

Key Points
-
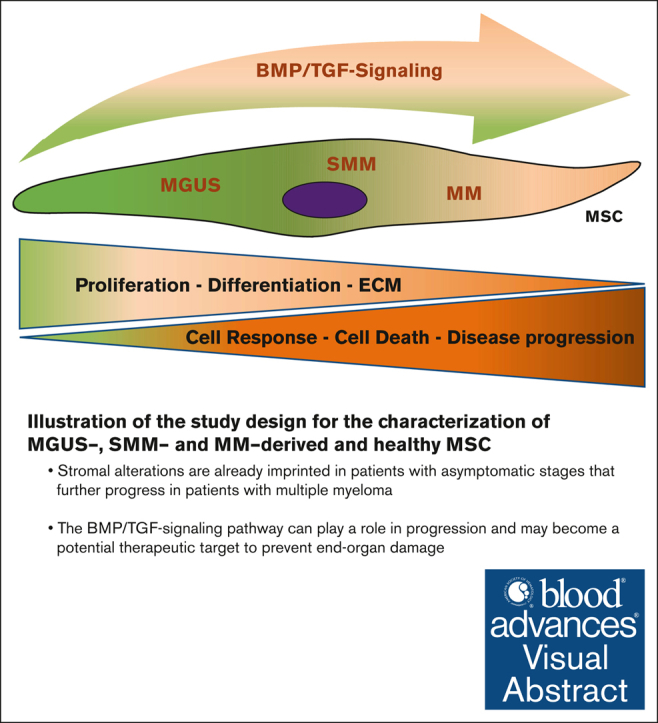
•
Stromal alterations are already imprinted in patients with asymptomatic stages that further progress in patients with MM.
-
•
The BMP/TGF-signaling pathway can play a role in progression and may become a potential therapeutic target to prevent end-organ damage.
Visual Abstract
Abstract
The hallmark of multiple myeloma (MM) is a clonal plasma cell infiltration in the bone marrow accompanied by myelosuppression and osteolysis. Premalignant stages such as monoclonal gammopathy of undetermined significance (MGUS) and asymptomatic stages such as smoldering myeloma (SMM) can progress to MM. Mesenchymal stromal cells (MSCs) are an integral component of the bone marrow microenvironment and play an important role in osteoblast differentiation and hematopoietic support. Although stromal alterations have been reported in MM contributing to hematopoietic insufficiency and osteolysis, it is not clear whether alterations in MSC already occur in MGUS or SMM. In this study, we analyzed MSCs from MGUS, SMM, and MM regarding their properties and functionality and performed messenger RNA sequencing to find underlying molecular signatures in different disease stages. A high number of senescent cells and a reduced osteogenic differentiation capacity and hematopoietic support were already present in MGUS MSC. As shown by RNA sequencing, there was a broad spectrum of differentially expressed genes including genes of the BMP/TGF-signaling pathway, detected already in MGUS and that clearly increases in patients with SMM and MM. Our data may help to block these signaling pathways in the future to hinder progression to MM.
Introduction
Multiple myeloma (MM) is characterized by an infiltration of clonal plasma cells in the bone marrow (BM) accompanied by end-organ damage such as hematopoietic insufficiency and/or osteolysis. The latter is a result of increased osteoclast and impaired osteoblast function.1, 2, 3 MM with indication for treatment is present when either end-organ damage is present, and thus, at least 1 of the CRAB (calcium elevation, renal insufficiency, anemia, and bone lesions) criteria is met, or clinical predictive biomarkers like 60% bone marrow plasma cells, involved:uninvolved serum free light chain ratio ≥100 or >1 focal lesions on MRI studies (SLiM-CRAB) are present that make progression of the disease highly likely.
Smoldering myeloma (SMM) does not clinically show CRAB or SLiM CRAB criteria and therefore does not need to be treated initially, but it can, with a significant probability of 10% per year, progress into a full blown MM.
This also applies to monoclonal gammopathie of undetermined significance (MGUS), which is present in ∼3% of those aged >50 years and has a probability of transformation into MM of ∼1% per year.4,5
The underlying mechanisms of how these asymptomatic or premalignant stages evolve to MM await further clarification, particularly because there are limited valid biomarkers reliably predicting the likelihood of progression.2,6,7 In the BM microenvironment, mesenchymal stromal cells (MSCs) are important components and are characterized by their capacity to differentiate into adipocytes, chondroblasts, and osteoblasts.8 MSCs are particularly relevant for the support of hematopoietic stem and progenitor cells with regard to proliferation, migration, and differentiation. These processes are governed and mediated via cross talk by direct cell-cell contact or secretion of a great variety of cytokines.9,10
Based on earlier findings of alterations in MSC in MM,11 we were interested in a better understanding of the mechanisms underlying disease progression from premalignant and asymptomatic stages to the final stage of MM. In addition, it is of major clinical interest to identify possible factors involved in progression, for example, the development of osteolysis, which can be blocked with already available drugs that are applied in other hematological diseases. To address this, we investigated for the first time MSCs from healthy donors (HD) and patients with MGUS, SMM, and MM toward functionality and performed RNA sequencing.
Material and methods
Patients, healthy controls, and cell preparation
BM samples were obtained from a total of 40 patients (MGUS, n = 11; SMM, n = 7; MM, n = 22). Healthy BM samples were derived from 41 healthy individuals (median age, 78 years; range, 33-90 years) undergoing orthopedic surgery. Detailed patient characteristics with typical features of the respective disease entity documented progression or pre-phase, fluorescence in situ hybridization data; high risk was defined according to revised international staging score12 and International Myeloma Working Group13 and C-reactive protein values, which could indicate inflammatory processes as potential confounders (Table 1).
Table 1.
