

Abstract
The dissemination of the Edmonston measles virus (Ed-MV) vaccine strain was studied with genetically modified mice defective for the alpha/beta interferon receptor and expressing human CD46 with human-like tissue specificity and efficiency. A few days after intranasal infection, macrophages expressing Ed-MV RNA were detected in the lungs, in draining lymph nodes, and in the thymus. In lymph nodes, large syncytia which stained positive for viral RNA and for macrophage surface marker proteins were found and apoptotic cell death was monitored. In the thymus, smaller syncytia which stained positive for macrophage and dendritic cell markers were detected. Thus, macrophages appear to be the main vectors for dissemination of MV infection in these mice; human macrophages may have a similar function in the natural host. We then compared the pathogenicities of two recombinant viruses lacking the C or V nonstructural proteins to that of the parental strain, Ed-MV. These viruses were less effective in spreading through the lymphatic system and, unlike Ed-MV, were not detected in the liver. After intracerebral inoculation the recombinant viruses caused lethal disease less often than Ed-MV and induced distinctive patterns of gliosis and inflammation. Ed-MV was reisolated from brain tissue, but its derivatives were not. C- and V-defective viruses should be considered as more-attenuated MV vaccine candidates.
A priority in measles virus (MV) research is the development of practical animal models for studying viral infection and evaluating recombinant MVs being developed as novel vaccines. Currently available vaccines, derived from the live attenuated strain Edmonston (Ed-MV), are safe and efficient and have progressively reduced measles-induced fatalities to less than 1 million per year (8). Their thorough application in developing countries may lead to measles eradication in the next decades, but new challenges are rising. First, a growing population of immunocompromised individuals may be at risk when inoculated with otherwise very safe vaccine strains (1). Second, recombinant MV expressing antigens of other pathogens are being developed in the hope of producing inexpensive multivalent vaccines (40). Candidate vaccine strains, more attenuated, multivalent, or both, are or will soon be in demand for testing. Moreover, MVs with altered cell tropism are being produced for applications in cytoreductive gene therapy (U. Schneider, A. Murphy, F. Bullogh, S. J. Russell, and R. Cattaneo, Abstract, Gene Ther. 6:S4, 1999), and their safety has to be tested in animals.
However, the only natural hosts for MV are humans. Primates, which can be used as experimental measles models (27, 44), are costly and in short supply. This and ethical considerations have driven the development of rodent models for MV infection. Due to restricted MV replication, however, only neuroadapted MV strains cause disease in adult mice, and solely when inoculated intracerebrally (25). Cotton rats, which were recently shown to be susceptible to intranasal MV infection (32), are genetically and immunologically poorly characterized compared to mice. Therefore, transgenic mice with enhanced susceptibility to MV infections were developed (5, 19). In particular, mice expressing in neurons the human regulator of complement activation CD46 are susceptible to intracerebral infection with Ed-MV (24, 26, 36). Alternatively, human thymus and liver implants grafted onto immunodeficient SCID mice have been successfully used to compare wild-type MV, Ed-MV, and Ed-MV-derived recombinant virus pathogenicities to human lymphatic tissue (2, 42).
More-attenuated candidate recombinant MV vaccine strains, which include C- and V-protein-defective viruses (34, 38), should still replicate in animals at levels which are high enough to efficiently induce immunity. C proteins (3), which are encoded in the genera Morbillivirus and Respirovirus, have auxiliary functions in viral replication (9, 42). V proteins (7), which are expressed in most members of the family Paramyxoviridae, are suspected to have a control role in viral RNA synthesis (22, 41). Infection of human liver and thymus implants of mice (42) and intranasal infection of cotton rats (43) indicated that in certain tissues C- and V-defective viruses replicate less efficiently than the parental Ed-MV strain. However, an analysis of the systemic replication of these viruses in an animal is not yet available.
The first detailed analysis of lymphatic organ spread and pathogenesis of Ed-MV and of two Ed-MV-derived viruses in an animal is presented here. This analysis was possible with genetically modified mice having a targeted mutation inactivating the interferon receptor type I gene and expressing human CD46 with the same tissue specificity and efficiency as humans (Ifnarko-CD46Ge mice) (30). In these animals, upon intranasal infection with a recombinant Ed-MV expressing a reporter gene, systemic spread was previously detected (30). We now show, using histology and a panel of cell surface markers, that macrophages may be the principal vectors disseminating Ed-MV from the lungs to peripheral organs. It is also shown that the organismal dissemination of the C- and V-defective viruses is strongly impaired and that upon intracerebral inoculation these viruses cause lethal disease less often than the parental strain.
MATERIALS AND METHODS
Mice and infections.
Five- to 7-week-old mice kept under optimal hygienic conditions and examined periodically for pathogens were infected. These animals have a targeted mutation inactivating the interferon receptor type I gene (31), and carry a 400-kb segment of the human genome covering the CD46 gene and its flanking sequences (20). Therefore, they express human CD46 with the same tissue specificity and efficiency as humans (30).
Intracerebral inoculations were done along the skull midline with a tuberculin syringe with a 27-gauge needle. The inoculum consisted of stock virus diluted in phosphate-buffered saline (PBS); the injection volume was 30 μl. For intranasal infections, 1 million PFU in a total volume of 50 μl was administered into both nares. For mock infection, the postnuclear supernatant of uninfected Vero cells was used. The animals were monitored daily for clinical symptoms.
Viruses.
The standard tagged Ed-MV derived from the Edmonston B strain (35) and recombinant Ed-MV strains defective for C or V nonstructural protein (C or V defective, respectively) were propagated as described elsewhere (34, 38). All virus stocks were produced and titrated on Vero (African green monkey kidney) cells.
Virus reisolation from infected tissue.
Tissues (brains and lungs) were removed aseptically, cut with a razor blade, and washed three times with PBS solution. Blocks of tissue (2 to 3 mm thick) were placed on a subconfluent monolayer of Vero cells in six-well culture plates, and the cocultures were incubated for 24 h at 37°C. The tissues and culture medium were removed, and after extensive washing, the Vero cells were incubated until syncytium formation was observed. If necessary, the cell cultures were transferred to T75 culture flasks after 4 days and monitored for syncytium formation during the next 9 days. To confirm virus isolation, the culture medium was removed and centrifuged for 5 min at 800 × g, and this cell-free supernatant was added to fresh indicator cells.
