

Abstract
Ciliary dysfunction causes a large group of developmental and degenerative human diseases known as ciliopathies. These diseases reflect the critical roles that cilia play in sensing the environment and in force generation for motility. Sensory functions include our senses of vision and olfaction. In addition, primary and motile cilia throughout our body monitor the environment allowing cells to coordinate their biology with the cells around them. This coordination is critical to organ development and maintenance, and ciliary dysfunction causes diverse structural birth defects and degenerative diseases. Defects in motility cause lung disease due to the failure of mucociliary clearance, male infertility due to the failure of sperm motility and the ability of sperm to move through the efferent ducts, and disturbances of the left-right axis due to a failure of nodal cilia to establish proper left-right cues.
Cilia Structure
Throughout the eukaryotic kingdom, cilia and flagella serve as sensory and force generating organelles (Bloodgood, 2010). To carry out these important functions, more than a thousand unique proteins are required (Pazour et al., 2005). The proteins are organized around a microtubule-based cytoskeleton or axoneme, which is covered by an extension of the plasma membrane. The axoneme serves as a scaffold to anchor and organize the proteins that carry out the motility and sensory functions of cilia. The typical axoneme consists of nine doublet microtubules arranged around the circumference of a cylinder (Figure 1). The central apparatus, which consists of a pair of singlet microtubules and associated proteins, is usually present in the center of the axoneme of motile cilia but is missing from non-motile cilia. These arrangements of microtubules are often described as 9+2 and 9+0. The ciliary membrane is continuous with the cell membrane but is a unique compartment and is enriched in specific receptors. In addition to thousands of olfactory receptors, at least 30 G-protein coupled receptors (GPCRs) are known to localize to cilia (Hilgendorf et al., 2016). The cytoplasm or matrix that surrounds the axoneme is distinct from the cellular cytoplasm (Witman et al., 1972) and is important to signaling as its small volume allows for rapid changes in second messenger concentrations (Delling et al., 2013).
Figure 1.
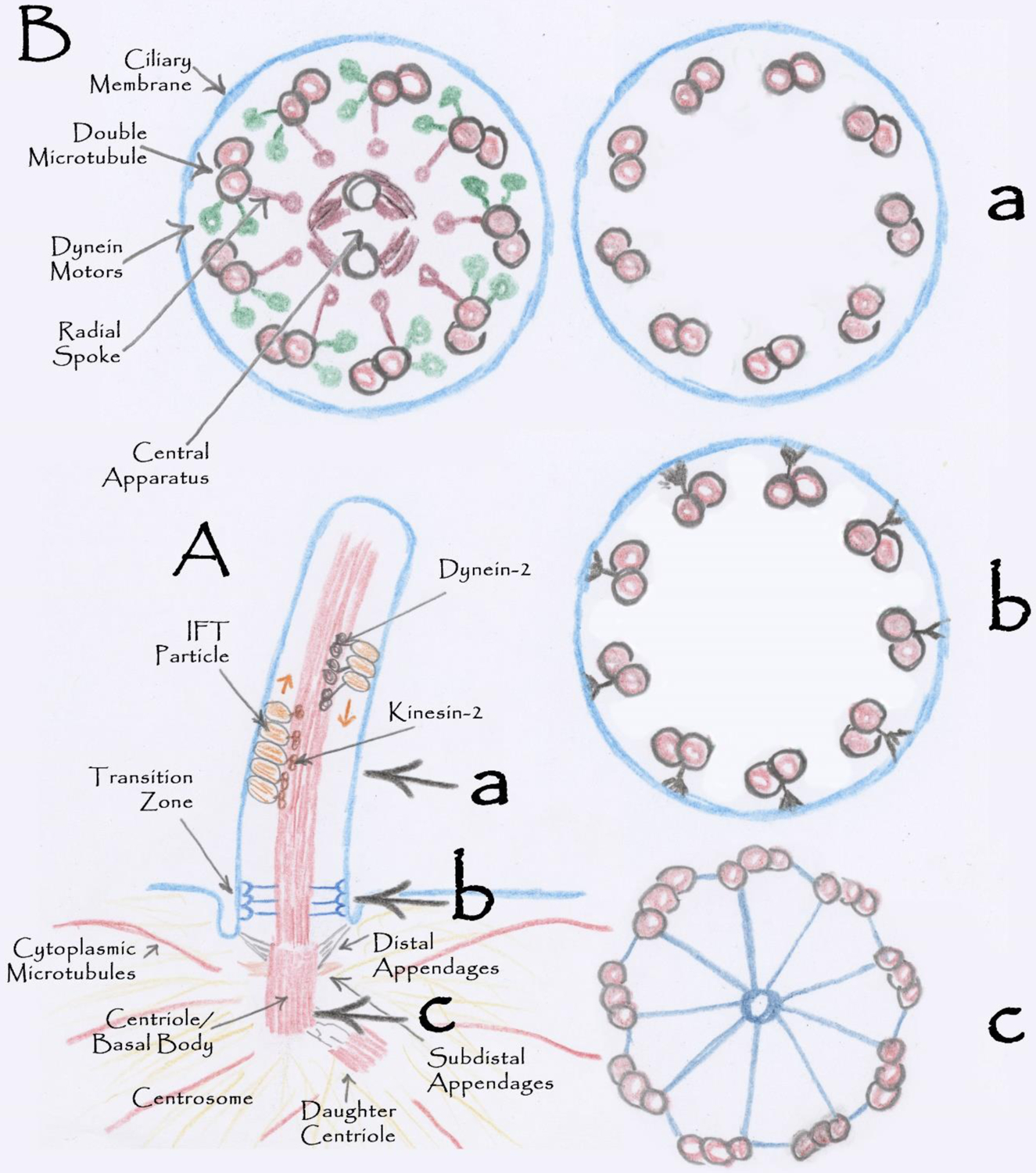
Cilia Structure
A. Structure of a typical mammalian cilium. B. Cross sections through the cilium at the levels marked a, b, and c. Ba shows a cross section through a motile 9+2 cilium on the left and a non-motile 9+0 on the right.