Patients demographics and clinical characteristics
| No. | % | |
|---|---|---|
| Patients no. | 40 | |
| Sex | ||
| Male | 22 | 55 |
| Female | 18 | 45 |
| MGUS no. | 11 | 27 |
| Median age, y (range) | 65 | (44-78) |
| Type | ||
| Heavy chain leading∗1 | 10 | 91 |
| Light chain leading∗2 | 1 | 9 |
| FISH | ||
| Standard risk | 3 | 27 |
| High risk§ | 2 | 18 |
| Missing | 6 | 55 |
| Mean CRP, mg/dL (range) | 0.65 | (0.1-2.8) |
| Documented SMM progression | 2 | 18 |
| SMM no. | 7 | 18 |
| Median age, y (range) | 60 | (51-80) |
| Type | ||
| Heavy chain leading∗3 | 6 | 86 |
| Light chain leading∗4 | 1 | 14 |
| FISH | ||
| Standard risk | 4 | 57 |
| High risk | 3 | 43 |
| Missing | 0 | 0 |
| Mean CRP, mg/dL (range) | 0.4 | (0.1-1.0) |
| Documented MGUS pre-phase | 2 | 29 |
| Documented MM progression | 5 | 71 |
| MM no. | 22 | 55 |
| Median age, y (range) | 58 | (50-76) |
| Type/Paraprotein | ||
| Heavy chain leading∗5 | 18 | 82 |
| Light chain leading∗6 | 4 | 18 |
| CRAB (see below) | ||
| Hyper calcaemia | 1 | 5 |
| Renal failure | 3 | 14 |
| Anaemia∗ | 8 | 36 |
| Bone lesions | 17 | 77 |
| SLiM CRAB (see below) | ||
| Sixty-percent bone marrow plasma cells† | 11 | 50 |
| Light chain ratio ≥100 | 6 | 27 |
| >1 focal lesions on MRI studies | 1 | 5 |
| FISH | ||
| Standard risk | 11 | 50 |
| High risk | 9 | 41 |
| Missing | 2 | 9 |
| Mean CRP, mg/dL (range) | 0.56 | (0.1-3.7) |
| Documented SMM or MGUS pre-phase | 8 | 36 |
Scatter Plot for Infiltration [%], Scatter Plot for hemoglobin (HB) [g/dl] of MGUS, SMM and MM patients included in this study at time of diagnosis shown on the right side.
MGUS, monoclonal gammopathy of undetermined significance; SMM, smoldering myeloma; MM, multiple myeloma; CRP, C-reactive protein; FISH, fluorescence in situ hybridization.
∗1IgG kappa n = 7, IgG lambda n = 2, IgM lambda n = 1.
∗2LC kappa n = 1.
∗3IgG kappa n = 3, IgG lambda n = 2, IgA kappa n = 1.
∗4LC kappa n = 1.
∗5IgG kappa n = 9, IgG lambda n = 4, IgA kappa n = 3, IgA lambda n = 1, IgG lambda n = 1.
∗6LC kappa n = 3, LC lambda n = 1.
Plot shows the HB values of the individual patients.
Plot shows the infiltration rate by plasma cells of the individual patients.
According to revised-international staging score and International Myeloma Working Group: del(17p), t(4;14), t(14;16), +1q21.
MSCs were derived from the mononuclear cell (MNC) fraction, after density gradient centrifugation of BM aspirates of these specimens, and were directly cultured as previously described.14,15 All experiments were carried out using MSC derived from passages 3 to 4. The study was approved by our local institutional review board (approval number: 4777) and all patients gave written informed consent.
Cell culture conditions and reagents
Healthy MSCs were cultured in Dulbecco's modified eagle medium low glucose supplemented with 30% fetal bovine serum and 1% penicillin/streptomycin/L-glutamine (PSG; all from Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany), given in the legends of the respective figures.
CM and coculture
To generate cell line–derived conditioned media (CM), 2.7 × 105/cm2 cells of the MM cell line INA-6 (purchased DSMZ) were cultivated in RPMI with 10% fetal bovine serum and 1% PSG (all from Sigma Aldrich, Taufkirchen Germany) as previously described. To generate patient-derived CM, 2 × 105/cm2 BM-derived MNC from patients with MGUS, SMM, or MM and from healthy controls were used freshly or from short time frozen storage in liquid nitrogen and thawed afterward. Cell number and cell viability were measured by CASY cell counter system and showed no differences in viability. Cells were then cultivated and further cocultivated with healthy MSCs as previously described.15,16 To investigate effects on the transforming growth factor beta 1 (TGFB1) signaling, we used the active adenosine triphosphate (ATP)-competitive transforming growth factor beta receptor 1 (TGF-βRI) (ALK5) inhibitor SD208 (0.5 μM; Biotechne/R&D systems, Minneapolis, MN), diluted in DMSO (10 mM). Healthy MSC were cocultured with respective CM with and without SD208 (DMSO served as control) for 3 days and subsequently after 14 days of osteogenic induction, Alizarin Red staining visualized osteogenic differentiation. DMSO served as control.
Phenotypic characterization of MGUS-, SMM- and MM-derived MSCs
Primarily patients-derived MSCs were characterized regarding their morphology and growth properties by light microscopy. Quantification of growth was determined as described elsewhere.14,15
Differentiation properties
Differentiation assays into osteoblasts, chondroblasts, and adipocytes were performed on Passage 3. Adipogenic and chondrogenic differentiation were induced as previously described.14 For osteogenic differentiation, Dulbecco's modified eagle medium low glucose were supplemented with dexamethasone (10-7 M), ascorbic acid (50 μg/mL), and β-glycerolphosphate (10 mM; all Sigma-Aldrich) and, after 14 days of differentiation, visualized by Alizarin Red staining. All images were visualized using Axiovert 25 microscope (Zeiss, Jena, Germany; 5 objective Zeiss CP-Achromat 5 Ph0, for native and osteogenic differentiated MSC or a 10 objective-Zeiss CP-Achromat 10 Ph1, for adipogenic differentiation, Zeiss EC Plan-Neofluar, 2.5×/0.075, for chondrogenic differentiation) and digitalized with the SPOT Software (Diagnostic Instruments Inc, Sterling Heights, MI).
Long-term culture-initiating cells assay
A total of 0.8 × 106 to 1.2 × 106 MSCs were cultivated on 96-well plates (Costar, Corning) and irradiated with 30 Gray using Gulmay RS225 X-ray equipment. Subsequently, 6 × 103 healthy CD34+ cells were plated on these MSC feeder layers and then further proceeded using the same conditions and reagents as in our previous work.14,15
Cellular senescence assay by β-galactosidase staining
β-galactosidase activity is an indicator for cellular senescence and was determined by using the Cellular Senescence Detection Kit (Biolabs, San Diego, CA), following manufacturer’s instructions. Cells were visualized under light microscope and senescent cells reflected blue staining through β-galactosidase activity were assessed.
