

Abstract
Upon interacting with its receptor, poliovirus undergoes conformational changes that are implicated in cell entry, including the externalization of the viral protein VP4 and the N terminus of VP1. We have determined the structures of native virions and of two putative cell entry intermediates, the 135S and 80S particles, at ∼22-Å resolution by cryo-electron microscopy. The 135S and 80S particles are both ∼4% larger than the virion. Pseudoatomic models were constructed by adjusting the beta-barrel domains of the three capsid proteins VP1, VP2, and VP3 from their known positions in the virion to fit the 135S and 80S reconstructions. Domain movements of up to 9 Å were detected, analogous to the shifting of tectonic plates. These movements create gaps between adjacent subunits. The gaps at the sites where VP1, VP2, and VP3 subunits meet are plausible candidates for the emergence of VP4 and the N terminus of VP1. The implications of these observations are discussed for models in which the externalized components form a transmembrane pore through which viral RNA enters the infected cell.
Poliovirus, like related picornaviruses, is a small nonenveloped virus consisting of a plus-sense RNA genome enclosed within a protein shell or capsid (reviewed in reference 53). The capsid consists of 60 copies each of four proteins (VP1, VP2, VP3, and VP4) arranged on an icosahedral lattice. VP1, VP2, and VP3 have similar wedge-shaped cores, each an eight-stranded beta-barrel (the strands are designated B, I, D, G, C, H, E, and F), but each protein has unique loops connecting the strands and unique N and C termini (31). VP4 is small, myristylated (15), and has an extended structure.
The wedge-shaped cores of the subunits form the closed protein shell of the virion, with five copies of VP1 packing around the fivefold axes and VP2 and VP3 alternating around the threefold axes of the particle. The virion is stabilized by interactions among the wedge-shaped cores and by a network—on the inner surface of the protein shell—that is formed by VP4 and the N-terminal extensions of VP1, VP2, and VP3. The high-resolution structure of an empty capsid assembly intermediate shows that formation of the internal network is dependent on a late proteolytic cleavage of the capsid protein precursor, VP0, to yield VP4 and VP2 (5). This “maturation cleavage” is associated with the encapsidation of the viral RNA and is required for virion stability. Unfortunately, the viral RNA (which lacks the icosahedral symmetry of the protein coat) is not visible in the high-resolution structure of the virus, and its role in stabilizing the virus structure is unknown.
Despite extensive chemical and molecular characterization, the mechanism of picornavirus cell entry is known only in outline (14, 48, 49, 51–53, 59). In order to initiate a productive infection, the viral RNA must be externalized, cross a membrane, and be delivered to the cytoplasm. Infection begins with binding of the virion to a receptor that is a member of the immunoglobulin superfamily (37, 45), whereupon the virion undergoes an irreversible rearrangement to form the 135S, or A, particle. The 135S particle has a lower sedimentation coefficient (135S versus 160S for poliovirus [52]) and altered antigenic and proteolytic properties. This conformational change results in the externalization of VP4 and the N terminus of VP1—components of the stabilizing network on the inner surface of the 160S capsid. The externalized N terminus of VP1 is predicted to form an amphipathic helix and has been shown to allow the 135S particle to attach to synthetic membranes in vitro (24). This suggests that the maturation cleavage of VP0 may serve to trap the virion in a metastable state that is primed to undergo receptor-mediated conformational changes that are necessary for subsequent virus-cell interactions (14, 59). Analogous roles have been proposed for the maturation cleavages of the glycoproteins of influenza virus and other enveloped viruses (12). Later, the 135S particle releases its RNA in converting to the 80S (or H) particle—the putative end state. Although several lines of evidence support the proposition that the 135S particle is indeed a cell entry intermediate (20, 24, 28, 51), its status as such has been questioned (22, 48, 49) (see Discussion).
To explore these transitions and their implications for cell entry, we have examined the 160S, 135S, and 80S particles by cryo-electron microscopy (cryo-EM), taking advantage of the property that similar if not identical particles are produced by heating 160S virions at ∼50°C in hypotonic medium containing divalent cations (20, 58). We determined their structures at ∼22-Å resolution by three-dimensional (3-D) image reconstruction. These maps were used to obtain pseudoatomic models of the altered particles by appropriate rigid-body movements of VP1, VP2, and VP3 from their positions in the virion (cf. reference 55), which are known from the high-resolution crystal structure (31). We proceeded on the reasonable assumption that the core domains of the capsid proteins remain unchanged in all three forms of the virus. This made it possible to interpret the cryo-EM reconstructions in much greater detail than their nominal resolution would imply. The resulting models are consistent with a multistep model for cell entry involving RNA translocation through a membrane channel formed by the externalized N termini of VP1.
MATERIALS AND METHODS
Preparation of viral particles.
Virus was grown and purified by differential centrifugation and CsCl density gradient fractionation as described previously (20, 54). Altered particles (135S and 80S) were prepared by heating purified virions for 3 min at 50°C (135S) or 10 min at 55°C (80S) in hypotonic buffers containing Ca2+ (20 mM Tris, 2 mM CaCl2 [pH 7.5]) (20; C. N. Hiremath, unpublished data).
EM.
Samples of 160S, 135S, and 80S particles at 0.1 to 0.6 mg of protein/ml were frozen and imaged as described previously (9, 63). Size calibration standards were mixed with some specimens before freezing (see below). Philips EM400, CM120, and CM200 electron microscopes (FEI, Mahwah, N.J.), all equipped with Gatan (Pleasanton, Calif.) 626 cryoholders, were used to record micrographs at magnifications of ×38,000 to 60,000. Some fields were imaged twice at different focal settings (63). The micrographs were screened by optical diffraction to assess defocus, drift, and stigmation and by eye for suitable distributions of particles.
3-D reconstruction.
Micrographs were scanned on microdensitometers: Perkin-Elmer (Norwalk, Conn.) 1010MG at 18 to 24 μm/step or Zeiss (Englewood, Colo.) SCAI at 7 μm/step (Zeiss scans were computationally reduced to 14 or 21 μm/step). Particle images were extracted, processed, and normalized as described previously (8, 18). Icosahedral reconstructions were determined by standard procedures (26). For the 135S and 80S data, initial models for model-based orientation and origin determination (3) were determined by common-line methods (11, 26). The initial model for the 160S reconstruction was the 135S reconstruction. Resolution was estimated by use of a reliability index (62) computed by comparing structure factors from two reconstructions, each calculated from half of the data. Eigen values (19) were ≥10 for the 160S, 135S, and 80S reconstructions, indicating an adequate diversity of views.
Correction of CTF.
The final reconstructions were computed from images that were corrected for contrast transfer function (CTF) effects as described by Zlotnick et al. (63) but modified as follows. Fourier amplitudes between spatial frequencies of 0 and the first CTF minimum were multiplied by a factor intended to restore 50% of the difference between the theoretical and the actual (CTF-attenuated) amplitudes. This correction gave a 160S reconstruction in which a single contour level optimally fit the atomic structure. To avoid overcorrection of low amplitudes and division-by-zero errors, the divisor was set to 0.1 if the amplitude of the combined (sum of multiple images in a focal series) or uncombined theoretical CTF—at spatial frequency ν—was less than 0.1 maximum amplitude. Focal pairs were combined for the 160S (1.1- and 1.5-μm underfocus) and 135S (1.3- and 1.9-μm underfocus) images as part of the correction process (63). 80S images (1.2-μm underfocus) were also corrected, although focal pairs were not available.
Size calibration.
The crystal structure of the 160S capsid (31) was used as a size standard. The 160S reconstruction was calibrated against it both by fitting the atomic coordinates into variously sized reconstructions and assessing each fit by an R-factor or phase residual (see below), and by computing a 22-Å-resolution density map of the 160S capsid from the coordinates and scaling it to the 160S reconstruction by 3-D cross-correlation (13). The two methods gave consistent results.
Once the 160S reconstruction had been calibrated, 160S particles were used as an internal standard for the 80S particles, which, being empty, are distinguishable. Similarly, 80S particles were used to calibrate 135S particles. In each case, the 160S, 80S, and 135S particles were prepared separately and mixed, and the mixed specimens were imaged and then separately reconstructed. Scale factors were determined by comparing radial density plots by least-squares (6) and 3-D density maps by cross-correlation (13). Radial density plots were computed from an aligned average of the two-dimensional particle images and from the 3-D reconstructions (6). Except for the scaling of the 22-Å X-ray map to the 160S reconstruction, all comparisons were made with images and reconstructions that had not been corrected for CTF effects.
