

Abstract
High Mobility Group Box 1 (HMGB1) is a ubiquitous, highly conserved nuclear and cytosolic protein that has diverse biological roles depending on its cellular location and posttranslational modifications. The HMGB1 is localized in the nucleus but can be translocated to the cytoplasm to modulate the intracellular signaling and eventually secreted outside the cells. It is widely established that HMGB1 plays a key role in inflammation; however, the role of HMGB1 in the cardiovascular diseases is not well understood. In this review, we will discuss the latest reports on the pathophysiological link between HMGB1 and cardiovascular complications, with special emphasis on the inflammation. Thus, the understanding of the role of HMGB1 may provide new insights into developing new HMGB1-based therapies.
Keywords: HMGB1, inflammation, cardiovascular, diabetes, angiogenesis
INTRODUCTION
High Mobility Group Box 1 is a non-histone DNA-binding protein and a member of the High Mobility Group (HMG) protein superfamily. HMG proteins were identified for the first time in 1973 by Goodwin and named according to their high mobility in polyacrylamide gel electrophoresis [1]. HMG proteins are divided in 3 groups: HMGA, HMGB, and HMGN [1]. HMGB1 belongs to the HMGB group, constitutively expressed in the nucleus of almost every cell type, highly conserved in mammalians species, and it is the most studied. HMGB1 knockout mice die shortly after birth due to a severe hypoglycemia, highlighting the importance of HMGB1 in survival [2]. HMGB1 can be actively secreted by activated immune cells including macrophages, monocytes, dendritic cells, epithelial cells, or passively released by dying or injured cells [3–5].
HMGB1 is organized into 3 functional domains, two positively charged DNA-binding domain called Box-A and Box-B and a negatively charged C-terminal acidic domain [6]. Boxes A and B have opposite biological properties: Box A is anti-inflammatory, while Box B is pro-inflammatory and consists of two binding sites for Toll-like receptor 4 (TLR4) and receptor for advanced glycation end products (AGE) [7]. HMGB1 has two nuclear localization sequences (NLS) located in Box-A and between Box-B and the C-terminal region [7]. The acetylation of the lysine residues presents in the NLS leads to the HMGB1 translocation from nucleus to the cytosol and its release. Due to its ability to translocate from the nucleus to the cytosol, HMGB1 can be found in the cytosol, the mitochondria, and on cell membrane [8]. In the nucleus, HMGB1 plays a major role in DNA repair, DNA replication, and chromatin structure. In the cytoplasm, HMGB1 acts as a signaling regulator, alarmin, and pro-inflammatory cytokine [9].
It has been reported that significant amounts of extracellular HMGB1 released after cellular activation (monocytes, macrophages, and dendritic cells) through the increased translocation of HMGB1 from the nucleus to cytoplasm or cell death leads to inflammation [10]. The translocation of HMGB1 from nuclear to cytoplasm appears to depend on its acetylation and methylation on lysine residues [11, 12]. Reports showed that not only HMGB1 is released in response to inflammatory stimuli but HMGB1 also triggers a variety of signaling pathways and activate immune cells to release proinflammatory cytokines and chemokines [13]. HMGB1 acts as an inflammatory cytokine under many conditions such as ischemia [14, 15] and seems to depend on the location, the redox state, and the associated signaling pathway involved [16, 17]. HMGB1 has several receptors such the receptors for RAGE, TLR9, TLR4, TLR2, CD24, CXCR4, and others. Only TLR4 and RAGE were extensively studied and confirmed by many investigators [16, 17] RAGE is one of the receptors that binds to HMGB1 and activates smooth muscle cells, immune cells, and the upregulation of RAGE and TLR4 on cells surface. Interestingly, HMGB1 interacts with both TLR4 and RAGE but only its interaction with TLR4 is required for the release of cytokines from macrophages [18].
CARDIAC INFLAMMATION
Cardiac Hypertrophy
Evidence is accumulating that chronic inflammation plays an important role in cardiac diseases. HMGB1 has been associated with many cardiac complications [19, 20]. Inflammation is involved in the progression of pressure overload–induced cardiac hypertrophy and heart failure; however, the mechanisms have not yet been fully determined [21]. It has been reported that HMGB1 expression was increased, during cardiac hypertrophy induced by pressure overload, accompanied with its translocation from the nucleus to the cytoplasm [22]. Moreover, exogenous HMGB1 induces hypertrophy in neonatal ventricular myocytes through calcineurin mechanism [23] while overexpression of nuclear HMGB1 prevents pressure overload–induced cardiac hypertrophy through inhibition of DNA damage mechanism [24]. Human biopsy samples showed a decrease in nuclear HMGB1 expression in failing hearts. The authors also found that nuclear HMGB1 was acetylated and translocated from the nucleus to the cytoplasm during cardiac remodeling. Moreover, cardiac hypertrophy induced by transverse aortic constriction (TAC) was attenuated in HMGB1 transgenic mice suggesting a novel approach to target nuclear HMGB1 in preventing cardiac hypertrophy and improving survival rate in a pressure overload heart failure model [24].
Myocardial Infarction
In a rodent model of myocardial infarction, exogenous HMGB1 has a beneficial effect on post infarct cardiac remodeling via the induction of myocardial regeneration through the induction of resident cardiac c-kit + cell proliferation and differentiation mechanism [25]. The overexpression of HMGB1 seems to protect the heart against myocardial infarction [26]. However, other reports showed that HMGB1 treatment increased infarct size, while mice lacking RAGE were protected from myocardial ischemia reperfusion injury [19].
Myocarditis
Recent reports showed that the immune system plays an important role in the pathology of heart failure [27]. Myocarditis, the leading cause of heart failure in young patients, is defined as an inflammation of the myocardium often leading to cardiomyopathy [28]. The exact mechanism involved in the induction of myocarditis and its progression is not yet fully understood. Studies showed that auto-antibodies to cardiac troponin-I are found in patients with acute coronary syndrome pointing to an early induction of an autoimmune response to troponin-I in these patients [29]. Goser et al. are the first to establish a mouse model in which immunization with murine cardiac troponin-I-induced myocardial inflammation, cardiac fibrosis, and heart failure [30]. Using the same murine model of troponin-I-induced experimental autoimmune myocarditis, Bangert et al. confirmed Goser et al.’s findings that troponin-I immunization induced myocardial inflammation, fibrosis, heart failure, and an increase in serum and myocardial HMGB1 [31]. Interestingly, the inhibition of HMGB1 by glycyrrhizin or anti-HMGB1 antibody reduced troponin-I-induced myocardial inflammation. Moreover, cardiac overexpression of HMGB1-induced cardiac inflammation in both wildtype and RAGE knockout mice suggests that the pro-inflammatory effect of HMGB1 is independent of RAGE-dependent mechanism. Importantly, RAGE knockout mice, immunized with troponin-I, were protected from cardiac inflammation supporting the concept that HMGB1-induced inflammation is independent of RAGE. Moreover, the authors found that HMGB1 and soluble RAGE levels were elevated in patients with myocarditis suggesting the clinical relevance of HMGB1 and RAGE in cardiac inflammation [31]. Additionally, HMGB1 could contribute to autoimmune myocarditis via a mechanism involving the stimulation of macrophage reprogramming toward a pro-inflammatory M1-like phenotype [32].