Enzyme-linked Immunosorbent Assay
Filtered conditioned media from MSC were harvested in passage 3 and frozen at –20°C. Human CXCL12/SDF-1 alpha Enzyme-Linked Immunosorbent Assay–Quantikine was performed according to the manufacturers' instructions (R&D systems, Minneapolis, Minnesota).
Quantitative real-time PCR
RNA was isolated using the RNeasy Micro Kit or Mini Kit (Qiagen, Hilden, Germany) with the optional DNase digestion, according the manufacturer's instruction.
All quantitative real-time PCR were performed in duplicates on a StepOne Plus Realtime PCR Cycler using SYBR Green PCR Master Mix (Applied Biosystems, Life Technologies, Carlsbad, CA). All primer sequences can be provided on request. GAPDH serves as reference control, and differences in the expression on messenger RNA level were calculated with the ΔCT method.
RNA-sequencing
DNase digested total RNA samples used for transcriptome analyses were quantified (Qubit RNA HS Assay, Thermo Fisher Scientific Inc, Waltham, MA) and quality measured by capillary electrophoresis using the fragment analyzer and the “Total RNA Standard Sensitivity Assay” or the bioanalyzer assay “Eukaryote Total RNA Pico” (Agilent Technologies, Inc, Santa Clara, CA). The library preparation was performed according to the manufacturer's protocol using the VAHTS universal V6 RNA-seq library prep kit (Illumina, San Diego, CA). Briefly, 10 ng total RNA was used for poly(A) RNA selection, fragmentation, complementary DNA generation, adapter ligation, strand selection, and library amplification. Bead purified libraries were normalized and finally sequenced on the NextSeq 2000 system (Illumina Inc, San Diego, CA) with a read setup of 1 × 150bp. The bcl2fastq tool was used to convert the bcl files to fastq files as well for adapter trimming and demultiplexing as previously described.16
Bioinformatical analysis
Data analyses on fastq files were conducted with CLC Genomics Workbench (version 20.0.3, QIAGEN, Venlo, The Netherlands). Reads of all probes were adapter trimmed and quality trimmed (using the default parameters: bases below Q13 were trimmed from the end of the reads, ambiguous nucleotides maximal 2). Mapping was done against the Homo sapiens (hg38) (Mai 25, 2017) genome sequence. After grouping of samples (3 biological replicates each) according to their respective experimental condition, multigroup comparisons were made and statistically determined using the empirical analysis of differential gene expression (version 1.1, cutoff = 5). The resulting P values were corrected for multiple testing by false discovery rate (FDR) correction. A P value ≤ .05 was considered significant. The data were further evaluated with the Ingenuity-Pathway analysis software (Qiagen Inc 2020, Venlo, The Netherlands) using the core analysis with default parameters (jFCj ≥ 1.5; P ≤ .01; P [FDR] ≤ .05).
After quality control, the sequencing raw data were trimmed by removing the first 12 bases from each read. Alignment was performed using the program STAR v2.7.2a17 against the reference human genome GRCh38.97 (hg38). Alignment was performed as a 2-pass experiment, which means that in a first step new splice junctions were detected, which were added in the second step to the initial reference. The analysis pipeline was adapted from an NIH protocol.
After applying the program featureCounts (1.6.5) to the aligned reads, DESeq2 (1.24.0) was used to calculate the differential expression of patients-derived samples in contrast to healthy individuals. Genes with an FDR q-value <0.05 were considered differentially expressed. StringTie (2.0.3)18 was applied to calculate fragments per kilobase of transcript per million mapped reads (FPKM values). Principal component analysis plots were created in R using the FactoMineR (2.3) and pheatmap (1.0.12) packages. For gene set enrichment analysis (GSEA) (Broad Institute, Boston, MA), the fragments per kilobase of transcript per million mapped reads values obtained from StringTie (2.0.3) for the different samples and the healthy controls were compared either with the gene sets contained in the Molecular Signature database or with self-made gene lists. The parameter metric was set to Signal2Noise, and 1000 gene set–based permutations were applied to all analyses.
Gene Ontology analysis was performed with differentially expressed genes using the panther overrepresentation test, which is accessible via the AmiGO 2 webpage (https://amigo.geneontology.org/amigo). A Fisher's exact test was performed, and the results were Bonferroni-corrected as previously reported.16
Statistical analysis
Statistical analyses were performed using Prism 8.4.3 (GraphPad Software Inc, La Jolla, CA). For interindividual comparison, the 2-sided unpaired Student t test was used, whereas for intraindividual analysis, the Wilcoxon signed rank test was used. For all experiments, means and standard error of the mean (SEM) are given. Statistical significance was established at P ≤ .05 (∗ P ≤ 0.05; ∗∗ P ≤ .01; ∗∗∗ P ≤ .001; ∗∗∗∗ P ≤ .0001).
Results
MSC transition from MGUS to SMM to MM - RNA-sequencing revealed a genetic signature
RNA-sequencing data were obtained for MSC from 5 healthy donors, 4 patients with MGUS, 4 patients with SMM, and 5 patients with MM (median age in years: HC, 72; MGUS, 65; SMM, 58; and MM, 57). Detailed patient characteristics of the samples we used for RNA sequencing with additional information such as documented progression or documented preliminary phase are shown in Figure 1A. Principal component analysis illustrated the distribution of the groups we analyzed, showing a closer relationship between MGUS and SMM MSC (Principal components 1 vs 3, Figure 1B, blue and dark blue dots). In comparison to MSC from normal donors, RNA-sequencing data revealed 1539 differentially expressed genes in MGUS-derived MSCs, 1513 in SMM-derived MSCs, and 4696 in MM-derived MSCs (FDR q-value ≤ 0.05). The most overlapping differentially expressed genes of 747 were found between MGUS and SMM MSC (Figure 1C).