Atomic modeling.
Alpha-carbon atomic models of poliovirus capsid proteins VP1, VP2, and VP3 (31) were fitted individually as rigid bodies to the 160S, 135S, and 80S reconstructions, using the program FRODO (33). Prior to fitting the 135S and 80S reconstructions, VP4 and the N terminus of VP1 were removed from the model. Other extended loops and termini were removed if their structures clearly depended on contact with another subunit. In the truncated models, the fivefold symmetric beta-tube formed by the intertwined N termini of VP3 (residues 1 to 12) was treated as a separate object.
Automated rigid-body refinements were carried out by a modification of protocols developed for crystallographic refinement (32). A box of density, which generously enclosed a single protomer, was extracted from each reconstruction. 3-D Fourier transformation of the box, treating it as if it were periodic, yielded a set of complex-valued pseudostructure factors (∼Fo) that served as a standard for the rigid-body refinement. The phases of this standard remained constant throughout refinement and were unaffected by the atomic model.
Model-based electron density values for all lattice points in the “protomer box” were calculated by using a standard five-term summed-Gaussian approximation for the density attributable to each atom within the protomer box. To compensate for the lack of RNA in the model, density values in the RNA and solvent areas of the reconstructions were identified by using inner and outer radial masks (Table 1) and were corrected to match the solvent area's average prior to refinement. In 10-Å buffer zones around the mask edges, there was a linear transition between modified and unmodified density values. The model-based pseudo-diffraction data were scaled linearly to the reference Fourier coefficients in shells of resolution. Refinements evaluated the fit of the model to the map with either the standard crystallographic R-factor or an amplitude-independent scoring function such as <ΔΦ>, the mean |F|-weighted absolute phase difference (see Table 1 for definitions). Between iterations, coordinates for the individual proteins were allowed to move as rigid bodies translationally and rotationally.
TABLE 1.
Rigid-body refinement data
| Parameter | Value
|
|||||
|---|---|---|---|---|---|---|
| 160S
|
135S | 80S | P1/Mc
|
|||
| Complete | Truncated | Complete | Truncated | |||
| Masking radii (Å) | ||||||
| Inner radius | 109 | 109 | 115 | 109 | ||
| Outer radius | 162 | 162 | 164.5 | 166 | ||
| Agreement statisticsa | ||||||
| Overall R | 0.309 | 0.406 | 0.284 | 0.344 | ||
| <ΔΦ> (°) | 25.9 | 30.6 | 21.7 | 25.9 | ||
| Interchain contactsb | ||||||
| No. where distance < 4Å | 4 | 33 | 43 | 9 | 4 | 2 |
| No. where 4Å < distance < 7Å | 499 | 178 | 171 | 176 | 522 | 208 |
| r.m.s. differences (versus P1/M) (Å) | ||||||
| VP1 | 0.6 | 2.2 | 6.1 | 4.7 | ||
| VP2 | 0.6 | 0.5 | 8.7 | 3.7 | ||
| VP3 | 0.5 | 1.6 | 6.7 | 4.5 | ||
| r.m.s. differences (versus 135S) (Å) | ||||||
| VP1 | 2.6 | |||||
| VP2 | 8.5 | |||||
| VP3 | 5.2 | |||||
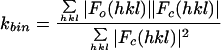 |
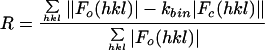 |
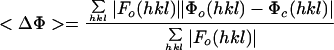 |
The number of contacts with alpha carbons from neighboring polypeptide chains provides a very approximate assessment of the completeness and plausibility of the rigid-body model.
P1/M is 2.9-Å-resolution X-ray crystal structure (31).
RESULTS
Structural analysis. (i) EM.
Poliovirus 160S, 135S, and 80S particles appear roundish when viewed in either negative stain or vitreous ice (Fig. 1A to C), although in some views straight edges are seen, particularly for the 135S and 80S particles. Negative staining typically showed differences in internal stain penetration for the three particles: 160S, little or none; 135S, partial; and 80S, heavy. Density maps of the 160S, 135S, and 80S particles (Fig. 2D and 3A) were computed from 123, 120, and 262 images, respectively. Their resolutions are 22, 22, and 23 Å, respectively. The sizes of the 135S and 80S reconstructions were calibrated against the known 160S structure (31) (Fig. 1D and E).
FIG. 1.

Electron micrographs of poliovirus 160S (A), 135S (B), and 80S (C) particles. In each case, the left-hand micrograph shows negatively stained material and the right hand micrograph shows a frozen hydrated preparation. 135S preparations usually contain a small percentage of empty 80S particles (20), e.g., the arrow in panel B. In size calibration experiments with frozen hydrated specimens, 160S was mixed with 80S (D) and 80S with 135S (E). Bar = 300 Å.
FIG. 2.

(A to C) Depiction of the 160S poliovirus particle as visualized by X-ray crystallography (31). One fivefold, one threefold, and two twofold symmetry axes are labeled. (A) Surface rendering of the outer surface, after the structure was limited to 22 Å by the procedure described (7). In panels B and C, the location of one protomer—consisting of one copy of VP1 (blue), VP2 (yellow), VP3 (red), and VP4 (green; shown only in panel C)—is marked. The five protomers that associate around a fivefold axis form a “pentamer.” In panels A and C, prominent surface features are labeled, namely, the flat top of one mesa (solid arrowhead), one arm of a mesa (shaded arrowhead), and one propeller blade tip (open arrowhead). Three VP1 loops form the top of the fivefold mesa, two portions of VP1 form a mesa arm, and portions of VP1 and VP2 form the blade tip. (D to G) Surface renderings of the 160S, 135S, and 80S cryo-EM reconstructions. Panels D and E show views of the 160S (top), 135S (middle), and 80S (bottom) particles along a twofold symmetry axis: stereo views of the outer surface are shown (D), as well as the inner surfaces (E). In panel E, RNA density was excised from the 160S and 135S structures with a spherical mask (r = 109 Å) so that the RNA-capsid contact regions appear smoothly spherical. Two twofold (lens shape), one threefold (triangle), and one fivefold (pentagon) symmetry axes in the central plane are shown. (F and G) Details of the outer surface viewed along the threefold (F) and fivefold (G) symmetry axes: 160S (left), 135S (middle), and 80S (right). Scale bar for panels A, D, E, F, and G = 100 Å.
FIG. 3.

