

Abstract
Purpose:
Personalized vaccines targeting multiple neoantigens (nAgs) are a promising strategy for eliciting a diversified antitumor T-cell response to overcome tumor heterogeneity. NOUS-PEV is a vector-based personalized vaccine, expressing 60 nAgs and consists of priming with a nonhuman Great Ape Adenoviral vector (GAd20) followed by boosts with Modified Vaccinia Ankara. Here, we report data of a phase Ib trial of NOUS-PEV in combination with pembrolizumab in treatment-naïve patients with metastatic melanoma (NCT04990479).
Patients and Methods:
The feasibility of this approach was demonstrated by producing, releasing, and administering to 6 patients 11 of 12 vaccines within 8 weeks from biopsy collection to GAd20 administration.
Results:
The regimen was safe, with no treatment-related serious adverse events observed and mild vaccine-related reactions. Vaccine immunogenicity was demonstrated in all evaluable patients receiving the prime/boost regimen, with detection of robust neoantigen-specific immune responses to multiple neoantigens comprising both CD4 and CD8 T cells. Expansion and diversification of vaccine-induced T-cell receptor (TCR) clonotypes was observed in the posttreatment biopsies of patients with clinical response, providing evidence of tumor infiltration by vaccine-induced neoantigen-specific T cells.
Conclusions:
These findings indicate the ability of NOUS-PEV to amplify and broaden the repertoire of tumor-reactive T cells to empower a diverse, potent, and durable antitumor immune response. Finally, a gene signature indicative of the reduced presence of activated T cells together with very poor expression of the antigen-processing machinery genes has been identified in pretreatment biopsies as a potential biomarker of resistance to the treatment.
Translational Relevance.
This study demonstrated the feasibility of a novel personalized neoantigen-based vaccine that in combination with pembrolizumab is safe and able to generate robust tumor-specific T cells capable of trafficking to the tumor. The data presented here support the use of a viral vector-based vaccine platform as a valuable therapeutic option with a potential to be used in a wide range of disease settings and indications, aiming to extend immunotherapy efficacy to patients.
Introduction
The field of immunotherapy has revolutionized the treatment of many tumors with the development and approval of immune checkpoint inhibitors (CPI). The activity of CPI relies mostly on the reinvigoration of T cells against tumor antigens, which are spontaneously induced by the tumor. Tumor neoantigens (nAgs) are considered a central target of such spontaneous antitumor T-cell responses and several studies have shown that cytotoxic CD8 T cells targeting tumor neoantigens play a critical role in mediating the response to checkpoint blockade (1, 2). Neoantigens are mutated self-antigens arising from somatic mutations unique to the tumor cells and not expressed on normal cells. As such, they are recognized by the immune system as nonself and capable of triggering a strong and tumor-specific immune response (3). These features make them an attractive target for cancer vaccines. The advance of next-generation sequencing (NGS) techniques has provided opportunities to rapidly identify these tumor-specific mutations in each individual patient, making the approach of personalized nAgs-based vaccines feasible. Personalized neoantigen vaccines aim to expand and broaden the repertoire of tumor antigen-specific T-cell responses by boosting tumor-induced T cells and by priming de novo T-cell responses against additional cancer nAgs, thus increasing the potency and breadth of antitumor immunity. In this scenario, the combination of neoantigen cancer vaccines with CPI may improve antitumor efficacy through complementary mechanisms of action (4). The choice of the vaccine platform, and associated number of nAgs that can be vaccinated, is key to elicit a proper magnitude, quality, and breadth of T-cell responses to achieve effective and durable antitumor responses. Indeed, various vaccine platform approaches are being pursued in the field of personalized vaccines (PEV; ref. 5). Viral vector-based vaccines represent an attractive choice as antigen delivery system. In particular, heterologous prime/boost with adenoviruses derived from nonhuman Great Apes (GAd), followed by Modified Vaccinia Ankara (MVA), is a powerful vaccine platform that has demonstrated robust and durable T-cell response in humans, both in the context of the infectious disease field and more recently also in cancer (6, 7). Moreover, these viral vectors have the capability to encode large gene inserts and, therefore, in the context of neoantigen vaccines, to target many neoantigens simultaneously, with the potential advantage of covering heterogeneity and curtailing tumor immune escape. Preclinical studies in various murine tumor models have demonstrated the synergy between anti-PD-1 therapy and viral vectored vaccines encoding tumor neoantigens, to eradicate large established tumors (8–10). Recently, vaccination based on GAd and MVA vectors has been also employed to target shared neoantigens in metastatic patients with mismatch repair deficiency/microsatellite instability (dMMR/MSI; ref. 9). More specifically, results from a phase I trial combining anti-PD-1 antibody with GAd20 and MVA encoding shared tumor neoantigens in patients with metastatic colorectal cancer, gastric, and gastro-esophageal MSI tumors have been recently published. The results of the trial demonstrated that the combination treatment was safe, highly immunogenic, and showed promising early signs of clinical efficacy (9).
Here we report translational results from a phase Ib, first in human trial (NCT04990479) in patients with metastatic malignant melanoma. The study evaluated feasibility, safety, immunogenicity, and antitumor activity of NOUS-PEV PEV in combination with the PD-1 blocking antibody pembrolizumab. We examined the induction of neoantigen-specific T-cell responses before and after vaccination, and showed expansion after treatment of vaccine-induced T-cell clonotypes recognizing neoantigens in the tumors of vaccinated patients. Moreover, the analysis of transcriptomic data obtained from pretreatment biopsies allowed the identification of signatures potentially associated with resistance to the combo treatment. Overall, these results support the potential of this personalized viral-based prime/boost vaccine as a promising immunotherapeutic approach to enhance antitumor responses and the use of biomarkers to inform on the responsiveness to combined vaccine and anti-PD-1 treatment.
Patients and Methods
Study design
This is a multicenter, open-label, phase I clinical study (NCT04990479), designed to evaluate safety, tolerability, and immunogenicity, and to detect any preliminary evidence of antitumor activity of a PEV based on GAd-PEV priming and MVA-PEV boosting, combined with standard of care (SoC) first-line immunotherapy using an anti-PD-1 checkpoint inhibitor in metastatic non–small cell lung cancer (NSCLC) and unresectable or metastatic melanoma. The representativeness of the study population is described in Supplementary Table S1. The PEV vaccines are prepared on an individual basis, following a tumor biopsy performed at the time of screening and subsequent NGS analysis, to identify patient-specific tumor mutations. Both neoantigen-encoding genetic vaccines are administered intramuscularly using one prime with GAd-PEV and three boosts with MVA–PEV in combination with the licensed programmed death receptor-1 (PD-1)-blocking antibody pembrolizumab in patients. The first vaccination with GAd occurs on the day of the 4th administration of pembrolizumab (week 10); MVA is administered as three boosts, every 3 weeks, with the first administration 3 weeks post-GAd vaccination. GAd and MVA vaccines are administered at the dose ranges of 5 × 1010 to 2 × 1011 vp and 1 × 108 to 3 × 108 ifu, respectively. Further details can be found at https://clinicaltrials.gov/study/NCT04990479. This study was conducted in accordance with ethical principles that have their origin in the Declaration of Helsinki and conducted in adherence to the study Protocol, Good Clinical Practices as defined in Title 21 of the US Code of Federal Regulations Parts 11, 50, 54, 56, and 312, as well as the International Conference on Harmonization E6 guideline for Good Clinical Practice and applicable regulatory requirements. The study was approved by UZ Leuven Ethics Committee (Study Reference S64578) and by Research Ethics Committee of Fundacion Jimenez Diaz. Written informed consent was obtained from each subject.
