer. Nat Rev Cancer. 2003;3(10):733‐744. [DOI] [PubMed] [Google Scholar]
- 48. Yu Q, Zhang M, Ying Q, et al. Decrease of AIM2 mediated by luteolin contributes to non‐small cell lung cancer treatment. Cell Death Dis. 2019;10(3):218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Landre T, Justeau G, Assié JB, et al. Anti‐PD‐(L)1 for KRAS‐mutant advanced non‐small‐cell lung cancers: a meta‐analysis of randomized‐controlled trials. Cancer Immunol Immunother. 2022;71(3):719‐726. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1.