

Significance
Mitochondria play a central role in muscle metabolism and function. In skeletal muscles, a unique family of iron–sulfur proteins, termed CISD/NEET proteins, support mitochondrial function and maintenance. The abundance of these proteins declines with age leading to muscle degeneration and atrophy. Here, we reveal that one of these proteins, CISD3/MiNT, is important for supporting the biogenesis and function of mitochondrial respiratory complex I, essential for muscle function, and that it interacts with one of the subunits of this complex and donates its cluster to it. Pharmacological and/or genetic interventions that target CISD3/MiNT could therefore impact multiple muscle degeneration syndromes, aging, and related conditions.
Keywords: mitochondria, NEET/CISD proteins, NDUFV2, complex I, Duchenne muscular dystrophy
Abstract
Mitochondria play a central role in muscle metabolism and function. A unique family of iron–sulfur proteins, termed CDGSH Iron Sulfur Domain-containing (CISD/NEET) proteins, support mitochondrial function in skeletal muscles. The abundance of these proteins declines during aging leading to muscle degeneration. Although the function of the outer mitochondrial CISD/NEET proteins, CISD1/mitoNEET and CISD2/NAF-1, has been defined in skeletal muscle cells, the role of the inner mitochondrial CISD protein, CISD3/MiNT, is currently unknown. Here, we show that CISD3 deficiency in mice results in muscle atrophy that shares proteomic features with Duchenne muscular dystrophy. We further reveal that CISD3 deficiency impairs the function and structure of skeletal muscles, as well as their mitochondria, and that CISD3 interacts with, and donates its [2Fe-2S] clusters to, complex I respiratory chain subunit NADH Ubiquinone Oxidoreductase Core Subunit V2 (NDUFV2). Using coevolutionary and structural computational tools, we model a CISD3–NDUFV2 complex with proximal coevolving residue interactions conducive of [2Fe-2S] cluster transfer reactions, placing the clusters of the two proteins 10 to 16 Å apart. Taken together, our findings reveal that CISD3/MiNT is important for supporting the biogenesis and function of complex I, essential for muscle maintenance and function. Interventions that target CISD3 could therefore impact different muscle degeneration syndromes, aging, and related conditions.
CDGSH/NEET proteins belong to a unique class of highly conserved iron–sulfur [2Fe-2S] proteins (1–3). Their iron–sulfur binding domain, coordinated by 3Cys-1His (part of the conserved CDGSH domain; C-X-C-X2-(S/T)-X3-P-X-C-D-G-(S/A/T)-H), is redox-sensitive and involved in many different [2Fe-2S] cluster and/or electron transfer reactions (4–8). In humans, three different genes encode for NEET/CDGSH proteins (termed CDGSH Iron Sulfur Domain-containing, or CISD/NEET proteins). These are CISD1/mitoNEET, localized to the outer mitochondrial membrane, CISD2/NAF-1, localized to the outer endoplasmic reticulum (ER), mitochondria, and ER-mitochondria associated membranes (MAM), and CISD3/MiNT, localized inside the mitochondria. Although multiple studies have shown that CISD1 and CISD2 are involved in the progression of different human pathologies, including cancer, diabetes, neurodegeneration, obesity, and cardiovascular diseases (1–3, 9–17), as well as the genetic disease Wolfram syndrome 2 (caused by CISD2 deficiency) (18–20), very little is known about the function of CISD3/MiNT.
In contrast to CISD1 and CISD2 that are membrane-embedded symmetric homodimers (each monomer containing one [2Fe-2S] cluster and one transmembrane α-helix), CISD3 is a soluble monomeric protein that folds into a pseudodimer that contains two, not perfectly symmetric, [2Fe-2S] clusters (1–4, 21). Recent studies support a model in which CISD3 can transfer its [2Fe-2S] clusters from within the mitochondria to CISD1, that is outside the mitochondria, and that CISD1 can transfer these clusters to CISD2 and/or Anamorsin, constituting a [2Fe-2S] shuttle from within the mitochondria to the cytosol (22–25). In support of this suggested pathway are the findings that disruption of any of the three CISD proteins in cancer cells causes an elevation in the levels of mitochondrial labile iron and reactive oxygen species (ROS), that is accompanied by the activation of a ferroptotic and/or apoptotic cell death pathways (2, 4, 25–27). A recent study has also revealed that targeting CISD3 function in cancer cells induces glutaminolysis and causes cystine deprivation-induced ferroptosis (28). Interestingly, the protein levels of CISD3 (as well as CISD1 and CISD2) are elevated in many different cancer cells to prevent the activation of ferroptosis and/or apoptosis that may be triggered due to the overaccumulation of ROS and/or labile iron in mitochondria (part of the iron/ROS “addiction” of many cancers) (2, 29, 30).
Studies of CISD3 function in Caenorhabditis elegans (C. elegans) revealed that this protein is required for maintaining normal germline structure and function by regulating physiological germline apoptosis (through the canonical programmed cell death pathway) (31, 32). CISD3 mutations in C. elegans consequently result in germline abnormalities, associated with an irregular stem cell niche, and disrupted formation of bivalent chromosomes (31). As the phenotypes associated with CISD3 in C. elegans were also found to be linked with mitochondrial dysfunction, including disruption of the mitochondrial network within the germline (31, 32), they further support a role for CISD3 in maintaining and/or promoting mitochondrial function in cells with a high energy demand and/or ROS levels (such as stem cells). Remarkably, although CISD3 is conserved in animal cells, some fungi, and bacteria, it does not appear to be present in higher plants (33, 34).
Although the role of CISD3 in cancer cells and C. elegans has been previously studied (4, 22, 28, 31), very little is known about the function of CISD3 in the development and physiological maintenance of animal cells and tissues. To identify a role for CISD3 in animal development and normal physiological functions, we generated a transgenic global deletion (knock-out; KO) mice model for CISD3 (Cisd3−/−). Our analysis of Cisd3−/− mice revealed that CISD3 is essential for the maintenance and function of skeletal muscles, and that deficiency in CISD3 results in muscle atrophy that shares proteomic features with Duchenne muscular dystrophy (DMD). In addition, we show that deficiency in CISD3 impairs the function and structure of skeletal muscle mitochondria and that CISD3 interacts with, and donates its clusters to, complex I mitochondrial respiratory chain subunit NDUFV2 (NADH:Ubiquinone Oxidoreductase Core Subunit V2; encoded by the nuclear gene NDUFV2). These results were further supported by a coevolutionary analysis (35–37) of CISD3 and NDUFV2 protein interactions, based on Direct Coupling Analysis (DCA) (38) and Structure Based Modelling (SBM) (39), that identified coevolving residues indicative of interactions between specific residues within and between these proteins and used to predict a structure of the CISD3–NDUFV2 complex, conducive for [2Fe-2S] cluster transfer (38, 40). Taken together, our findings reveal that CISD3 has an important function within the mitochondria, supporting the biogenesis/function of complex I, and that this function is essential for muscle maintenance and function. Interventions that target CISD3 function in muscles could therefore impact different muscle degeneration syndromes, aging, and related conditions.
Results
Characterization of CISD3 Knockout Mice.
C57BL/6J wild-type (WT) and Cisd3−/− male and female mice were grown under controlled conditions for 44 to 47 wk. Compared to WT, Cisd3−/− male and female mice were significantly smaller (Fig. 1 A–C and SI Appendix, Fig. S1) and had a life span of about 60 wk (about half of WT mice). As the CISD/NEET protein family was shown to be functionally linked (2, 22, 25), we tested the expression of all CISD proteins in the quadriceps muscle of WT and Cisd3−/− mice. This analysis confirmed that Cisd3−/− mice lacked the CISD3 protein and revealed that the deficiency in CISD3 caused a significant decrease in the abundance of the CISD1 protein, as well as a significant increase in the abundance of the CISD2 protein (Fig. 1D). This finding suggests that the CISD protein network is linked and that changes in the levels of one CISD protein (i.e., CISD3) alter the levels of the two other members of this family (i.e., CISD1 and CISD2).
Fig. 1.

Generation and characterization of Cisd3−/− mice. (A) Targeting of exon 2 of Cisd3 using a flox strategy. (B) PCR analysis of wild-type (WT), floxed, and the knockout (KO) alleles of Cisd3 in transgenic mice. (C) Representative images of a WT and a Cisd3−/− mice (Left) and box-and-whisker plots showing the weight of WT and Cisd3−/− mice at age 44 to 47 wk old (Right; SI Appendix, Fig. S1). (D) Representative protein blots (Left) and box-and-whisker plots showing quantitative analysis (Right) for the levels of CISD1, CISD2, and CISD3 proteins in WT and Cisd3−/− mice. (E and F) Box-and-whisker plots showing the amount of time wild-type (WT) or Cisd3−/− mice can hang from a wheel (E) or a rod (F). Results are shown for male and female separately and presented as box-and-whisker plots and include all data points of six different animals (three different males and three different females) from each group (WT and Cisd3−/− mice). Two-way ANOVA followed by a Tukey test was used to calculate statistical significance. White box and white square, WT male mice; gray box and black square, Cisd3−/− male mice; white box and white circle, WT female mice; gray box and black circle, Cisd3−/− female mice. Abbreviations: CISD, CDGSH Iron Sulfur Domain; KO, knock out; WT, wild type.
Deficiency in CISD3 Affects Skeletal Muscle Structure and Function.
When handling the Cisd3−/− mice, it was found that compared to WT, Cisd3−/− male and female mice were physically weaker and had a reduced grip strength. We therefore measured their grip strength by using a wheel and rod hang tests (41). Compared to male and female WT mice, both male and female Cisd3−/− mice displayed a lower ability to keep their own weight while hanging from a wheel or a rod (Fig. 1 E and F). This finding suggested that compared to WT, the muscle strength of male and female Cisd3−/− mice is weaker and does not allow them to hang from a wheel or a rod for the same length of time as WT mice can.