MV-specific immunostaining was done with a monoclonal anti-N antibody (1:500; courtesy of E. Norrby) and a goat anti-mouse antibody coupled to alkaline phosphatase (AP). The color substrate was a nitroblue tetrazolium–5-bromo-4-chloro-3-indolylphosphate (BCIP) chromogen (Boehringer Mannheim).
Histology, immunohistochemistry, and in situ hybridization.
Mice were euthanized with CO2, and the appropriate organs were removed. The tissues were either immersed in Hanks balanced salt solution and snap frozen in liquid nitrogen or fixed in 4% PBS-buffered formaldehyde and subsequently embedded in paraplast by standard procedures. For general histological analysis, sections were deparaffinized and stained with hematoxylin-eosin (HE) staining solution.
For the staining of cell differentiation markers, frozen tissue sections were cut in a cryostat at 2- to 3-μm thickness, fixed in acetone for 10 min, and stored at −70°C. Rehydrated tissue sections were incubated with primary rat anti-mouse monoclonal antibodies against major histocompatibility complex MHC class II (M5/114; American Type Culture Collection, Manassas, Va.), CD45RABC/B220 (RA3-6B2; PharMingen, San Diego, Calif.), CD4 (YTS 191), CD8 (YTS 169), F4/80 macrophages (A3-1; American Type Culture Collection), splenic marginal metallophilic macrophages (MOMA 1; Biomedicals AG, Augst, Switzerland), follicular dendritic cells (4C11), and interdigitating dendritic cells (NLDC-145; Biomedicals AG). CD11c was stained with primary monoclonal hamster antibodies (N418). Epithelial-cell-specific immunostaining was done with monoclonal mouse anti-cytokeratin 19 antibodies (Amersham International, Amersham, United Kingdom). Primary antibodies were revealed by sequential incubation with AP-labeled species-specific secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, Pa.). For the staining of F4/80 macrophages on paraplast-embedded tissues, deparaffinized sections were digested with 0.1% pronase E for 2.5 min and preincubated with 2% fetal calf serum in Tris-buffered saline. F4/80 antibodies were applied followed by peroxidase-labeled goat anti-rat antibodies (Jackson). The signal was amplified by the addition of biotinylated tyramide in the presence of H2O2. This leads to the peroxidase-catalyzed covalent deposition of multiple biotin moieties, which serve as binding sites for the subsequently added avidin-biotin-AP complexes (Dako AS, Glostrup, Denmark). AP was visualized with naphthol AS-BI (6-bromo-2-hydroxy-3-naphtholic acid-2-methoxy anilide) phosphate and new fuchsin (Sigma Chemical Co., St. Louis, Mo.) as a substrate, yielding a red reaction product. Sections were counterstained with hemalum. Astrocytes were stained on paraffin sections with mouse-specific polyclonal anti-glial fibrillary acidic protein (GFAP) antibodies (Calbiochem, San Diego, Calif.) and biotinylated species-specific secondary antibodies. A colorimetric reaction was performed with the avidin-biotin-peroxidase detection kit (Vector Laboratories, Burlingame, Calif.) with diaminobenzidine as a substrate, yielding a brown reaction product.
Detection of MV N mRNA in situ was performed as described previously (30). Briefly, digoxigenin-labeled N RNA probe (30 pg/μl) was added to prehybridized paraffin sections under appropriate conditions. The hybridized probe was immunologically detected with a digoxigenin-nucleic acid detection kit (Boehringer Mannheim).
Pathologic findings in tissue sections.
For estimation of brain pathological signs, samples were graded without knowledge of the experimental groups; each group consisted of six animals. Both hemispheres from each brain were analyzed over sagittal or coronal tissue sections.
For estimation of lung pathological changes, at least four lung lobes per animal were graded for the presence of inflammation, hemorrhage, and hyperemia (six mice in each experimental group). The following scale was used for reference: 0, no abnormalities; and 1, weak; 2, moderate; and 3, strong pathological changes.
RESULTS
MV replication and pathogenesis in lungs.
Intranasal inoculation of Ifnarko-CD46Ge mice with Ed-MV results in respiratory infection and prominent lung tissue inflammation (30). In an attempt to verify if fusion of lung cells occurs, we examined regions characterized by increased cellularity 3 days after infection with 106 PFU of Ed-MV. Indeed, multinucleated giant cells were occasionally observed (Fig. 1A and B); Ed-MV-specific mRNA was detected in cells forming syncytia as well as in neighboring cells (Fig. 1C).
FIG. 1.

Lung pathology in mice infected intranasally with Ed-MV. (A) HE staining; increased cellular density and indistinct multinuclear giant cells (frame and asterisks). (B) Enlargement of the frame in panel A. (C) In situ hybridization for MV N mRNA in the tissue section consecutive to that shown in panel A. (D and E) Colocalization of an MV N mRNA signal (D) and F4/80-specific immunostaining on consecutive tissue sections (E). (F and I) Immunohistochemical analysis of the cells infiltrating the lungs: the stainings are for CD4 (F), CD8 (G), F4/80 (H), and NLDC-145 (I). Magnification: A and C, ×400; B, ×1,250; D and E, ×1,000; F to I, ×250.
To determine which cells infiltrate the lungs and which ones are infected, we stained consecutive lung sections with antibodies against CD4, CD8, F4/80, MOMA 1, NLCD-145, and CD11c. Fig. 1D shows a group of cells localized in the alveolar wall. These cells strongly express Ed-MV mRNA, and since the consecutive section (Fig. 1E) identifies them as F4/80 positive, many of these cells may be macrophages. Infected F4/80-positive cells were also found in respiratory bronchioles, but less frequently. Similar results were obtained with MOMA 1 antibodies. In addition, Ed-MV mRNA was detected in the lung epithelial cells, as identified by immunostaining with a cytokeratin antibody (data not shown), and in the endothelial cells of the blood vessels, as recognized by their characteristic morphology, in agreement with previous observations in primates (27). Ed-MV mRNA was not detected in the lungs of mice infected with UV-inactivated virus. Thus, in the lungs Ed-MV replicates in epithelial cells, endothelial cells, and macrophages.