Vaccine vector generation
PEV design was performed according to a pipeline aimed at analyzing patient next-generation sequencing data collected from tumor and blood to select the best neopeptides suitable for the inclusion in viral vectors. NGS data were produced at CeGAT GBMH. Genomic DNA was fragmented and used for Illumina library construction. Exonic regions were captured in solution using the Twist Human Core Kit. Paired-end sequencing (2 × 100 bp) was performed with the Novaseq600 Genome Analyser (Illumina) at a target depth of 30 Gb. RNA was fragmented and the sequencing library was prepared using SMART-Seq mRNA Stranded Kit. Sequencing was performed with the Novaseq6000 Genome Analyser (Illumina) at a target depth of 6 Gb (2 × 100 bp). The patient HLA typing and the prioritization of somatic variants encoding neopeptides were performed as described previously (11).
The transgene design was performed according to a procedure that minimizes the formation of predicted junctional epitopes that may be generated by the juxtaposition of two adjacent neoantigen peptides. A custom tool was developed specifically for this purpose. The synthetic cassette aminoacid sequence was finally retrotranscribed by applying an inverse translation scheme with a minimization of penalty score associated to the presence in the sequence of forward/reverse repeats or 5TNT motifs and homopolymers stretches greater than 6bp.
Synthetic transgenes expression cassettes were generated via Gibson assembly (New England Biolabs) of double-strand DNA fragments obtained from different providers (Integrated DNA Technologies, GeneART, Thermo Fisher Scientific; Doulix; Officinae Bio; Eurofins Genomics); fragments were assembled into acceptor shuttle plasmids containing TetO-CMV promoter (with two Tet Operator repeats) and BGH polyA (GAd-PEV transgenes) or P7.5 promoter (MVA-PEV transgenes).
For generation of GAd-PEV vectors, transgene cassettes were transferred into a GAd20 BAC construct by recombineering in SW102 Escherichia coli cells. GAd20 BAC contains the genome of a Great Ape Adenovirus (serotype group C) deleted in E1 and E3 regions, whereas the E4 region is replaced with Ad5 E4 ORF6. Positive clones, selected and characterized by PCR, restriction digestion, and transgene DNA sequencing, were used for vector rescue by transfection in M9 cells [human embryonic kidney (HEK) 293 derivative]. Resulting GAd-PEV vectors were amplified in suspension M9 cells and purified by anion exchange chromatography (9). The GMP production was conducted by Reithera Sri.
For generation of MVA-PEV vectors, transgene cassettes were transferred into MVA Deletion III locus by homologous recombination in AGE1.CR.pIX cells. Recombinant vectors were isolated by a combination of marker gene swapping and florescence-activated cell sorting as described previously (12). A limiting dilution step was performed to ensure clonality of the obtained MVA-PEV vectors, followed by amplification in AGE1.CR.pIX cells. The GMP production was conducted by Reithera Srl.
Peripheral blood mononuclear cell preparation
Venous blood samples from patients were collected in lithium-heparin Vacutest blood collection tubes (Kima) at different time points and cryopreserved at the clinical sites before shipment to the central lab for immunogenicity assessment. To preserve peripheral blood mononuclear cell (PBMC) functionality, isolation and freezing procedures were performed within a maximum of 8 hours from blood collection. The isolation of PBMCs was performed using Leucosep Bio-One Polypropylene Tube (prefilled; Greiner, Merck) according to the instructions of the manufacturer. After that, PBMCs were counted and frozen with freezing media [10% DMSO (Sigma- Aldrich) and 90% FBS (Defined, HyClone], then placed at −80°C for 1 to 3 days before shipment to the central laboratory. Cryopreserved cells were thawed in thawing media [RPMI1640 (Gibco), CTL wash supplement (Immunospot), 1% L-glutamine (Gibco), and 50 U/μL of Benzonase (Merck Millipore)]. Cell counts and viability were assessed using the Guava Via Count reagents (Luminex) and module on the Guava Muse Cell Analyzer (Luminex).
Ex vivo IFNγ ELISpot
The frequency of IFNγ producing T cells was measured by ex vivo ELISpot-forming cell assay after antigen-specific stimulation, as previously described (9). PBMCs were resuspended in R10, stimulated with a set of peptides designed to cover the NOUS-PEV patient specific vaccine sequence, and arranged into 6 peptide pools (P1 to P6). Cells were plated at 2 × 105 cells per well in ELISpot plates (Human IFN-γ ELISpot PLUS kit, Mabtech) and incubated for 18 to 20 hours in the presence of the peptides in a 37°C, humidified CO2 incubator. At the end of incubation, the ELISpot assay was developed according to the manufacturer's instructions. Spontaneous cytokine production (background) was assessed by incubating PBMCs with the medium only plus the peptide diluent DMSO (negative control) (Sigma-Aldrich), whereas CEFX (JPT), a pool of known peptide epitopes for a range of human leukocyte antigen (HLA) subtypes and different infectious agents, was used as positive control. Results are expressed as SFC/106 PBMCs in stimulating cultures after subtracting the DMSO (Sigma-Aldrich) background. A response was considered positive if (i) the number of SFC/106 cells was greater than 50 and (ii) higher than three times the background DMSO value. A subject is defined as a responder if reactivity to at least 1 of the peptide pools is induced after vaccination.
In vitro expansion of neoantigen-specific T cells
For in vitro expansion of antigen-specific T cells, PBMCs were cultured in RPMI1640 medium (Gibco) supplemented with L-glutamine (Gibco), HEPES (Gibco), penicillin/streptomycin (Gibco), and 10% heat-inactivated human serum (Defined, HyClone), as described previously (9). Cells (4 × 106 per well) in 1-ml volume were stimulated in a 24-well plate with individual (4 μg/mL) or peptide pool (each peptide at 4 μg/mL) in the presence of IL7 (330 U/mL; PeproTech). On day 3, low-dose IL2 (10 U/mL; PeproTech) was added. Half-medium change and supplementation of IL2 were performed at days 3, 7, and 10. After 12 days, PBMCs were collected and resuspended in complete medium without cytokines and peptides for overnight resting at 37°C. The day after, T-cell response against peptides of interest was tested by IFNγ ELISpot assay.