To study the effects of CISD3 deficiency on skeletal muscle of mice, we focused on the quadriceps muscle (SI Appendix, Fig. S2A) and compared its cellular and subcellular structure between 44- and 47-wk-old WT and Cisd3−/− male and female mice using light and transmission electron microscopy (TEM). Staining of muscle sections with Hematoxylin and Eosin (H&E) did not reveal significant differences between WT and Cisd3−/− mice, except for a larger number of centralized nuclei in muscle cells of male Cisd3−/− mice compared to male WT mice (SI Appendix, Figs. S2B and S3). TEM analyses of muscle tissue from male and female WT and Cisd3−/− mice revealed that the muscular sarcomere (i.e., the distance between the muscle Z-lines) of male Cisd3−/− mice is significantly shorter compared to that of WT mice (SI Appendix, Fig. S2C). A shorter Z-line distance was previously shown to correlate with muscle atrophy (42, 43) supporting our reduced muscle strength results obtained with the rod and wheel hanging tests of Cisd3−/− mice (Fig. 1 E and F).
As deficiency in the levels of CISD/NEET proteins correlates with enhanced levels of iron in cells (26, 44), we used the Perls’ Prussian blue iron stain method to measure the levels of iron in muscle sections from WT and Cisd3−/− mice. This analysis revealed that muscle tissue from male and female Cisd3−/− mice contained higher levels of iron compared to male and female muscle sections from WT mice (SI Appendix, Fig. S2D).
Deficiency in CISD3 Affects the Structure and Function of Skeletal Muscle Mitochondria.
Previous studies demonstrated that deficiency in CISD3 (as well as CISD1 or CISD2) results in mitochondrial structural abnormalities and increased mitochondrial damage in cancer cells (4, 26, 45). We therefore studied the structure of mitochondria in male and female Cisd3−/− mice using TEM. This analysis revealed that compared to male and female WT mice, mitochondria from male and female Cisd3−/− mice are abnormal and appear swollen with damaged crista (Fig. 2A).
Fig. 2.

Impaired structure and function of mitochondria from skeletal muscles of Cisd3−/− mice. (A) Representative transmission electron microscope (TEM) images of mitochondria from wild-type (WT) and Cisd3−/− mice (Top) and box-and-whisker plots showing quantification of mitochondrial damage (Bottom). (B–E) Bar graphs for basal (B), maximal (C), and spare (D) respiration, and glycolytic activity (E) of skeletal muscle fibers from WT and Cisd3−/− mice (measured with an XFe24 Seahorse apparatus). Results are shown for male mice and presented as box-and-whisker plots and include all data points of six different animals from each group (WT and Cisd3−/− mice). Two-way ANOVA followed by a Tukey test was used to calculate statistical significance. White box and white square, WT male mice; Gray box and black square, Cisd3−/− male mice; white box and white circle, WT female mice; Gray box and black circle, Cisd3−/− female mice. Abbreviations: CISD, CDGSH Iron Sulfur Domain; ECAR, Extracellular Acidification Rate; KO, knock out; OCR, Oxygen Consumption Rate; TEM, transmission electron microscope; WT, wild type.
To test whether the damage to mitochondria in Cisd3−/− mice resulted in suppressed respiration, we used an XFe24 Seahorse apparatus to measure respiration and glycolysis in muscle fibers from WT and Cisd3−/− mice. As the phenotype of the Cisd3 deletion appeared to be more severe in male mice (e.g., Fig. 1E and SI Appendix, Figs. S2C and S3), we focused our studies henceforth on male mice. In agreement with the analysis of mitochondrial structure (Fig. 2A), and compared to muscle fibers from WT male mice, the basal respiration, maximal respiration, and spare respiratory capacity of muscle fibers from Cisd3−/− mice were significantly reduced (Fig. 2 B–D). In contrast, the glycolytic capacity of muscle fibers from Cisd3−/− mice was higher than that of male WT fibers (Fig. 2E).
Proteomics Analysis of Skeletal Muscles from Cisd3−/−Mice.
To determine the molecular function of CISD3 in mice skeletal muscles, we conducted a comparative proteomics analysis of quadriceps muscle tissue from 44- to 47-wk-old WT and Cisd3−/− male mice (SI Appendix, Fig. S2A and Dataset S1). Our analysis identified a total of 2,545 proteins (Dataset S2). Of these, 804 proteins (32%) had a significant change in expression (up or down) in Cisd3−/− mice compared to WT (Fig. 3A and Datasets S1 and S2). Of the 804 proteins, 650 proteins (26%) decreased in their expression, while the expression of 154 proteins (6%) was elevated (Fig. 3A and Dataset S2).
Fig. 3.

Proteomics analysis of skeletal muscles from wild-type and Cisd3−/− mice. (A) A diagram showing the number of proteins altered in Cisd3−/− mice compared to the wild type (WT) in quadricep muscle tissues of 44-wk-old mice. (B) Subcellular localization and gene ontology (GO) annotation of proteins altered in Cisd3−/− mice compared to WT. (C) Venn diagrams showing the overlap between proteins altered in Cisd3−/− mice and proteins altered in mice model systems for Duchenne Muscular Dystrophy (DMD), Huntington Disease (HD; R6/2), and Multiple Sclerosis (MS; EAE). (D) Same as in (C) but for mitochondrial proteins. (E–G) Changes in protein abundance in Cisd3−/− mice are shown for the tricarboxylic acid cycle (TCA; E), fatty acid oxidation (F), and glycolysis/gluconeogenesis (G) pathways. Results are shown for male mice and presented as mean ± SD of three different animals from each group (WT and Cisd3−/− mice). Two-way ANOVA followed by a Tukey test was used to calculate statistical significance. Abbreviations: CISD, CDGSH Iron Sulfur Domain; DMD, Duchenne muscular dystrophy; EAE, experimental allergic encephalomyelitis; HD, Huntington disease; GO, gene ontology; GO, gene ontology; KO, knock out; MS, multiple sclerosis; WT, wild type; Cs, citrate synthase; Acly, ATP-citrate synthase; Aco2, Aconitase 2; Idh, Isocitrate dehydrogenase [NADP]; Ogdh, Oxoglutarate Dehydrogenase; Suclg, Succinate--CoA ligase; Pc, pyruvate carboxylase; Sdh, Succinate dehydrogenase [ubiquinone]; Fh, Fumarate hydratase; Mdh, Malate dehydrogenase; A-KG, α-Ketoglutarate; Acaa2, 3-ketoacyl-CoA thiolase; Acsl5, Long-chain-fatty-acid—CoA ligase 5; Acad11, Acyl-CoA dehydrogenase family member 11; Acadl, Long-chain specific acyl-CoA dehydrogenase; Echs1, Enoyl-CoA hydratase; Fbp1, Fructose-1,6-bisphosphatase 1; Pck, Phosphoenolpyruvate carboxykinase; Pfkfb1, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 1; Pdk, Pyruvate dehydrogenase (acetyl-transferring)] kinase; Aldoart1, Fructose-bisphosphate aldolase; Mpc, Mitochondrial pyruvate carrier; Vdac3, Voltage-dependent anion-selective channel protein 3, F2.6BP, Fructose 2,6-bisphosphate; DHAP, Dihydroxyacetone phosphate.
Annotation of subcellular localization of the 804 proteins (significantly altered in their expression in Cisd3−/− mice compared to WT) revealed that over 40% of these proteins localized to the cytosol and over 20% localized to mitochondria (Fig. 3B). Gene ontology (GO) analysis of the 804 proteins identified lipid and carbohydrate metabolism, cytoskeleton, oxidative stress, aging, hypoxia, iron homeostasis, and gluconeogenesis among the most abundant proteins in this group (Fig. 3B and Dataset S3). These findings are in agreement with our analysis of mitochondrial structure and function, supporting the observed reduction in respiration, coupled with the enhanced rate of glycolysis in Cisd3−/− mice (Fig. 2 B–E), as well as the overall damage to mitochondria (Fig. 2A).
To determine the degree of similarity between the proteomic changes occurring in Cisd3−/− mice and changes in protein expression occurring in mice models of different diseases, associated with muscle atrophy/dystrophy, we compared the changes in the proteome of Cisd3−/− mice to that of DMD (46), Huntington disease (47), and multiple sclerosis (MS) (48). Interestingly, over 29% of proteins altered in Cisd3−/− mice were also altered in DMD mice, while only 21 and 14% of proteins altered in Cisd3−/− mice were also altered in Huntington diseases and MS mice, respectively (Fig. 3C and Dataset S4). When the comparison of proteome similarity between Cisd3−/−, DMD, Huntington, and MS mice models was restricted to mitochondrial proteins, the overlap between Cisd3−/− mice, DMD, Huntington, and MS was 66, 22, and 11%, respectively (Fig. 3D and Dataset S5). The high overlap between Cisd3−/− and DMD (at the total and mitochondrial protein levels; Fig. 3 C and D) could suggest that CISD3 and DYSTROPHIN deficiency share some similarities with respect to their effect(s) on mitochondrial function and proteome alterations. In this respect, it is interesting to note that the abundance of several proteins involved in the DYSTROPHIN muscle complex (NOS1, DAG1, and Sgcd/Delta-sarcoglycan), was significantly reduced in Cisd3−/− male mice (SI Appendix, Fig. S4; DYSTROPHIN itself was not significantly altered in Cisd3−/− mice).
Changes in the Abundance of Proteins Associated with Metabolic Pathways in Cisd3−/− Mice.