Heart Failure and Cardiac Fibrosis
Interestingly, high level of HMGB1 has been observed in patients with heart failure [33] suggesting the potential importance of HMGB1 in heart failure. Interestingly, cardiac-specific deletion of HMGB1 using c-TNT-CRE mouse system resulted in cardiomyopathy which further led to heart failure and impaired body growth [34]. The deletion of HMGB1 in cardiomyocyte was associated with low blood glucose and changes in inflammatory genes, glucocorticoid receptor’s function, and glycolipid metabolism [34]. These findings might explain why the deletion of HMGB1 in mice die prematurely of severe hypoglycemia [2]. Cardiac fibrosis is another feature of heart disease. In a mouse model of cardiac fibrosis induced by isoproterenol infusion, Wu et al. showed that the interaction between HMGB1 and TLR2 contributes to cardiac fibrosis via the suppression of autophagy in cardiac fibroblasts [35]. The authors also found that Glycyrrhizin acid, an inhibitor of HMGB1, alleviates cardiac fibrosis through the abrogation of HMGB1-TLR2 interaction [35]. Together, the understanding of HMGB1 mechanism in heart diseases is important to develop a potential therapy to prevent and/or stop the progression of heart diseases.
VASCULAR INFLAMMATION
Kawasaki Disease
Kawasaki disease (KD) is a juvenile autoimmune acute vasculitis disease and is the leading cause of pediatric cardiac disease [36]. It is well-established that KD affects small and medium arteries, especially coronary arteries leading to myocardial infarction, aneurysm, and stenosis. Whether HMGB1 plays a critical role in KD-induced vasculitis was the interest of many studies. First, it has been reported that children with KD exhibit a high level of HMGB1 in comparison to healthy children [37]. In line with these findings, Qian et al. confirmed that the expression of HMGB1/sRAGE/NFκB axis was elevated in the serum of children with KD as well as in an animal model of KD [38]. The authors also found that HMGB1, RAGE, and NFκB were localized in the endothelial cells of coronary arteries supporting the concept that endothelial cell inflammation is a key factor in coronary artery remodeling, aneurysm, and coronary vasculitis [38]. It is well-established that HMGB1 can mediate the pro-inflammatory response of endothelial cells and induce apoptotic, autophagic, and pyroptotic cell death, which is a form of proinflammatory cell death that combines features of both apoptosis and necrosis and can be mediated by inflammasome-dependent caspase-1 activation leading to the activation of Gasdermin D, IL-1β, and IL-18. Interestingly, previous studies showed that HMGB1 was able to induce macrophage pyroptosis via RAGE signaling. Also, recent studies using a mouse model of KD as well as in vitro data elucidated that HMGB1 released by immune cells triggered HMGB1-RAGE signaling pathway in endothelial cells leading to the activation of cathepsin B and consequently endothelial cells pyroptosis via NLRP3 inflammasome. These results indicate the importance of endothelial cell inflammation-induced pyroptosis in the pathology of KD HMGB1-dependent mechanism [39]. These findings were supported by recent study showing that HMGB1 plays a critical role in the development of KD pathogenesis [40]. Although the role of HMGB1 in KD-induced vasculitis is well described, further studies should delineate the full mechanism by which HMGB1 is involved in this vascular pathology for a potential therapeutic approach.
Angiogenesis
Mitola et al. were the first to document that extracellular HMGB1 induces angiogenesis in endothelial cells [41]. Using an in vitro model of angiogenesis, the authors showed that HMGB1 stimulates chemotaxis, increases endothelial cell motility, and triggers the formation of endothelial cell sprouting. Supporting the concept about the role of HMGB1 in angiogenesis, HMGB1 also induced neovascularization in a chick embryo CAM model. Interestingly, blocking RAGE inhibits the capacity of HMGB1 to induce endothelial proliferation as well as neovascularization in the chick embryo CAM. These data suggest that HMGB1-induced angiogenesis is mediated by RAGE mechanism. However, HMGB1 can bind to LTRs and Syndecan-1, both known to be involved in angiogenesis [42, 43], which may suggest the potential mechanism that LTRs and Syndecan-1 are signaling pathways to HMGB1 in the regulation of angiogenesis.
It is well documented that HMGB1 promotes recovery from muscle ischemia [44, 45]. In fact, HMGB1 is required for endothelial tube formation in vitro and increases vascular density in ischemic skeletal muscles [44]. A link between autophagy, inflammasome, and HMGB1 release has been proposed as a mechanism to promote muscle recovery from ischemic injury [46, 47]. Xu et al. confirmed that HMGB1 is prominently expressed in muscle from patients with peripheral artery disease and that in the setting of ischemia, the inhibition of autophagy by chloroquine regulates the mobilization of HMGB1 from the nucleus into the extracellular space promoting a better recovery from ischemic injury [47].
Recently Lan et al. [48] proposed that the internalization of HMGB1 in endothelial cells is a new mechanism by which HMGB1 promotes angiogenesis mediated by dynamin and RAGE and the release of VEGF. The authors also validated their finding in vivo using a murine model of hind limb ischemia-induced angiogenesis showing that HMGB1 internalization in endothelial cells occurred in vivo. Although, the study supports the role of HMGB1 in ischemia-induced angiogenesis and sheds light on a new mechanism by which HMGB1 stimulates angiogenesis through its internalization by endothelial cells, the authors only used human umbilical vein endothelial cells (HUVECs) as an in vitro model for angiogenesis. HUVECs are isolated from large vein and might have different signaling for angiogenesis when compared to endothelial cells from the microcirculation [48]. Therefore, HMGB1 internalization–induced angiogenesis still needs to be investigated in endothelial cells isolated from the microcirculation. While the involvement of RAGE activation in HMGB1-induced angiogenesis is widely known, the role of RAGE in angiogenesis is controversial. For instance, Lan et al. [48] showed that blood flow measurements were identical between C57BL6 and RAGE knockout mice after hind limb ischemia. Other investigators showed that RAGE knockout mice exhibit an increase in the perfusion ratio and number of capillaries when compared to C57BL6 mice [49].
Another important aspect worth to mention is the internalization of HMGB1 in endothelial cells during arteriogenesis or collateral growth in response to ischemia. Activated vascular smooth muscle cells (VSMCs) are also known to produce HMGB1 [50]. Arteriogenesis is a process that requires VSMC migration and proliferation dictated by HMGB1. Therefore, would VSMCs also use HMGB1 internalization as a mechanism to promote arteriogenesis? What is the status of HMGB1 internalization in pathological condition such as ischemic diseases?