TCR-seq analysis
T-cell receptor (TCR) Vβ sequencing (TCR-seq) was performed on blood at baseline and post-vaccination, and on IVS expanded T cells. DNA from bulk PBMCs or stimulated specific T cells was prepared using a Qiagen DNA Blood and Tissue Extraction Kit according to the manufacturer's instructions. Samples were quantified using Nanodrop 2000 (Thermo Fisher Scientific). DNA was extracted from each collected sample and sequenced at CeGaT GMBH. Raw NGS data were analyzed with MiXCR 2.1.11 (13), applying default parameters. The repertoire detected in tumor at baseline and after vaccination was determined was assessed from the RNA-seq data using the MiXCR tool and applying the standard parameters reported in the RNA-seq workflow of the manual. The derived CDR3 sequences were further analyzed by tracking the expression of clonotypes that were shared between pre-/post-vaccination tumor biopsies and in vitro samples stimulated with NeoAg7 minimal 9-mer mutated peptide.
Gene expression analysis
Differentially expressed genes were estimated by comparing treated versus untreated with a consensus of four different methods: DESeq2 (RRID:SCR_015687; ref. 14), edgeR (RRID:SCR_012802; ref. 15), limma with Voom correction (16), and NOISeq (17). A count matrix reporting the number of reads mapping to each gene was determined by using the Rsubread (PMID: 30783653) package and gene expression was expressed as transcripts per kilobase million (TPM). The Benjamini–Hochberg correction was applied to the list of differentially expressed genes identified by each method. Only genes identified by three out of the four methods, with a difference of log2FC of at least ±1 and a corrected P value ≤0.05 were retained. IFNG/IMS signature was computed as described in Cui and colleagues (18); R software was used to implement the signature computation script.
Clonality analysis
Analysis of clonality of detected mutations was performed by using allele-specific copy-number analysis of tumors (ASCAT) software (19) to detect copy-number variation in tumor exome sequencing data, followed by PyClone (20) to estimate the clonality of each somatic mutation, as described previously (9). All the parameters were kept to default except for the input of ASCAT that was created with a custom tool that filters only the SNPs with at least 30% of mutation allele frequency in blood. The list of filtered SNPs was then passed to the AlleleCounter tool to prepare the input for ASCAT.
Data availability
Exome and RNA-sequencing (RNA-seq) data generated in this study have been deposited in Sequence Read Archive under the accession code PRJNA1081187. All the other data of this study are available in the article or upon request from the corresponding author.
Results
Production of GAd and MVA for personalized vaccination is feasible in a short turnaround time
This study was open to treatment-naïve subjects with unresectable stage III/IV cutaneous melanoma and/or PD ligand 1 (PD-L1) ≥50% stage IV non–small cell lung cancer. The study endpoints included the evaluation of safety as the primary endpoint and evaluation of tumor response according to RECIST v1.1 as secondary endpoint. Immunogenicity and additional exploratory pharmacodynamics measures and correlates of vaccine-induced immune response were evaluated as exploratory endpoints. As of the data cutoff of June 12, 2023, 7 patients with unresectable stage III/IV cutaneous melanoma were enrolled in the trial; 6 received the vaccination, whereas 1 patient (Pt 7) progressed prior to any vaccination (demographics and disease characteristics for each patient are shown in Table 1). Following pembrolizumab induction treatment (three cycles), NOUS-PEV vaccine was administered intramuscularly with the following scheme: a single priming dose of GAd20 encoding 60 tumor-specific nAgs concomitantly with the fourth pembrolizumab infusion at week 10, followed by 3 boosts with MVA, each 3 weeks apart. Baseline and on treatment tumor biopsies were collected at screening and after the first MVA administration, respectively (Fig. 1A).
Table 1.
Demographic and disease features of the NOUS-PEV study population.
| Patient ID | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| Age | 64 | 72 | 77 | 88 | 74 | 62 | 72 |
| Sex | Male | Male | Male | Female | Female | Male | Male |
| ECOG | Grade 0 | Grade 0 | Grade 1 | Grade 1 | Grade 1 | Grade 1 | Grade 0 |
| BRAF mutation | Negative | Positive | Positive | Negative | Not applicable | Negative | |
| Disease status | Stage III | Stage IV | Stage III | Stage III | Stage III | Stage III | Stage IV |
| BOR | PR | PR | PD | SD | PR | CR | PD |
| Sum of RECIST measurements at baseline | 12 mm | 100 mm | 26 mm | 10 mm | 31 mm | 46 mm | 81 mm |
| No. of target lesions | 1 | 3 | 1 | 1 | 1 | 2 | 4 |
| No. of nontarget lesions | 1 | 2 | 1 | 1 | 1 | 3 | 2 |
Abbreviation: ECOG, Eastern Cooperative Oncology Group.
Figure 1.

NOUS-PEV neoantigen vaccines: trial design, feasibility, and selection of encoded neoantigens. A, Schematic outlining the NOUS-PEV trial design, vaccine production, and treatment schedule. B, Achieved (triangles) and expected (green dotted line) turnaround time (TAT) to manufacture and release GAd20 and MVA-PEV. The TAT is calculated in days, from when the biopsy is available for the nucleic acid extraction till the day the vaccine is released. C, Total number of neopeptides detected in NOUS-PEV patients. Each bar represents the total number of neopeptides encoded by nonsynonymous somatic mutations detected in baseline tumor biopsies. Black and gray bars indicate the patients’ mutation subjected or not to VENUS prioritization algorithm, respectively. D, Quality of neopeptides included in NOUS-PEV. The pie chart displays the total number of candidate neopeptides targeted by NOUS-PEV. In green, blue, and salmon are indicated respectively, the selected neopeptides showing one, two, or three of the parameters defined “good” according to the following thresholds (TPM of the RNA carrying the mutation ≥1; MHC class I predicted binding IC50 ≤ 500 nmol/L; mutation allele frequency >50%). In gray are the other neopeptides not included in the previous three categories. The bar plot indicates the detail of candidate neopeptides for each individual patient.
To produce each personalized neoantigen GAd20 and MVA vaccine, tumor mutations were first identified by whole-exome and RNA-sequencing of baseline patient's tumor biopsy and matched normal cells from blood, as outlined in Fig. 1A. Neoantigens were then bioinformatically predicted and selected using a previously published algorithm, called VENUS (11). About 60 tumor specific neopeptides were selected for inclusion in a codon-optimized artificial gene cloned into GAd20 and MVA vectors. Eleven of 12 vaccines were successfully manufactured and released, according to GMP and EU Pharmacopoeia guidelines, demonstrating the feasibility of this viral vector-based personalized vaccination approach. More specifically, the turnaround time (TAT), defined as the time for vaccine generation and release starting from the day of the tumor biopsy analysis, was an average of 59 days (SD ±4) for GAd20 (expected 56 days), and 81 days (SD ±3) for MVA (expected 81 days; Fig. 1B).