Figure 1.


RNA sequencing analysis of MSCs derived from patients with MGUS, SMM, and MM revealed a genetic signature in the respectively stages. (A) Patient characteristics of included samples in RNA sequencing analysis. Table shows clinical parameters including prestages and/or progression status of MGUS (n = 4), SMM (n = 4), and MM (n = 5) samples in comparison to healthy (n = 5) samples. (B) Principal component analysis of MGUS, SMM and MM MSC. PC1 vs PC3 is shown. Green squares depict healthy MSC, dark blue squares depict MGUS MSC, light blue squares depict SMM MSC, and red squares depict MM MSC. (C) Venn diagrams of all differentially expressed genes, unique genes, and overlapping genes (FDR q-value ≤ 0.05) between the groups to be compared. (D) Top: graphical summary of representative overlapping genes from all 3 groups and between the groups to be compared. Several genes from that total overlap such as TGFB1, BMP8B, or WNT5B are represented by red framing. Unique overlapping genetic signature between MGUS and SMM MSC (such as TGFBR3L, RPSA or RPS27A) or the unique overlapping genetic signature between MGUS and MM (such as CDKN2A, CDKN2B) or between SMM and MM MSC (such as TGFR1, WNT2 or SFRP2) are represented by black framing. Bottom: graph table of the overlapping 296 differentially expressed genes by contrasting MGUS, SMM and MM MSC (FDR q-value ≤ 0.05).
By contrasting MGUS and MM or SMM and MM MSCs, 505 and 549 genes were revealed differentially expressed, respectively (Figure 1C).
Subtracting the 296 genes that were overlapping differentially expressed by all 3 groups, MGUS and SMM MSCs have the highest number of an overlapping signature, with 451 differentially expressed genes only detected in this comparison. These genes include, for example, TGFBR3L, CD274, DKK2, MMP9, or S100A9 (Figure 1D; supplemental Figure 1). An overlap of 253 differentially expressed genes were found only between SMM and MM MSC (Figure 1D; supplemental Figure 1), which includes TGFBR1, WNT2, S100A6, S100A4, or TNFRSF25 (Death Receptor3). Moreover, the lowest number of 209 overlapping differentially expressed genes were found by comparing only MGUS and MM MSC, with, for example, the cell cycle markers CDKN2A and CDKN2B or NOTCH1, which were only detected in this comparison (Figure 1D; supplemental Figure 1). The aforementioned 296 differentially expressed genes that were overlapping in all 3 MSC groups, includes genes from the BMP/TGF (BMP8B, TGFB1) or WNT-signaling Pathway (WNT5B, FZD8) and downstream targets of these pathways that are involved in skeletal development (Integrin Binding Sialoprotein; IBSP, Msh Homeobox 2; MSX2), proliferation (TNF receptor superfamily member 19; TNFRSF19), or extracellular matrix (ELN; Elastin) (Figure 1D). This was confirmed by gene ontology analysis, demonstrating enriched cell processes such as proliferation, skeletal system, or apoptosis in all 3 groups contrasted to the healthy samples. Moreover, cell processes such as mesenchymal differentiation or mesenchymal development arises in SMM and remain to the progression in myeloma (Figure 2).
Figure 2.

Gene Ontology (GO) of MGUS, SMM and MM MSC contrasted to healthy individuals revealed contributed cell processes Representative overview of GO representing contributed processes such as skeletal system, cell communication or apoptotic processes in MGUS MSC. (A) Developmental processes, cell death, or mesenchymal cell differentiation were enriched in SMM (B) and in MM MSC processes such as DNA repair, mesenchyme development, or regulation of cell-matrix adhesion (C) were strongly enriched. For all GO terms, fold enrichment and P-values of overrepresented and underrepresented processes are given.
RNA-sequencing revealed a unique genetic signature in MSC transition from MGUS to SMM to MM
Apart from that overlapping signature of all 3 MSC groups, we also focused on genes that were uniquely expressed in the MSC for that entity alone. MGUS MSCs exhibit a unique signature of 583 differentially expressed genes such as MAPK7, FZD9, FZD7, TFGB1l1, and TGFB2 and downstream targets such as SPP1 (Secreted Phosphoprotein 1, Osteopontin) (Figure 3A, supplemental Table 1). A unique signature of 513 differentially expressed genes in SMM MSC revealed CCND2 (Cyclin D2), CDK15 (Cyclin dependent kinase 15), or WNT7B, EMILIN1, or CXCL2 (Figure 3A; supplemental Table 1).
Figure 3.

Ingenuity pathway analysis and GSEA in MSC from patients with MGUS, SMM and MM revealed a unique genetic signature in MSC transition. (A) Table overview of unique differentially expressed genes from DESeq2 output such as MAPK7, SPP1 or FGF11 in MGUS MSC, CCND2, WNT7B or SESN2 in SMM MSC as well as several CDK's, MAPK14 or BMP2K in MM MSC within the comparison of all 3 groups (FDR q-value ≤0.05). (B) Ingenuity pathway analysis predicted the top 5 most probable upstream regulators in MGUS, SMM and MM MSC. (C) GSEA contrasting healthy samples to MGUS, SMM and MM samples. The results confirmed a strong enrichment for the BMP/TGFB-signaling pathway signature. Neither TNFA/NFKB nor WNT are strongly enriched. MM MSC to healthy MSC confirmed a strong enrichment of a gene set also for TNF/p38 and Estrogen signaling signature. For all plots, the normalized enrichment score, P-values and (FDR q-value) are given.
MM MSCs exhibit the highest number of unique differentially expressed genes of 3938. Here, MAPK14 (p38), SMAD5, SMAD6, TGFA, TGFB3, TGFBR3, WNT9A, or several genes involved in cell cycle such as cyclin dependent kinases (CDKs) (4, 6, 7, 10, 12, and 19) were detected (Figure 3A; supplemental Table 1). For additional information regarding ribosomal, mitochondrial and apoptotic processes see GSEA results in supplemental Figure 3.