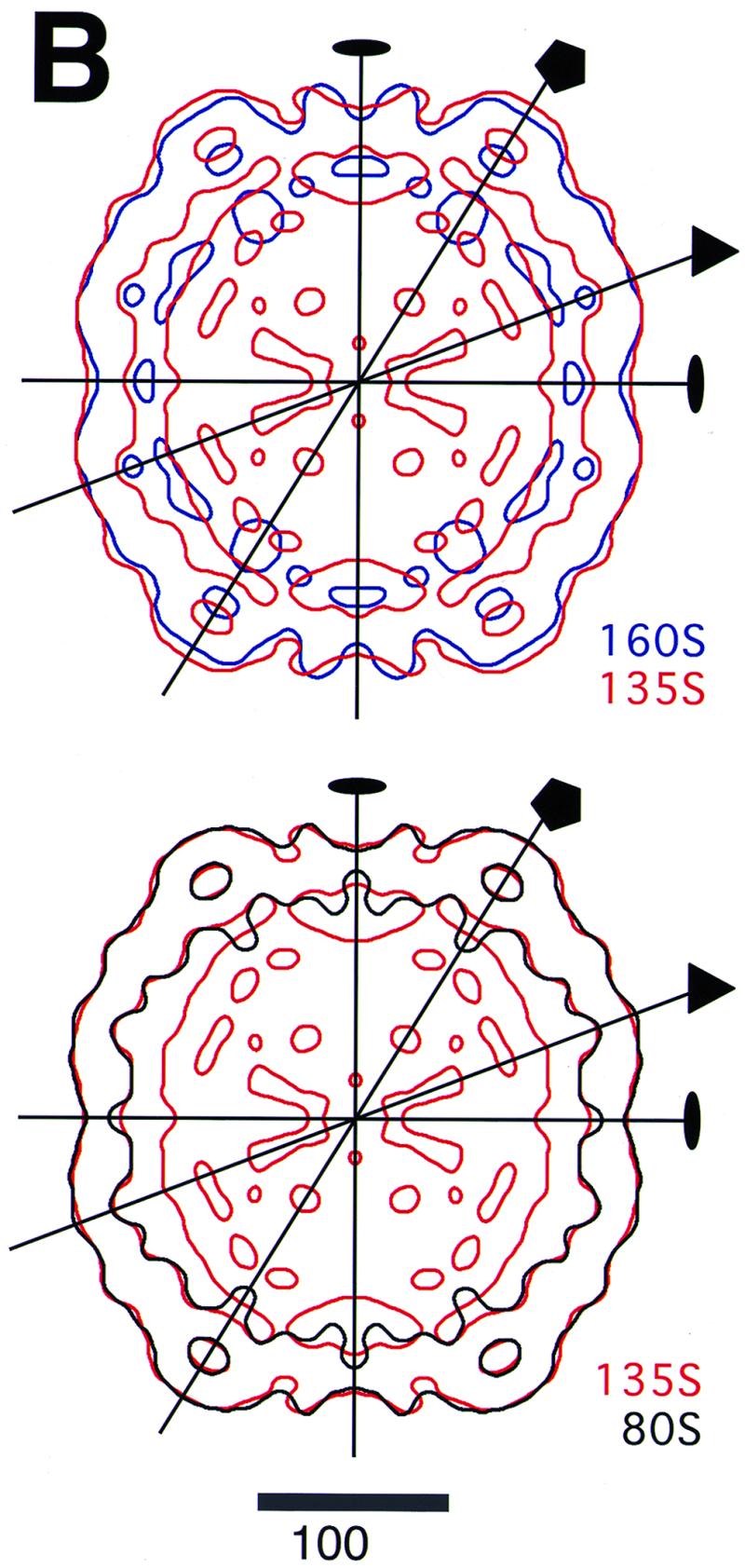
Central planar sections of the poliovirus 160S, 135S, and 80S reconstructions. Some symmetry axes are labeled. (A) Sections perpendicular to the twofold (left), fivefold (center), and threefold (right) symmetry axes. The arrows indicate the outer edge of RNA density in 160S and 135S. The arrowheads point to bubbles—regions of low density—in the middle of the capsid shell on the fivefold axes. (B) Single-level contour plots with the twofold axis normal to the page: blue, 160S; red, 135S; black, 80S. The plots are superimposed to show size relationships. The contour selected is the same level selected in Fig. 2 for the surface renderings. Bars = 100 Å.
(ii) Pseudoatomic modeling.
The 135S and 80S reconstructions were used to derive pseudoatomic models of these particles by adjusting the positions and orientations of the beta-barrel domains of VP1, VP2, and VP3 (Fig. 4) from their locations in the 160S virion. At the current resolution of the reconstructions, the density does not provide sufficient constraints to allow refinement of the internal structures of the capsid protein subunits. Therefore, the core structures were fitted as rigid bodies into the cryo-EM maps by a combination of visual adjustment and quantitative refinement. To assess the reliability of this procedure, the three core domains, as well as VP4 and nontruncated VP1, VP2, and VP3, were fitted to the 160S reconstruction. The quality of each fit was assessed in terms of crystallographic parameters and by tabulating the number of short-range contacts (indicating possible collisions between subunits) and medium-range contacts (indicating favorable packing interactions between subunits) (Table 1). These statistics, along with visual assessment of the fit of the models to the density (Fig. 5), indicate that the rigid-body approximation is justified.
FIG. 4.
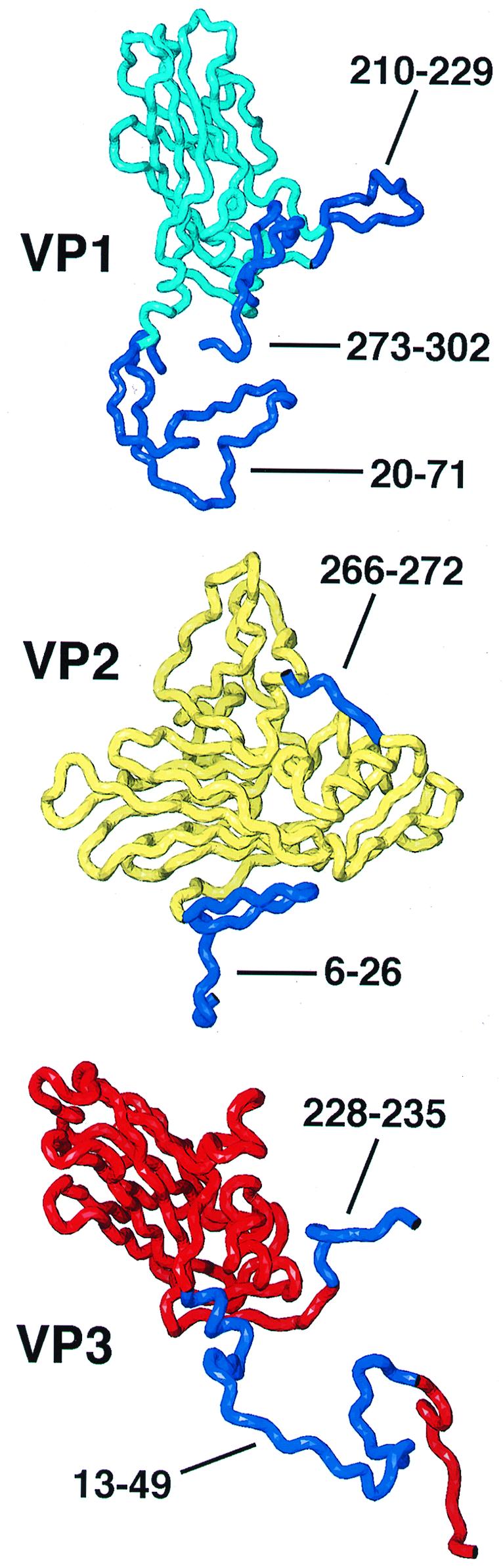
Polypeptide chains included in the truncated rigid-body models are shown in cyan (residues 72 to 209 and 230 to 272 of VP1), yellow (residues 72 to 265 of VP2), or red (residues 1 to 12 and 50 to 227 of VP3). Residues omitted from the model are dark blue and are labeled. The narrow end of each beta-barrel is on the top (VP1) or left (VP2 and VP3), and the wide end is opposite.
FIG. 5.

The capsid proteins VP1, VP2, and VP3 are arranged differently in each particle. (A) 160S; (B) 135S; (C) 80S. Side views of the respective models are shown in identical orientations, both alone and superimposed on a contour of the corresponding reconstructions. Five copies of residues 1 to 12 (the VP3 beta-tube) are included. In this orientation, the outer surface of the capsid faces upward. Selected fivefold and threefold axes are labeled. VP1 is cyan; VP2 is yellow; and VP3 is red. In 160S, VP4 is green. Bubbles of solvent-level density on the fivefold axis are labeled with gray arrows. (D) On the bottom, the different positions and orientations of the major capsid proteins are shown: dark blue, 160S; green, 135S; and magenta, 80S. Each capsid protein is represented in stereo by its principal axes that intersect at the center of mass. The axes were calculated from truncated alpha-carbon models, with the VP3 beta-tube (residues 1 to 12) excluded. The end of the long axes labeled VP1, VP2, or VP3 corresponds to the narrow end of the wedge-shaped beta-barrel. A truncated 160S protomer is shown in stereo in the same orientation to indicate the vantage point.
(iii) Comparison of Cryo-EM and X-ray structures of the 160S virion.
Initial 160S reconstructions (not shown) agreed well with the crystal structure on the outer surface of the virion but less well in other parts of the capsid—most notably within the protein shell and at the inner surface, where there appeared to be missing protein density. To address this problem, our correction of the cryo-EM images for effects of the CTF was adjusted to include a correction of low-resolution terms. This measure largely rectified the discrepancies. For consistency, a similar CTF correction was applied to both the 135S and 80S data.