The total number of nAgs for the analyzed tumor biopsies is shown in Fig. 1C. For 1 patient (Pt 6), the tumor biopsy displayed a total number of 34 neoantigens. Therefore, in this case, the 34 neoantigens were all encoded in the NOUS-PEV vaccine and not subject to the VENUS selection (11), as the cumulative length of the sequence corresponding to the candidate neoantigens was lower than the maximum capability of the gene expression cassette. For the remaining patients, the best subset of neoantigens were all selected by VENUS, which ranked the total detected nAgs and selected the top ranked mutations up to the maximum capability allowed by our viral vectors (∼60 25-mer neoantigens; ref. 8). An overall number of 398 of 1591 identified nAgs were selected by the prioritization algorithm. The selected nAgs were retrospectively classified according to arbitrary defined thresholds for the three parameters used by VENUS for neoAgs selection: (i) the likelihood of presentation by MHC-I; (ii) the expression and (iii) the allele frequency of the somatic mutations encoding nAgs as detailed in Fig. 1D. The nAgs present in the vaccines were enriched for having high scores in these three parameters, with 92% of the selected nAgs (365 of 398) displaying at least one “good” parameter; 52% had at least two out of three “good” parameters (Fig. 1D).
NOUS-PEV is safe and induces T-cell responses against neoantigens in all evaluable patients
The vaccination was safe and well tolerated. No treatment-related serious adverse events (SAE) or Grade 3 and 4 (CTCAE) AEs occurred. AEs related to NOUS-PEV (attributed by investigator to NOUS -PEV alone or NOUS-PEV and pembrolizumab) were reported in four (66.7%) patients, all of which were Grade 1 or 2. The most common AEs related to NOUS-PEV were localized injection site reactions (50%, n = 3; Table 2).
Table 2.
Treatment-Related AEs (TRAE) in patients treated with NOUS-PEV and pembrolizumab.
| Grade 1/2 | |
|---|---|
| Preferred term | n (%) |
| NOUS-PEV treatment-related AEsa | |
| Injection site reaction | 3 (50) |
| Fatigue | 1 (17) |
| Headache | 1 (17) |
| Hyperthyroidism | 1 (17) |
| Influenza-like illness | 1 (17) |
| Injection site erythema | 1 (17) |
| Injection site hypersensitivity | 1 (17) |
| Musculoskeletal stiffness | 1 (17) |
| Pruritus | 1 (17) |
| Pembrolizumab treatment-related AEsb | |
| Pruritus | 4 (67) |
| Diarrhea | 3 (50) |
| Fatigue | 3 (50) |
| Dyspnea | 2 (33) |
| Hyperthyroidism | 2 (33) |
| Hypothyroidism | 2 (33) |
| Injection site reaction | 2 (33) |
| Lipase increased | 2 (33) |
| Blood iron decreased | 1 (17) |
| Cough | 1 (17) |
| Dermatitis acneiform | 1 (17) |
| Dry mouth | 1 (17) |
| Dysgeusia | 1 (17) |
| Memory impairment | 1 (17) |
| Myalgia | 1 (17) |
| Nausea | 1 (17) |
| Papule | 1 (17) |
| Photopsia | 1 (17) |
| Rhabdomyolysis | 1 (17) |
| Vitiligo | 1 (17) |
Note: Summary of number (n) and percentage (%) of treatment-related AEs (TRAE). AEs were coded using MedDRA version 23.1. No patients had TRAE with CTCAE grade 3 or higher. Two patients had CTCAE grade 3/4 SAEs (1 patient had vertigo and 1 patient had hydronephrosis) all considered to be unrelated to the treatment.
aNOUS-PEV–related AEs include events attributed by the investigator to NOUS-PEV alone as well as events attributed to both NOUS-PEV and pembrolizumab.
bAEs related to pembrolizumab include events attributed by the investigator to pembrolizumab alone and events attributed to both NOUS-PEV and pembrolizumab.
Of the 6 patients who received vaccination and pembrolizumab, the best overall responses (BOR) observed were one complete response (CR), three partial responses (PR), one stable disease (SD), and one disease progression (PD; Table 1).
Vaccine immunogenicity was an exploratory endpoint of the study and was evaluated by an IFNγ ex vivo enzyme-linked immunosorbent spot (ELISpot) assay using 6 patient-specific peptide pools (∼10 peptides per pool) covering the entire vaccine sequence (Fig. 2A). For the assessment of immune response, peripheral blood mononuclear cells (PBMC) were collected at different time points before and after vaccination. Stimulation of PBMCs with patient-specific peptide pools resulted in IFNγ T-cell responses post-vaccination in 100% of evaluable patients who had received the prime/boost regimen (n = 4; Fig. 2B). Interestingly, immune response was not detected in Pt 5, who did not receive GAd20 but only MVA. None of the patients had a positive ex vivo IFNγ ELISpot response to their cognate neoantigen peptide pools after pembrolizumab, prevaccination administration (Fig. 2B). Total response at peak after vaccination reached a mean of ∼700 spot-forming cells (SFC)/million PBMCs. Responses were directed against different peptide pools, with the induction of a polytope response to the vaccine neoantigens (Fig. 2C). Antigen-specific expansion of T cells after in vitro stimulation (IVS) resulted in additional positive responses by PBMC samples collected after vaccination against pools with undetectable ex vivo response (Supplementary Fig. S1). Depletion of CD8 cells from PBMC showed that vaccine induced T-cell responses included both CD4 and CD8 T cells (Fig. 2D). Assessment of individual CD4 and CD8 T-cell specificity against each peptide was not feasible for all patients and for all the peptide pools, given the considerable number of encoded neoantigens coupled to the limitation in sample availability. Vaccine-induced T-cell responses were long lasting, as we found that cellular immunity was still detected 7 months after vaccination (Fig. 2E).
Figure 2.

NOUS-PEV elicits a potent neoantigen T-cell response. A, Schematic overview of ex vivo IFNγ ELISpot assay to measure immune response on PBMCs stimulated with 6 patient-specific peptide pools P1 to P6 (∼10 peptides per pool) covering the entire vaccine sequence. B, T-cell response measured in NOUS-PEV–vaccinated patients by ex vivo IFNγ ELISpot on PBMCs collected pre- and post-vaccination. Numbers of IFNγ spot-forming cells (SFC) per 106 PBMCs are shown for 4 patients receiving the full regimen GAd and MVA prime/boost (solid dots), and 1 receiving MVA-PEV (empty dot), comparing the baseline (post-pembrolizumab) responses versus the post-vaccination response at peak. Two-tailed Mann–Whitney test was performed (*, P < 0.05). C, T-cell response measured in NOUS-PEV evaluable patients receiving the full regimen GAd/MVA (Pt 1, 2, 4, 6). Shown are the ex vivo immune responses measured post-pembrolizumab versus the post-vaccine immune responses against the peptide pools (P1–P6) covering the entire vaccine sequence for 4 patients (Pt 1, 2, 4, 6). Graphs show mean SFC ± SEM per 106 PBMCs for triplicate ELISpot wells. Pools showing positive after ex vivo and in vitro restimulation (IVS) cultures are indicated with a “+” symbol. Pie charts on the top represent the frequency of peptide pools inducing CD8 and CD4 responses on the total pools eliciting a positive response by ex vivo/IVS ELISpot for each patient (NA, not available). D,Ex vivo IFNγ ELISpot on PBMCs before or after depletion of CD8+ T cells in the presence of patient-specific neopeptide pools. “CD4” and “CD8” indicate the subtype-specific CD4 and CD8 T-cell responses. An unpaired t test was used to detect significant differences between the groups (pre- and post-CD8 depletion); ****, P < 0.0001; **, P < 0.01; *, P < 0.05. E, Representative wells from IFNγ ELISpot assay of PBMCs analyzed 7 months post-vaccination. The dimethylsulfoxide (DMSO) wells represent the negative control, whereas a pool of viral peptides (CEFX) was used as positive control.