To further investigate the proteomic changes associated with CISD3 deficiency, we examined changes in protein expression associated with different metabolic pathways (Fig. 3 E–G). This analysis revealed that the expression of many key proteins involved in the TCA cycle is suppressed in Cisd3−/− mice (Fig. 3E). The expression of several proteins involved in fatty acid oxidation, supplying Acetyl-CoA to the TCA pathway, was also suppressed (Fig. 3F). In addition, and in agreement with the enhanced glycolytic activity of muscle fibers from Cisd3−/− mice (Fig. 2E), the expression of several proteins involved in glycolysis was enhanced, while the expression of several proteins involved in gluconeogenesis (reversal of glycolysis) was suppressed (Fig. 3G). The findings presented in Fig. 3 E–G suggest that CISD3 deficiency causes major alterations in mitochondrial metabolism and function that could result in the suppression of respiration (also supported by the reduced respiratory activity displayed by muscle fibers from Cisd3−/− mice; Fig. 2 B–D). We therefore focused our attention on the abundance of proteins involved in the mitochondrial respiratory electron transfer chain.
Comparative analysis of complex I, II, III, IV, and V protein levels between WT and Cisd3−/− mice revealed that CISD3 deficiency resulted in a rebalancing process between different subunits of these complexes (Fig. 4A and Dataset S2). Expression of several proteins, including key [Fe-S] proteins in complexes I and II, was reduced, while expression of other proteins associated with these complexes was enhanced (Fig. 4A and Dataset S2). Considering the reduction in the level of key proteins associated with the TCA cycle and Acetyl-CoA metabolism, involved in electron transfer to complexes I and II, in Cisd3−/− mice (Fig. 3 E–G), and the overall reduction in respiration (Fig. 2 B–D and Dataset S2), this finding suggests that CISD3 could play an important role in maintaining proper metabolic reactions associated with respiration in muscles. Bearing in mind the soluble nature of CISD3 (4), its potential to transfer clusters to acceptor proteins (4, 22), and the fact that many of the proteins altered in their expression in complex I were localized to the mitochondrial matrix-exposed part of this complex (Fig. 4A), we hypothesized that CISD3 is involved in donating [2Fe-2S] clusters/electrons to a subunit(s) of this complex (e.g., the [2Fe-2S] subunit of complex I, NDUFV2).
Fig. 4.

Identification of proteins associated with CISD3 in skeletal muscles and cluster transfer between CISD3 and NDUFV2. (A) Changes in protein abundance in Cisd3−/− mice are shown for complex I, II, and III/IV/V proteins. (B) Identification of proteins that associate with CISD3 in vivo in skeletal muscles following immunoprecipitation assays performed with skeletal muscles from wild-type (WT) and Cisd3−/− mice. Protein identification was performed by proteomics analysis. (C and D) Protein gel (C) and protein blot with antibody against CISD3 (D) showing in vitro protein–protein interaction between CISD3 and NDUFV2. Arrow indicates the location of the protein–protein complex between CISD3 and NDUFV2. Identification of proteins found in the band indicated by arrows in (D) using proteomics analysis is shown below (D). (E) Cluster transfer from holo-CISD3 to apo-NDUFV2 visualized by changes in spectra of the two proteins following coincubation. Results are shown for male mice. Abbreviations: CISD, CDGSH Iron Sulfur Domain; Ndufv2, NADH:ubiquinone oxidoreductase core subunit V2; IB, Immunoblotting; IBAQ, intensity-based absolute quantification. mt-Nd3, NADH-ubiquinone oxidoreductase chain 3; Ndufa, NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit; Ndufb10, NADH dehydrogenase [ubiquinone] 1 beta subcomplex subunit 10; Ndufs, NADH:ubiquinone oxidoreductase core subunit; Ndufv, NADH:ubiquinone oxidoreductase core subunit V; Sdh, Succinate dehydrogenase [ubiquinone]; Uqcr, Cytochrome b-c1 complex subunit; Cox, Cytochrome c oxidase subunit 5A; Mtatp, ATP synthetase protein; Co-ip, Coimmunoprecipitation; SN, Supernatant; MW, Molecular Weight; CISD, CDGSH Iron Sulfur Domain; KO, knock out; WT, wild type.
Identification of Proteins Associated with CISD3 in Mice Skeletal Muscles.
To identify the different proteins that interact via protein–protein interactions with CISD3 in mice skeletal muscles, we conducted an immunoprecipitation study using quadriceps muscle tissues from WT and Cisd3−/− mice and an antibody against CISD3/MiNT (Fig. 1D) (4, 22). This analysis revealed that several proteins that are part of complex I (NDUFS2, NDUFV1, NDUFV2, NDUFA11, and NDUFS1) and complex II (SDHA and SDHB) associate with CISD3 in vivo in quadriceps muscles of WT (but not in Cisd3−/− mice; Fig. 4B and Dataset S6). Of these, the levels of NDUFA11, NDUFS1, and SDHB were reduced in their expression in Cisd3−/− mice (Fig. 4A). In addition, among the proteins identified as interacting with CISD3 in vivo, at least two contained [2Fe-2S] clusters: NDUFV2 and SDHB, of which NDUFV2 was part of the matrix-exposed part of complex I (Fig. 4A). We therefore focused on the potential interaction of CISD3 with NDUFV2.
In Vitro Interaction and [2Fe-2S] Cluster Transfer between CISD3 and NDUFV2.
To further study the potential interaction between CISD3 and NDUFV2 (Fig. 4B), we cloned the cDNAs of CISD3 and NDUFV2, overexpressed these proteins in bacteria, and purified them for in vitro studies. Coincubation of purified holo-CISD3 and holo-NDUFV2 proteins, followed by immunoprecipitation and proteomics analysis, revealed that the two proteins physically interact in vitro (Fig. 4 C and D and Dataset S7). As β-mercaptoethanol prevented this interaction (Fig. 4 C and D), it is possible that it involves the formation of disulfide bridges within one or both proteins and/or between the two different proteins. To examine whether the physical interaction between CISD3 and NDUFV2 (Fig. 4 B–D) results in a cluster-transfer reaction (between CISD3 and NDUFV2), we studied the potential of CISD3 to transfer its [2Fe-2S] clusters to NDUFV2. Coincubation of purified holo-CISD3 with purified apo-NDUFV2 resulted in the transfer of the [2Fe-2S] clusters of CISD3 to NDUFV2 (evident by the shift in the spectrum of apo- to holo-NDUFV2; Fig. 4E). For a cluster transfer reaction to occur, the distance between the two cluster binding sites of CISD3 and NDUFV2 needs to be in the 10 to 15 Å range (25).
Computational Modeling of the Interaction between CISD3 and NDUFV2.
To further explore the potential interaction between CISD3 and NDUFV2, we designed a workflow to utilize coevolutionary information derived from combinations of paired sequences from the families of the two proteins (Fig. 5A). Given that both CISD3 and NVDUFV2 MSAs consisted of a non-negligible percentage of sequences with very similar domain architectures within each organism, the problem of matching pairs of sequences became relevant. To address this issue, a paralog matching algorithm called Progressive Paralog Matching algorithm (PPM) (49) was used. DCA (38) provides a coevolutionary metric needed to match interacting paralog proteins using PPM and reveals direct couplings of residues ranked on the basis of Direct Information (DI), which we refer to as Direct Information pairs. DI pairs were plotted along with native contacts to evaluate accuracy in classifying a residue pair as interacting and were used for subsequent protein complex prediction with SBM simulations (Fig. 5). This simulation produced a complex that was stable at around 8 Å of distance between the evolutionary constraints of the two proteins and was then optimized through Amber Minimization to remove any steric clashes (Fig. 5C and SI Appendix, Fig. S6A). The resulting complex was highly indicative of interaction between CISD3 and NDUFV2; many distances between coevolving residues were 8 to 12 Å apart (SI Appendix, Table S1). The distance between [2Fe-2S] clusters within the complex between the two different proteins was around 15.7 Å, and the complex was oriented in a way that cluster transfer appears viable (Fig. 5C and Movie S1). Additionally, some of the DI pairs from DCA did not correspond with the native contacts, as in the case of NDUFV2, which may contribute to a dynamical conformation where its two globular domains come closer. However, further exploration of this idea is warranted before any meaningful conclusions can be made. The model was further validated through Protein Frustration Analysis. Frustratometer Server results revealed a large number of minimally frustrated contacts along the interface of the proteins (SI Appendix, Fig. S5A). Furthermore, AlphaFold Multimer (50) produced a similar complex as the SBM+DCA simulation, where coevolving residues were close to each other; a similar interface was produced, and a cluster distance of about 11.8 Å was obtained (SI Appendix, Figs. S6B and S8 and Table S2 and Movie S2). The fact that two models, both of which use evolutionary information and are known to produce reasonable orientations for protein complexes, produced a consistent interface provides support that this complex might be functionally relevant.
Fig. 5.

Coevolutionary analysis of the CISD3–NDUFV2 complex. (A) A general pipeline for extraction and analysis of coevolutionary data: seed sequences for CISD3 and NDUFV2 are used to generate their respective MSAs through phmmer software. Entries from one MSA are paired to entries in the other MSA via PPM and the pairs are concatenated horizontally. The full MSA of concatenated pairs is used in Direct Coupling Analysis to generate ranked DI contacts. A subset of the top DI contacts is used in future simulations and visualizations. (B) Contact map with residue pairs plotted as grid points. DI pairs of intraprotein contacts (CISD3: purple; NDUFV2: orange) and interprotein contacts (CISD3–NDUFV2: dark red) are represented with monomeric native contacts (gray) and SBM+DCA contacts (blue). (C) Visualization in UCSF Chimera (51) of the Amber minimized SBM+DCA model. From the top 50, the top 10 proximal interprotein DI pairs are connected with pseudobonds (green). The minimal distance between 2Fe-2S clusters is approximately 15.7 Å, shown as a black, dashed line. Abbreviations: CISD, CDGSH Iron Sulfur Domain; NDUFV2, NADH:ubiquinone oxidoreductase core subunit V2; DI, direct information; HMMER, biosequence analysis using profile hidden Markov models; MSA, multiple sequence alignment.