Atherosclerosis
Early report indicated that HMGB1 plays a critical role in the development of atherosclerosis through pro-inflammatory factors and immune cell mechanism [51]. Recent reports showed that mice lacking endothelial HMGB1 are protected from developing atherosclerosis, a major cardiovascular disease, which includes inflammation [52]. The authors demonstrated that transcytosis of low-density lipoprotein across endothelial cell, critical in the development of atherosclerosis, depends on HMGB1 role in the nucleus by prolonging the half-life of sterol regulatory element-binding protein 2 essential to the production of scavenger receptor class B type 1 [52]. The role of HMGB1 in atherosclerosis is supported by studies from Dr. Zhang’s laboratory [53]. The authors demonstrated that inhibiting the HMGB1 with glycyrrhizic acid reduced the intimal thickness of carotid artery and plaque formation in rat with diabetic atherosclerosis [53]. The protective effect of HMGB1 inhibition against atherosclerosis is through the inhibition of macrophages and inflammation [53]. In a recent study, it has been reported that HMGB1 is a direct target of miR-141–5p, which is downregulated in atherosclerosis [54]. The overexpression of miR-141–5p decreases the inflammation and abnormal proliferation and migration of VSMCs in the setting of atherosclerosis through the reduction in HMGB1 and NFkB expression [54]. As described in the Introduction, HMGB1 binds to a variety of receptors including the TLR4, which are involved in the development of atherosclerosis [55]. The inhibition of HMGB1 protects against atherosclerosis by blunting the production of pro-inflammatory cytokines and upregulating PPARg/LXRa-ABCA1 through TLR4 [55]. Together, the two studies [48, 52] indicate that HMGB1 is needed for the induction of angiogenesis in the setting of ischemia but its effect in atherosclerosis is still controversial. This aspect is worth to mention in order to understand the versatile role of HMGB1 in vascular pathologies. The data reported by Lan et al. [48] shed new lights into a novel mechanism of action of HMGB1 in endothelial cells to stimulate angiogenesis. Further studies are needed to unravel the mechanism by which HMGB1 from other cells such as microvascular endothelial cells and VSMCs, and macrophages to stimulate angiogenesis/arteriogenesis. The reported findings open new avenues to enhance our understanding about the beneficial effect of HMGB1 and the potential therapeutic approach in the setting of cardiovascular diseases.
DIABETES
Diabetes is an endemic, chronic disease, and its prevalence is increasing worldwide. During the past decade, inflammation emerges as a key feature of obesity and type 2 diabetes. In fact, obesity, metabolic syndrome (MetS), and diabetes are characterized by low-grade chronic inflammation and an increase in HMGB1 release [56, 57]. Several studies have shown the relation between HMGB1 and diabetes mellitus (DM). Diabetic patients exhibit higher serum HMGB1 compared to non-diabetic patients and been shown to be associated with glucose metabolism and body mass index [58]. In a recent case–control study, children with type 1 diabetes exhibit higher level of HMGB1 compared to control group suggesting the possible involvement of HMGB1 in the etiology of type 1 diabetes [59]. Moreover, Yan et al. showed an increase in HMGB1 serum level in type 2 diabetic patients with coronary arteries disease (CAD) versus type 2 diabetic without CAD [58]. These data indicate that HMGB1 could be a common mechanism involved in the development of type 1 and type 2 diabetes.
Moreover, using a model of middle cerebral artery occlusion-reperfusion (MCAO/Re) Wang et al. showed that HMGB1 secretion increased in the brain of diabetic compared to non-diabetic rat [15]. The authors also found that ischemia induces an increase in the secretion of HMGB1 in diabetic rats highlighting the importance of targeting HMGB1 as a therapy for post ischemic injury in diabetic patients [15]. Interestingly, HMGB1 level is increased in muscle tissue and serum of patients with peripheral artery disease (PAD). Moreover, HMGB1 plasma levels were correlated with the severity of PAD [60, 61]. Conversely, HMGB1 seems to be involved in tissue repair after peripheral ischemia [62]. In diabetic animal models of hind limb ischemia, HMGB1 protein expression was lower in the ischemic tissues and the administration of HMGB1 restored the blood flow recovery and improved neovascularization through VEGF mechanism [62]. Furthermore, the secretion of HMGB1 from activated macrophages enhanced the release of VEGF, TNFα, and IL-8 promoting the mobilization of endothelial progenitor cells to stimulate angiogenesis [62]. Moreover, hyperglycemia stimulates HMGB1 release leading to vascular dysfunction via ROS (32, 40) and RAGE-NFκB axis [63].
Diabetic cardiomyopathy is a chronic heart complication seen in diabetic patients that occurs independently from coronary artery diseases and hypertension. The treatment of cardiac fibroblasts, macrophages, and cardiomyocytes with high glucose level led to the induction of HMGB1 expression and an increase in the NFκB binding activity, IL-6, and TNFα level [63]. Moreover, these in vitro data were validated in a murine model of type 1 diabetes induced by streptozotocin and subjected to myocardial infarction. After myocardial infarction, mice exhibit higher levels of HMGB1 in the myocardium and in the circulation. Interestingly, HMGB1 blockade significantly reduced post myocardial infarction remodeling and cardiac damages through the involvement of ERK1/2, JNK, and NFκB signaling [63]. In line with these findings, the authors conducted a clinical pilot study in diabetic patients with a history of myocardial infarction. HMGB1 plasma levels were higher in diabetic patients compared to healthy controls and were negatively correlated with cardiac performance [63]. Together, these data indicate that HMGB1 is an important mechanism in the etiology of diabetes and in the cardiovascular complications related to diabetes.
Vascular calcification is a common vascular complication associated with age and diabetes, and characterized by the transformation of vascular smooth muscle cells to osteo/chondrocyte cells [64]. Studies in animal models and patients documented the association between HMGB1 and aortic valve calcification [65]. Patients with aortic valve calcification have an increase in tissue and plasma level of HMGB1 [65] as well as an accumulation of HMGB1 extracellularly in areas associated with macrophage infiltration and calcification in calcific aortic valve stenosis [66]. Recent reports showed that HMGB1 mediates high glucose-induced calcification in VSMCs of saphenous vein via the activation of NFκB and the induction of BMP2 expression in VSMC [67]. A recent study in a mouse model of type 1 diabetes showed that diabetes induced HMGB1 secretion via the induction of endoplasmic reticulum (ER) stress leading to vascular calcification [68]. Interestingly, the authors found that the inhibition of HMGB1 and ER stress alleviates vascular calcification in Streptozotocin-induced diabetic mice. Moreover, AGEs induce the translocation and secretion of HMGB1 from VSMCs through the ER stress. Runx2 is a transcription factor involved in the pathology of vascular calcification [69–71]. In fact, HMGB1 has been shown to upregulate the expression of Runx2 in VSMCs via the ER stress [68] suggesting the implication of HMGB1 in cardiovascular calcification through ER stress–dependent mechanism. TGFβ/BMP signaling are important players in calcification. Interestingly, recent findings showed that HMGB1 regulates the expression or release of TGFβ and BMP2 suggesting its importance in cardiovascular calcification. Inflammation, oxidative stress, and autophagy signaling were documented as mechanisms by which HMGB1 promotes cardiovascular calcification [64]. All these data indicate that HMGB1 is not just important in inflammation-induced pathology but also critical in structural remodeling.