The outer surface of the 160S particle is dominated by star-shaped mesas on the fivefold axes and propeller-like structures centered on the threefold axes (Fig. 2). Each mesa is formed by five copies of VP1 and is surrounded by a depression called the “canyon.” The five “arms” of the mesa are separated by indentations. The outer surface of the mesa is formed by the BC, HI, DE, and EF loops of VP1. The propeller hub corresponds to the narrow ends of the beta-barrels of VP2 and VP3, while the prominent blade tip is formed by the EF loop of VP2 flanked by the GH loop and C terminus of VP1. Three small bumps surround the threefold axes and are formed by the HI loops of VP2. Within the capsid, large “bubbles” of solvent-level density are enclosed at the fivefold axes between VP1 and a “plug” of density from intertwined N termini of VP3 (Fig. 5A). Smaller bubbles of solvent-level density at the threefold axes are surrounded by N termini of VP2 and C termini of VP4 (Fig. 5A). The virion has outer diameters of 272 (±1), 303 (±1), and 327 (±2) Å at the twofold, threefold, and fivefold axes, respectively (Fig. 2E and 3B).
(iv) Fit of X-ray coordinates into cryo-EM map.
By visual criteria, the 160S atomic model fits snugly into the cryo-EM reconstruction (Fig. 5A). In both structures, the thicknesses of the protein shells are similar, and solvent bubbles are seen at fivefold and threefold axes (Fig. 2E and 5A). Only a few residues (e.g., 50 to 54 and 60 to 64 of VP4) lie outside the capsid boundaries. As expected, the full coordinate set fits the reconstruction significantly better than the truncated version (Table 1).
(v) RNA structure.
Poliovirus RNA is not icosahedrally ordered and hence was not observed in the crystal structure at high resolution. In contrast, density corresponding to the RNA is observed in the reconstruction (Fig. 3A). Although icosahedral symmetry is (spuriously) imposed on the RNA, the cryo-EM map does retain valid information about its radial density distribution and its points of contact with the surrounding capsid. RNA is quite densely packed throughout the interior of the virion. The outermost parts of RNA follow the inner surface of the capsid, resulting in apparent invaginations in the RNA at the fivefold axes where the capsid protein protrudes inwards. In a rendering of the inner surface in which densities were trimmed at a radius of 109 Å (Fig. 2E, top), RNA and protein densities appear to merge around each fivefold axis and in finger-like regions that extend towards threefold axes. Unfortunately, it is not possible to describe the protein RNA interactions in any more detail due to the artificially imposed symmetry, the limited resolution, and the presence of a negative density ripple (which is caused by series termination) between the protein shell and the RNA.
The 135S particle. (i) Gross features.
Although the 135S particle is generally similar to the virion in morphology, it differs in several respects (Fig. 2 and 3). Its mesa is broader and has a more rounded profile. Its propeller has a triangular plateau that fits between the blade tips instead of three bumps at the center, and its blade tips are relatively swollen. The capsid is thinner and more angular. Small bumps are found on the inner surface adjacent to the twofold axes. In cross-section, the bubble on the fivefold axis is flanked by dense regions on the inner and outer edges, including strong density for the VP3 beta-tube on the inner side of the bubble. The bubble at the threefold axis is weaker than before on its inner surface, presumably due to externalization of VP4 and rearrangements of the N terminus of VP2. The 135S capsid is (4 ± 1)% larger than the virion at the twofold and fivefold axes but is only (1 ± 1)% larger on the threefold axis.
In a central section of the 160S reconstruction (Fig. 3A, top left), strong finger-like features extend outward from either side of the vertical twofold axis. They represent a slice through twofold-related EF loops of VP2—the propeller blade tips. In the corresponding 135S sections (Fig. 3A), these features are fainter and the angle between them is wider, reflecting a shift of the EF loops relative to the section plane.
The overall distribution of RNA is changed significantly in the 135S particle, compared to the virion (cf. Fig. 3A). The RNA has moved closer to the inner capsid edge near the fivefold axis, and invaginations in the RNA distribution are seen at twofold instead of fivefold axes.
(ii) Pseudoatomic model.
Nearly all parts of the truncated VP1, VP2, and VP3 subunits fit comfortably within the 135S envelope (Fig. 5B). The R-factor and phase difference between the model and reconstruction were lower than in the 160S controls, but the number of close contacts was higher (Table 1). The modeling indicated root mean square (r.m.s.) shifts in subunit positions of 6.1 to 8.7 Å (Table 1). To a first approximation, the 160S-to-135S transition can be described as follows. The wide ends of all three beta-barrels move outwards while the narrow ends (the ends near the fivefold and threefold axes) undergo much smaller shifts (Fig. 5D). VP1 and VP2 also undergo significant rotations about their principal axes. These concerted movements increase the angle that VP1 makes with VP2 and VP3 and cause the protomer (Fig. 2B and C) to appear flatter in profile (Fig. 5B), accounting for the more angular appearance of the 135S particle (Fig. 3A) and (together with the loss of VP4) contributing to the observed thinning of the shell.
According to the pseudoatomic models, the 160S-to-135S transition is accompanied by the opening of gaps between core domains. One such gap is located between adjacent VP1 subunits (Fig. 6B). Like an opening umbrella, the wide ends of the VP1 beta-barrels move apart to leave gaps between adjacent copies. These gaps extend to the base of the canyon and end near the point at which the VP1, VP2, and VP3 subunits of a single protomer meet. This gap lies immediately above the point where the N-terminal extension of VP1 joins the B strand of the VP1 beta-barrel, and is wide enough to allow the extrusion of the N-terminal extension of VP1 and perhaps VP4. Density that is not accounted for by the truncated model extends above this gap, filling the creases between VP1 beta-barrels and connecting to additional unoccupied density at the top of the fivefold mesa (Fig. 5B). A second gap, which extends from the twofold axis towards the threefold axis, disrupts the contacts that VP2 normally makes with symmetry-related copies of VP2 and VP3 from neighboring pentamers (Fig. 2B and C and 6B). Additional unexplained density is present at the base of the canyon and at inward protrusions beneath the inner surface of VP2 near the twofold axis.
FIG. 6.

Close-up views of the capsids, as seen from outside, for 160S (A), 135S (B), and 80S (C). Only the core domains of the capsid proteins are shown. Fivefold and threefold axes are labeled. In each case, one protomer is shown in color: cyan, VP1; yellow, VP2; and red, VP3. Symmetry-related copies of the same proteins are dark blue. These models predict larger gaps (arrows) between subunits in the 135S and 80S capsids than in 160S.
The 80S particle. (i) Gross features.
Apart from its lack of RNA, the 80S particle is generally similar to the 135S particle (Fig. 2 and 3), both in dimensions and appearance. Surface renderings bring out some differences. The arms of the fivefold mesa appear less substantial than those of the 135S capsid, and the canyon around the mesa is less pronounced. The threefold propeller has a distinctive swirl with a small peak at the threefold axis, and the blades are smaller than in the 135S particle. The inner surface of the capsid has inward protrusions visible at the twofold and fivefold axes.
(ii) Pseudoatomic model.
In their modeled positions, the truncated VP1, VP2, and VP3 subunits (Fig. 4) fit well into the 80S reconstruction (Fig. 5C and Table 1). In the 135S-to-80S transition, the shifts of VP2 and VP3 which occur in the 160S-to-135S transition are reversed, but incompletely (Fig. 5C and D). Again, the wide end of each subunit moves more than the narrow end. In contrast, the VP1 subunit shifts only slightly from its position and orientation in the 135S particle. The largest difference in VP1 involves a rotation that causes the wider end of the beta-barrel to move slightly inward. Nevertheless, both ends of the VP1 beta-barrel remain farther from the particle center in the 80S state than in the 160S virion.