Discussion
CISD3/MiNT was proposed to function as part of an iron–sulfur cluster relay mechanism, transferring [2Fe-2S] clusters from within the mitochondria to CISD1 (localized on the OMM), and from there, via CISD2, to the cytosolic iron–sulfur cluster (ISC) biogenesis complex of the cytosol (i.e., CIA; via Anamorsin and perhaps other proteins) (2, 22, 24, 25). However, very little is currently known about the function of CISD3 within the mitochondria. In addition, most studies of CISD3 function were focused on its role in regulating ferroptotic and apoptotic cell death in cancer cells and C. elegans (4, 28, 31, 32, 52). In contrast, very little is known about the physiological role of CISD3 in mammalian cells and tissues. Here we show that CISD3 plays a key role in the maintenance and function of skeletal muscles, supporting respiration and other mitochondrial metabolic pathways via interacting with different subunits of complexes I and II (Figs. 2–5). We further show that CISD3 can physically interact with and transfer its clusters to NDUFV2, an [2Fe-2S]-containing protein that is localized to the mitochondrial matrix-exposed part of complex I (Figs. 4 and 5 and SI Appendix, Figs. S5 and S6). Deficiency in CISD3 therefore causes reduced respiration, enhanced glycolysis, mitochondrial structural damage, and muscle atrophy (Figs. 1 and 2 and SI Appendix, Fig. S2).
In cancer cells, all three CISD/NEET proteins were shown to function in a similar role, regulating mitochondrial iron and ROS levels and suppressing apoptotic and ferroptotic cell death (2, 3, 26, 53). As described above, they were also proposed to function as part of an [2Fe-2S] relay that transfers Fe-S clusters from within the mitochondria to the cytosol (22). In contrast, while all three CISD/NEET proteins were found to play a role in muscle maintenance and function (11, 12, 44, 54–56) (Figs. 1–6 and SI Appendix, Figs. S1 and S2), they appear to have different functions in muscle tissues. CISD2 was shown to regulate calcium signaling and autophagy in skeletal muscles, as well as to protect muscle cells from overaccumulation of iron (12, 44, 57), CISD1 was shown to protect heart muscle cells and tissues from iron and ROS accumulation, and ferroptosis activation (11, 58), and the present study reveals that CISD3 plays a key role in supporting the mitochondrial respiratory electron transport chain by providing [2Fe-2S] cluster to NDUVF2 (Figs. 2–6). This role for CISD3 demonstrates that under physiological conditions, CISD/NEET proteins may mediate a variety of cellular roles that differ depending on their subcellular localization. While CISD2 that is localized to the OMM, ER, and MAM, is involved in regulating apoptosis, ferroptosis, calcium signaling, and autophagy via interactions with key signaling proteins found associated with these different membrane systems, CISD1 that is localized to the OMM, and CISD3 that is localized inside the mitochondria, are primarily involved in regulating the flow of [2Fe-2S] clusters from the mitochondria to the cytosol, thereby controlling mitochondrial iron and ROS levels and suppressing ferroptosis. In addition, our study reveals that CISD3 supports respiration from within the mitochondria by providing [2Fe-2S] clusters to NDUFV2 (Figs. 2–6).
Fig. 6.
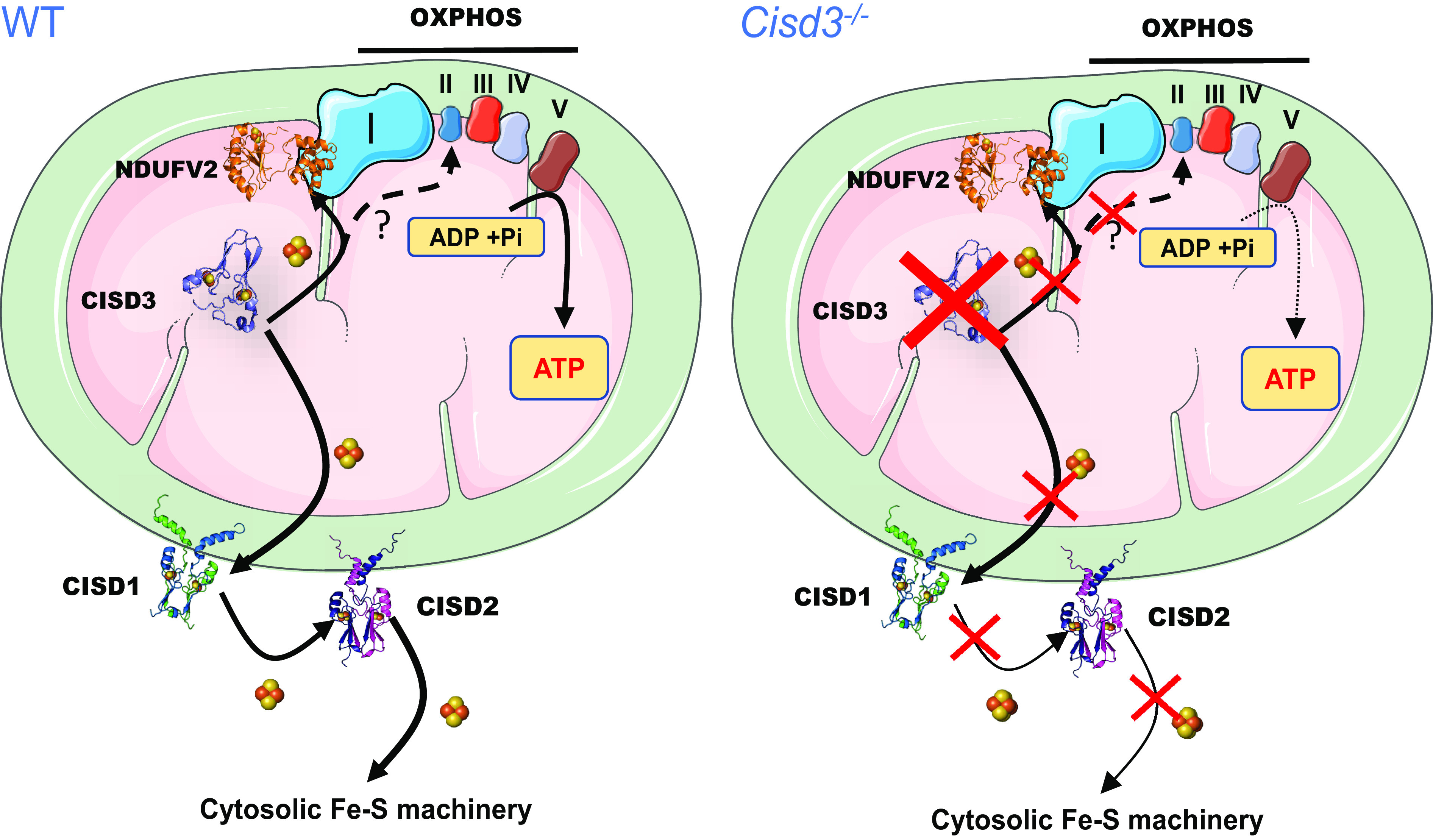
A model for the function of CISD3 in mitochondria from skeletal muscles of mice. CISD3 is shown to donate its [2Fe-2S] clusters to NDUFV2 (and perhaps other 2Fe-2S proteins of complexes I and II), thereby supporting the function of the mitochondrial respiratory chain in muscle mitochondria and enabling enhanced metabolic activity. Based on previous studies in cancer cells (22, 23), CISD3 is also proposed to function as a part of a [2Fe-2S] cluster relay that transfer clusters from within the mitochondria to the cytosol. In the absence of CISD3 the function of the mitochondrial respiratory chain is reduced, and mitochondria are more prone to iron overload and structural damage. Abbreviations: CISD, CDGSH Iron Sulfur Domain; NDUFV2, NADH:ubiquinone oxidoreductase core subunit V2; OXPHOS, oxidative phosphorylation; ROS, reactive oxygen species.
NDUFV2 encodes a 27 kDa subunit of the complex I N module (59, 60). Mutations in NDUFV2 were reported to cause hypertrophic cardiomyopathy and encephalopathy, as well as Leigh syndrome, a severe neurological and respiratory disorder that typically results in mortality (due to respiratory failure) at ages 2 to 3 (61–63). Our findings that CISD3 physically interacts with, and transfers its clusters to, NDUFV2 (Figs. 4 and 5 and SI Appendix, Figs. S5 and S6), and is important for supporting overall respiration and mitochondrial integrity (Fig. 2), could implicate CISD3 in some of the different conditions described above, associated with NDUFV2 deficiency, as well as in additional mitochondrial complex I deficiencies, such as fatal neonatal lactic acidosis, leukoencephalopathy, hepatopathy, cardiomyopathy, childhood‐onset mitochondrial encephalomyopathy, and stroke‐like episodes (63–65). The potential involvement of CISD3 in these and other presentations associated with mitochondrial/complex I function should be explored in future studies, as CISD3 could become a molecular marker for some of these conditions.