Diabetic Retinopathy
Diabetic retinopathy is a common complication of diabetes mellitus and the leading cause of blindness in adults. Increasing evidence has indicated that HMGB1 plays a central role in the pathogenesis of diabetic retinopathy [72, 73]. HMGB1 expression was found to be upregulated in the serum of patients with diabetic retinopathy with a positive correlation between serum and vitreous levels of HMGB1 [74]. In vitro studies using human retinal endothelial cells, Chip sequencing, and luciferase assay showed the interaction between IκB-α and HMGB1 and demonstrated that HMGB1 was involved in the pathogenesis of diabetic retinopathy via the NFκB pathway [75]. Moreover, Abu El-Asrar et al. showed a link between HMGB1 and different marker of oxidative stress in the pathogenesis of inflammation and neovascularization in diabetic retinopathy [76]. The exchange protein activated by cAMP (Epac) has been shown to regulate HMGB1 expression in retinal vasculature through AMPK and SIRT1 pathway in the setting of diabetes [77–79]. Moreover, the inhibition of the release of HMGB1 using Glycyrrhizin reduces HMGB1 levels in the retinas as well as diabetic-induced damage to the retinas associated with reduced reactive oxygen species, TNFα and IL1β [80]. These data indicate the importance of targeting HMGB1 to stop the progression of diabetes-induced retinopathy.
THERAPEUTIC TARGET FOR CARDIOVASCULAR COMPLICATIONS
Coronary artery diseases (CAD) are the leading cause of morbidity and mortality worldwide. Diabetes mellitus is considered to be an independent risk factor in the development of CAD. Metformin, an anti-diabetic drug, has been shown to lower cardiovascular complications in diabetic patients [81]. Previous study showed that metformin has anti-inflammatory properties due to the inhibition of pro-inflammatory cytokine production as well as the inhibition of HMGB1 release likely through AMPK-mechanism [82]. Recent study reported that metformin inhibits HMGB1 translocation from the nucleus to the cytosol and therefore limiting HMGB1 in the nucleus and blocking its release through the direct binding of metformin to the C-terminal acidic tail of HMGB1 [82–84]. Moreover, other studies strengthen the concept that metformin protects the heart against hyperglycemia-induced injury via the inhibition of HMGB1/RAGE expression [85]. These studies opened new avenues to understand how metformin inhibits inflammation by targeting HMGB1, which will be clinically significant. Glycyrrhizin, a natural anti-inflammatory compound, found in licorice that inhibits HMGB1 activities via a direct binding to HMGB1 [86] has been shown to protect against ischemia reperfusion injury in rat brains through the reduction of the release of HMGB1 into the extracellular space as well as blocking its activity [87]. Moreover, Glycyrrhizin inhibits HMGB1 expression and its signaling pathways in diabetic retinopathy [88, 89]. Recent reports by Dandona et al. showed that insulin infusion in type 1 diabetic patients suppressed HMGB1 expression in monocytes [90]. Resveratrol, an antioxidant compound, has been found to prevent the occurrence of diabetic cardiomyopathy by decreasing HMGB1 expression in the left ventricular myocardium of type 1 diabetic rats [91, 92]. Astilbin, another antioxidant, has been shown to provide cardio-protection against ischemia reperfusion injury in diabetic rat through the manipulation of HMGB1/NFκB axis [93]. ACE inhibitors also inhibit cardiac inflammation via the reduction of the release of HMGB1 [94]. It is important and clinically relevant to understand the molecular mechanism of induction as well as the release of HMGB1 in diseases associated with inflammation for an efficient potential therapy.
CONCLUSION
The detrimental and beneficial effect of HMGB1 in the setting of different cardiovascular diseases is very intriguing (Scheme 1). The translocation of HMGB1 from nucleus to cytoplasm plays a key role in inflammation. We believe that in order to develop an efficient therapeutic strategy targeting HMGB1, the approach should modulate the level of the HMGB1 in a cell-specific manner with a special focus on the localization of HMGB1. Further studies are necessary to understand the mechanisms by which extracellular and nuclear HMGB1 affects cardiac and vascular inflammation and repair/regeneration and how it can be manipulated to maximize its therapeutic potential in cardiovascular diseases. Summary of HMGB1 roles and mechanisms in cardiovascular complications are represented in Scheme 1 and Table 1.
Scheme 1.
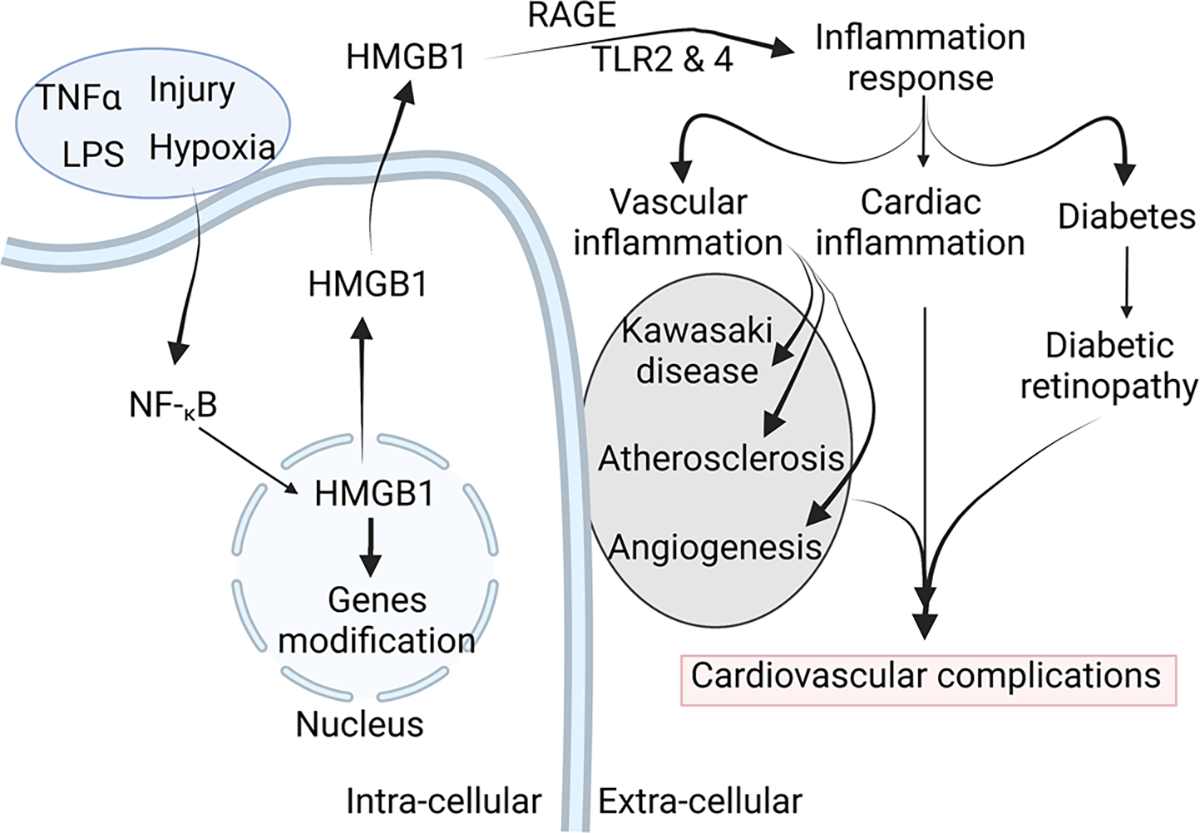
Schema Graphical representation illustrating the role of HMGB1 in cardiovascular complications
Table 1.