To compare the replication and pathogenesis of the C- and V-defective strains to that of parental Ed-MV, we infected intranasally groups of six mice with the three inocula. Either four or all five lung lobes of each animal were analyzed, and pathological changes were graded on a three-point scale. In Ed-MV infections, as described before (30), pathological signs were strongest 6 days after infection (grade 3) and then declined. C- and V-defective viruses induced lower levels of pathology than Ed-MV (grades 2.5 and 2, respectively), and the strongest pathological signs were observed 12 days after infection (data not shown).
The composition of the lung infiltrates was then examined immunohistochemically. Six days after infection the majority of infiltrating cells were CD4-positive lymphocytes (Fig. 1F) and F4/80-positive macrophages (Fig. 1H). CD8-positive lymphocytes (Fig. 1G) were also present at significant levels. In contrast, NLDC-145-positive cells (Fig. 1I) and B220-positive B lymphocytes (data not shown) were detected at low levels. Furthermore, the prominent up-regulation of MHC class II was detected after standard MV infection on cells throughout the lung in general (data not shown). Thus, at this time after infection T-cell-driven cellular immune responses are activated.
MV dissemination in lymphatic organs and in liver.
Next, we characterized histologically the systemic spread of Ed-MV and of the mutant viruses. Previously, documentation of MV systemic spread in these mice was based exclusively on the detection of enzymatic activity of a recombinant virus expressing a reporter gene (chloramphenicol acetyltransferase) in peripheral blood lymphocytes, spleen, and liver (30). Three days after Ed-MV infection, histological analysis revealed the existence of large syncytia in the tracheobronchial lymph nodes of infected animals (Fig. 2A and B). By in situ hybridization, Ed-MV transcription in such syncytia was confirmed (Fig. 2C). It is remarkable that in many instances, including the example shown, more than 50 nuclei were detected in a single planar section. In animals sacrificed 6 days post-Ed-MV infection, pathological signs were considerably less strong.
FIG. 2.

MV-induced pathology in lymphatic organs and liver. (A to D) Syncytia in lymph nodes 3 days postinfection. (A and B) HE staining; the asterisk and the frame indicate multinuclear giant cells. (C) In situ hybridization for MV N mRNA. (D) F4/80-specific immunostaining in consecutive tissue sections. (E and F) HE staining of apoptotic cells (arrows) in the lymph nodes: pycnotic nuclei in a syncytium (E) and in the tissue parenchyma (F). (G to J) Cytopathic effects in the thymus 3 days (G and H) and 12 days (I and J) after MV intranasal inoculation. (G to I) Fused cells by HE staining (G), F4/80 immunostaining (H and I), and CD11c immunostaining (J). (K and L) Liver tissue by HE-staining (K) and MV N mRNA-specific in situ hybridization of the consecutive section (L). Virus strains: panels A to D and F to L, Ed-MV; panel E, C-defective virus. Magnification: panel A, ×80; panels B to E, ×1,250; panels F to J, ×1,100; panels K and L, ×400.
Ed-MV-induced syncytia were also detected in the other lymphatic organs: frequently in the thymus (Fig. 2G to J) and rarely in the spleen (data not shown). Mice infected with C-defective virus also developed syncytia in draining tracheobronchial lymph nodes (Fig. 2E). However, these were smaller and appeared less frequently. In contrast, there was no evidence of syncytium formation in animals infected with V-defective virus; only occasionally were single infected cells observed. Interestingly, we monitored apoptotic cell death, characterized by nuclear condensation and DNA fragmentation, in syncytia (Fig. 2E) and in the tissue parenchyma near syncytia (Fig. 2F). These data indicate that MV-induced apoptosis (10) occurs in lymph nodes of Ifnarko-CD46Ge mice.
Next, we asked which cellular markers can be detected by immunohistochemical analysis of the syncytia. On the tissue section consecutive to that used to detect viral RNA (Fig. 2C), strong reactivity with the macrophage marker F4/80 was monitored (Fig. 2D), as with MOMA 1 (data not shown). Other cell markers reacted weakly or were negative. The same analysis was repeated on thymic tissue sections: in that organ, not only the F4/80 macrophage marker (Fig. 2H and I) but also the CD11c (Fig. 2J) and NLCD-145 markers (data not shown) reacted with infected syncytia. Thus, cells of different origins, including dendritic cells, may contribute to these syncytia. MV-positive cells and syncytia were detected in the thymuses of animals sacrificed 3 (Fig. 2G and H) to 12 (Fig. 2I and J) days after infection. Additionally, rare single cells of epithelial-reticular-type morphology expressed MV antigen (data not shown). Altogether, these data suggest that pulmonary macrophages may carry the virus from the lungs to the regional lymph nodes. Macrophages and dendritic cells may be involved in Ed-MV infection of the thymus.
Analysis of the lymphatic organs was completed by examining the spleen, where MV replication was found to be limited. Nevertheless, in a majority of mice 12 days after infection the cellular composition and tissue organization revealed significant stimulation of B220-positive B cells and increased populations of F4/80-positive and MOMA 1-positive macrophages and of 4C11-positive follicular dendritic cells (data not shown). These events may reflect the stimulation of immune responses by antigen presentation. We then asked if the stimulation of the B-cell areas correlated with the establishment of a humoral anti-MV response. Indeed MV-specific immunoglobulin M (IgM), IgG1, and IgG2a fractions were detected 14 and 28 days after infection; Ed-MV induced slightly higher antibody titers than the C- and V-defective mutants (data not shown). Similarly, virus neutralization tests revealed higher titers for Ed-MV than for the two defective mutants (data not shown).
The next question was whether C- and V-defective viruses could spread to a nonlymphatic organ. Since in these mice Ed-MV replication was previously detected in the liver (30), we examined that organ. In liver tissue of Ed-MV-infected animals viral mRNA was detected (Fig. 2L). However, the livers of animals infected with the C- or V-defective mutants were negative (data not shown). Thus, the dissemination of C- and V-defective viruses is considerably more restricted than that of parental Ed-MV.