A common thread that appears to be shared by all three CISD/NEET proteins is their involvement in supporting enhanced metabolic activity of cells. This theme is evident in the important role CISD/NEET proteins play in disorders such as diabetes, cancer, and neurodegeneration, as well as in their role in muscle tissues and the germline of C. elegans (1–3, 66, 67). Interestingly, among the three CISD proteins, CISD3 is the most ancient and can be found in archaea and bacteria, as well as different eukaryotic cells and animals (33, 34, 68). Our findings that CISD3 plays an important role in supporting respiration in muscle tissues could shed light on the evolution of this protein and the role it played in the origin of the more “modern” CISD proteins. Once mitochondria became a part of eukaryotic cells, the function of CISD3 as a cluster donor to respiratory complexes could have been expanded via gene duplications to CISD1 and CISD2 (34), to support [2Fe-2S] metabolism in the cytosol, and CISD1/2 acquired additional cytosolic roles related to coordinating mitochondrial-cytosolic interactions. The fact that CISD3 is generally absent in plants (33, 34, 68), could suggest that once the chloroplast was acquired by eukaryotic cells, a different protein could have overtaken the role of CISD3, or that CISD1/2 that were already present in these early cells became the potential progenitors of the plant CISD proteins, as exemplified by the Arabidopsis thaliana AtNEET protein (8, 69, 70). The fact that our Cisd3−/− mice survived to 60 wk strongly suggests that additional molecular mechanisms can support, or at least partially replace, the function of CISD3 in mammalian cells (perhaps similar to the mechanisms replacing the function of this protein in plants). However, once the energetic demand from cells becomes high (as in the skeletal muscles of Cisd3−/− mice, or in cancer cells), over time, these alternative mechanisms may not be sufficient to replace CISD3 function, and mitochondrial integrity and function is hindered. In future studies, the identity of the alternative mechanisms that replace CISD3 function should be addressed as they may be highly important for manipulating the enhanced metabolic state of cells in the presence or absence of CISD3.
The similarity in proteomic patterns between Cisd3−/− mice and a mice model for DMD (Fig. 3) is intriguing. Duchenne and Becker muscular dystrophies (DMD and BMD) are caused by a mutation in DYSTROPHIN, a protein that links cytoskeletal F-actin with the extracellular matrix (71–74). Muscles without DYSTROPHIN are more sensitive to damage, resulting in progressive loss of muscle tissue and function (75). It is possible that the similarity between Cisd3−/− and DMD mice is due to the enhanced muscle damage in Cisd3−/− mice (a result of mitochondrial dysfunction and structural impairment; Fig. 2). Alternatively, it could be due to the decrease in DYSTROPHIN complex proteins in Cisd3−/− mice (SI Appendix, Fig. S4). It is also possible that at least part of the muscle damage in DMD mice is caused by respiratory complex failure, that is partially similar to the phenotype observed in Cisd3−/− mice (Fig. 2). Production and analyses of double Cisd3−/−/Dystrophin−/− mice may help unravel this question. Overall, the similarity between DMD and Cisd3−/− model mice supports a role for mitochondrial respiration impairment in DMD and could highlight broad avenues for the proposed gene therapy of DMD (75, 76). In future studies, it would also be interesting to generate muscle tissue-specific knockouts for CISD3/MiNT so that the deficiency of CISD3 in other tissues (e.g., nerve tissues) would be distinguished from the effects of CISD3 deficiency in muscles.
Several different drugs were shown to stabilize or destabilize the clusters of CISD1 and CISD2 proteins, including pioglitazone, used in the treatment of diabetes (28, 77–80). Due to the high similarity in structure between the different CISD proteins (1, 3), and considering the findings revealed by this work regarding the role of CISD3 in muscle function and performance (Fig. 6), it might be interesting to test the effect of some of these drugs on muscle performance in control mice. Such studies could be important in aging mice, as the levels of the different CISD proteins in muscle tissues decline with age (15, 54, 55, 57, 66, 67). Specific drugs could also be developed to target CISD3, and these could impact muscle degeneration syndromes, aging, and different sport applications.
Conclusions
Taken together, our study reveals a distinct physiological role for CISD3/MiNT as an [2Fe-2S] cluster donor important for supporting the biogenesis and function of complex I, essential for muscle maintenance and function. Interventions that target CISD3 could therefore impact different muscle degeneration syndromes, aging, and related conditions.
Materials and Methods
Production of Cisd3−/− Mice.
All animals used in this study were housed under pathogen-free barrier conditions in an animal care facility and received humane care in compliance with the Principles of Laboratory Animal Care formulated by the National Society of Medical Research and the Guide for the Care and Use of Laboratory Animals (https://www.ncbi.nlm.nih.gov/books/NBK54050/). All experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of Missouri (https://research.missouri.edu/acqa/acuc). C57BL/6NTac-Cisd3em1(floxed ex2)Davis heterozygous mice were generated by the University of Missouri Animal Modelling Core (https://research.missouri.edu/animal-modeling) and bred to homozygous status. C57BL/6NTac-Cisd3em1(floxed ex2)Davis mice were then bred to B6.C-Tg(CMV-cre)1Cgn/J mice from Jackson Laboratory (stock 006054) (81), and homozygous Cisd3−/− mice were generated (Fig. 1 A and B). The following primer sequences were used for genotyping the mice (Fig. 1 A and B): forward primer 5′- AGGACAGCCACCTGATTTCAAGC-3′, reverse primer 5′- CTAACCGTTAGCGTCAGAGATCAGG-3′, which produced a wild-type band of 304 bp and a mutant band of 162 bp (Fig. 1 A and B).
Cisd3−/− Mice Phenotyping.
The physical condition, body weight, and general condition of the animals were recorded on a weekly basis, until the day of the termination. Weight was measured by placing the mice in an empty sterilized pipet tip container, and data were recorded on Ohaus PS121 portable balance (SI Appendix, Fig. S1). The grip strength of control and Cisd3−/− mice was measured using a homemade wheel which consisted of a metal ball bearing roller on which a circular 2 mm thick loop was suspended on a support stand (25 cm of diameter at a height of 66 cm, SI Appendix, Fig. S7A). Mice were allowed to hang from the bottom of the wheel and timed until they fell off into a soft container. A 2 mm thick straight brass rod was also held by a support stand horizontally and time until mice fell off from it into a soft container was determined (SI Appendix, Fig. S7B). Male and female C57BL/6 J wild-type (Control) and Cisd3−/− mice were killed at the age of 47 weeks. Mice were placed under anesthesia using 4% isoflurane, killed, and subjected to quadriceps tissue collection.
Protein Blot Analysis.
Quadriceps tissue was collected, immediately flash-frozen in liquid nitrogen, and stored at −80 °C. Muscle tissues were lysed by grinding to a fine powder in liquid nitrogen and then resuspended in 20 mm Tris pH 8.0, 150 mm NaCl, 1% Triton 100-X, 10% glycerol, 5 mm EDTA, 5 ng mL−1 aprotinin, and 5 mm PMSF and kept on ice for 2 h with vortexing every 30 min. Following centrifugation at 20,000 g for 15 min, the supernatant was denatured with Laemmli sample buffer, as described in ref. 44. Protein content was determined by The Pierce 660 nm Protein Assay (catalog number 1861426). Following SDS-PAGE separation of equal amounts of proteins, proteins were transferred to nitrocellulose blots and incubated with antibodies (in-house produced) against CISD3, CISD2 and CISD (4, 22–27, 44, 45), and β-actin (Abcam). Peroxidase-conjugated Affinity Pure goat anti-rabbit IgG from Jackson ImmunoResearch Laboratories (Jackson ImmunoResearch) was used as secondary antibody.
H&E Staining of Muscle Tissue.
Quadriceps tissue was collected and snap-frozen by immersing it in liquid 2-methyl butane. After a quick evaporation time, the frozen tissues were completely covered with optimal cutting temperature (OCT) embedding compound in a cryomold and then immersed in liquid nitrogen until completely frozen. Twelve-micron sections were prepared using a cryostat (Leica CM1520) and subjected to H&E staining (82) at the Histopathology Department of the Veterinary Medical Diagnostic Laboratory (University of Missouri; https://vmdl.missouri.edu/).
TEM Analysis of Muscle Tissue.
Quadriceps tissue was collected, sliced into 1 mm sections, and fixed in 2% paraformaldehyde and 2% glutaraldehyde in 100 mM sodium cacodylate buffer pH 7.35. Tissue slices were then processed and analyzed by TEM at the Electron Microscopy Core Facility, University of Missouri (https://emc.missouri.edu/; see SI Appendix, Materials and Methods for more details).
Perls’ Prussian Blue Staining of Muscle Tissues.
Quadriceps tissues were fixed in 4% paraformaldehyde, processed for paraffin mounting, and sectioned (12 µm). Slides prepared from each sample were subjected to iron staining using Mallory’s iron stain (Perls’ Prussian blue) method (83). Slides were deparaffinized and rehydrated with distilled water; then, potassium ferrocyanide/HCL solution was added for 10 min. Percent (%) of stained cells per field were calculated in the Cisd3−/− group and compared to control. Data were acquired on an Invitrogen EVOS XL Core Configured Cell Imager with Mechanical Stage. Results are shown for males and females and presented as mean ± SD for six different animals (three different males and three different females) from each group (control and Cisd3−/−), as described in ref. 44.
Muscle Fiber Isolation.
Full quadriceps muscle tissues were collected, rinsed in PBS, and incubated in collagenase A (4 mg/mL) prepared in Dulbecco’s Modified Eagle Medium (D-MEM) high glucose, no sodium pyruvate or phenol red (Invitrogen, Waltham, MA, 21063-029), FBS (2%), and collagenase A (4 mg/mL; Sigma-Aldrich, COLLA-RO) pH 7.4 for 30 min. The fibers were separated using a wide bore p1000 pipette to yield single quadriceps myofibers in D-MEM Medium (Invitrogen, 21063-029), FBS (2%). The separated myofibers were then plated in an Agilent Seahorse XF24 Islet capture Microplates and covered with a grid to reduce the movement of the fibers and incubated for 1 h in an incubator at 37 °C and 5% CO2.
Seahorse Analysis of Muscle Fibers from Control and Cisd3−/− Mice.