The Mechanism of HMGB1 in Cardiovascular Complications
| HMGB1 | Involvement | Signaling/Markers/Mechanism | Reference |
|---|---|---|---|
|
| |||
| Cytosol | Cardiac hypertrophy and heart failure | Increase HMGB1 expression and translocation from nucleus to cytoplasm and intercellular space | [22] |
| Not specify | Cardiac hypertrophy | Calcineurin | [23] |
| Nucleus to cytoplasm | Cardiac remodeling | HMGB1 acetylation and translocation from the nucleus to the cytoplasm | [24] |
| Not specify | Myocardial regeneration | Resident cardiac c-kit + cell proliferation and differentiation | [25] |
| Not specify | Myocardial inflammation | Troponin-I | [31] |
| Not specify | Autoimmune myocarditis | Pro-inflammatory M1 | [32] |
| Not specify | Cardiac fibrosis | TLR2 and autophagy | [35] |
| Not specify | Kawasaki disease | sRAGE/NFκB | [38] |
| Not specify | Kawasaki disease | RAGE/cathepsin B/NLRP3 inflammasome | [39] |
| Not specify | Angiogenesis | RAGE/LTRs/Syndecan-1 | [42, 43] |
| Not specify | Angiogenesis | Dynamin/RAGE/VEGF | [48] |
| Not specify | Angiogenesis | VEGF/TNFα/IL-8 | [62] |
| Nucleus | Atherosclerosis | Sterol regulatory element-binding protein 2/receptor class B type 1 | [52] |
| Not specify | Diabetic atherosclerosis | Macrophages and inflammation inhibition | [53] |
| Not specify | Atherosclerosis | TLR-VPPARγ/LXRa–ABCAl | [55] |
| Not specify | Vascular dysfunction in diabetes | ROS/RAGE/NFkB | [32, 40, 63] |
| Not specify | Myocardial infarction remodeling | ERK1/2/JNK/NFkB | [63] |
| Not specify | VSMC calcification | HMGB1 regulates the expression of TGFβ, BMP2, and autophagy, activates NFκB, and upregulates Runx2 and oxidative stress | [64, 67, 68] |
| Not specify | Diabetic retinopathy | NFkB/reactive oxygen species/TNFα and IL1β | [75, 80] |
FUNDING
This work is supported by the NIH-R01-HL150014 (KM), NIH-R01-HL151616 (KM), and AHA-19AIREA34380220 (KM).
Footnotes
Competing Interests The authors declare no competing interests.
Ethics Approval and Consent to Participate (Human Ethics, Animal Ethics or Plant Ethics) Not applicable.
Disclaimer All authors certify that they have no affiliations with or involvement in any organization or entity with any financial interest or non-financial interest in the subject matter or materials discussed in this manuscript.
REFERENCES
- 1.Goodwin GH, Sanders C, and Johns EW. 1973. A new group of chromatin-associated proteins with a high content of acidic and basic amino acids. European Journal of Biochemistry 38: 14–19. [DOI] [PubMed] [Google Scholar]
- 2.Calogero S, Grassi F, Aguzzi A, Voigtländer T, Ferrier P, Ferrari S, et al. 1999. The lack of chromosomal protein Hmg1 does not disrupt cell growth but causes lethal hypoglycaemia in newborn mice. Nature Genetics 22: 276–280. [DOI] [PubMed] [Google Scholar]
- 3.Tang D, Kang R, Xiao W, Wang H, Calderwood SK, and Xiao X. 2007. The anti-inflammatory effects of heat shock protein 72 involve inhibition of high-mobility-group box 1 release and proinflammatory function in macrophages. The Journal of Immunology 179: 1236–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scaffidi P, Misteli T, and Bianchi ME. 2002. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418: 191–195. [DOI] [PubMed] [Google Scholar]
- 5.Wakabayashi A, Shimizu M, Shinya E, and Takahashi H. 2018. HMGB1 released from intestinal epithelia damaged by cholera toxin adjuvant contributes to activation of mucosal dendritic cells and induction of intestinal cytotoxic T lymphocytes and IgA. Cell Death & Disease 9: 631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang H, Antoine DJ, Andersson U, and Tracey KJ. 2013. The many faces of HMGB1: Molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. Journal of Leukocyte Biology 93: 865–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang H, Wang H, Chavan SS, and Andersson U. 2015. High Mobility Group Box Protein 1 (HMGB1): The prototypical endogenous danger molecule. Molecular Medicine 21 (Suppl 1): S6–s12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stumbo AC, Cortez E, Rodrigues CA, Henriques M, Porto LC, Barbosa HS, et al. 2008. Mitochondrial localization of non-histone protein HMGB1 during human endothelial cell-Toxoplasma gondii infection. Cell Biology International 32: 235–238. [DOI] [PubMed] [Google Scholar]
- 9.Malarkey CS, and Churchill ME. 2012. The high mobility group box: The ultimate utility player of a cell. Trends in Biochemical Sciences 37: 553–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wahid A, Chen W, Wang X, and Tang X. 2021. High-mobility group box 1 serves as an inflammation driver of cardiovascular disease. Biomedicine and Pharmacotheraphy 139:111555. [DOI] [PubMed] [Google Scholar]
- 11.Magna M, and Pisetsky DS. 2014. The role of HMGB1 in the pathogenesis of inflammatory and autoimmune diseases. Molecular Medicine 20: 138–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu B, Antoine DJ, Kwan K, Lundbäck P, Wähämaa H, Schierbeck H, et al. 2014. JAK/STAT1 signaling promotes HMGB1 hyperacetylation and nuclear translocation. Proceedings of the National Academy of Sciences of the United States of America 111: 3068–3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andersson U, Wang H, Palmblad K, Aveberger AC, Bloom O, Erlandsson-Harris H, et al. 2000. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. Journal of Experimental Medicine 192: 565–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim JB, Sig Choi J, Yu YM, Nam K, Piao CS, Kim SW, et al. 2006. HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. Journal of Neuroscience 26: 6413–6421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang C, Jiang J, Zhang X, Song L, Sun K, and Xu R. 2016. Inhibiting HMGB1 reduces cerebral ischemia reperfusion injury in diabetic mice. Inflammation 39: 1862–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andersson U, Yang H, and Harris H. 2018. Extracellular HMGB1 as a therapeutic target in inflammatory diseases. Expert Opinion on Therapeutic Targets 22: 263–277. [DOI] [PubMed] [Google Scholar]
- 17.Andersson U, Yang H, and Harris H. 2018. High-mobility group box 1 protein (HMGB1) operates as an alarmin outside as well as inside cells. Seminars in Immunology 38: 40–48. [DOI] [PubMed] [Google Scholar]