As a consequence of these movements, the gaps between adjacent VP1 subunits become smaller, and the gaps between neighboring pentamers (Fig. 2B and C) are almost entirely repaired (Fig. 6C). At the same time, the drop of the wider ends of the VP2 and VP3 beta-barrels to a lower radius opens a new gap at the interface that they share with VP1, which fails to drop significantly. In the 80S particle this gap lies immediately above the point where the N terminus of VP1 leaves the beta-barrel. There is unfilled density at the base of the canyon above this gap (Fig. 5C). Other unfilled density is found on top of the fivefold mesa and near the twofold axis beneath the inner surface of VP2 (Fig. 5C). As in the 135S model, the excluded portions of the subunits (Fig. 4) probably occupy the unfilled density. For example, likely candidates for the unfilled density at the base of the canyon are the N terminus of VP1, the GH loop of VP1, the C terminus of VP1, and the C terminus of VP3. The unfilled density on the inner surface of VP2 is also present in the 135S particle and may be occupied by the N terminus of VP2.
DISCUSSION
Comparison of cryo-EM and X-ray renditions of the virion.
The 160S reconstruction matches well with the crystal structure (31), by both visual appraisal (Fig. 2A and D and 5A) and quantitative criteria (Table 1). To obtain an optimal match between the cryo-EM and X-ray structures, we found it necessary to restore the low-resolution terms of the reconstruction, which are attenuated in the phase-contrast imaging mode (46) used in cryo-EM. Since attenuation of low frequencies is essentially an “edge sharpening,” it may have little adverse effect when the goal is simply to define molecular envelopes. However, restoration of these frequencies appears to be more important when the goal is to depict the distribution of density throughout the molecular volume, as in the present study. Thus, our initial 160S reconstructions (calculated without restoring the low-resolution terms) agreed well with the crystal structure on the outer surface, but to achieve a satisfactory fit elsewhere, it was necessary to effect a 50% restoration of the low-resolution terms. However, it is important to note that the pseudoatomic modeling process was sufficiently robust for the outcome to be essentially unaffected by this CTF correction.
Sedimentation coefficient change: 160S to 135S.
If, apart from the 5% mass loss due to the exit of VP4, the change in sedimentation coefficient were due solely to a change in particle size, we would expect a radial expansion of 10 to 12%. This calculation is based on the Svedberg and Stokes-Einstein equations for spherical particles and employed published values for the sedimentation coefficients, the virion mass (8.43 MDa), partial specific volume (0.685 ml/g), percent RNA (31.6), and VP4 subunit mass (7.5 kDa) (31, 52, 53). The observed expansion is significantly smaller at 4%. This discrepancy implies that other factors contribute to the observed change in sedimentation coefficient. These factors may include a small increase in partial specific volume (up to 3%) or additional changes in the diffusion coefficient due to (i) the nonuniform expansion (Fig. 3), (ii) increased drag from the exposed N terminus of VP1, or (iii) increased flexibility (that would allow the particles to deform in the gradient).
Plate tectonic model of the 135S and 80S transitions.
Our modeling experiments with the 135S and 80S density maps revealed rigid-body displacements of up to 8.7 Å for the VP1, VP2, and VP3 core domains (Table 1) as well as changes in their orientations (Fig. 5D). Similar fits for the 160S reconstruction (Fig. 5A and Table 1) showed only small shifts (0.5 to 2.2 Å) and provided a significance baseline for the shifts detected in the 135S and 80S models. By this standard, their movements of 3.7 to 8.7 Å (Table 1) are highly significant. Further confidence in this conclusion is given by the fact that the quality of fit of the 135S and 80S models is similar to that of the 160S complete fit and slightly better than the 160S truncated fit (Table 1). Given the scale of these movements, it follows that the intersubunit interactions undergo major revision in the transitions to the 135S and 80S states.
In the models, gaps are generated between the core domains. Although not evident in the reconstructions at ∼22-Å resolution, the gaps are large enough in the case of the 135S particle to allow passage of an extended polypeptide chain. As such, they may provide conduits for externalization of VP4 and the N termini of VP1.
Externalization of internal components.
Viral capsids are dynamic structures, and the exposure of internal components (such as VP4 and the N terminus of VP1) during conformational transitions is not unique to poliovirus. Indeed, exposure of the N-terminal extensions of capsid protein subunits occurs upon expansion of simple plant viruses (30). Similarly, during maturation of some bacteriophage (34) and herpesvirus (4) procapsids, internal scaffold proteins are expelled, creating space for the incoming DNA. In the radical conformational change that accompanies maturation and stabilization of the phage T4 capsid, epitopes are transferred between the inner and outer surfaces (36, 56).
In addition to irreversible changes, virus particles undergo reversible alterations as well. Studies with poliovirus have demonstrated that VP4 and the N-terminal extension of VP1 are transiently and reversibly exposed when the virus is incubated at physiological temperature (40). More recent studies with limited proteolysis and mass spectrometry have shown that Flock House virus and rhinovirus undergo similar apparently reversible conformational changes that externalize normally internal components (10, 39).
How do VP4 and the N termini of VP1 exit the particle?
The observation that irreversible exposure of the VP1 N terminus in 135S particles correlates with their ability to attach to membranes suggests that this peptide may insert into the cell membrane during entry (24). It has been proposed that VP4, the N terminus of VP1, and the viral RNA all exit the particle through a channel at the fivefold axes (29, 47, 53). This model is attractive in that five copies of the VP1 N terminus would be exposed around the axis, poised for membrane insertion and pore formation. However, two observations from the 135S reconstruction argue strongly against the fivefold axis as the exit site for the N terminus of VP1. (i) The plug formed by the N termini of VP3 is present in the 135S particle. (ii) There is insufficient density and, perhaps more importantly, insufficient room on the inner surface of the fivefold mesa to accommodate five copies of the N terminus of VP1 without invading the well-defined bubble.
The data presented are consistent with an alternative model in which the N terminus of VP1 exits near the base of the canyon (23, 38) and follows the surface to the top of the fivefold mesa. Thus, in our pseudoatomic models, VP1 undergoes an umbrella-like movement in which the loops at the narrow end of VP1 serve as pivot points for a radial swing of the wide end of the subunit. This movement opens gaps in the interfaces between fivefold-related copies of VP1 and between VP1 and fivefold-related copies of VP3 at the base of the canyon. The nature of these changes is consistent with mutational data that implicate the interfaces disrupted by these gaps and the loops of VP1 near the fivefold axis as determinants of particle stability and of the ability to undergo the 160S-to-135S transition (16, 17, 23, 41–43, 60). In principle, egress of the N terminus of VP1 from a point at the base of the canyon might pose problems for membrane insertion, and in particular for coinsertion of several copies in close enough proximity to form a pore. However, when the model is superimposed on the 135S reconstruction there is unfilled density in the crease between neighboring VP1 beta-barrels (Fig. 6B) that may correspond to an extended segment of the N terminus of VP1 as it follows the surface of the fivefold mesa toward the top.
The well-characterized expansions of two T=3 plant viruses, tomato bushy stunt virus (50) and cowpea chlorotic mottle virus (55), results in disruption of analogous interfaces. In the case of tomato bushy stunt virus the holes opened up by the disruption of the analogous interfaces have been suggested to be the sites of extrusion of portions of the N-terminal extensions of the capsid subunits. Note, however, that in contrast to the expanded forms of the plant viruses, where the holes are sufficiently large to be visualized at low resolution, the gaps between VP1 subunits are barely large enough to allow passage of an extended polypeptide chain. This suggests that at some point during the 160S-to-135S transition the holes are larger. Indeed, the observation that the N terminus of VP1 and portions of VP4 are transiently and reversibly externalized when viruses are incubated at physiological temperatures (40) is consistent with this concept.
How does RNA exit the virion?
The reconstructions do not directly indicate the manner of RNA exit from the 135S particle, as we see no holes in the symmetrical 135S or 80S reconstruction (Fig. 2). This implies that during the 135S-to-80S transition the particle contains holes of sufficient size for RNA to exit. A model in which holes are opened by dislodging protein subunits from the 135S or 80S particle is deemed improbable in light of the resistance of the 135S particle to RNase (20) and the limited sensitivity of the 80S particle to proteases (25). In principle, the holes might be created by symmetric expansion, producing multiple equivalent holes (as in the expansion of tomato bushy stunt virus [50] and cowpea chlorotic mottle virus [55]). Alternatively, an individual hole could result from an asymmetric local expansion. In either case there must be a factor that determines which symmetry equivalent hole is used or which symmetry equivalent site undergoes local expansion. Otherwise the RNA would be extruded through multiple sites and could not be fully released. The factor may be structural (e.g., some portion of the RNA or perhaps the VPg protein which is covalently linked to the 5′ end of the RNA) or kinetic (once expansion or release is initiated at one site, subsequent release is fast). The proximity of the membrane could provide the symmetry-breaking factor. However, the ability of the RNA to be released by heating in vitro argues that membrane proximity need not play an essential role in determining the exit portal.