For mitochondrial measurements, 1 h before the experiment, the fibers were placed in an incubator at 37 °C with no CO2, and the medium was replaced with XF Base media (Agilent) with glucose (10 mM), sodium pyruvate (1 mM), and L-glutamine (2 mM) (Gibco), pH 7.4 at 37 °C. Fiber oxygen consumption rate (OCR) was measured during the Seahorse Mito Stress assay (Agilent), with the addition of oligomycin (0.65 µM), carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP; 1.5 µM), and antimycin A and rotenone (1 μM each). Measured parameters were defined as follows: 3 min mix, 3 min wait, 3 min measurement, repeated three to four times at basal and after each addition. For glycolysis measurements, 1 h before the analysis, the fibers were placed in an incubator at 37 °C with no CO2, and the medium was replaced with XF Base media (Agilent), with sodium pyruvate (1 mM) and L-glutamine (2 mM) (Gibco), and without glucose at pH 7.4 at 37 °C). Fiber extracellular acidification rate (ECAR) was measured during a Seahorse Glycolysis Stress assay (Agilent) with the addition of glucose (10 mM), oligomycin (2 µM), and 2-Deoxy-D-glucose (2-DG: 50 µM). Measured parameters were defined as follows: 3 min mix, 3 min wait, 3 min measurement, repeated three to four times at basal and after each addition.
Proteomic Analysis.
Quadriceps tissue was collected, immediately flash-frozen in liquid nitrogen, and stored at −80 °C. Muscle tissues were lysed by grinding to a fine powder in liquid nitrogen and resuspending in 20 mM Tris pH 8.0, 150 mm NaCl, 1% Triton 100-X, 10% glycerol, 5 mm EDTA, 5 ng mL−1 aprotinin, and 5 mM PMSF and kept on ice for 2 h with mixing every 30 min. Following centrifugation at 20,000 g for 15 min, the supernatant was denatured with Laemmli sample buffer, as described in ref. 38. The proteins were precipitated with cold acetone and stored at −20 °C overnight. Protein pellets were recovered by centrifugation at 16,000 g for 10 min at 4 °C, washed with 80% acetone, and subjected to proteomics analysis at the Proteomics Core Facility, University of Missouri (https://research.missouri.edu/gehrke-proteomics-center; see SI Appendix, Materials and Methods for more details).
Proteomic Data Analysis.
For comparative analysis, we employed Spectronaut version 14.1 (Biognosys) and utilized the default search unless otherwise indicated. All data were searched against the reviewed mouse proteome (UniProt UP000000589), using trypsin/LysC as the digestion enzymes. Cysteine carbamidomethylation was set as a fixed modification, while methionine oxidation and acetylation at the N terminus were selected as variable modifications. A maximum of two missed cleavages and up to three variable modifications were allowed. The FDR cutoff was established at 1%, while the precursor peptide and q-value cutoffs were set at 0.2 and 0.01, respectively. In addition, protein q-value experiment and run-wide cutoffs were determined to be 0.01 and 0.05, respectively. Protein quantification was performed by filtering precursors using a q-value approach, using imputation with the background signal. The prototypicity filter was set to only protein group-specific peptides. Protein quantification was performed using MaxLFQ based on the area of MS2, with cross-run normalization enabled. Differential abundance testing was performed using unpaired t tests and postanalysis based on both MS level on Spectronaut. In our analysis, we used a significance threshold of q value < 0.05, and an absolute average log2 (fold change) > 0.58 to identify significant candidates. Proteomics data were deposited in the Pride ProteomeXchange database (https://www.ebi.ac.uk/pride/) under the identifier PXD042593.
Mitochondria Isolation.
Quadriceps muscle tissues were collected, flash-frozen in liquid nitrogen, and ground to powder in liquid nitrogen using a mortar and pestle. The powder was dissolved in an isolation buffer (IB) containing 0.22 M mannitol, 0.07 M sucrose, 0.002 M Tris-HCL, 0.001 M EDTA, 0.4% BSA, and 20 mM Hepes at pH 7,5 and homogenized using a glass pestle in 1.7 mL Microtubes (Axygen) with 10 strokes. The sample was then centrifuged 10 min at 800 g, and the pellet was discarded. The supernatant was then centrifuged for 10 min at 14,000 g. The supernatant was discarded, and the pellet containing the mitochondrial fraction was resuspended in IB.
Coimmunoprecipitation.
A day before the assay, 25 µL protein A magnetic beads S1425S (New England Biolabs) were washed three times in PBS and incubated overnight with 2 to 3 µg of antibody directed against CISD34 at 4 °C with gentle mix. On the day of the assay, freshly isolated purified mitochondria were washed and pelleted for 10 min at 14,000 g. The mitochondria were resuspended in a lysis buffer containing 1% digitonin, 0.1% Noniped40, 0.15 M NaCl, 0.001 M EDTA, 0.05 M Hepes pH 7.5, and 0.00125 M dithiobis(succinimidyl propionate) (DSP) for 1 h at 4 °C with gentle mix. The mitochondria were then centrifuged at 14,000 g, the supernatant containing the protein extract was collected, and protein content was determined with The Pierce 660 nm Protein Assay (catalog number 1861426). Then, 4 mg/mL protein was incubated with the coated beads with a gentle mix for four 4 h at 4 °C. The beads were there precipitated and washed five times with PBS and analyzed by MS and protein blots.
Protein Expression and Purification.
CISD3 (residues 36 to 127) and NDUFV2 (residues 32 to 249) cDNAs were inserted in the expression vector pet-28a+ (Novagen). Both proteins were expressed in Escherichia coli BL21-RIL grown in lysogeny broth supplemented with 30 μg/mL kanamycin and 34 μg/mL chloramphenicol. At an O.D. 600 nm of 0.6, the cells were supplemented with 0.75 mM FeCl3 (only for NDUFV2 protein), and protein expression was activated using 0.25 mM of isopropyl ß-D-1-thiogalactopyranoside. Cell growth proceeded for an additional 12 h at 37 °C. NDUFV2 was purified from lysed cells using Ni-agarose and size exclusion chromatography as described (22, 77). CISD3 was purified by two consecutive rounds of ion exchange chromatography followed by size exclusion chromatography as described (4). Apo-NDUFV2 was obtained by incubating the purified protein with EDTA and ferricyanide in the following molar ratio, 1:50:20; until the protein became colorless, and the protein was then dialyzed against 20 mM Tris pH 8.0 and 100 mM NaCl (84).
Protein–Protein Interaction.
First, 100 µM Holo-CISD3 and 100 µM Holo-NDUFV2, prepared as described above, were incubated in 20 mM Tris pH 8.0 and 100 mM NaCl for 10 min, and the mixture was then denatured with or without β-mercaptoethanol for 6 h. Then, the samples were separated using a 15% SDS-acrylamide gel. The band corresponding to the molecular weight of the complex CISD3–NDUFV2 was excised and analyzed by mass spectrometry.
[2Fe-2S] Cluster Transfer Assay.
Apo-NDUFV2 (250 µm) was prereduced in the presence of 5 mM Na-dithionite and 5 mM Na2-EDTA, pH 8.0, for 60 min. Apo-NDUFV2 was then incubated with holo-CISD3 (250 µm) in 50 mM Tris pH 8.0, 100 mM NaCl, 5 mM DTT, and 5 mM EDTA, and [2Fe–2S] cluster transfer was analyzed by absorption spectroscopy from 380 nm to 700 nm at 37 °C as described in ref. 4. Data were acquired using a BioTek Synergy H1 Multimode Reader (Agilent).
Data Extraction and Processing.
Seed sequences for the two proteins, CISD3 (UniProt ID: P0C7P0) and NDUFV2 (UniProt ID: P19404), were obtained in FASTA format from UniProt. The entire sequence for both proteins was used, including the protein domain portions, as input parameters in Profile HMMER (phmmer) to produce associated HMM files and Multiple Sequence Alignment (MSA) FASTA files. Each MSA consisted of an alignment of the respective seed sequence and homologous sequences belonging to the same protein family from multiple organisms. Both MSAs were refined by eliminating sequences exceeding a certain number of maximum gaps, with refinements excluding approximately 5% of the original number of entries. Protein structures of CISD3 (PDB: 6AVJ, chain A) and NDUFV2 (PDB: 5XTD, chain O) were obtained as PDB files from RCSB. The MSAs of both proteins were truncated to match the residue sequence and length of their respective PDB file. The atoms and residues of PDB files were renumbered where CISD3 residues ranged from 1 to 92 and NDUFV2 residues ranged from 93 to 304.
Computational Modeling of the Interaction between CISD3 and NDUFV2.
Protein concatenation procedure and direct coupling analysis (38, 49), structure-based model molecular dynamics simulation (39), alignment of individual crystal structures to the SBM+DCA predicted complex (85), protonation and amber minimization (86, 87), and protein frustratometer analysis and AlphaFold multimer structure prediction (50, 88, 89) are described in detail in SI Appendix, Materials and Methods.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Dataset S07 (XLSX)
Movie of a GROMACS-generated trajectory of the CISD3-NDUFV2 complex from the SBM+DCA simulation with coevolutionary constraints.
Video of the trajectory of the CISD3-NDUFV2 complex depicting a morph from the Amber minimized SBM+DCA predicted complex (red-blue) to the Amber minimized AlphaFold Multimer CISD3-NDUFV2 complex (green-pink).