- 18.Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, et al. 2010. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proceedings of the National Academy of Sciences of the United States of America 107: 11942–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andrassy M, Volz HC, Igwe JC, Funke B, Eichberger SN, Kaya Z, et al. 2008. High-mobility group box-1 in ischemia-reperfusion injury of the heart. Circulation 117: 3216–3226. [DOI] [PubMed] [Google Scholar]
- 20.Oozawa S, Mori S, Kanke T, Takahashi H, Liu K, Tomono Y, et al. 2008. Effects of HMGB1 on ischemia-reperfusion injury in the rat heart. Circulation Journal 72: 1178–1184. [DOI] [PubMed] [Google Scholar]
- 21.Higashikuni Y, Tanaka K, Kato M, Nureki O, Hirata Y, Nagai R, et al. 2013. Toll-like receptor-2 mediates adaptive cardiac hypertrophy in response to pressure overload through interleukin-1β upregulation via nuclear factor κB activation. Journal of the American Heart Association 2:e000267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang L, Liu M, Jiang H, Yu Y, Yu P, Tong R, et al. 2016. Extracellular high-mobility group box 1 mediates pressure overload-induced cardiac hypertrophy and heart failure. Journal of Cellular and Molecular Medicine 20: 459–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Su FF, Shi MQ, Guo WG, Liu XT, Wang HT, Lu ZF, et al. 2012. High-mobility group box 1 induces calcineurin-mediated cell hypertrophy in neonatal rat ventricular myocytes. Mediators of Inflammation 2012:805149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Funayama A, Shishido T, Netsu S, Narumi T, Kadowaki S, Takahashi H, et al. 2013. Cardiac nuclear high mobility group box 1 prevents the development of cardiac hypertrophy and heart failure. Cardiovascular Research 99: 657–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Limana F, Germani A, Zacheo A, Kajstura J, Di Carlo A, Borsellino G, et al. 2005. Exogenous high-mobility group box 1 protein induces myocardial regeneration after infarction via enhanced cardiac C-kit+ cell proliferation and differentiation. Circulation Research 97: e73–83. [DOI] [PubMed] [Google Scholar]
- 26.Kitahara T, Takeishi Y, Harada M, Niizeki T, Suzuki S, Sasaki T, et al. 2008. High-mobility group box 1 restores cardiac function after myocardial infarction in transgenic mice. Cardiovascular Research 80: 40–46. [DOI] [PubMed] [Google Scholar]
- 27.Kaya Z, Afanasyeva M, Wang Y, Dohmen KM, Schlichting J, Tretter T, et al. 2001. Contribution of the innate immune system to autoimmune myocarditis: A role for complement. Nature Immunology 2: 739–745. [DOI] [PubMed] [Google Scholar]
- 28.Heymans S 2006. Inflammation and cardiac remodeling during viral myocarditis. Ernst Schering Research Found Workshop 197–218. [DOI] [PubMed] [Google Scholar]
- 29.Eriksson S, Hellman J, and Pettersson K. 2005. Autoantibodies against cardiac troponins. New England Journal of Medicine 352: 98–100. [DOI] [PubMed] [Google Scholar]
- 30.Göser S, Andrassy M, Buss SJ, Leuschner F, Volz CH, Ottl R, et al. 2006. Cardiac troponin I but not cardiac troponin T induces severe autoimmune inflammation in the myocardium. Circulation 114: 1693–1702. [DOI] [PubMed] [Google Scholar]
- 31.Bangert A, Andrassy M, Müller AM, Bockstahler M, Fischer A, Volz CH, et al. 2016. Critical role of RAGE and HMGB1 in inflammatory heart disease. Proceedings of the National Academy of Sciences of the United States of America 113: E155–E164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Su Z, Zhang P, Yu Y, Lu H, Liu Y, Ni P, et al. 2016. HMGB1 facilitated macrophage reprogramming towards a proinflammatory M1-like phenotype in experimental autoimmune myocarditis development. Science and Reports 6: 21884. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Kohno T, Anzai T, Naito K, Miyasho T, Okamoto M, Yokota H, et al. 2009. Role of high-mobility group box 1 protein in post-infarction healing process and left ventricular remodelling. Cardiovascular Research 81: 565–573. [DOI] [PubMed] [Google Scholar]
- 34.Yu P, Liu M, Zhang B, Yu Y, Su E, Xie S, et al. 2020. Cardiomyocyte-restricted high-mobility group box 1 (HMGB1) deletion leads to small heart and glycolipid metabolic disorder through GR/PGC-1α signalling. Cell Death Discov 6: 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu RN, Yu TY, Zhou JC, Li M, Gao HK, Zhao C, et al. 2018. Targeting HMGB1 ameliorates cardiac fibrosis through restoring TLR2-mediated autophagy suppression in myocardial fibroblasts. International Journal of Cardiology 267: 156–162. [DOI] [PubMed] [Google Scholar]
- 36.Uehara R, and Belay ED. 2012. Epidemiology of Kawasaki disease in Asia, Europe, and the United States. Journal of Epidemiology 22: 79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoshina T, Kusuhara K, Ikeda K, Mizuno Y, Saito M, and Hara T. 2008. High mobility group box 1 (HMGB1) and macrophage migration inhibitory factor (MIF) in Kawasaki disease. Scandinavian Journal of Rheumatology 37: 445–449. [DOI] [PubMed] [Google Scholar]
- 38.Qian B, Huang H, Cheng M, Qin T, Chen T, and Zhao J. 2020. Mechanism of HMGB1-RAGE in Kawasaki disease with coronary artery injury. European Journal of Medical Research 25: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jia C, Zhang J, Chen H, Zhuge Y, Chen H, Qian F, et al. 2019. Endothelial cell pyroptosis plays an important role in Kawasaki disease via HMGB1/RAGE/cathespin B signaling pathway and NLRP3 inflammasome activation. Cell Death & Disease 10: 778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ueno K, Nomura Y, Morita Y, and Kawano Y. 2021. Prednisolone suppresses the extracellular release of HMGB-1 and associated inflammatory pathways in Kawasaki disease. Frontiers in Immunology 12:640315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mitola S, Belleri M, Urbinati C, Coltrini D, Sparatore B, Pedrazzi M, et al. 2006. Cutting edge: Extracellular high mobility group box-1 protein is a proangiogenic cytokine. The Journal of Immunology 176: 12–15. [DOI] [PubMed] [Google Scholar]
- 42.Frantz S, Vincent KA, Feron O, and Kelly RA. 2005. Innate immunity and angiogenesis. Circulation Research 96: 15–26. [DOI] [PubMed] [Google Scholar]
- 43.Yuan K, Hong TM, Chen JJ, Tsai WH, and Lin MT. 2004. Syndecan-1 up-regulated by ephrinB2/EphB4 plays dual roles in inflammatory angiogenesis. Blood 104: 1025–1033. [DOI] [PubMed] [Google Scholar]