Distribution and pathogenesis of MV mutants in the brain.
To further assess the pathogenicity of the C- and V-defective viruses, we inoculated them into the brains of Ifnarko-CD46Ge mice. Figure 3 illustrates animal survival following injection with 105 (Fig. 3A) or 3,000 (Fig. 3B) PFU of C- or V-defective virus or Ed-MV. At a high dose, only small differences in the lethalities of the three inocula were evident (Fig. 3A). However, when less virus was injected, the differences were pronounced: the mortality for both C- and V-defective virus dropped to about 50%, whereas the mortality for Ed-MV remained about 90% (Fig. 3B). Before dying, all animals showed clinical signs of neural disease.
FIG. 3.

Survival of Ifnarko-CD46Ge mice after intracerebral inoculation with three different MV strains. Solid circles, Ed-MV; open circles, V defective; triangles, C defective. (A) 105 PFU; (B) 3,000 PFU. The numbers of animals in each group were as follows: panel A, 19 Ed-MV, 12 C defective, and 11 V defective; panel B, 19 Ed-MV, 20 C defective, and 20 V defective. p.i., postinfection.
We then compared the spread of the three viruses in brain tissue. Previous studies showed that GFAP, which marks the reactive astrocyte response to central nervous system (CNS) injury in general, is a good indicator of Ed-MV replication (6, 30). We verified that the same is true for the C- and V-defective mutants. As shown for the C-defective virus, there was indeed a significant colocalization of inflammatory infiltrations within the brain parenchyma (Fig. 4B), gliosis (Fig. 4C), and viral RNA expression (Fig. 4D). From the comparison of the viral RNA expression pattern with the other stainings, it was also evident that virus replication occurred in the brain parenchyma in both neurons and glial cells.
FIG. 4.

Brain pathology 14 days after intracerebral inoculation with the C-defective virus (3,000 PFU). (A) Mouse CNS regions with the most prominent pathological changes. C, cortex; CC, corpus callosum; CPU, caudatus/putamen; OAL, olfactorius anterior lateralis; LV, lateral ventricles; HI, hippocampus; P, pons; MO, medulla oblongata; SC, spinal cord. (B to D) Histological sections showing inflammatory infiltration (HE staining) (B), gliosis (GFAP immunostaining) (C), and viral RNA expression (MV N-specific in situ hybridization) (D) in the caudatus/putamen region. The asterisks on each panel indicate the same area of the consecutive sections. Magnification, ×250.
We then monitored reactive astrocytes 6 days postinfection in the brains and in the cervical regions of the spinal cords of animals infected with 3,000 PFU of Ed-MV or of the C- or V-defective viruses (Table 1). Levels of gliosis were graded in six animals per group on the following scale: no, few, many, or clusters of reactive astrocytes. The eight regions listed in Table 1 (localization in Fig. 4A) showed significant pathology. In four regions, the cortex, hippocampus, caudatus/putamen, and olfactorius anterioris lateralis, the extent of gliosis observed in Ed-MV infections markedly exceeded that observed in C- and V-defective virus infections. In the corpus callosum and ventricles no significant difference in the extents of gliosis induced by the three viral strains was observed. Interestingly, in the posterior regions of the pons and medulla oblongata, and in the spinal cord, gliosis was more pronounced for C- and V-defective virus than in standard MV infections. Thus, histological analysis revealed mutant-specific distribution patterns of gliosis in different CNS regions.
TABLE 1.
Reactive astrocytes in the CNS of Ifnarko-CD46Ge mice inoculated with three different MV strainsa
| Brain region | Virus | Amt of reactive astrocytes
|
|||
|---|---|---|---|---|---|
| Clusters | Many | Few | None | ||
| Cortex | Ed-MV | xxx | xxx | ||
| C defective | x | xxxxx | |||
| V defective | xxxxx | x | |||
| Corpus callosum | Ed-MV | xxx | xx | x | |
| C defective | x | xx | xxx | ||
| V defective | xx | x | xxx | ||
| Hippocampus | Ed-MV | xxxx | xx | ||
| C defective | x | xxx | xx | ||
| V defective | xxxx | xx | |||
| Caudatus/putamen | Ed-MV | xxxx | xx | ||
| C defective | xx | xxxx | |||
| V defective | xxx | xx | x | ||
| Olfactorius anterior lateralis | Ed-MV | xxxxx | x | ||
| C defective | xxx | xx | x | ||
| V defective | x | xxxx | x | ||
| Ventricles | Ed-MV | xx | xxxx | ||
| C defective | xxx | xxx | |||
| V defective | x | xxx | x | x | |
| Pons and medulla oblongata | Ed-MV | xxx | xxx | ||
| C defective | x | xx | xxx | ||
| V defective | xx | xxx | x | ||
| Spinal cordb | Ed-MV | xxxx | |||
| C defective | xxxxx | ||||
| V defective | xx | xx | |||
Six animals per group; each x represents one animal.
The spinal cords of only four animals infected with Ed-MV and V-defective virus and five animals infected with C-defective virus were examined.
To evaluate the cellular compositions of the extensive lymphocytic infiltrations monitored by HE staining (Fig. 4B), cell-specific immunohistology was performed. In infections with Ed-MV, very abundant CD4-positive T cells and F4/80-positive macrophage and microglia cells infiltrating both perivascular regions and the brain parenchyma were detected. CD8-positive T cells were significantly less abundant, whereas B220-positive cells were detected at a very low level (data not shown). The cell infiltrates in the brains of mice succumbing to C- or V-defective virus infections had the same cellular composition as those in brains of animals succumbing to Ed-MV infections. In mock-infected brains, only a few F4/80-positive cells were detected without T- or B-cell infiltration.
Ed-MV can be reisolated from mouse tissues.
We previously reported some success in recovering Ed-MV by disrupting brain tissue of infected mice and cocultivating it with Vero cells (30). We show here that virus can be more efficiently reisolated from the brains of Ifnarko-CD46Ge mice injected with Ed-MV by another technique involving the coculture of organ blocks with permissive cells.