Acknowledgments
This work was supported by the NIH grant GM111364 (to R.M.), the NSF-Binational Science Foundation (BSF) Grant NSF-MCB 1613462 (to R.M.) and BSF Grant 2015831 (to R.N.), and The University of Missouri. J.N.O. is supported by the Center for Theoretical Biological Physics sponsored by the NSF (Grant PHY-2019745), by the Welch Foundation (Grant C-1792), and by NSF-PHY-2210291. J.N.O. is a CPRIT Scholar in Cancer Research sponsored by the Cancer Prevention and Research Institute of Texas. F.M. and S.M. acknowledge support from NSF CAREER (Grant MCB-1943442) and the NIH NIGMS Grant R35GM133631 (to F.M.). We thank the Animal Modeling, Proteomics, Histopathology, and Electron Microscopy Research Core Facilities at the University of Missouri, Columbia.
Author contributions
R.N., C.M.-A., F.M., J.N.O., and R.M. designed research; L.R., O.K., H.-B.M., T.T.N., S.M., R.S.A., and D.G. performed research; L.R., O.K., and R.M. contributed new reagents/analytic tools; R.N., L.R., O.K., H.-B.M., T.T.N., S.M., R.S.A., D.G., C.M.-A., F.M., J.N.O., and R.M. analyzed data; and R.N., C.M.-A., F.M., J.N.O., and R.M. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
Reviewers: M.L.P., University of California San Diego; and P.D.L.R., Ecole polytechnique federale de Lausanne Faculte des sciences de base.
Contributor Information
José N. Onuchic, Email: jonuchic@rice.edu.
Ron Mittler, Email: mittlerr@missouri.edu.
Data, Materials, and Software Availability
Protemics data have been deposited in ProteomeXchange (PXD042593) (90).
Supporting Information
References
- 1.Nechushtai R., et al. , The balancing act of NEET proteins: Iron, ROS, calcium and metabolism. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 1867, 118805 (2020). [DOI] [PubMed] [Google Scholar]
- 2.Mittler R., et al. , NEET proteins: A new link between iron metabolism, reactive oxygen species, and cancer. Antioxid. Redox Signal. 30, 1083–1095 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tamir S., et al. , Structure–function analysis of NEET proteins uncovers their role as key regulators of iron and ROS homeostasis in health and disease. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 1853, 1294–1315 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Lipper C. H., et al. , Structure of the human monomeric NEET protein MiNT and its role in regulating iron and reactive oxygen species in cancer cells. Proc. Natl. Acad. Sci. U.S.A. 115, 272–277 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tamir S., et al. , A point mutation in the [2Fe-2S] cluster binding region of the NAF-1 protein (H114C) dramatically hinders the cluster donor properties. Acta Crystallogr. D Biol. Crystallogr. 70, 1572–1578 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conlan A. R., et al. , Crystal structure of Miner1: The redox-active 2Fe-2S protein causative in wolfram syndrome 2. J. Mol. Biol. 392, 143–153 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paddock M. L., et al. , MitoNEET is a uniquely folded 2Fe-2S outer mitochondrial membrane protein stabilized by pioglitazone. Proc. Natl. Acad. Sci. U.S.A. 104, 14342–14347 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nechushtai R., et al. , Characterization of Arabidopsis NEET reveals an ancient role for NEET proteins in iron metabolism. Plant Cell 24, 2139–2154 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geldenhuys W. J., et al. , MitoNEET (CISD1) knockout mice show signs of striatal mitochondrial dysfunction and a Parkinson’s disease phenotype. ACS Chem. Neurosci. 8, 2759–2765 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kusminski C. M., Park J., Scherer P. E., MitoNEET-mediated effects on browning of white adipose tissue. Nat. Commun. 5, 1–14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Furihata T., et al. , Cardiac-specific loss of mitoNEET expression is linked with age-related heart failure. Commun. Biol. 4, 1–11 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang N. C., et al. , Bcl-2-associated autophagy regulator Naf-1 required for maintenance of skeletal muscle. Hum. Mol. Genet. 21, 2277–2287 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Huang Y.-L., et al. , Cisd2 slows down liver aging and attenuates age-related metabolic dysfunction in male mice. Aging Cell 20, e13523 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang C.-H., Kao C.-H., Chen Y.-F., Wei Y.-H., Tsai T.-F., Cisd2 mediates lifespan: Is there an interconnection among Ca2+ homeostasis, autophagy, and lifespan? Free Radic. Res. 48, 1109–1114 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Chen Y.-F., et al. , Cisd2 deficiency drives premature aging and causes mitochondria-mediated defects in mice. Genes Dev. 23, 1183–1194 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu C.-Y., et al. , A persistent level of Cisd2 extends healthy lifespan and delays aging in mice. Hum. Mol. Genet. 21, 3956–3968 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Li R., et al. , CDGSH iron sulfur domain 2 over-expression alleviates neuronal ferroptosis and brain injury by inhibiting lipid peroxidation via AKT/mTOR pathway following intracerebral hemorrhage in mice. J. Neurochem. 165, 426–444 (2023). [DOI] [PubMed] [Google Scholar]
- 18.Loncke J., Vervliet T., Parys J. B., Kaasik A., Bultynck G., Uniting the divergent Wolfram syndrome–linked proteins WFS1 and CISD2 as modulators of Ca2+ signaling. Sci. Signal 14, 6165 (2021). [DOI] [PubMed] [Google Scholar]
- 19.Rosanio F. M., et al. , Wolfram syndrome type 2: A systematic review of a not easily identifiable clinical spectrum. Int. J. Environ. Res. Public Health 19, 835 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Danielpur L., et al. , GLP-1-RA corrects mitochondrial labile iron accumulation and improves β-cell function in Type 2 wolfram syndrome. J. Clin. Endocrinol. Metab. 101, 3592–3599 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Grifagni D., Silva J. M., Cantini F., Piccioli M., Banci L., Relaxation-based NMR assignment: Spotlights on ligand binding sites in human CISD3. J. Inorg. Biochem. 239, 112089 (2023). [DOI] [PubMed] [Google Scholar]
- 22.Karmi O., et al. , A VDAC1-mediated NEET protein chain transfers [2Fe-2S] clusters between the mitochondria and the cytosol and impacts mitochondrial dynamics. Proc. Natl. Acad. Sci. U.S.A. 119, e2121491119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lipper C. H., et al. , Redox-dependent gating of VDAC by mitoNEET. Proc. Natl. Acad. Sci. U.S.A. 116, 19924–19929 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lipper C. H., et al. , Cancer-related NEET proteins transfer 2Fe-2S clusters to Anamorsin, a protein required for cytosolic iron-sulfur cluster biogenesis. PLoS One 10, e0139699 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karmi O., et al. , Interactions between mitoNEET and NAF-1 in cells. PLoS One 12, e0175796 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sohn Y.-S., et al. , NAF-1 and mitoNEET are central to human breast cancer proliferation by maintaining mitochondrial homeostasis and promoting tumor growth. Proc. Natl. Acad. Sci. U.S.A. 110, 14676–14681 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karmi O., et al. , Disrupting CISD2 function in cancer cells primarily impacts mitochondrial labile iron levels and triggers TXNIP expression. Free Radic. Biol. Med. 176, 92–104 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Y., et al. , CISD3 inhibition drives cystine-deprivation induced ferroptosis. Cell Death Dis. 12, 1–15 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manz D. H., et al. , Iron and cancer: Recent insights. Ann. N Y Acad. Sci. 1368, 149–161 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salnikow K., Role of iron in cancer. Semin. Cancer Biol. 76, 189–194 (2021). [DOI] [PubMed] [Google Scholar]
- 31.King S. D., Gray C. F., Song L., Mittler R., Padilla P. A., The mitochondrial localized CISD-3.1/CISD-3.2 proteins are required to maintain normal germline structure and function in Caenorhabditis elegans. PLoS One 16, e0245174 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.King S. D., et al. , The cisd gene family regulates physiological germline apoptosis through ced-13 and the canonical cell death pathway in Caenorhabditis elegans. Cell Death Differ. 26, 162–178 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sengupta S., et al. , Phylogenetic analysis of the CDGSH iron-sulfur binding domain reveals its ancient origin. Sci. Rep. 8, 4840 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Inupakutika M. A., et al. , Phylogenetic analysis of eukaryotic NEET proteins uncovers a link between a key gene duplication event and the evolution of vertebrates. Sci. Rep. 7, 42571 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malinverni D., Jost Lopez A., De Los Rios P., Hummer G., Barducci A., Modeling Hsp70/Hsp40 interaction by multi-scale molecular simulations and coevolutionary sequence analysis. Elife 6, e23471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weigt M., White R. A., Szurmant H., Hoch J. A., Hwa T., Identification of direct residue contacts in protein-protein interaction by message passing. Proc. Natl. Acad. Sci. U.S.A. 106, 67–72 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dos Santos R. N., Morcos F., Jana B., Andricopulo A. D., Onuchic J. N., Dimeric interactions and complex formation using direct coevolutionary couplings. Sci. Rep. 5, 13652 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morcos F., et al. , Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc. Natl. Acad. Sci. U.S.A. 108, E1293–E1301 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Noel J. K., et al. , SMOG 2: A versatile software package for generating structure-based models. PLoS Comput. Biol. 12, e1004794 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tamir S., et al. , An integrated strategy reveals the protein interface between the cancer targets Bcl-2 and NAF-1. Proc. Natl. Acad. Sci. U.S.A. 111, 5177–5182 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jeneson J. A. L., et al. , Treadmill but not wheel running improves fatigue resistance of isolated extensor digitorum longus muscle in mice. Acta Physiol. 190, 151–161 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Tassinari V., et al. , Atrophy, oxidative switching and ultrastructural defects in skeletal muscle of the ataxia telangiectasia mouse model. J. Cell Sci. 132, jcs223008 (2019). [DOI] [PubMed] [Google Scholar]