- 44.Sachdev U, Cui X, Hong G, Namkoong S, Karlsson JM, Baty CJ, et al. 2012. High mobility group box 1 promotes endothelial cell angiogenic behavior in vitro and improves muscle perfusion in vivo in response to ischemic injury. Journal of Vascular Surgery 55: 180–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sachdev U, Cui X, and Tzeng E. 2013. HMGB1 and TLR4 mediate skeletal muscle recovery in a murine model of hindlimb ischemia. Journal of Vascular Surgery 58: 460–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundbäck P, et al. 2012. Novel role of PKR in inflammasome activation and HMGB1 release. Nature 488: 670–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu J, Cui X, Li J, Koutakis P, Pipinos I, Tzeng E, et al. 2018. Chloroquine improves the response to ischemic muscle injury and increases HMGB1 after arterial ligation. Journal of Vascular Surgery 67: 910–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lan J, Luo H, Wu R, Wang J, Zhou B, Zhang Y, et al. 2020. Internalization of HMGB1 (High Mobility Group Box 1) promotes angiogenesis in endothelial cells. Arteriosclerosis, Thrombosis, and Vascular Biology 40: 2922–2940. [DOI] [PubMed] [Google Scholar]
- 49.Hansen LM, Gupta D, Joseph G, Weiss D, and Taylor WR. 2017. The receptor for advanced glycation end products impairs collateral formation in both diabetic and non-diabetic mice. Laboratory Investigation 97: 34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Inoue K, Kawahara K, Biswas KK, Ando K, Mitsudo K, Nobuyoshi M, et al. 2007. HMGB1 expression by activated vascular smooth muscle cells in advanced human atherosclerosis plaques. Cardiovascular Pathology 16: 136–143. [DOI] [PubMed] [Google Scholar]
- 51.Kanellakis P, Agrotis A, Kyaw TS, Koulis C, Ahrens I, Mori S, et al. 2011. High-mobility group box protein 1 neutralization reduces development of diet-induced atherosclerosis in apolipo-protein e-deficient mice. Arteriosclerosis, Thrombosis, and Vascular Biology 31: 313–319. [DOI] [PubMed] [Google Scholar]
- 52.Ghaffari S, Jang E, Naderinabi F, Sanwal R, Khosraviani N, Wang C, et al. 2021. Endothelial HMGB1 is a critical regulator of LDL transcytosis via an SREBP2-SR-BI axis. Arteriosclerosis, Thrombosis, and Vascular Biology 41: 200–216. [DOI] [PubMed] [Google Scholar]
- 53.Zhao Y, Li W, and Zhang D. Gycyrrhizic acid alleviates atherosclerotic lesions in rats with diabetes mellitus. Molecular Medicine Report 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li Y, Li H, Chen B, Yang F, and Hao Z. 2021. miR-141–5p suppresses vascular smooth muscle cell inflammation, proliferation, and migration via inhibiting the HMGB1/NF-kappaB pathway. Journal of the Biochemical Molecular Toxicology 35:e22828. [DOI] [PubMed] [Google Scholar]
- 55.Roshan MH, Tambo A, and Pace NP. 2016. The Role of TLR2, TLR4, and TLR9 in the pathogenesis of atherosclerosis. International Journal of the Inflammation 2016: 1532832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pahwa R, Adams-Huet B, and Jialal I. 2017. The effect of increasing body mass index on cardio-metabolic risk and biomarkers of oxidative stress and inflammation in nascent metabolic syndrome. Journal of Diabetes and Its Complications 31: 810–813. [DOI] [PubMed] [Google Scholar]
- 57.Pahwa R, and Jialal I. 2016. The role of the high-mobility group box1 protein-Toll like receptor pathway in diabetic vascular disease. Journal of Diabetes and Its Complications 30: 1186–1191. [DOI] [PubMed] [Google Scholar]
- 58.Yan XX, Lu L, Peng WH, Wang LJ, Zhang Q, Zhang RY, et al. 2009. Increased serum HMGB1 level is associated with coronary artery disease in nondiabetic and type 2 diabetic patients. Atherosclerosis 205: 544–548. [DOI] [PubMed] [Google Scholar]
- 59.Marjanac I, Lovrić R, and Barbić J. 2019. Serum levels of the high-mobility group box 1 protein (HMGB1) in children with type 1 diabetes mellitus: Case-control study. Central-European Journal of Immunology 44: 33–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Giovannini S, Tinelli G, Biscetti F, Straface G, Angelini F, Pitocco D, et al. 2017. Serum high mobility group box-1 and osteoprotegerin levels are associated with peripheral arterial disease and critical limb ischemia in type 2 diabetic subjects. Cardiovascular Diabetology 16: 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oozawa S, Sano S, and Nishibori M. 2014. Usefulness of high mobility group box 1 protein as a plasma biomarker in patient with peripheral artery disease. Acta Medica Okayama 68: 157–162. [DOI] [PubMed] [Google Scholar]
- 62.Biscetti F, Straface G, De Cristofaro R, Lancellotti S, Rizzo P, Arena V, et al. 2010. High-mobility group box-1 protein promotes angiogenesis after peripheral ischemia in diabetic mice through a VEGF-dependent mechanism. Diabetes 59: 1496–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Volz HC, Seidel C, Laohachewin D, Kaya Z, Müller OJ, Pleger ST, et al. 2010. HMGB1: The missing link between diabetes mellitus and heart failure. Basic Research in Cardiology 105: 805–820. [DOI] [PubMed] [Google Scholar]
- 64.Chen Q, Wang ZY, Chen LY, and Hu HY. 2017. Roles of High Mobility Group Box 1 in cardiovascular calcification. Cellular Physiology and Biochemistry 42: 427–440. [DOI] [PubMed] [Google Scholar]
- 65.Wang B, Wei G, Liu B, Zhou X, Xiao H, Dong N, et al. 2016. The role of High Mobility Group Box 1 protein in interleukin-18-induced myofibroblastic transition of valvular interstitial cells. Cardiology 135: 168–178. [DOI] [PubMed] [Google Scholar]
- 66.Passmore M, Nataatmadja M, Fung YL, Pearse B, Gabriel S, Tesar P, et al. 2015. Osteopontin alters endothelial and valvular interstitial cell behaviour in calcific aortic valve stenosis through HMGB1 regulation. European Journal of Cardio-Thoracic Surgery 48: e20–e29. [DOI] [PubMed] [Google Scholar]
- 67.Wang Y, Shan J, Yang W, Zheng H, and Xue S. 2013. High mobility group box 1 (HMGB1) mediates high-glucose-induced calcification in vascular smooth muscle cells of saphenous veins. Inflammation 36: 1592–1604. [DOI] [PubMed] [Google Scholar]