Ingenuity pathway analysis predicted TGFB1 (Transforming Growth Factor-β1) or CG (Calycosin-7-O-β-d-glucopyranoside) in MGUS and SMM derived MSC and DAP3 (death associated protein 3) or ESR1 (estrogen receptor 1) in MM derived MSCs as potential top 5 upstream regulators (Figure 3B).
BMP/TGF-signaling as potential key pathway contributing to MSC transition from MGUS to SMM to MM
As predicted by Ingenuity pathway analysis, gene set enrichment analysis (GSEA) confirmed a clear transition to affected BMP/TGF-signaling in all 3 MSC groups. The bone morphogenetic protein 2 (BMP2) is a major regulator for osteogenesis and can be stimulated by estrogen signaling. We observed a significantly shifted enrichment score of signatures describing BMP2 and TGFB1 signaling and target genes already in the analysis with MGUS-derived MSC contrasted to healthy samples, while the gene set for TNFA/NFKB signaling was not affected in GSEA. The BMP2 signature was clearly conserved when contrasting healthy and SMM MSC while WNT signature was not strongly affected. GSEA results from MM MSC contrasted to healthy revealed a strong enrichment of genes related to TGFB1 signaling signature and expanded with a strong enriched TNF/p38 and Estrogen-signature (Figure 3C). BMP's, TGF und ESR are known to be involved in several cell processes such as osteogenesis and multiple myeloma progression and we already have seen in MGUS MSC a marked enrichment of genes that could be assigned to multiple myeloma in the later course of progression. This was confirmed by a strong enrichment of processes such as hematopoiesis, osteogenesis or cell cycle regulation by GSEA analysis in all 3 groups (supplemental Figure 2).
Differential gene expression translates to functional alterations
To address the question whether programmatic alterations detected on molecular level underlying the transition from MGUS to MM are also seen in MSCs concerning their functionality, we performed different in vitro assays.
Typical characteristics of in vitro MSCs are a spindle type morphology and the formation of fibroblast colonies (Colony Forming Unit-Fibroblast; CFU-F). We already found a significantly reduced CFU-F activity of MGUS-derived MSC (mean, MGUS: 31.94; HC: 91.2, P-value: 0.002). SMM-derived MSC showed a 2.6-fold reduced CFU-F activity (mean, SMM: 34.6; HC: 91.2, P-value: 0.01) and MM-derived MSC showed the most reduced CFU-F activity (mean, MM: 22.7; HC: 91.2 P-value: ≤0.0001). We observed a heterogeneous morphology in MGUS-derived MSC, ranging from no relevant to single alterations, while a more disorganized and broad shaping structure in SMM-derived MSC was observed which was even more apparent in MM derived MSC (Figure 4A). Cellular senescence was up to 1.7-fold increased in MGUS- and SMM-derived MSC (mean, MGUS: 15.2%, SMM 13.2%, HC: 8.7%), whereas it was 5.9-fold increased in MM-derived MSC compared to healthy MSC (mean, MM: 51.8%, HC: 8.7%, P ≤ .0001, Figure 4B) and in line with this, RNA sequencing data show an upregulation of CDKN2A (p16), as a marker for senescent cells (Figure 4C).
Figure 4.

Alteration in morphology and reduced growth capacity of MGUS, SMM and MM MSC. (A) Bar charts of CFU-F were normalized to 1 × 107 plated BM-MNC and representative micrograph of the phenotype of MGUS-, SMM- and MM-derived MSC with scale bars indicating 100 μm. (B) Bar charts of the number of senescent cells in native MGUS-, SMM- and MM-derived MSC in passage 3 cells after the β-galactosidase staining and representative micrographs to visualize senescent cells in blue (scale bars indicating 100 μm). For all experiments results are expressed as mean ± SEM. Asterisks indicate P-values ∗P < .05, ∗∗P < .01, ∗∗∗P < .001. (C) Box plot of the TPM (Transcripts per Kilobase Million) values for CDKN2A (p16) from our RNA sequencing data of healthy (n = 5), MGUS (n = 4), SMM (n = 4) and MM (n = 5) MSC. Significances were included from the DESeq2 results of CDKN2A of MGUS, SMM and MM MSC contrasted to healthy MSC. Asterisks indicate P-values ∗P < .05, ∗∗P < .01, ∗∗∗P < .001, ∗∗∗∗P < .0001.
MGUS, SMM and MM derived MSC show a diminished osteogenic-chondrogenic differentiation capacity
In the next step, we investigated patients-derived MSC in their capacity to differentiate into the osteogenic, chondogenic, or adipogenic lineage. No relevant differences could be observed in adipogenic differentiation in MGUS, SMM or in MM MSC (Figure 5A), but the chondrogenic differentiation was reduced in all 3 groups, indicated by reduced area size of cartilage pellets and Safranin O staining (Figure 5B). Since we know about the clinical relevance of osteoblast impairment in MM, we were interested in the question whether osteogenic differentiation is already impaired in MGUS and SMM. We observed a significantly reduced osteogenic differentiation capacity already in MGUS and SMM derived MSC as well as in MM derived MSC in comparison to healthy MSC (mean, HC: 2.28, MGUS: 1.33, P-value: .02 SMM: 1.25, P-value: .03; MM: 1.3, P-value: .009, Figure 5C). The early osteogenic marker OSTERIX was significantly reduced in all groups and the late osteogenic factor OSTEOCALCIN was significantly reduced in MGUS and MM MSC, but not in SMM MSC. For the latter the limited size of the samples analyzed must be noted.
Figure 5.