Figure 5A shows MV-specific syncytium formation monitored after coculturing brain blocks obtained from mice inoculated with 105 PFU of Ed-MV and sacrificed 6 days postinfection. Figure 5B shows a negative control in which the same inoculum was used to infect a wild-type C57BL/6 mouse. The results of several reisolation experiments can be summarized as follows. Ed-MV was always recovered from both hemispheres of mice infected with 105 or more PFU, whereas when 3,000 PFU was inoculated, two of four reisolation attempts were successful. When 3,000 PFU of the C- or V-defective viruses was inoculated, none of four reisolation attempts were positive. These results are consistent with the less efficient propagation of the C- and V-defective viruses.
FIG. 5.
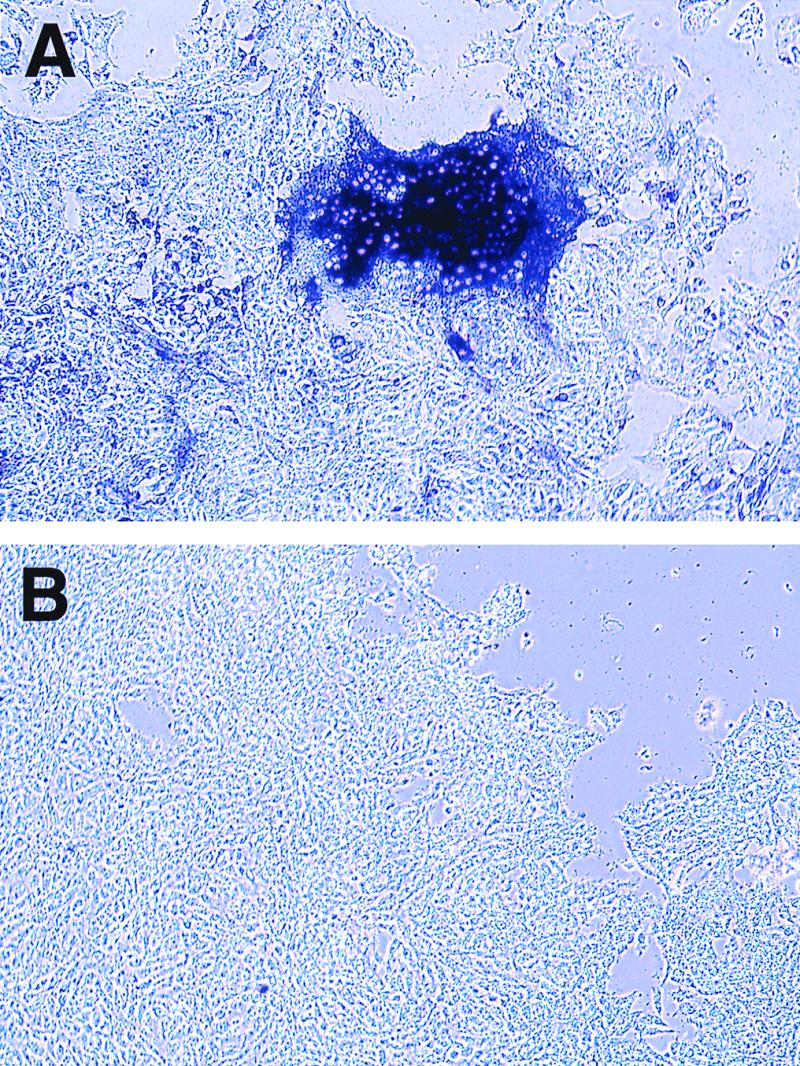
MV reisolation from infected Ifnarko-CD46Ge mice. The animals were infected, and the brains were removed and cocultured with Vero cells. MV N-specific immunostaining of brain cocultures from Ifnarko-CD46Ge mice (A) and C57BL/6 mice (B) is shown.
Finally, we applied the same virus recovery procedure to lung tissue of Ifnarko-CD46Ge mice infected intranasally with 1 million PFU of Ed-MV and sacrificed 6 days after infection: two of five reisolation attempts were successful, whereas no virus was recovered from control infections of C57BL/6 mice. These results are consistent with a cell-associated mode of propagation of MV in Ifnarko-CD46Ge mice.
DISCUSSION
Macrophages as vectors for MV dissemination.
To further characterize the infection of Ifnarko-CD46Ge mice with Ed-MV, their lungs and lymphatic organs were analyzed by histology. Ed-MV replication in the lungs was associated with multinucleated fused cells localizing in the alveolar walls. The most prominent specific pathology was in the tracheobronchial lymph nodes soon after infection: MV-RNA-positive syncytial cells containing several dozen nuclei were monitored, and apoptotic cell death was detected. In these large syncytia and in smaller ones in the thymus, macrophage surface markers were detected, suggesting that infected macrophages are the primary vehicles for dissemination of Ed-MV infections in the lymphatic organs of Ifnarko-CD46Ge mice. In the thymus, macrophage and dendritic cell markers were monitored on syncytia.
Precedents for cell-associated MV dissemination in human disease exist. MVs causing the lethal neural disease subacute sclerosing panencephalitis are often, if not always, assembly defective and therefore cannot propagate lytically (4). Is cell-associated propagation of significance in the initial stages of acute human illness? Many observations are consistent with this. First, measles viremia in humans and monkeys is quantified by determining the number of MV-infected peripheral blood mononuclear cells (13, 43, 45). Second, cultured monocytes and dendritic cells produce few infectious virus particles: maximum yields range between 1 infectious unit per 70 cells (17) and 1 infectious unit per 500 cells (14). Third, when monocytic-promyelocytic (U937) cells contact acceptor cells, a remarkably efficient infection is established (12). Fourth, MV infection of human endothelial cells is dramatically reduced in the absence of cell-cell contact (21). Fifth, in the lymphoid tissue of infected monkeys, virus budding or free viruses were not detected, but giant cells were (37). Thus, during acute human illness MV dissemination may occur mostly by fusion of MV-infected monocytic cells, including macrophages, with other cells.