The role of the receptor in cell entry.
There is a wealth of biochemical and genetic data that suggests that the interaction between poliovirus and its receptor induces conformational changes in the virus that are essential for cell entry (reviewed in reference 14). Thus, the receptor serves as both a “hook” that increases the local concentration of virions at the surface of the cell and the “unzipper” that initiates uncoating (49) by using some of the energy of receptor binding to facilitate conformational transitions (59). Interestingly, cells lacking the receptor but expressing the high-affinity Fc receptor can be infected by complexes of the virus with certain monoclonal antibodies (1). Clearly, the Fc receptor provides the hook that brings the virus-antibody complexes to the cell surface, but what provides the unzipper? A clue may come from the observation that only certain monoclonal antibodies are capable of facilitating infection. Either these antibodies facilitate a step that is downstream of binding or the antibodies which fail to facilitate infection block this step. Because the pathway of antibody-mediated infection has not yet been well characterized it is not clear whether this pathway parallels that of receptor-mediated infection.
Is the 135S particle an intermediate in cell entry?
While there is some consensus that receptor binding induces structural changes in the virion, the identities of the intermediates and the details of the entry pathway remain uncertain. Characterization of the cell entry pathway for poliovirus (and many other viruses) is complicated by the very high ratio of particles to PFU (typically 100 to 1,000 particles/PFU). Since most optical and biochemical experiments sample the bulk population of particles, one is never certain whether the measurements are relevant to productive infection. For example, biochemical studies that show significant levels of native virions in clathrin-coated vesicles early in infection have been interpreted as evidence that the virus enters the cell via classical clathrin-mediated endocytosis (61). However, in a more recent experiment in which infection was assayed, cells expressing a dominant-negative dyamin mutant showed that entry via clathrin-mediated endocytosis is not obligatory for poliovirus infection (21).
Similarly, the failure to detect a possible intermediate under certain conditions does not prove that it is not an intermediate. In a steady-state process such as cell entry, an intermediate will accumulate only if it is upstream of a rate-limiting step in the pathway. If the rate-limiting step is upstream, the intermediate will be consumed as fast as it is produced and may not be detected. Dove and Racaniello recently characterized the early stages of infection by cold-adapted mutants of poliovirus. They showed that the cold-adapted virus did not accumulate 135S when grown at 25°C and concluded that the 135S particle was therefore not a true intermediate (22; see also references 48 and 49). However, the receptor-mediated production of the 135S particle is known to be very slow at low temperatures (27) and may have become rate limiting at 25°C. In contrast, some step immediately downstream is rate limiting at physiological temperatures. Indeed, studies with neutral red to probe the kinetics of RNA release (with infection being assayed) suggest that RNA release is rate limiting at physiological temperatures (35). Since the receptor alone appears to be unable to convert 135S to 80S (2, 27), this second step may require an additional, as yet uncharacterized trigger.
Several lines of evidence support designation of the 135S particle as an intermediate in cell entry. (i) The 135S particle (but not the 80S particle) binds to liposomes (24). (ii) 135S particles can form ion channels in synthetic bilayers without acidification (57). (iii) The cell-associated fraction of 135S is the dominant form of the virus early in infection and subsequently chases into the 80S form (24). (iv) Ligands (including antiviral drugs) that bind in a pocket in the capsid protein VP1 prevent the accumulation of 135S particles and result in the loss of infectivity (28, 44, 51). (v) Viral mutants that reversed the effects of a mutant poliovirus receptor were more labile to 135S conversion (60). (vi) The 135S particle is infectious in a receptor-independent manner (20). Although the per particle infectivity of the 135S particle is several orders of magnitude lower than that of virus, it should be noted that the receptor-independent infection by 135S particles did not benefit from a locally high concentration of virions at the cell membrane which is induced by receptor binding. Indeed, recent experiments show that the infectivity of the 135S particle can be raised to levels that are comparable to virions when the 135S particles are first bound to appropriate antibodies and the complexes are used to infect cells expressing the Fc receptor (M. Chow, personal communication). We argue that the evidence (especially the infectivity of the 135S particle) suggests that the 135S particle is either an intermediate or is sufficiently similar to an intermediate to serve as a template for developing (ultimately testable) models for viral entry and uncoating.
A revised pore model for translocation of RNA into the infected cell.
Taken together with prior information, the present observations lead us to propose (Fig. 7) a modification of previous models for poliovirus cell entry (29, 38, 53). The virion initially attaches by binding to the receptor, triggering the conformational change to the 135S state. In this transition, VP4, together with the N termini of VP1, is extruded from the bottom of the canyon and arranged around the outside of the mesa (near the fivefold axis), where five copies of the predicted amphipathic helix at the N terminus of VP1 (24, 29) would be ideally positioned for membrane insertion. The N-terminal myristate of VP4—and perhaps other regions of VP4—also may be imbedded in the membrane and may facilitate the insertion of the VP1 N termini into the membrane. To open a channel at the fivefold axis by local or symmetric expansion, a transmembrane pore is formed and the VP3 plug is moved out of the way (perhaps like a float valve). RNA then exits the particle and enters the cytoplasm. During this process, the 80S particle is formed by shifts of the VP1, VP2, and VP3 subunits and the fivefold plug is restored.
FIG. 7.

A possible mechanism for transferring RNA across the cell membrane. VP1, VP2, VP3, and VP4 are colored cyan, yellow, red, and green, respectively. In the crystal structure of the virion (upper right), the beta-tube of VP3 (red) forms a plug at the fivefold axis that separates the virus interior from the outer surface. Attachment of the 160S particle (upper left) to the poliovirus receptor (three gray circles) triggers conversion to the 135S form (lower left). Upon conversion, cell attachment is mediated by externalized VP4 (green tubes) and the N termini of VP1 (blue tubes). The N termini emerge from the bottom of the canyon and extend along the sides of the fivefold mesa towards the apex. Once the N-terminal helices of VP1 have inserted into the membrane, they rearrange to form a pore (lower right). To permit the RNA (purple tube) to pass through the pore into the cytoplasm, it would be necessary for the VP3 beta-tube (red rectangle) to shift on its 40-residue tether (red tube) and for the VP1 barrels to splay farther apart.
ACKNOWLEDGMENTS
We thank R. Grant for a 135S preparation and A. Zlotnick and M. Chow for helpful discussions.
This work was supported in part by NIH grant AI20566 (to J.M.H.) and by an NSF grant for High Performance Computing and Communication (NSF grant MCB 9527181 to G. Wagner). S.C. was supported by a postdoctoral fellowship from the Wellcome Trust. The Harvard Center for Structural Biology is supported by the Giovanni Armenise-Harvard Foundation for Advanced Scientific Research.