TCR neoantigen-specific T-cell repertoire is expanded and diversified after NOUS-PEV vaccination
Clonal dynamics of antitumor TCR clonotypes before and after treatment were next investigated. More specifically, the intratumoral TCR repertoire from bulk RNA-seq in matched pre- and posttreatment tumors was analyzed in 4 patients whose pre- and posttreatment biopsies were available (Pt 1, Pt 2, Pt 4, and Pt 5). Three out of 4 of these patients (Pt 1, Pt 2, and Pt 4) received the vaccine prime and boost regimen, whereas Pt 5 only received MVA and did not mount a detectable positive immune response in periphery post-vaccination (Fig. 2B). Analyses of intratumoral lymphocyte populations showed an increase of T cells measured as number of TCRβ copies of nearly three-fold on average in posttreatment biopsies versus baseline in 3 patients who received prime and boost NOUS-PEV vaccine (Fig. 3A and B). Interestingly, the intratumoral TCR repertoire of Pt 5, in the absence of full regimen vaccination, did not show expansion of T-cell repertoire as observed for the other 3 patients, suggesting that full regimen heterologous prime boost vaccination is required to achieve T-cell expansion, in keeping with the lack of T-cell response measured in the periphery. Consistent with the increase of the number of T-cell clones after NOUS-PEV, a more diverse TCR repertoire was also found in the three prime and boost vaccinated patients, suggesting amplification and diversification of antitumor T-cell responses posttreatment (Fig. 3C).
Figure 3.

Increase of intratumoral T cells post-NOUS-PEV treatment with vaccine-induced T cells infiltrating tumor. A, Expansion and diversification of TCRβ repertoire in pre- and posttreatment tumor biopsies in four NOUS-PEV patients (3 PR;1 SD). Each bar indicates the abundance of individual TCRβ clones detected in the total tumor RNAs extracted from the biopsies collected at the two time points (details in Patients and Methods). B, Total number of TCRβ counts detected in tumor biopsies estimated by summing up the abundance of the individual clones reported in A. The solid lines represent NOUS-PEV patients receiving the full vaccine regimen GAd/MVA; the dashed line represents the patient (Pt 5) who only received MVA-PEV. C, Number of individual clones with different TCRβ CDR3 detected in the patients’ tumor biopsies (*, P ≤ 0.05, paired t test).
To investigate the migration of individual neoantigen-specific vaccine-induced CD8 T cells from the peripheral blood to the tumor, we focused on Pt 1, who showed a strong CD8 T-cell response detected ex vivo with PBMC samples post-vaccination. In this patient, elicitation and increase of vaccine-induced neoantigen T cells over time coincided with the deepening of the clinical response (Fig. 4A). Deconvolution of pool 1 in this patient revealed that the immune response was directed against one strong CD8 neopeptide, mutated NOL9 (named NeoAg7). NeoAg7 mutation was found in about 70% of tumor cells according to the mutation clonality analysis in the baseline biopsy, with a good predicted binding of the nonamer sequence RDLSIFSYL to the patient haplotypes HLA-B*37:01 (Fig. 4B). In vitro expansion of PBMCs collected after pembrolizumab (week 10) and vaccine treatment (week 14), in the presence of the NeoAg7 peptide, led to the expansion of NeoAg7-specific CD8 T cells as shown by the higher number of IFNγ+ SFCs (Fig. 4C). Unstimulated (DMSO only) cells from the same cultures served as negative control. Expanded NeoAg7-specific CD8 T cells were then subjected to TCRβ sequencing and compared with unexpanded T cells from the same cell culture conditions (week 14 PBMC). Neoantigen 7-specific TCR clonotypes were then tracked in the patient tumor biopsies (baseline and on-treatment). Six NeoAg7 reactive TCRβ clones expanded after IVS were detected in the tumor biopsies by bulk RNA analysis, with five clones exclusively detected in the posttreatment biopsy (Fig. 4D).
Figure 4.

Vaccine-induced neoantigen T cells migrate into the tumor. A, T-cell response assessed by ex vivo ELISpot versus percentage changes in target lesion size (sum of target lesion measurements evaluated per RECIST v1.1.) from baseline are displayed for NOUS-PEV Pt 1. B, Deconvolution of T-cell response by ex vivo ELISpot against individual peptides of the immunogenic pool 1. The immunogenic NeoAg7 is shown in black. The unpaired t test was used to make comparisons (****, P < 0.0001). C, Top, schematic representation of the IVS protocol to expand vaccine induced neoantigen specific T cells against NeoAg7 peptide. Bottom, T-cell responses in Pt 1 measured by IFNγ ELISpot assay after IVS with NeoAg7 peptide. Tested PBMCs were collected after pembrolizumab (week 10) and after vaccination (week 14). The unpaired t test was used to make comparisons (***, P < 0.001). D, Expansion and diversification of TCRβ repertoire in pre- and posttreatment tumor biopsies of Pt 1. Each bar is a TCRβ individual clone; the clonotypes specific for NeoAg7 detected on the tumor biopsies of Pt 1 are shown as black bars.
Together, these results indicate that neoantigen-specific CD8 T cells induced by NOUS-PEV vaccine expand and diversify upon treatment. Moreover, the presence of clonotypes from neoantigen-specific CD8 T cells in tumor tissue that were also detected in PBMCs provides proof-of-concept that vaccine-induced neoantigen-specific T cells in the periphery are able to traffic and infiltrate into the tumor.