Reduced differentiation capacity and hematopoietic support of MGUS, SMM and MM MSC. (A) Representative micrographics of lipid vacuoles after Oil Red O staining of adipogenic differentiated MGUS-, SMM- and MM-derived MSC after 21 days of differentiation. Scale bars indicating 100 μm are shown. (B) Bar charts of measured cartilage area of chondrogenic differentiated MGUS-, SMM- and MM-derived MSC after 21 days of differentiation, stained with Safranin O and representative micrographics of orange proteoglycan after staining. Scale bars indicating 100 μm are shown. Chondrogenic differentiation capacity was graded according to the area of chondrogenic pellets and according to microscopic analysis. (C) Osteogenic differentiation was induced for 14 days and stained with Alizarin Red. Differences of the osteogenic potential between healthy MSC and MGUS-, SMM- and MM-derived MSC were quantified and are shown in bar charts. Representative micrographics of healthy and MGUS-, SMM- and MM-derived MSC with scale bars indicating 100 μm. For quantification, adipogenic, chondrogenic and osteogenic differentiation capacity was graded according to microscopic analysis of staining intensity as: 0 = absent; 1 = weak; 2 = moderate; 3 = intensive as previously described.14 (D) mRNA expression level of osteogenic factors as OSTERIX and OSTEOCALCIN were measured by quantitative Realtime-PCR in healthy MSC and MGUS-, SMM- and MM-derived MSC. (E) Hematopoietic Support of MGUS, SMM and MM MSC. Left side: Bar charts showing LTC-IC frequency of healthy CD34+ HSPC cultured on healthy-, MGUS-, SMM- or MM-derived MSC. Colonies were calculated under light microscope and visualized in graph. Right side: mRNA expression level of CXCL12 was measured by quantitative Realtime-PCR in healthy MSC, MGUS-, SMM- and MM-derived MSC. CXCL12 protein level in conditioned media (CM) from healthy (n = 5), MGUS (n = 5), SMM (n = 6), MM (n = 4) were assessed by Enzyme-Linked Immunosorbent Assay. For all experiments, results are expressed as mean ±SEM. Asterisks display P-values ∗P ≤ .05; ∗∗ P ≤ .01; ∗∗∗ P ≤ .001.
Impaired hematopoietic support is already diminished in MGUS- and SMM-derived MSC and progresses in MM-derived MSC
To investigate hematopoietic supporting capacity of MGUS-, SMM- and MM-derived MSC we performed Long Term Culture Initiating Cell (LTC-IC) assays. Already MGUS and SMM-derived MSC exhibited a twofold reduced hematopoietic supporting capacity and in line with the clinical presentation we observed a 12-fold reduced hematopoietic supporting capacity in MM-derived MSC. On a molecular level, the expression of CXCL12, also known as Stromal-derived factor-1 (SDF-1), a crucial player for several cell processes such as migration of stem cells, bone formation, inflammation, or cancer association, was significantly downregulated in MGUS, SMM and MM derived MSC (Figure 5E). This was validated on protein level by measuring SDF-1 (CXCL12) in supernatants of cultured MGUS, SMM, MM MSC (Figure 5E).
Soluble factors from TGF beta family as potential candidates to induce the observed alterations
Finally we wanted to test whether these observed MSC alterations are inducible and whether this could be responsible due to soluble factors. For this we co-cultured healthy MSC with supernatants from MGUS, SMM, and MM patients. Inducible effects on morphology and growth properties of healthy MSC after exposure to supernatants from MGUS, SMM, and MM patients could be observed, similar to those observed in patients-derived MSC from our aforementioned functional analysis (Figure 6A). Previous data from our group 19 show, that TGF could be a potential candidate for inducing alterations in healthy MSC as representative shown in Figure 6B. Furthermore, the database OncoDB shows, that a dysregulated expression of TGFB1, BMP8B and BMP2 is noted in other mature B-cell neoplasia like diffuse large B-cell lymphoma (DLBC, 48 samples), comparable to those expression data in our Multiple Myeloma MSC (Figure 6C) and may suggest a potential contribution to disease progression to multiple myeloma. Assuming TGFβ1 as a soluble factor in CM derived from patients' cells have the potential to induce the observed alterations in healthy MSC we wondered whether blocking by the active ATP-competitive transforming growth factor-β receptor 1 (TGF-βRI) (ALK5) inhibitor SD208 can revert the effects.
Figure 6.

Blocking of the potential candidate TGFB in supernatants derived from MGUS, SMM and MM restores differentiation capacity of co-cultured healthy MSC. (A) Study design for coculture of healthy MSC with conditioned media (CM) from MGUS, SMM, and MM patients. MSC were cultured with CM from healthy, MGUS, SMM, or MM MNC for 3 days. Representative micrographics of the phenotype of healthy MSC after exposure to supernatants from MGUS-, SMM-, and MM-derived MNC for 3 days with scale bars indicating 100 μm. Bar charts of cell number after exposure to supernatants from MGUS, SMM, and MM MNC as well as the MM cell line INA-6. (B) Healthy MSC were treated 3 days with TGFB1 and representative pictures show phenotype of untreated healthy control and after exposure to TGFB1. Scale bars indicating 100 μm. (C) Expression TPM-values (transcripts per kilobase million) from our RNA sequencing data for TGFB1, BMP8B, and BMP2 from our MGUS, SMM, and MM MSC samples in comparison to 48 patients samples with diffuse large B-cell lymphoma from the database OncoDB. (D) Coculture of healthy MSC with CM from healthy, MGUS, SMM, and MM MNC. To analyze and inhibit potential TGFB1 inducing effects, the potent ATP-competitive TGF-βRI inhibitor SD208 was added to CM from healthy, MGUS, SMM, and MM-derived MNC. DMSO containing CM served as control group. Healthy MSC were co-cultured with respective CM for 3 days. Representative micrographs of the morphology of healthy MSC cocultured with CM supplemented with DMSO or SD208 are shown. Inner pictures show Alizarin Red staining of 14 days osteogenic induced healthy MSC after the period of 3 days of exposure to respective condition (CM MGUS, SMM, MM + DMSO, or +SD208). Scale bars indicating 100 μm.
Therefore, we incubated healthy MSC with CM from patients-derived MNCs (MGUS, SMM, or MM) and blocked TGFB1 signaling by SD208 (Figure 6D). In line with our previous findings, coculture with patient derived MNC lead to a dysregulated phenotype, reduced cell number, and diminished osteogenic differentiation capacity of healthy MSCs. The addition of SD208 restores the phenotype and osteogenic differentiation capacity (Figure 6D).