REFERENCES
- 1.Arita M, Horie H, Nomoto A. Interaction of poliovirus with its receptor affords a high level of infectivity to the virion in poliovirus infections mediated by the Fc receptor. J Virol. 1999;73:1066–1074. doi: 10.1128/jvi.73.2.1066-1074.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arita M, Koike S, Aoki J, Horie H, Nomoto A. Interaction of poliovirus with its purified receptor and conformational alteration in the virion. J Virol. 1998;72:3578–3586. doi: 10.1128/jvi.72.5.3578-3586.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baker T S, Cheng R H. A model-based approach for determining orientations of biological macromolecules imaged by cryo-electron microscopy. J Struct Biol. 1996;116:120–130. doi: 10.1006/jsbi.1996.0020. [DOI] [PubMed] [Google Scholar]
- 4.Baker T S, Newcomb W W, Booy F P, Brown J C, Steven A C. Three-dimensional structures of maturable and abortive capsids of equine herpesvirus 1 from cryoelectron microscopy. J Virol. 1990;64:563–573. doi: 10.1128/jvi.64.2.563-573.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Basavappa R, Syed R, Flore O, Icenogle J P, Filman D J, Hogle J M. Role and mechanism of the maturation cleavage of VP0 in poliovirus assembly: structure of the empty capsid assembly intermediate at 2.9 Å resolution. Protein Sci. 1994;3:1651–1669. doi: 10.1002/pro.5560031005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belnap D M, Grochulski W D, Olson N H, Baker T S. Use of radial density plots to calibrate image magnification for frozen-hydrated specimens. Ultramicroscopy. 1993;48:347–358. doi: 10.1016/0304-3991(93)90110-j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belnap D M, Kumar A, Folk J T, Smith T J, Baker T S. Low-resolution density maps from atomic models: how stepping 'back' can be a step 'forward.' J. Struct Biol. 1999;125:166–175. doi: 10.1006/jsbi.1999.4093. [DOI] [PubMed] [Google Scholar]
- 8.Belnap D M, Olson N H, Cladel N M, Newcomb W W, Brown J C, Kreider J W, Christensen N D, Baker T S. Conserved features in papillomavirus and polyomavirus capsids. J Mol Biol. 1996;259:249–263. doi: 10.1006/jmbi.1996.0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Booy F P, Newcomb W W, Trus B L, Brown J C, Baker T S, Steven A C. Liquid-crystalline, phage-like packing of encapsidated DNA in herpes simplex virus. Cell. 1991;64:1007–1015. doi: 10.1016/0092-8674(91)90324-r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bothner B, Dong X F, Bibbs L, Johnson J E, Siuzdak G. Evidence of viral capsid dynamics using limited proteolysis and mass spectrometry. J Biol Chem. 1998;273:673–676. doi: 10.1074/jbc.273.2.673. [DOI] [PubMed] [Google Scholar]
- 11.Castón J M, Belnap D M, Steven A C, Trus B L. A strategy for determining the orientations of refractory particles for reconstruction from cryo-electron micrographs with particular reference to round, smooth-surfaced, icosahedral viruses. J Struct Biol. 1999;125:209–215. doi: 10.1006/jsbi.1999.4085. [DOI] [PubMed] [Google Scholar]
- 12.Chen J, Wharton S A, Weissenhorn W, Calder L J, Hughson F M, Skehel J J, Wiley D C. A soluble domain of the membrane-anchoring chain of influenza virus hemagglutinin (HA2) folds in Escherichia coli into the low-pH-induced conformation. Proc Natl Acad Sci USA. 1995;92:12205–12209. doi: 10.1073/pnas.92.26.12205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng R H, Reddy V S, Olson N H, Fisher A J, Baker T S, Johnson J E. Functional implications of quasi-equivalence in a T=3 icosahedral animal virus established by cryo-electron microscopy and X-ray crystallography. Structure. 1994;2:271–282. doi: 10.1016/s0969-2126(00)00029-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chow M, Basavappa R, Hogle J M. The role of conformational transitions in poliovirus pathogenesis. In: Chiu W, Garcea R, Burnett R, editors. Structural biology of viruses. New York, N.Y: Oxford University Press; 1997. pp. 157–186. [Google Scholar]
- 15.Chow M, Newman J F, Filman D, Hogle J M, Rowlands D J, Brown F. Myristylation of picornavirus capsid protein VP4 and its structural significance. Nature. 1987;327:482–486. doi: 10.1038/327482a0. [DOI] [PubMed] [Google Scholar]
- 16.Colston E, Racaniello V R. Soluble receptor-resistant poliovirus mutants identify surface and internal capsid residues that control interaction with the cell receptor. EMBO J. 1994;13:5855–5862. doi: 10.1002/j.1460-2075.1994.tb06930.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Colston E M, Racaniello V R. Poliovirus variants selected on mutant receptor-expressing cells identify capsid residues that expand receptor recognition. J Virol. 1995;69:4823–4829. doi: 10.1128/jvi.69.8.4823-4829.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Conway J F, Trus B L, Booy F P, Newcomb W W, Brown J C, Steven A C. The effects of radiation damage on the structure of frozen hydrated HSV-1 capsids. J Struct Biol. 1993;111:222–233. doi: 10.1006/jsbi.1993.1052. [DOI] [PubMed] [Google Scholar]
- 19.Crowther R A, Amos L A, Finch J T, DeRosier D J, Klug A. Three dimensional reconstructions of spherical viruses by Fourier synthesis from electron micrographs. Nature. 1970;226:421–425. doi: 10.1038/226421a0. [DOI] [PubMed] [Google Scholar]
- 20.Curry S, Chow M, Hogle J M. The poliovirus 135S particle is infectious. J Virol. 1996;70:7125–7131. doi: 10.1128/jvi.70.10.7125-7131.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeTulleo L, Kirchhausen T. The clathrin endocytic pathway in viral infection. EMBO J. 1998;17:4585–4593. doi: 10.1093/emboj/17.16.4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dove A W, Racaniello V R. Cold-adapted poliovirus mutants bypass a postentry replication block. J Virol. 1997;71:4728–4735. doi: 10.1128/jvi.71.6.4728-4735.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Filman D J, Syed R, Chow M, Macadam A J, Minor P D, Hogle J M. Structural factors that control conformational transitions and serotype specificity in type 3 poliovirus. EMBO J. 1989;8:1567–1579. doi: 10.1002/j.1460-2075.1989.tb03541.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fricks C E, Hogle J M. Cell-induced conformational change in poliovirus: externalization of the amino terminus of VP1 is responsible for liposome binding. J Virol. 1990;64:1934–1945. doi: 10.1128/jvi.64.5.1934-1945.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fricks C E, Icenogle J P, Hogle J M. Trypsin sensitivity of the Sabin strain of type 1 poliovirus: cleavage sites in virions and related particles. J Virol. 1985;54:856–859. doi: 10.1128/jvi.54.3.856-859.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fuller S D, Butcher S J, Cheng R H, Baker T S. Three-dimensional reconstruction of icosahedral particles—the uncommon line. J Struct Biol. 1996;116:48–55. doi: 10.1006/jsbi.1996.0009. [DOI] [PubMed] [Google Scholar]
- 27.Gómez Yafal A, Kaplan G, Racaniello V R, Hogle J M. Characterization of poliovirus conformational alteration mediated by soluble cell receptors. Virology. 1993;197:501–505. doi: 10.1006/viro.1993.1621. [DOI] [PubMed] [Google Scholar]
- 28.Grant R A, Hiremath C N, Filman D J, Syed R, Andries K, Hogle J M. Structures of poliovirus complexes with anti-viral drugs: implications for viral stability and drug design. Curr Biol. 1994;4:784–797. doi: 10.1016/s0960-9822(00)00176-7. [DOI] [PubMed] [Google Scholar]
- 29.Hadfield A T, Lee W, Zhao R, Oliveira M A, Minor I, Rueckert R R, Rossmann M G. The refined structure of human rhinovirus 16 at 2.15 Å resolution: implications for the viral life cycle. Structure. 1997;5:427–441. doi: 10.1016/s0969-2126(97)00199-8. [DOI] [PubMed] [Google Scholar]
- 30.Harrison S C, Sorger P K, Stockley P G, Hogle J, Altman R, Strong R K. Mechanism of RNA virus assembly and disassembly. In: Brinton M A, Rueckert R R, editors. Positive strand RNA viruses. New York, N.Y: Alan R. Liss, Inc.; 1987. pp. 379–395. [Google Scholar]