Evaluation of biomarkers predictive for clinical response to NOUS-PEV and anti-PD-1 combination treatment
Transcriptomic analysis of baseline biopsies from patients treated with CPI allows the identification of potential mechanisms underlying tumor response and resistance (21). Recently, Cui and colleagues have shown that the ratio of IFNγ gene signature to an immunosuppression signature (IMS) is a good predictor of the clinical response to CPI immunotherapy in patients with metastatic melanoma (18). This ratio was evaluated on a public dataset of tumor RNA expression data in patients with melanoma treated with CPI in parallel with our NOUS-PEV patient cohort. In this analysis, patients of the benchmark cohort were divided into three categories: responding patients (R; including CR and PR); patients with stable disease (SD); and patients in progression (PD). We noticed that none of the patients with a clinical response in the public cohort had an IFNγ/IMS ratio lower than −1, different from the PD or SD patients (Fig. 5A). NOUS-PEV patients were thus arbitrarily separated into two subgroups, one with a ratio IFN-γ/IMS below −1 (Pt 1, 3, 4, and 7) and the second group with a ratio higher than −1 (Pt 2, 5, 6; Fig. 5A). According to this analysis, patients 2, 5, and 6 fall under the group of predicted good responders to the pembrolizumab monotherapy, whereas patients 1, 3, 4, and 7 were predicted as subjects very unlikely to respond to pembrolizumab. Indeed, Pt 3 and Pt 7 rapidly progressed with Pt 7 progressing even before receiving the vaccine. Instead, Pt 1 showed an increase of tumor volume before vaccination at the first CT scan but then a deep and durable response after vaccination. We then investigated additional signatures aiming to identify factors that could characterize and diversify Pt 1 in clinical response from the others with PD.
Figure 5.
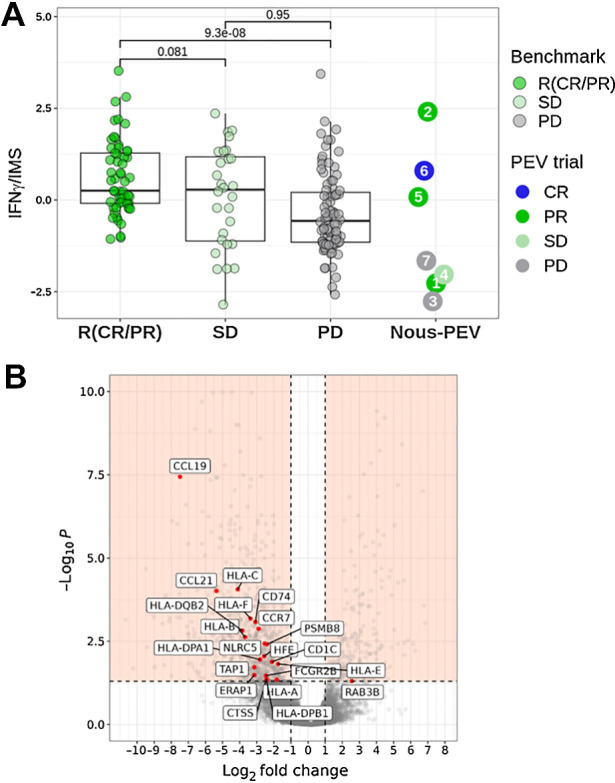
Analysis of potential biomarkers predictive for antitumor response. A, Ratio of the IFNγ signature score to the IMS score predictive of PD-1 blockade (18) estimated according to RNASEQ gene expression values detected in pretreatment tumor biopsies. The values of the signature estimated in NOUS-PEV patients were compared with a dataset of 172 patients with melanoma (57 CR/PR; 28 SD; 87 PD) treated with anti-PD-1 monotherapy and retrieved from four published studies (18, 31–33). B, Volcano plot of differentially expressed genes detected by analyzing the RNASEQ of pretreatment biopsies of patients in progression (PD; n = 2) versus the ones collected from responder patients (n = 3; median log2 FC < −1 or >1; Benjamini–Hochberg corrected P value <0.05 according to a consensus of four different methods. Details in Patients and Methods). The plot highlights in red a subset of 21 antigen present machinery-related genes (APM) downregulated in NOUS-PEV PD patients.
Loss/downregulation of MHC-I expression, as well as other key components of the MHC-I antigen processing machinery (APM), is a frequent immune evasion mechanism exploited by many cancers to escape CTL-mediated tumor cell killing. As low APM also hampers the outcome of CPI and other immunotherapies including vaccines, we investigated the expression levels of key APM genes in NOUS-PEV responders (R; Pt 1, Pt 2, Pt 6) and PD patients (Pt 3 and Pt 7). The SD patient, being only one at the moment, was excluded from this analysis. Volcano plot depiction of differentially expressed genes between NOUS-PEV PD versus R patients highlighted a significant downregulation in the set of APM genes, such as NLRC5, TAP1, HLA PSMB8 genes, in the PD group (Fig. 5B).
Discussion
Tumor neoantigens represent the optimal target for the generation and amplification of immune responses by therapeutic vaccination combined with CPI treatment. In this manuscript, we report the feasibility and immunogenicity of NOUS-PEV assessed in a phase Ib study in patients with metastatic melanoma. NOUS-PEV is an individualized neoantigen vaccine based on heterologous GAd20/MVA prime boost, in combination with pembrolizumab. We demonstrated that this personalized vaccine approach relying on two different viral vectors is feasible, given the fact that 90% of the NOUS-PEV products were successfully manufactured. Moreover, in all cases, vaccines were administered to the patients according to the predefined timeline with a median vaccine release of about 8 weeks from biopsy. Manufacturing speed is particularly relevant in a setting of metastatic disease but also in curatively resected disease to avoid unnecessary treatment delay and maximize the opportunity for clinical benefit. To date, depending on the choice of vaccine platform, the manufacturing time reported in the field typically ranges from 8 to 16 weeks (22), positioning this approach among the more rapidly manufactured ones so far. Combination of NOUS-PEV with pembrolizumab was safe, with no treatment-related SAEs reported for any of the treated patients, and with only mild reactions to the vaccines, consistent with the results of other studies of heterologous prime/boost vaccine schedules incorporating GAd and MVA (7, 23). The vaccine approach presented here allows targeting an unprecedented number of neoantigens, with a strategy of polyvalent vectors encoding 60 patient-specific neoantigens. Indeed, antigen loss and tumor heterogeneity represent important challenges to the effectiveness and durability of the antitumor response and several evidences indicate that the targeting of multiple neoantigens may enhance the rate of efficacy (8).