Ed-MV can establish persistent infections even in standard (non-CD46 transgenic) cultured mouse macrophages (16). Moreover, in mouse macrophage cell lines expressing human CD46, MV infection causes immediate cytopathic effects with extensive syncytium formation (23) but it also induces a long-term antiviral state (18) which may account for inefficient, if any, MV replication in macrophages of another CD46 transgenic mouse (19).
Do macrophages disseminate acute MV infections in humans? This possibility has often been discussed, but experimental evidence for it is sparse. Monocytes, the macrophage precursors, are the major MV target cells in humans (11). Interestingly, immature myelomonocytic cells do support productive virus infection, whereas MV release is inhibited in mature macrophages, in spite of the availability of considerable amounts of viral proteins (17). These facts do not imply that macrophages are less important than monocytes for MV dissemination: restriction of particle release may favor accumulation of viral glycoproteins at the cell surface and thus cell-cell fusion. Moreover, in patients with acute measles and in the lymphoid tissue of MV-infected rhesus monkeys macrophage-like multinucleated giant cells were found (27, 29, 39). Thus, even if cultivated human macrophages do not release virus, circulating macrophages may disseminate MV infection by cell-cell fusion.
More-attenuated vaccine strains.
Biologically attenuated Ed-MV is a safe vaccine, but very rarely it causes disease in immunocompromised patients (1, 28). Since inactivated MV vaccines are not effective in preventing disease (33), more-attenuated derivatives of available replicating strains may be needed to vaccinate such patients. Could the Ed-MV-derived C- and/or V-defective viruses be those more-attenuated vaccines? Even if only primate experiments can give a definitive answer to that question, a classification of the relative pathogenicity of recombinant MV can now be attempted based on results obtained in other in vivo systems.
Using thymus and liver implants engrafted into SCID mice, Valsamakis et al. (42) observed that the C-defective virus reached slightly lower titers than parental Ed-MV but its pathogenicity for the human implant tissues appeared to be maintained. The V-defective virus reached titers similar to those of the parental strain but with slower replication kinetics, and its pathogenicity was diminished. Replication of the V-defective virus was also examined in cotton rats, where viral titers in the lungs were reduced less than 10-fold (41).
The lung pathology induced in Ifnarko-CD46Ge mice by C- and V-defective viruses was only slightly reduced compared to that of Ed-MV, but systemic spread of the C-defective virus was markedly reduced: smaller virus-induced syncytia were detected in draining lymph nodes of infected animals and none in the thymuses. Even more strikingly, the V-defective virus did not induce any overt pathology in lymphatic organs of Ifnarko-CD46Ge mice. Replication of neither the C- nor the V-defective viruses was detected in the liver, and these viruses were less lethal than the parental strain when inoculated intracerebrally. Thus, the C- and V-defective viruses are indeed considerably more attenuated than Ed-MV in animals.
Can these viruses be expected to have similar attenuation characteristics in humans? Ifnarko-CD46Ge mice cannot rely on a functional alpha/beta interferon system, and thus data obtained for these animals will underestimate the effects of viral proteins counteracting the interferon system. Interestingly, one of the several C proteins of another paramyxovirus, Sendai virus, does prevent establishment of the antiviral state by counteracting interferon induction (15). If the only MV C protein has similar characteristics, the virulence of C-defective viruses may be overestimated. Nevertheless, our data indicate that silencing the MV C protein does result in attenuation in vivo and thus suggest that this protein has at least one other function.
Results obtained with Ifnarko-CD46Ge mice also allow comparison of the pathogenicities of different classes of MV mutants. In particular, following intracerebral inoculation with 3,000 PFU of viruses with small alterations in the cytoplasmic tails of the envelope proteins (6) or with a mutated fusion protein activation sequence (25a), lethality of both classes of virus was reduced to near zero compared to about 50% with C- and V-defective viruses. Thus, it appears that certain alterations of the envelope proteins have more profound consequences for Ed-MV pathogenicity in Ifnarko-CD46Ge mice than complete silencing of C and V protein expression.
Finally, the characterization of the immune responses to different recombinant MVs showed not only neutralizing antibodies but also early and significant infiltration of CD4 and CD8 T lymphocytes in tissues where MV replicates and stimulation of B-cell areas in the spleen. Thus, strong humoral and cellular immune responses are generated during experimental infection of Ifnarko-CD46Ge mice. In the perspective of vaccine development, it is now important to further define the immune response generated to Ed-MV in our practical experimental system.
ACKNOWLEDGMENTS
This work was supported by grants 31-29343.90 (START) and 31-45900.95 of the Schweizerischer Nationalfonds to R.C. and by the Siebens and Mayo Foundations. The salary of Branka Mrkic was provided in part by grant 3786.1 of the Commission for Technology and Innovation and grant 31475.95 of the Schweizerischer Nationalfonds to M.A.B.
We thank Marianne König and Lenka Vlk for technical assistance and Anthea Murphy for helpful comments on the manuscript.