- 43.Li W., et al. , Dysfunction of mitochondria and deformed gap junctions in the heart of IL-18-deficient mice. Am. J. Physiol. Heart Circ. Physiol. 311, H313–H325 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Karmi O., et al. , The [2Fe-2S] protein CISD2 plays a key role in preventing iron accumulation in cardiomyocytes. FEBS Lett. 596, 747–761 (2022). [DOI] [PubMed] [Google Scholar]
- 45.Holt S. H., et al. , Activation of apoptosis in NAF-1-deficient human epithelial breast cancer cells. J. Cell Sci. 129, 155–165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Westering T. L. E., et al. , Mutation-independent proteomic signatures of pathological progression in murine models of duchenne muscular dystrophy. Mol. Cell Proteomics 19, 2047–2068 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Skotte N. H., et al. , Integrative characterization of the R6/2 mouse model of Huntington’s disease reveals dysfunctional astrocyte metabolism. Cell Rep. 23, 2211–2224 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Hasan M., et al. , Quantitative proteome analysis of brain subregions and spinal cord from experimental autoimmune encephalomyelitis mice by TMT-based mass spectrometry. Proteomics 19, 1800355 (2019). [DOI] [PubMed] [Google Scholar]
- 49.Gueudré T., Baldassi C., Zamparo M., Weigt M., Pagnani A., Simultaneous identification of specifically interacting paralogs and interprotein contacts by direct coupling analysis. Proc. Natl. Acad. Sci. U.S.A. 113, 12186–12191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Evans R., et al. , Protein complex prediction with alphaFold-multimer. bioRxiv [Preprint] (2021). 10.1101/2021.10.04.463034 (Accessed 2 January 2024). [DOI]
- 51.Pettersen E. F., et al. , UCSF chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 52.Jiang H., et al. , FGF4 improves hepatocytes ferroptosis in autoimmune hepatitis mice via activation of CISD3. Int. Immunopharmacol. 116, 109762 (2023). [DOI] [PubMed] [Google Scholar]
- 53.Darash-Yahana M., et al. , Breast cancer tumorigenicity is dependent on high expression levels of NAF-1 and the lability of its Fe-S clusters. Proc. Natl. Acad. Sci. U.S.A. 113, 10890–10895 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yokokawa T., et al. , Exercise training increases CISD family protein expression in murine skeletal muscle and white adipose tissue. Biochem. Biophys. Res. Commun. 506, 571–577 (2018). [DOI] [PubMed] [Google Scholar]
- 55.Teng Y.-C., Wang J.-Y., Chi Y.-H., Tsai T.-F., Exercise and the Cisd2 prolongevity gene: Two promising strategies to delay the aging of skeletal muscle. Int. J. Mol. Sci. 21, 9059 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang Y.-L., et al. , Comparative proteomic profiling reveals a role for Cisd2 in skeletal muscle aging. Aging Cell 17, e12705 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yeh C.-H., et al. , Cisd2 is essential to delaying cardiac aging and to maintaining heart functions. PLoS Biol. 17, e3000508 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Habener A., et al. , MitoNEET protects HL-1 cardiomyocytes from oxidative stress mediated apoptosis in an in vitro model of hypoxia and reoxygenation. PLoS One 11, e0156054 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pamplona R., Jové M., Mota-Martorell N., Barja G., Is the NDUFV2 subunit of the hydrophilic complex I domain a key determinant of animal longevity? FEBS J. 288, 6652–6673 (2021). [DOI] [PubMed] [Google Scholar]
- 60.Hirst J., Mitochondrial complex I. Annu. Rev. Biochem. 82, 551–575 (2013). [DOI] [PubMed] [Google Scholar]
- 61.Tang X., et al. , Compound heterozygous mutations of NDUFV1 identified in a child with mitochondrial complex I deficiency. Genes Genomics 44, 691–698 (2022). [DOI] [PubMed] [Google Scholar]
- 62.Kishita Y., et al. , Genome sequencing and RNA-seq analyses of mitochondrial complex I deficiency revealed Alu insertion-mediated deletion in NDUFV2. Hum. Mutat. 42, 1422–1428 (2021). [DOI] [PubMed] [Google Scholar]
- 63.Fiedorczuk K., Sazanov L. A., Mammalian mitochondrial complex I structure and disease-causing mutations. Trends Cell Biol. 28, 835–867 (2018). [DOI] [PubMed] [Google Scholar]
- 64.Zanette V., et al. , NDUFV1 mutations in complex I deficiency: Case reports and review of symptoms. Genet. Mol. Biol. 44, e20210149 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Scheffler I. E., Mitochondrial disease associated with complex I (NADH-CoQ oxidoreductase) deficiency. J. Inherit. Metab. Dis. 38, 405–415 (2015). [DOI] [PubMed] [Google Scholar]
- 66.Shen Z.-Q., et al. , CISD2 maintains cellular homeostasis. Biochim. Biophys. (BBA) Acta Mol. Cell Res. 1868, 118954 (2021). [DOI] [PubMed] [Google Scholar]
- 67.Chen Y.-F., et al. , A role for the CISD2 gene in lifespan control and human disease. Ann. N Y Acad. Sci. 1201, 58–64 (2010). [DOI] [PubMed] [Google Scholar]
- 68.Lin J., Zhang L., Lai S., Ye K., Structure and molecular evolution of CDGSH iron-sulfur domains. PLoS One 6, e24790 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Su L.-W., et al. , Purification and biochemical characterization of Arabidopsis At-NEET, an ancient iron-sulfur protein, reveals a conserved cleavage motif for subcellular localization. Plant Sci. 213, 46–54 (2013). [DOI] [PubMed] [Google Scholar]
- 70.Zandalinas S. I., et al. , Expression of a dominant-negative AtNEET-H89C protein disrupts iron–sulfur metabolism and iron homeostasis in Arabidopsis. Plant J. 101, 1152–1169 (2020). [DOI] [PubMed] [Google Scholar]
- 71.Mercuri E., Bönnemann C. G., Muntoni F., Muscular dystrophies. Lancet 394, 2025–2038 (2019). [DOI] [PubMed] [Google Scholar]
- 72.Flanigan K. M., Duchenne and Becker muscular dystrophies. Neurol. Clin. 32, 671–688 (2014). [DOI] [PubMed] [Google Scholar]
- 73.Salari N., et al. , Global prevalence of Duchenne and Becker muscular dystrophy: A systematic review and meta-analysis. J. Orthop. Surg Res. 17, 1–12 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pascual-Morena C., et al. , Epileptic disorders in Becker and Duchenne muscular dystrophies: A systematic review and meta-analysis. J. Neurol. 269, 3461–3469 (2022). [DOI] [PubMed] [Google Scholar]
- 75.Duan D., Goemans N., Takeda S., Mercuri E., Aartsma-Rus A., Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 7, 1–19 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Elangkovan N., Dickson G., Gene therapy for Duchenne muscular dystrophy. J. Neuromuscul. Dis. 8, S303–S316 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Marjault H.-B., et al. , An anti-diabetic drug targets NEET (CISD) proteins through destabilization of their [2Fe-2S] clusters. Commun. Biol. 5, 1–9 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bai F., et al. , The Fe-S cluster-containing NEET proteins mitoNEET and NAF-1 as chemotherapeutic targets in breast cancer. Proc. Natl. Acad. Sci. U.S.A. 112, 3698–3703 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee S., Seok B. G., Lee S.-J., Chung S. W., Inhibition of mitoNEET attenuates LPS-induced inflammation and oxidative stress. Cell Death Dis. 13, 1–9 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Marjault H.-B., et al. , Structure-based screening reveals a ligand that stabilizes the [2Fe-2S] clusters of human mitoNEET and reduces ovarian cancer cell proliferation. J. Phys. Chem. B 126, 9559–9565 (2022). [DOI] [PubMed] [Google Scholar]
- 81.Schwenk F., Baron U., Rajewsky K., A cre -transgenic mouse strain for the ubiquitous deletion of loxP -flanked gene segments including deletion in germ cells. Nucleic Acids Res. 23, 5080–5081 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Drury R. A. B., Theory and practice of histotechnology. J. Clin. Pathol. 34, 1406 (1981). [Google Scholar]
- 83.Gössl M., et al. , Segmental heterogeneity of vasa vasorum neovascularization in human coronary atherosclerosis. JACC Cardiovasc. Imaging 3, 32–40 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kennedy M. C., Beinert H., The state of cluster SH and S2- of aconitase during cluster interconversions and removal. A convenient preparation of apoenzyme. J. Biol. Chem. 263, 8194–8198 (1988). [PubMed] [Google Scholar]
- 85.The PyMOL Molecular Graphics System, Version 2.5.1 Schrödinger, LLC.
- 86.Anandakrishnan R., Aguilar B., Onufriev A. V., H++ 3.0: Automating p K prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 40, W537–W541 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Maghsoud Y., Dong G. C., Cisneros A., Computational characterization of the inhibition mechanism of Xanthine oxidoreductase by topiroxostat. ACS Catal. 13, 6023–6043 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gonzalo Parra R., et al. , Protein Frustratometer 2: A tool to localize energetic frustration in protein molecules, now with electrostatics. Nucleic Acids Res. 44, W356–W360 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jumper J., et al. , Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nguyen T. T., CISD3 is required for Complex I function, mitochondrial integrity, and skeletal muscle maintenance. PRIDE. https://www.ebi.ac.uk/pride/. Deposited 31 May 2023. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Dataset S07 (XLSX)
Movie of a GROMACS-generated trajectory of the CISD3-NDUFV2 complex from the SBM+DCA simulation with coevolutionary constraints.
Video of the trajectory of the CISD3-NDUFV2 complex depicting a morph from the Amber minimized SBM+DCA predicted complex (red-blue) to the Amber minimized AlphaFold Multimer CISD3-NDUFV2 complex (green-pink).
Data Availability Statement
Protemics data have been deposited in ProteomeXchange (PXD042593) (90).