- 68.Chen Z, Li R, Pei LG, Wei ZH, Xie J, Wu H, et al. 2021. High-mobility group box-1 promotes vascular calcification in diabetic mice via endoplasmic reticulum stress. Journal of Cellular and Molecular Medicine 25: 3724–3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sun Y, Byon CH, Yuan K, Chen J, Mao X, Heath JM, et al. 2012. Smooth muscle cell-specific runx2 deficiency inhibits vascular calcification. Circulation Research 111: 543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lin ME, Chen T, Leaf EM, Speer MY, and Giachelli CM. 2015. Runx2 expression in smooth muscle cells is required for arterial medial calcification in mice. American Journal of Pathology 185: 1958–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Byon CH, Javed A, Dai Q, Kappes JC, Clemens TL, Darley-Usmar VM, et al. 2008. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. Journal of Biological Chemistry 283: 15319–15327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nebbioso M, Lambiase A, Armentano M, Tucciarone G, Bonfiglio V, Plateroti R, et al. 2020. The complex relationship between diabetic retinopathy and High-Mobility Group Box: a review of molecular pathways and therapeutic strategies. Antioxidants (Basel) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Steinle JJ 2020. Role of HMGB1 signaling in the inflammatory process in diabetic retinopathy. Cell Signal 73:109687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.El-Asrar AM, Nawaz MI, Kangave D, Geboes K, Ola MS, Ahmad S, et al. 2011. High-mobility group box-1 and biomarkers of inflammation in the vitreous from patients with proliferative diabetic retinopathy. Molecular Vision 17: 1829–1838. [PMC free article] [PubMed] [Google Scholar]
- 75.Liang WJ, Yang HW, Liu HN, Qian W, and Chen XL. 2020. HMGB1 upregulates NF-kB by inhibiting IKB-α and associates with diabetic retinopathy. Life Sciences 241:117146. [DOI] [PubMed] [Google Scholar]
- 76.Abu El-Asrar AM, Alam K, Garcia-Ramirez M, Ahmad A, Siddiquei MM, Mohammad G, et al. 2017. Association of HMGB1 with oxidative stress markers and regulators in PDR. Molecular Vision 23: 853–871. [PMC free article] [PubMed] [Google Scholar]
- 77.Jiang Y, Liu L, Curtiss E, and Steinle JJ. 2017. Epac1 blocks NLRP3 inflammasome to reduce IL-1β in retinal endothelial cells and mouse retinal vasculature. Mediators of Inflammation 2017: 2860956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jiang Y, Liu L, and Steinle JJ. 2018. Epac1 deacetylates HMGB1 through increased IGFBP-3 and SIRT1 levels in the retinal vasculature. Molecular Vision 24: 727–732. [PMC free article] [PubMed] [Google Scholar]
- 79.Jiang Y, and Steinle JJ. 2020. Epac1 requires AMPK phosphorylation to regulate HMGB1 in the retinal vasculature. Investigative Ophthalmology & Visual Science 61: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu L, Jiang Y, and Steinle JJ. 2019. Epac1 and glycyrrhizin both inhibit HMGB1 levels to reduce diabetes-induced neuronal and vascular damage in the mouse retina. Journal of the Clinical Medicine 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Luo F, Das A, Chen J, Wu P, Li X, and Fang Z. 2019. Metformin in patients with and without diabetes: A paradigm shift in cardiovascular disease management. Cardiovascular Diabetology 18: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tsoyi K, Jang HJ, Nizamutdinova IT, Kim YM, Lee YS, Kim HJ, et al. 2011. Metformin inhibits HMGB1 release in LPS-treated RAW 264.7 cells and increases survival rate of endotoxaemic mice. British Journal of the Pharmacology 162:1498–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Horiuchi T, Sakata N, Narumi Y, Kimura T, Hayashi T, Nagano K, et al. 2017. Metformin directly binds the alarmin HMGB1 and inhibits its proinflammatory activity. Journal of Biological Chemistry 292: 8436–8446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yang H, Wang H, and Andersson U. 2020. Targeting inflammation driven by HMGB1. Frontiers in Immunology 11: 484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang T, Hu X, Cai Y, Yi B, and Wen Z. 2014. Metformin protects against hyperglycemia-induced cardiomyocytes injury by inhibiting the expressions of receptor for advanced glycation end products and high mobility group box 1 protein. Molecular Biology Reports 41: 1335–1340. [DOI] [PubMed] [Google Scholar]
- 86.Mollica L, De Marchis F, Spitaleri A, Dallacosta C, Pennacchini D, Zamai M, et al. 2007. Glycyrrhizin binds to high-mobility group box 1 protein and inhibits its cytokine activities. Chemistry & Biology 14: 431–441. [DOI] [PubMed] [Google Scholar]
- 87.Gong G, Xiang L, Yuan L, Hu L, Wu W, Cai L, et al. 2014. Protective effect of glycyrrhizin, a direct HMGB1 inhibitor, on focal cerebral ischemia/reperfusion-induced inflammation, oxidative stress, and apoptosis in rats. PLoS One 9:e89450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mohammad G, Siddiquei MM, Othman A, Al-Shabrawey M, and Abu El-Asrar AM. 2013. High-mobility group box-1 protein activates inflammatory signaling pathway components and disrupts retinal vascular-barrier in the diabetic retina. Experimental Eye Research 107: 101–109. [DOI] [PubMed] [Google Scholar]
- 89.Abu El-Asrar AM, Siddiquei MM, Nawaz MI, Geboes K, and Mohammad G. 2014. The proinflammatory cytokine high-mobility group box-1 mediates retinal neuropathy induced by diabetes. Mediators of Inflammation 2014:746415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dandona P, Ghanim H, Green K, Sia CL, Abuaysheh S, Kuhadiya N, et al. 2013. Insulin infusion suppresses while glucose infusion induces Toll-like receptors and high-mobility group-B1 protein expression in mononuclear cells of type 1 diabetes patients. American Journal of Physiology, Endocrinology and Metabolism 304: E810–E818. [DOI] [PubMed] [Google Scholar]
- 91.Delucchi F, Berni R, Frati C, Cavalli S, Graiani G, Sala R, et al. Resveratrol treatment reduces cardiac progenitor cell dysfunction and prevents morpho-functional ventricular remodeling in type-1 diabetic rats. PLoS One 7:e39836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wu H, Chen Z, Xie J, Kang LN, Wang L, and Xu B. 2016. High Mobility Group Box-1: A missing link between diabetes and its complications. Mediators of Inflammation 2016: 3896147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Diao H, Kang Z, Han F, and Jiang W. 2014. Astilbin protects diabetic rat heart against ischemia-reperfusion injury via blockade of HMGB1-dependent NF-κB signaling pathway. Food and Chemical Toxicology 63: 104–110. [DOI] [PubMed] [Google Scholar]
- 94.Kikuchi K, Tancharoen S, Ito T, Morimoto-Yamashita Y, Miura N, Kawahara K, et al. 2013. Potential of the angiotensin receptor blockers (ARBs) telmisartan, irbesartan, and candesartan for inhibiting the HMGB1/RAGE axis in prevention and acute treatment of stroke. International Journal of Molecular Sciences 14: 18899–18924. [DOI] [PMC free article] [PubMed] [Google Scholar]