REFERENCES
- 1.Angel J B, Walpita P, Lerch R A, Sidhu M S, Masurekar M, DeLellis R A, Noble J T, Snydman D R, Udem S A. Vaccine-associated measles pneumonitis in an adult with AIDS. Ann Intern Med. 1998;129:104–106. doi: 10.7326/0003-4819-129-2-199807150-00007. [DOI] [PubMed] [Google Scholar]
- 2.Auwaerter P G, Kaneshima H, McCune J M, Wiegand G, Griffin D E. Measles virus infection of thymic epithelium in the SCID-hu mouse leads to thymocyte apoptosis. J Virol. 1996;70:3734–3740. doi: 10.1128/jvi.70.6.3734-3740.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bellini W J, Englund G, Rozenblatt S, Arnheiter H, Richardson C D. Measles virus P gene codes for two proteins. J Virol. 1985;53:908–919. doi: 10.1128/jvi.53.3.908-919.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Billeter M A, Cattaneo R. Molecular biology of defective measles virus persisting in the human central nervous system. In: Kingsbury D W, editor. The paramyxoviruses. New York, N.Y: Plenum Press; 1991. pp. 323–345. [Google Scholar]
- 5.Blixenkrone-Moller M, Bernard A, Bencsik A, Sixt N, Diamond L E, Logan J S, Wild T F. Role of CD46 in measles virus infection in CD46 transgenic mice. Virology. 1998;249:238–248. doi: 10.1006/viro.1998.9301. [DOI] [PubMed] [Google Scholar]
- 6.Cathomen T, Mrkic B, Spehner D, Drillien R, Naef R, Pavlovic J, Aguzzi A, Billeter M A, Cattaneo R. A matrix-less measles virus is infectious and elicits extensive cell fusion: consequences for propagation in the brain. EMBO J. 1998;17:3899–3908. doi: 10.1093/emboj/17.14.3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cattaneo R, Kaelin K, Baczko K, Billeter M A. Measles virus editing provides an additional cysteine-rich protein. Cell. 1989;56:759–764. doi: 10.1016/0092-8674(89)90679-x. [DOI] [PubMed] [Google Scholar]
- 8.Clements C J, Cutts F T. The epidemiology of measles: thirty years of vaccination. In: ter Meulen V, Billeter M A, editors. Measles virus. Berlin, Germany: Springer-Verlag; 1995. pp. 13–33. [DOI] [PubMed] [Google Scholar]
- 9.Escoffier C, Manie S, Vincent S, Muller C P, Billeter M, Gerlier D. Nonstructural C protein is required for efficient measles virus replication in human peripheral blood cells. J Virol. 1999;73:1695–1698. doi: 10.1128/jvi.73.2.1695-1698.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Esolen L M, Park S W, Hardwick J M, Griffin D E. Apoptosis as a cause of death in measles virus-infected cells. J Virol. 1995;69:3955–3958. doi: 10.1128/jvi.69.6.3955-3958.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Esolen L M, Ward B J, Moench T R, Griffin D E. Infection of monocytes during measles. J Infect Dis. 1993;168:47–52. doi: 10.1093/infdis/168.1.47. [DOI] [PubMed] [Google Scholar]
- 12.Firsching R, Buchholz C J, Schneider U, Cattaneo R, ter Meulen V, Schneider-Schaulies J. Measles virus spread by cell-cell contacts: uncoupling of contact-mediated receptor (CD46) downregulation from virus uptake. J Virol. 1999;73:5265–5273. doi: 10.1128/jvi.73.7.5265-5273.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Forthal D N, Aarnaes S, Blanding J, de la Maza L, Tilles J G. Degree and length of viremia in adults with measles. J Infect Dis. 1992;166:421–424. doi: 10.1093/infdis/166.2.421. [DOI] [PubMed] [Google Scholar]
- 14.Fugier-Vivier I, Servet-Delprat C, Rivailler P, Rissoan M C, Liu Y J, Rabourdin-Combe C. Measles virus suppresses cell-mediated immunity by interfering with the survival and functions of dendritic and T cells. J Exp Med. 1997;186:813–823. doi: 10.1084/jem.186.6.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcin D, Latorre P, Kolakofsky D. Sendai virus C proteins counteract the interferon-mediated induction of an antiviral state. J Virol. 1999;73:6559–6565. doi: 10.1128/jvi.73.8.6559-6565.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldman M B, Buckthal D J, Picciotto S, O'Bryan T A, Goldman J N. Measles virus persistence in an immortalized murine macrophage cell line. Virology. 1995;207:12–22. doi: 10.1006/viro.1995.1047. [DOI] [PubMed] [Google Scholar]
- 17.Helin E, Salmi A A, Vanharanta R, Vainionpaa R. Measles virus replication in cells of myelomonocytic lineage is dependent on cellular differentiation stage. Virology. 1999;253:35–42. doi: 10.1006/viro.1998.9460. [DOI] [PubMed] [Google Scholar]
- 18.Hirano A, Yang Z, Katayama Y, Korte-Sarfaty J, Wong T C. Human CD46 enhances nitric oxide production in mouse macrophage in response to measles virus infection in the presence of gamma interferon: dependence on the CD46 cytoplasmic domains. J Virol. 1999;73:4776–4785. doi: 10.1128/jvi.73.6.4776-4785.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Horvat B, Rivailler P, Varior-Krishnan G, Cardoso A, Gerlier D, Rabourdin-Combe C. Transgenic mice expressing human measles virus (MV) receptor CD46 provide cells exhibiting different permissivities to MV infections. J Virol. 1996;70:6673–6681. doi: 10.1128/jvi.70.10.6673-6681.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hourcade D, Garcia A D, Post T W, Taillon-Miller P, Holers V M, Wagner L M, Bora N S, Atkinson J P. Analysis of the human regulators of complement activation (RCA) gene cluster with yeast artificial chromosomes (YACs) Genomics. 1992;12:289–300. doi: 10.1016/0888-7543(92)90376-4. [DOI] [PubMed] [Google Scholar]
- 21.Hummel K B, Bellini W J, Offermann M K. Strain-specific differences in LFA-1 induction on measles virus-infected monocytes and adhesion and viral transmission to endothelial cells. J Virol. 1998;72:8403–8407. doi: 10.1128/jvi.72.10.8403-8407.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kato A, Kiyotani K, Sakai Y, Yoshida T, Nagai Y. The paramyxovirus, Sendai virus, V protein encodes a luxury function required for viral pathogenesis. EMBO J. 1997;16:578–587. doi: 10.1093/emboj/16.3.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Korte-Sarfaty J, Pham V D, Yant S, Hirano A, Wong T C. Expression of human complement regulatory protein CD46 restricts measles virus replication in mouse macrophages. Biochem Biophys Res Commun. 1998;249:432–437. doi: 10.1006/bbrc.1998.9173. [DOI] [PubMed] [Google Scholar]