- 31.Hogle J M, Chow M, Filman D J. Three-dimensional structure of poliovirus at 2.9 Å resolution. Science. 1985;229:1358–1365. doi: 10.1126/science.2994218. [DOI] [PubMed] [Google Scholar]
- 32.Jacobson D H, Hogle J M, Filman D J. A pseudo-cell based approach to efficient crystallographic refinement of viruses. Acta Crystallogr. 1996;D52:693–711. doi: 10.1107/S0907444996001060. [DOI] [PubMed] [Google Scholar]
- 33.Jones T A. Interactive computer graphics: FRODO. Methods Enzymol. 1985;115:157–171. doi: 10.1016/0076-6879(85)15014-7. [DOI] [PubMed] [Google Scholar]
- 34.King J, Casjens S. Catalytic head assembling protein in virus morphogenesis. Nature. 1974;251:112–119. doi: 10.1038/251112a0. [DOI] [PubMed] [Google Scholar]
- 35.Kirkegaard K. Mutations in VP1 of poliovirus specifically affect both encapsidation and release of viral RNA. J Virol. 1990;64:195–206. doi: 10.1128/jvi.64.1.195-206.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kistler J, Aebi U, Onorato L, ten Heggeler B, Showe M K. Structural changes during the transformation of bacteriophage T4 polyheads: characterization of the initial and final states by freeze-drying and shadowing Fab-fragment-labelled preparations. J Mol Biol. 1978;126:571–590. doi: 10.1016/0022-2836(78)90059-1. [DOI] [PubMed] [Google Scholar]
- 37.Koike S, Horie H, Ise I, Okitsu A, Yoshida M, Iizuka N, Takeuchi K, Takegami T, Nomoto A. The poliovirus receptor protein is produced both as membrane-bound and secreted forms. EMBO J. 1990;9:3217–3224. doi: 10.1002/j.1460-2075.1990.tb07520.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lentz K N, Smith A D, Geisler S C, Cox S, Buontempo P, Skelton A, DeMartino J, Rozhon E, Schwartz J, Girijavallabhan V, O'Connell J, Arnold E. Structure of poliovirus type 2 Lansing complexed with antiviral agent SCH48973: comparison of the structural and biological properties of the three poliovirus serotypes. Structure. 1997;5:961–978. doi: 10.1016/s0969-2126(97)00249-9. [DOI] [PubMed] [Google Scholar]
- 39.Lewis J K, Bothner B, Smith T J, Siuzdak G. Antiviral agent blocks breathing of the common cold virus. Proc Natl Acad Sci USA. 1998;95:6774–6778. doi: 10.1073/pnas.95.12.6774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Q, Gómez Yafal A, Lee Y M-H, Hogle J, Chow M. Poliovirus neutralization by antibodies to internal epitopes of VP4 and VP1 results from reversible exposure of these sequences at physiological temperature. J Virol. 1994;68:3965–3970. doi: 10.1128/jvi.68.6.3965-3970.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liao S, Racaniello V. Allele-specific adaptation of poliovirus VP1 B-C loop variants to mutant cell receptors. J Virol. 1997;71:9770–9777. doi: 10.1128/jvi.71.12.9770-9777.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Macadam A J, Arnold C, Howlett J, John A, Marsden S, Taffs F, Reeve P, Hamada N, Wareham K, Almond J, Cammack N, Minor P D. Reversion of the attenuated and temperature-sensitive phenotypes of the Sabin type 3 strain of poliovirus in vaccinees. Virology. 1989;172:408–414. doi: 10.1016/0042-6822(89)90183-9. [DOI] [PubMed] [Google Scholar]
- 43.Martin A, Wychowski C, Couderc T, Crainic R, Hogle J, Girard M. Engineering a poliovirus type 2 antigenic site on a type 1 capsid results in a chimaeric virus which is neurovirulent for mice. EMBO J. 1988;7:2839–2847. doi: 10.1002/j.1460-2075.1988.tb03140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McSharry J J, Caliguiri L A, Eggers H J. Inhibition of uncoating of poliovirus by arildone, a new antiviral drug. Virology. 1979;97:307–315. doi: 10.1016/0042-6822(79)90342-8. [DOI] [PubMed] [Google Scholar]
- 45.Mendelsohn C L, Wimmer E, Racaniello V R. Cellular receptor for poliovirus: molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell. 1989;56:855–865. doi: 10.1016/0092-8674(89)90690-9. [DOI] [PubMed] [Google Scholar]
- 46.Misell D L. Image analysis, enhancement, and interpretation. In: Glauert A M, editor. Practical methods in electron microscopy. Vol. 7. Amsterdam, The Netherlands: Elsevier/North-Holland; 1978. [Google Scholar]
- 47.Mosser A G, Rueckert R R. WIN 51711-dependent mutants of poliovirus type 3: evidence that virions decay after release from cells unless drug is present. J Virol. 1993;67:1246–1254. doi: 10.1128/jvi.67.3.1246-1254.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Racaniello V R. Early events in poliovirus infection: virus-receptor interactions. Proc Natl Acad Sci USA. 1996;93:11378–11381. doi: 10.1073/pnas.93.21.11378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Racaniello V R. The poliovirus receptor: a hook, or an unzipper? Structure. 1996;4:769–773. doi: 10.1016/s0969-2126(96)00083-4. [DOI] [PubMed] [Google Scholar]
- 50.Robinson I K, Harrison S C. Structure of the expanded state of tomato bushy stunt virus. Nature. 1982;297:563–568. [Google Scholar]
- 51.Rossmann M G, Greve J M, Kolatkar P R, Olson N H, Smith T J, McKinlay M A, Rueckert R R. Rhinovirus attachment and cell entry. In: Chiu W, Garcea R, Burnett R, editors. Structural biology of viruses. New York, N.Y: Oxford University Press; 1997. pp. 105–133. [Google Scholar]
- 52.Rueckert R R. On the structure and morphogenesis of picornaviruses. In: Fraenkel-Conrat H, Wagner R R, editors. Comprehensive virology. Vol. 6. New York, N.Y: Plenum; 1976. pp. 131–213. [Google Scholar]
- 53.Rueckert R R. Picornaviridae: the viruses and their replication. In: Fields B N, Knipe D M, Howley P M, editors. Fields virology. 3rd ed. Philadelphia, Pa: Lippincott-Raven; 1996. pp. 609–654. [Google Scholar]
- 54.Rueckert R R, Pallansch M A. Preparation and characterization of encephalomyocarditis (EMC) virus. Methods Enzymol. 1981;78:315–325. [PubMed] [Google Scholar]
- 55.Speir J A, Munshi S, Wang G, Baker T S, Johnson J E. Structures of the native and swollen forms of cowpea chlorotic mottle virus determined by X-ray crystallography and cryo-electron microscopy. Structure. 1995;3:63–78. doi: 10.1016/s0969-2126(01)00135-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Steven A C, Bauer A C, Bisher M E, Robey F A, Black L W. The maturation-dependent conformational change of phage T4 capsid involves the translocation of specific epitopes between the inner and the outer capsid surfaces. J Struct Biol. 1991;106:221–236. doi: 10.1016/1047-8477(91)90072-5. [DOI] [PubMed] [Google Scholar]
- 57.Tosteson M T, Chow M. Characterization of the ion channels formed by poliovirus in planar lipid membranes. J Virol. 1997;71:507–511. doi: 10.1128/jvi.71.1.507-511.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wetz K, Kucinski T. Influence of different ionic and pH environments on structural alterations of poliovirus and their possible relation to virus uncoating. J Gen Virol. 1991;72:2541–2544. doi: 10.1099/0022-1317-72-10-2541. [DOI] [PubMed] [Google Scholar]
- 59.Wien M W, Chow M, Hogle J M. Poliovirus: new insights from an old paradigm. Structure. 1996;4:763–767. doi: 10.1016/s0969-2126(96)00082-2. [DOI] [PubMed] [Google Scholar]
- 60.Wien M W, Curry S, Filman D J, Hogle J M. Structural studies of poliovirus mutants that overcome receptor defects. Nat Struct Biol. 1997;4:666–674. doi: 10.1038/nsb0897-666. [DOI] [PubMed] [Google Scholar]
- 61.Willingmann P, Barnert H, Zeichhardt H, Habermehl K-O. Recovery of structurally intact and infectious poliovirus type 1 from HeLa cells during receptor-mediated endocytosis. Virology. 1989;168:417–420. doi: 10.1016/0042-6822(89)90286-9. [DOI] [PubMed] [Google Scholar]
- 62.Winkelmann D A, Baker T S, Rayment I. Three-dimensional structure of myosin subfragment-1 from electron microscopy of sectioned crystals. J Cell Biol. 1991;114:701–713. doi: 10.1083/jcb.114.4.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zlotnick A, Cheng N, Conway J F, Booy F P, Steven A C, Stahl S J, Wingfield P T. Dimorphism of hepatitis B virus capsids is strongly influenced by the C-terminus of the capsid protein. Biochemistry. 1996;35:7412–7421. doi: 10.1021/bi9604800. [DOI] [PubMed] [Google Scholar]