High-magnitude vaccine-induced immune responses detected by ex vivo IFNγ ELISpot were elicited in all evaluable patients post-NOUS-PEV administration, with polytope responses measured against multiple peptides. These results are in line with those obtained in a different clinical trial using the same platform targeting frameshift (FSP) neoantigens shared across patients with dMMR and eliciting a potent and broad T-cell response in the vast majority of patients (9). Immune responses were both CD8 and CD4 T-cell responses, confirming the capability of this platform to induce both types of T lymphocytes, as also observed in preclinical models (8, 11). One patient (Pt 5) who only received MVA instead of the full vaccine regimen, did not mount T-cell responses against vaccine encoded neoantigens, indicating that the combined GAd/MVA approach is required to mount proper T-cell immunity. Of particular interest from the clinical point of view is patient 1, who showed stable disease at the first CT scan (after 3 pembrolizumab infusions) with dynamic target lesion growth, followed by tumor shrinkage only after vaccine administration, resulting in a confirmed PR. Deepening of tumor shrinkage coincided with the increase of the vaccine induced immune response detected in the periphery. The potential lack of response to anti-PD-1 of this patient was in line with the predictive gene signatures evaluated from pre-treatment tumor biopsy. Indeed, this patient displayed a very low ratio of IFNγ signature to IMS, previously demonstrated to be indicative of a low probability of responding to anti-PD-1 monotherapy (18). Deconvolution of immune response at the level of single peptides was possible in this patient and allowed the identification of immunogenic neoepitope inducing the CD8 T-cell response. Interestingly, the identified NeoAg (#7) is in the mutated nucleolar protein NOL9 and shares 100% sequence identity with a Lachnospiraceae peptide. Sequence homology between neoantigens and microbial peptides has been suggested to contribute to the induction of an immune response against neoantigens (24). Moreover, Lachnospiraceae belongs to the group of Bacteroides previously shown to correlate positively with the rescue of clinical response to anti-PD-1 by fecal transplant (25). Enrichment of T cells in the tumor post MVA was detected via TCR sequence in all three evaluable patients receiving the full GAd20/MVA vaccine regimen. This finding is indicative of the NOUS-PEV vaccine's ability to expand and diversify T cells in the tumor, consistently with the TCR repertoire changes observed by the very same platform in MSI patients treated with Nous-209 off-the-shelf vaccine (9). Moreover, in patient 1, TCR clonotypes identified as specific for NeoAg (#7) were tracked in the tumor biopsy and expanded and diversified after NOUS-PEV vaccination. These results provided further evidence of tumor infiltration by neoantigen-specific T-cell clones following vaccination. Enrichment of T cells in the tumor correlates with the kinetics of T-cell expansion in the periphery post-vaccination. Previous findings have shown that expansion of T cells in the tumor due to CPI activity occurs very early post treatment initiation and therefore may no longer be detected at the biopsy collection time selected in this study, 13 weeks post-CPI treatment initiation. Interestingly enrichment of T cells in the tumor was not observed in Pt 5 in clinical response, who did not receive GAd20 priming and had no detectable immune response to NOUS-PEV in the periphery.
To date, immunogenicity of neoantigen vaccines has been shown in patients with cancer to a varying extent using different technologies (26–28). A recent study in patients with surgically resected pancreatic ductal adenocarcinoma (PDAC) tumors treated adjuvantly with mRNA vaccine encoding 20 neoantigens, in combination with atezolizumab and mFOLFIRINOX, showed induction of T-cell responses in 50% of treated patients that correlates with delayed recurrence (28). Although a direct comparison between platforms is not feasible, as the clinical settings and indications targeted are different, it is interesting to note that immunogenicity of mRNA vaccine was detected in only half of the treated patients, whereas viral-based neoantigen vaccine elicited T-cell responses in all patients of this trial and in a previous study (9). Besides immunogenicity, initial promising results on the direct clinical activity of neoantigen vaccines are emerging, providing evidence that these therapeutics can yield meaningful clinical benefit to patients, as recently shown in a randomized phase II trial of personalized vaccine mRNA-4157 plus pembrolizumab versus pembrolizumab alone as adjuvant therapy in patients with high-risk melanoma (29). These recent results also highlight the potential of a personalized vaccine in earlier-stage disease, particularly following tumor surgery.
Finally, potential biomarkers for lack of clinical response to the combination of vaccine and CPI were also investigated. Tumor immunogenicity is determined not only by the tumor antigenicity itself, but also influenced by factors such as the tumor microenvironment, including antigen presentation efficiency. Defects in the APM genes may result in lack of antigen presentation and thereby lack of cytotoxic T-cell-mediated tumor elimination, even in the presence of vaccine-induced T cells. Expression of APM genes was found downregulated in the PD patients. Tertiary lymphoid structures (TLS) have been shown to play a key role in shaping the immune microenvironment of several tumor types, including melanoma (30). Interestingly, our analysis showed a downregulation of chemokine expression necessary for the induction of TLS, such as CCL19, CCL21, and CCR7, in PD patients, that may be relevant for effective treatment outcome, also in the case of combined vaccine and anti-PD-1 treatment.
We acknowledge several limitations of our study, in particular the small sample size and the absence of a control arm to estimate the role of vaccine-induced neoantigen-specific T cells in determining the disease outcome. Larger cohorts and randomized trials will be necessary to establish superior activity of vaccine and anti-PD-1 with anti-PD-1 alone. The expectation is that vaccination will improve the clinical outcome without increasing toxicity, which is in contrast to currently used CPI combinations. Another limitation is the availability of patients’ specimens, either blood or tumors, for deeper characterization of the immune responses at single-cell resolution by scRNA-seq, and detailed analysis of CD4 and CD8 responses.
In summary, our study demonstrated the feasibility of a novel personalized neoantigen-based vaccine that in combination with pembrolizumab was shown to be safe and able to generate tumor specific T cells capable of trafficking to the tumor. The data presented here support the use of this vaccine platform as a valuable therapeutic option with a potential to be used in a wide range of disease settings and indications.
Supplementary Material
Supplementary Figure 1. IFN-γ ELISpot response post-IVS
Supplementary Table 1. Representativeness of study participants
Acknowledgments
Funding for this study was provided by Nouscom Srl.
This article is featured in Highlights of This Issue, p. 2295
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Disclosures
A. D'Alise reports employment with Nouscom Srl. G. Leoni reports a patent for PCT/EP2019/081428 pending and employment with Nouscom Srl. G. Cotugno reports employment with Nouscom Srl. L. Siani reports employment with Nouscom Srl. R. Vitale reports employment with Nouscom Srl. V. Ruzza reports employment with Nouscom Srl. I. Garzia reports employment with Nouscom Srl. L. Antonucci reports employment with Nouscom Srl. E. Micarelli reports employment with Nouscom Srl. S. Gogov reports employment with Nouscom Srl and ownership of Nouscom Srl stock. A. Capone reports employment with Nouscom Srl. J. Martin-Liberal reports personal fees from Astellas, Bristol Myers Squibb, Sanofi, and Highlight Therapeutics; grants and personal fees from MSD, Novartis, Pierre Fabre, Pfizer, and Roche; and grants from Ipsen and Merck outside the submitted work. E. Calvo reports employment with START and HM Hospitales Group; leadership with START, PharmaMar, EORTC, Sanofi, BeiGene, Novartis, and Merus NV; stock/ownership with START and Oncoart Associated; honoraria from HM Hospitales Group; consulting or advisory roles with Nanobiotix, Janssen-Cilag, Roche/Genentech, TargImmune Therapeutics, Servier, BMS, Amunix, Adcendo, Anaveon, AstraZeneca/MedImmune, Chugai Pharma, MonTa, MSD Oncology, Nouscom, Novartis, OncoDNA, T-Knife, Elevation Oncology, PharmaMar, Ellipses Pharma, Syneos Health, Genmab, and Diaccurate; research funding from START; and other relationships with Foundation INTHEOS, Foundation PharmaMar, and CRIS Cancer Foundation. V. Moreno reports other support from Nouscom Srl during the conduct of the study and personal fees from BMS and AstraZeneca outside the submitted work. S.N. Symeonides reports grants from Cancer Research UK and Chief Scientist Office (Scottish Government) during the conduct of the study; grants and personal fees from MSD; grants from Verastem; and personal fees from Duke Street Bio, EUSA, Eisai, Ellipses, Eugit Therapeutics, Exscientia, Grey Wolf Therapeutics, Roche, Ipsen, and WCG outside the submitted work. E. Scarselli reports a patent for PCT/EP2019/081428 pending as well as employment with Nouscom Srl; E. Scarselli is also the founder of Nouscom Srl. No disclosures were reported by the other authors.