- 24.Lawrence D M, Vaughn M M, Belman A R, Cole J S, Rall G F. Immune response-mediated protection of adult but not neonatal mice from neuron-restricted measles virus infection and central nervous system disease. J Virol. 1999;73:1795–1801. doi: 10.1128/jvi.73.3.1795-1801.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liebert U G, Finke D. Measles virus infections in rodents. In: ter Meulen V, Billeter M A, editors. Measles virus. Berlin, Germany: Springer-Verlag; 1995. pp. 149–166. [DOI] [PubMed] [Google Scholar]
- 25a.Maisner, A., B. Mrkic, G. Herrler, M. Moll, M. A. Billeter, R. Cattaneo, and H.-D. Klenk. Recombinant measles virus requiring an exogenous protease for activation of infectivity. J. Gen. Virol., in press. [DOI] [PubMed]
- 26.Manchester M, Eto D S, Oldstone M B A. Characterization of the inflammatory response during acute measles encephalitis in NSE-CD46 transgenic mice. J Neuroimmunol. 1999;96:207–217. doi: 10.1016/s0165-5728(99)00036-3. [DOI] [PubMed] [Google Scholar]
- 27.McChesney M B, Miller C J, Rota P A, Zhu Y D, Antipa L, Lerche N W, Ahmed R, Bellini W J. Experimental measles. I. Pathogenesis in the normal and the immunized host. Virology. 1997;233:74–84. doi: 10.1006/viro.1997.8576. [DOI] [PubMed] [Google Scholar]
- 28.Mitus A. Attenuated measles vaccine in children with acute leukemia. Am J Dis Child. 1962;103:413–418. doi: 10.1001/archpedi.1962.02080020425051. [DOI] [PubMed] [Google Scholar]
- 29.Moench T R, Griffin D E, Obriecht C R, Vaisberg A J, Johnson R T. Acute measles in patients with and without neurological involvement: distribution of measles virus antigen and RNA. J Infect Dis. 1988;158:433–442. doi: 10.1093/infdis/158.2.433. [DOI] [PubMed] [Google Scholar]
- 30.Mrkic B, Pavlovic J, Rulicke T, Volpe P, Buchholz C J, Hourcade D, Atkinson J P, Aguzzi A, Cattaneo R. Measles virus spread and pathogenesis in genetically modified mice. J Virol. 1998;72:7420–7427. doi: 10.1128/jvi.72.9.7420-7427.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Müller U, Steinhoff U, Reis L F, Hemmi S, Pavlovic J, Zinkernagel R M, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 32.Niewiesk S, Eisenhuth I, Fooks A, Clegg J C, Schnorr J J, Schneider-Schaulies S, ter Meulen V. Measles virus-induced immune suppression in the cotton rat (Sigmodon hispidus) model depends on viral glycoproteins. J Virol. 1997;71:7214–7219. doi: 10.1128/jvi.71.10.7214-7219.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Norrby E, Oxman M N. Measles virus. In: Fields B N, et al., editors. Virology. New York, N.Y: Raven Press; 1990. pp. 1013–1044. [Google Scholar]
- 34.Radecke F, Billeter M A. The nonstructural C protein is not essential for multiplication of Edmonston B strain measles virus in cultured cells. Virology. 1996;217:418–421. doi: 10.1006/viro.1996.0134. [DOI] [PubMed] [Google Scholar]
- 35.Radecke F, Spielhofer P, Schneider H, Kaelin K, Huber M, Dotsch C, Christiansen G, Billeter M A. Rescue of measles viruses from cloned DNA. EMBO J. 1995;14:5773–5784. doi: 10.1002/j.1460-2075.1995.tb00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rall G F, Manchester M, Daniels L R, Callahan E M, Belman A R, Oldstone M B. A transgenic mouse model for measles virus infection of the brain. Proc Natl Acad Sci USA. 1997;94:4659–4663. doi: 10.1073/pnas.94.9.4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sakaguchi M, Yoshikawa Y, Yamanouchi K, Sata T, Nagashima K, Takeda K. Growth of measles virus in epithelial and lymphoid tissues of cynomolgus monkeys. Microbiol Immunol. 1986;30:1067–1073. doi: 10.1111/j.1348-0421.1986.tb03036.x. [DOI] [PubMed] [Google Scholar]
- 38.Schneider H, Kaelin K, Billeter M A. Recombinant measles viruses defective for RNA editing and V protein synthesis are viable in cultured cells. Virology. 1997;227:314–322. doi: 10.1006/viro.1996.8339. [DOI] [PubMed] [Google Scholar]
- 39.Sherman F E, Ruckle G. In vivo and in vitro cellular changes specific for measles. Pathology. 1958;65:587–599. [PubMed] [Google Scholar]
- 40.Singh M, Cattaneo R, Billeter M. A recombinant measles virus expressing hepatitis B virus surface antigen induces humoral immune responses in genetically modified mice. J Virol. 1999;73:4823–4828. doi: 10.1128/jvi.73.6.4823-4828.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tober C, Seufert M, Schneider H, Billeter M A, Johnston I C, Niewiesk S, ter Meulen V, Schneider-Schaulies S. Expression of measles virus V protein is associated with pathogenicity and control of viral RNA synthesis. J Virol. 1998;72:8124–8132. doi: 10.1128/jvi.72.10.8124-8132.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Valsamakis A, Schneider H, Auwaerter P G, Kaneshima H, Billeter M A, Griffin D E. Recombinant measles viruses with mutations in the C, V, or F gene have altered growth phenotypes in vivo. J Virol. 1998;72:7754–7761. doi: 10.1128/jvi.72.10.7754-7761.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Binnendijk R S, van der Heijden R W, van Amerongen G, UytdeHaag F G, Osterhaus A D. Viral replication and development of specific immunity in macaques after infection with different measles virus strains. J Infect Dis. 1994;170:443–448. doi: 10.1093/infdis/170.2.443. [DOI] [PubMed] [Google Scholar]
- 44.van Binnendijk R S, van der Heijden R W J, Osterhaus A D M E. Monkeys in measles research. In: ter Meulen V, Billeter M A, editors. Measles virus. Berlin, Germany: Springer-Verlag; 1995. pp. 135–148. [DOI] [PubMed] [Google Scholar]
- 45.Zhu Y D, Heath J, Collins J, Greene T, Antipa L, Rota P, Bellini W, McChesney M. Experimental measles. II. Infection and immunity in the rhesus macaque. Virology. 1997;233:85–92. doi: 10.1006/viro.1997.8575. [DOI] [PubMed] [Google Scholar]