Discussion
The hallmark of MM is a clonal plasma cell infiltration in the BM accompanied by end-organ damage such as hematopoietic insufficiency and/or osteolysis. MM is often considered to be preceded by a sequence of premalignant stages such as MGUS and asymptomatic stages such as SMM. Although there are studies showing this sequence within the malignant cell itself, it is unclear whether this sequence is also found in MSC.20 Some studies have addressed the pathophysiological contribution in disease development of MSC within the BM microenvironment but mainly focused on MM.11,21
In this study, we could show that alterations of MSC are already imprinted in MGUS and further progresses in SMM and finally in MM. Within this sequence, we were able to show both structural and functional alterations of MSC, with reduced osteogenic differentiation capacity and impaired hematopoietic support. In part, these alterations showed a significant progression to full blown MM. In line with this, PCR analyses showed reduced expression of CXCL12, which is known as a crucial player for several cell processes; for example, for migration of stem cells, bone formation, and inflammation.20,21 The deletion of CXCL12 in murine MSC leads to a dysregulated bone formation in the number of osteoclasts and osteoblasts and increased adipocytes.22
Physiologically TGF/BMP signaling plays a crucial role in several cell processes,19,22,23 and it has been shown to play a role in MM progression.11 Although the role of BMPs in the development of osteolysis in myeloma is controversial, blocking these signaling pathways has already been considered.24,25
RNA sequencing revealed that MSC from all 3 groups exhibited activation of BMP/TGF signaling. This may lead to dysregulation of downstream targets such as the osteogenic-chondrogenic transcription factor RUNX2, which in turn led to dysregulation of induction factors for osteogenesis such as OSTERIX or IBSP.26 With the knowledge of the osteoblast's crucial role in normal BM and MM, it has been reported that osteoblasts directly inhibit MM cells in vitro.27 We and other groups consistently show overrepresented TGFB-1, which was described in correlation with bone lesions in patients with MM and other WNT-regulators or reduced osteogenic differentiation.11,21,28, 29, 30 Blocking of TGB1-signaling by SD208 restores functionality of MSC as shown by our group and others for other entities.11,19
Surprisingly, osteogenic capacity of MGUS MSC was already diminished and reflected by a reduced OSTERIX and OSTEOCALCIN level.
This imbalance, with impaired osteoblastic differentiation as shown in this study and the clinically known activation of osteoclasts, may contribute to defective bone remodeling that is implicated in early disease stages.
In addition, immunologic dysfunction of MSCs became a greater focus of interest and has been reported by other groups, because activation of an immunosuppressive microenvironment by MSCs may contribute to the transition from MGUS to MM.29,31 Furthermore, TGFB plays a role here, which can create an immunosuppressive milieu and lead to drug resistance of modern immunotherapeutics.32 In addition, our group has already shown that extrinsic TGF-β is capable of generating functional deficits in MSC.19 In conclusion, our data lead to a broader understanding of the impact of the microenvironment on the progression of MGUS and SMM to MM, and MSCs are a potential therapeutic target. This would provide a treatment that not only targets the malignant clone and tumor-associated immunosuppression but also addresses the remodeling of the microenvironment by altered MSCs as a third pillar. We, therefore, consider blocking the BMP/TGF signaling pathway as a future approach to prevent end-organ damage and to avoid progression to MM.
Conflict-of-interest dislosure: The authors declare no competing financial interests.
Acknowledgments
The authors thank Dennis Sohn from the Laboratory of Molecular Radio-Oncology for technical assistance regarding LTC-IC assay.
This work was supported by the Leukämie Lymphom Liga e. V., Duesseldorf, Germany and a grant (2019-21) from the Research Committee of the Medical Faculty, Heinrich-Heine-University, Duesseldorf (S.G.), and the German Research Foundation (417677437/GRK2578).
The funders had no role in the design of the study, the collection, analyses, or interpretation of data, the writing of the manuscript, or in the decision to publish the results.
Authorship
Contribution: L.B., T.S., P.J., and S.G. contributed to conception and design, provision of study material or patients, collection and/or assembly of data, data analysis and interpretation, manuscript writing, and final approval of the manuscript; R.F. contributed to provision of study material or patients, collection and/or assembly of data, data analysis and interpretation, manuscript writing, and final approval of the manuscript; U.M. and R.Z. contributed to provision of study material or patients, collection and/or assembly of data, and final approval of the manuscript; A.K., B.S., P.P., T.W., K.K., and F.B. contributed to collection and/or assembly of data, data analysis and interpretation, and final approval of the manuscript; and S.D. and R.H. contributed to data analysis and interpretation, manuscript writing, and final approval of the manuscript.
Footnotes
P.J. and S.G. contributed equally to this study.
Parts of this study have been presented at the annual meeting of the German-Austrian-Suisse Society of Hematology and Oncology (DGHO), Vienna, Austria, October 2022.
RNA-seq expression data are deposited in NCBI Sequence Read Archive (SRA) with BioProject (ID PRJNA1015628).
Data are available upon reasonable request from corresponding authors, Paul Jäger (paulsebastian.jaeger@med.uni-duesseldorf.de) and Stefanie Geyh (stefanie.geyh@med.uni-duesseldorf.de).
The full-text version of this article contains a data supplement.
Contributor Information
Paul Jäger, Email: paulsebastian.jaeger@med.uni-duesseldorf.de.
Stefanie Geyh, Email: Stefanie.geyh@med.uni-duesseldorf.de.