Authors' Contributions
A.M. D'Alise: Conceptualization, data curation, supervision, investigation, visualization, methodology, writing–original draft. G. Leoni: Investigation, visualization, methodology. G. Cotugno: Investigation, methodology. L. Siani: Investigation, methodology. R. Vitale: Investigation, methodology. V. Ruzza: Investigation, methodology. I. Garzia: Formal analysis, investigation, methodology. L. Antonucci: Formal analysis, investigation, methodology. E. Micarelli: Investigation, methodology. V. Venafra: Investigation. S. Gogov: Supervision, investigation, writing–review and editing. A. Capone: Investigation. S. Runswick: Investigation, visualization, writing–original draft. J. Martin-Liberal: Investigation. E. Calvo: Investigation. V. Moreno: Investigation. S.N. Symeonides: Investigation, writing–review and editing. E. Scarselli: Supervision, investigation, visualization, writing–original draft, project administration. O. Bechter: Investigation, visualization, writing–original draft, writing–review and editing.
References
- 1. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015;348:124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015;348:69–74. [DOI] [PubMed] [Google Scholar]
- 3. Yarchoan M, Johnson BA 3rd, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer 2017;17:209–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hu Z, Ott PA, Wu CJ. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat Rev Immunol 2018;18:168–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Blass E, Ott PA. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat Rev Clin Oncol 2021;18:215–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sasso E, D'Alise AM, Zambrano N, Scarselli E, Folgori A, Nicosia A. New viral vectors for infectious diseases and cancer. Semin Immunol 2020;50:101430. [DOI] [PubMed] [Google Scholar]
- 7. Swadling L, Capone S, Antrobus RD, Brown A, Richardson R, Newell EW, et al. A human vaccine strategy based on chimpanzee adenoviral and MVA vectors that primes, boosts, and sustains functional HCV-specific T cell memory. Sci Transl Med 2014;6:261ra153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. D'Alise AM, Leoni G, Cotugno G, Troise F, Langone F, Fichera I, et al. Adenoviral vaccine targeting multiple neoantigens as strategy to eradicate large tumors combined with checkpoint blockade. Nat Commun 2019;10:2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. D'Alise AM, Brasu N, De Intinis C, Leoni G, Russo V, Langone F, et al. Adenoviral-based vaccine promotes neoantigen-specific CD8(+) T cell stemness and tumor rejection. Sci Transl Med 2022;14:eabo7604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. D'Alise AM, Leoni G, De Lucia M, Langone F, Nocchi L, Tucci FG, et al. Maximizing cancer therapy via complementary mechanisms of immune activation: PD-1 blockade, neoantigen vaccination, and tregs depletion. J Immunother Cancer 2021;9:e003480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Leoni G, D'Alise AM, Tucci FG, Micarelli E, Garzia I, De Lucia M, et al. VENUS, a novel selection approach to improve the accuracy of neoantigens' prediction. Vaccines (Basel) 2021;9:880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Di Lullo G, Soprana E, Panigada M, Palini A, Agresti A, Comunian C, et al. The combination of marker gene swapping and fluorescence-activated cell sorting improves the efficiency of recombinant modified vaccinia virus Ankara vaccine production for human use. J Virol Methods 2010;163:195–204. [DOI] [PubMed] [Google Scholar]
- 13. Bolotin DA, Poslavsky S, Mitrophanov I, Shugay M, Mamedov IZ, Putintseva EV, et al. MiXCR: software for comprehensive adaptive immunity profiling. Nat Methods 2015;12:380–1. [DOI] [PubMed] [Google Scholar]
- 14. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26:139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Law CW, Chen Y, Shi W, Smyth GK. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 2014;15:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tarazona S, Garcia-Alcalde F, Dopazo J, Ferrer A, Conesa A. Differential expression in RNA-seq: a matter of depth. Genome Res 2011;21:2213–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cui C, Xu C, Yang W, Chi Z, Sheng X, Si L, et al. Ratio of the interferon-gamma signature to the immunosuppression signature predicts anti-PD-1 therapy response in melanoma. NPJ Genom Med 2021;6:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Van Loo P, Nordgard SH, Lingjaerde OC, Russnes HG, Rye IH, Sun W, et al. Allele-specific copy number analysis of tumors. Proc Natl Acad Sci USA 2010;107:16910–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Roth A, Khattra J, Yap D, Wan A, Laks E, Biele J, et al. PyClone: statistical inference of clonal population structure in cancer. Nat Methods 2014;11:396–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Litchfield K, Reading JL, Puttick C, Thakkar K, Abbosh C, Bentham R, et al. Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell 2021;184:596–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Richard G, Princiotta MF, Bridon D, Martin WD, Steinberg GD, De Groot AS. Neoantigen-based personalized cancer vaccines: the emergence of precision cancer immunotherapy. Expert Rev Vaccines 2022;21:173–84. [DOI] [PubMed] [Google Scholar]
- 23. Sheehy SH, Duncan CJ, Elias SC, Choudhary P, Biswas S, Halstead FD, et al. ChAd63-MVA-vectored blood-stage malaria vaccines targeting MSP1 and AMA1: assessment of efficacy against mosquito bite challenge in humans. Mol Ther 2012;20:2355–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Balachandran VP, Luksza M, Zhao JN, Makarov V, Moral JA, Remark R, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017;551:512–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Davar D, Dzutsev AK, McCulloch JA, Rodrigues RR, Chauvin JM, Morrison RM, et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science 2021;371:595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017;547:217–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Palmer CD, Rappaport AR, Davis MJ, Hart MG, Scallan CD, Hong SJ, et al. Individualized, heterologous chimpanzee adenovirus and self-amplifying mRNA neoantigen vaccine for advanced metastatic solid tumors: phase 1 trial interim results. Nat Med 2022;28:1619–29. [DOI] [PubMed] [Google Scholar]
- 28. Rojas LA, Sethna Z, Soares KC, Olcese C, Pang N, Patterson E, et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature 2023;618:144–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dolgin E. Personalized cancer vaccines pass first major clinical test. Nat Rev Drug Discov 2023;22:607–9. [DOI] [PubMed] [Google Scholar]
- 30. Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020;577:561–5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. IFN-γ ELISpot response post-IVS
Supplementary Table 1. Representativeness of study participants
Data Availability Statement
Exome and RNA-sequencing (RNA-seq) data generated in this study have been deposited in Sequence Read Archive under the accession code PRJNA1081187. All the other data of this study are available in the article or upon request from the corresponding author.