Supplementary Material
References
- 1.Kyle RA. Multiple myeloma. N Engl J Med. 2004;351(18):1860–1873. doi: 10.1056/NEJMra041875. [DOI] [PubMed] [Google Scholar]
- 2.Kyle RA. Multiple myeloma. Blood. 2007;111(6):2962–2972. doi: 10.1182/blood-2007-10-078022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Medical Masterclass, e.b.J.F Haematology: multiple myeloma. Clin Med (Lond) 2019;19(1):58–60. doi: 10.7861/clinmedicine.19-1-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dispenzieri A, Katzmann JA, Kyle RA, et al. Prevalence and risk of progression of light-chain monoclonal gammopathy of undetermined significance: a retrospective population-based cohort study. Lancet. 2010;375(9727):1721–1728. doi: 10.1016/S0140-6736(10)60482-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kyle RA. Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med. 2006;354(13):1362–1369. doi: 10.1056/NEJMoa054494. [DOI] [PubMed] [Google Scholar]
- 6.Rodriguez-Otero P, Paiva B, San-Miguel JF. Roadmap to cure multiple myeloma. Cancer Treat Rev. 2021;100 doi: 10.1016/j.ctrv.2021.102284. [DOI] [PubMed] [Google Scholar]
- 7.Michels T, Petersen KE. Multiple myeloma: diagnosis and treatment. Am Fam Physician. 2017;95(6):373–383. [PubMed] [Google Scholar]
- 8.Dominici M, Le Blanc K, Mueller I, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8(4):315–317. doi: 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- 9.Anthony BA, Link DC. Regulation of hematopoietic stem cells by bone marrow stromal cells. Trends Immunol. 2014;35(1):32–37. doi: 10.1016/j.it.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505(7483):327–334. doi: 10.1038/nature12984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bruns I, Cadeddu RP, Brueckmann I, et al. Multiple myeloma-related deregulation of bone marrow-derived CD34(+) hematopoietic stem and progenitor cells. Blood. 2012;120(13):2620–2630. doi: 10.1182/blood-2011-04-347484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palumbo A, Avet-Loiseau H, Oliva S, et al. Revised International staging system for multiple myeloma: a report from International Myeloma Working Group. J Clin Oncol. 2015;33(26):2863–2869. doi: 10.1200/JCO.2015.61.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chng W J, Dispenzieri A, Chim C-S, et al. IMWG consensus on risk stratification in multiple myeloma. Leukemia. 2014;28(2):269–277. doi: 10.1038/leu.2013.247. [DOI] [PubMed] [Google Scholar]
- 14.Geyh S, Oz S, Cadeddu RP, et al. Insufficient stromal support in MDS results from molecular and functional deficits of mesenchymal stromal cells. Leukemia. 2013;27(9):1841–1851. doi: 10.1038/leu.2013.193. [DOI] [PubMed] [Google Scholar]
- 15.Geyh S, Rodríguez-Paredes M, Jäger P, et al. Functional inhibition of mesenchymal stromal cells in acute myeloid leukemia. Leukemia. 2016;30(3):683–691. doi: 10.1038/leu.2015.325. [DOI] [PubMed] [Google Scholar]
- 16.Jager P, Geyh S, Twarock S, et al. Acute myeloid leukemia-induced functional inhibition of healthy CD34+ hematopoietic stem and progenitor cells. Stem Cells. 2021;39(9):1270–1284. doi: 10.1002/stem.3387. [DOI] [PubMed] [Google Scholar]
- 17.Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33(3):290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Geyh S, Rodríguez-Paredes M, Jäger P, et al. Transforming growth factor beta1-mediated functional inhibition of mesenchymal stromal cells in myelodysplastic syndromes and acute myeloid leukemia. Haematologica. 2018;103(9):1462–1471. doi: 10.3324/haematol.2017.186734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Nieuwenhuijzen N, Spaan I, Raymakers R, Peperzak V. From MGUS to multiple myeloma, a paradigm for clonal evolution of premalignant cells. Cancer Res. 2018;78(10):2449–2456. doi: 10.1158/0008-5472.CAN-17-3115. [DOI] [PubMed] [Google Scholar]
- 21.Jurczyszyn A, Zebzda A, Czepiel J, et al. The analysis of the relationship between multiple myeloma cells and their microenvironment. J Cancer. 2015;6(2):160–168. doi: 10.7150/jca.10873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen G, Deng C, Li YP. TGF-beta and BMP signaling in osteoblast differentiation and bone formation. Int J Biol Sci. 2012;8(2):272–288. doi: 10.7150/ijbs.2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Batlle E, Massague J. Transforming growth factor-beta signaling in immunity and cancer. Immunity. 2019;50(4):924–940. doi: 10.1016/j.immuni.2019.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghosh-Choudhury N, Windle JJ, Koop BA, et al. Immortalized murine osteoblasts derived from BMP 2-T-antigen expressing transgenic mice. Endocrinology. 1996;137(1):331–339. doi: 10.1210/endo.137.1.8536632. [DOI] [PubMed] [Google Scholar]
- 25.Gooding S, Olechnowicz SWZ, Morris EV, et al. Transcriptomic profiling of the myeloma bone-lining niche reveals BMP signalling inhibition to improve bone disease. Nat Commun. 2019;10(1):4533. doi: 10.1038/s41467-019-12296-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bruderer M, Richards RG, Alini M, Stoddart MJ. Role and regulation of Runx2 in osteogenesis. Eur Cell Mater. 2014;28:269–286. doi: 10.22203/ecm.v028a19. [DOI] [PubMed] [Google Scholar]
- 27.Reagan MR, Ghobrial IM. Multiple myeloma mesenchymal stem cells: characterization, origin, and tumor-promoting effects. Clin Cancer Res. 2012;18(2):342–349. doi: 10.1158/1078-0432.CCR-11-2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Todoerti K, Lisignoli G, Storti P, et al. Distinct transcriptional profiles characterize bone microenvironment mesenchymal cells rather than osteoblasts in relationship with multiple myeloma bone disease. Exp Hematol. 2010;38(2):141–153. doi: 10.1016/j.exphem.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 29.Garcia-Gomez A, De Las Rivas J, Ocio EM, et al. Transcriptomic profile induced in bone marrow mesenchymal stromal cells after interaction with multiple myeloma cells: implications in myeloma progression and myeloma bone disease. Oncotarget. 2014;5(18):8284–8305. doi: 10.18632/oncotarget.2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tian E, Zhan F, Walker R, et al. The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N Engl J Med. 2003;349(26):2483–2494. doi: 10.1056/NEJMoa030847. [DOI] [PubMed] [Google Scholar]
- 31.Giallongo C, Tibullo D, Camiolo G, et al. TLR4 signaling drives mesenchymal stromal cells commitment to promote tumor microenvironment transformation in multiple myeloma. Cell Death Dis. 2019;10(10) doi: 10.1038/s41419-019-1959-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rana PS, Soler DC, Kort J, Driscoll JJ. Targeting TGF-beta signaling in the multiple myeloma microenvironment: steering CARs and T cells in the right direction. Front Cell Dev Biol. 2022;10 doi: 10.3389/fcell.2022.1059715. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.