The doublet microtubules are templated from the distal end of the basal body or mother centriole, which is embedded within the centrosome. In addition to templating the microtubules of the axoneme, the centriole and associated centrosome organize the interphase microtubule network and the mitotic spindle. The centriole is composed of nine triplet microtubules, two of which are continuous with the doublets of the axoneme. Distal and subdistal appendages project from the sides of the centriole. The distal appendages link the centriole and associated cilium to the membrane via transition fibers. The subdistal appendages project into the pericentriolar material. They are likely important for organizing the microtubule nucleating proteins and signaling components that are enriched at the base of cilia.
The base of the cilium contains a barrier that isolates the ciliary membrane from the rest of the plasma membrane and keeps the ciliary matrix distinct from the cellular cytoplasm. It appears that the barrier is largely achieved by the proteins of the transition zone and an associated septin ring (Hu et al., 2010), but lipid composition may also contribute (Kamiya and Witman, 1984; Vieira et al., 2006). The structure of the transition zone varies from species to species but typically contains a “Y” or champagne glass-shaped structure that connects the outer double microtubules to the membrane. At the membrane, the ends of the Y-shaped connectors are embedded in the bilayer and can be observed in freeze fracture electron microscopy as rows of transmembrane proteins known as the ciliary necklace (Gilula and Satir, 1972).
While all cilia share the features of a microtubule-based axoneme covered with membrane there are significant variations in structure throughout eukaryotes and within mammals (Figure 2). Microtubule number can vary from the nine doublets typically seen to as few as six and to as many as fourteen (Carvalho-Santos et al., 2011). Within mammals, primary cilia lose outer doublet microtubules as the axoneme extends distally from the centriole (Kiesel et al., 2020). Similarly, in photoreceptor rod and cone cells the 9+0 arrangement is observed in the connecting cilium but is lost once the microtubules extend past the region where the ciliary membrane is remodeled into disks for light detection (Lewis et al., 2023). Olfactory cilia share a 9+0 arrangement at the base but then lose doublets as they extend past the proximal segment and into the distal segment (Jenkins et al., 2009). In addition, the central apparatus is highly variable across species with some motile cilia having only a single central apparatus microtubule and others none at all (Zhao et al., 2020). In some cilia, additional accessory structures like the outer dense fibers, fibrous sheaths, and paraflagellar rods are present and critical to the specialized functions of these cilia.
Figure 2.
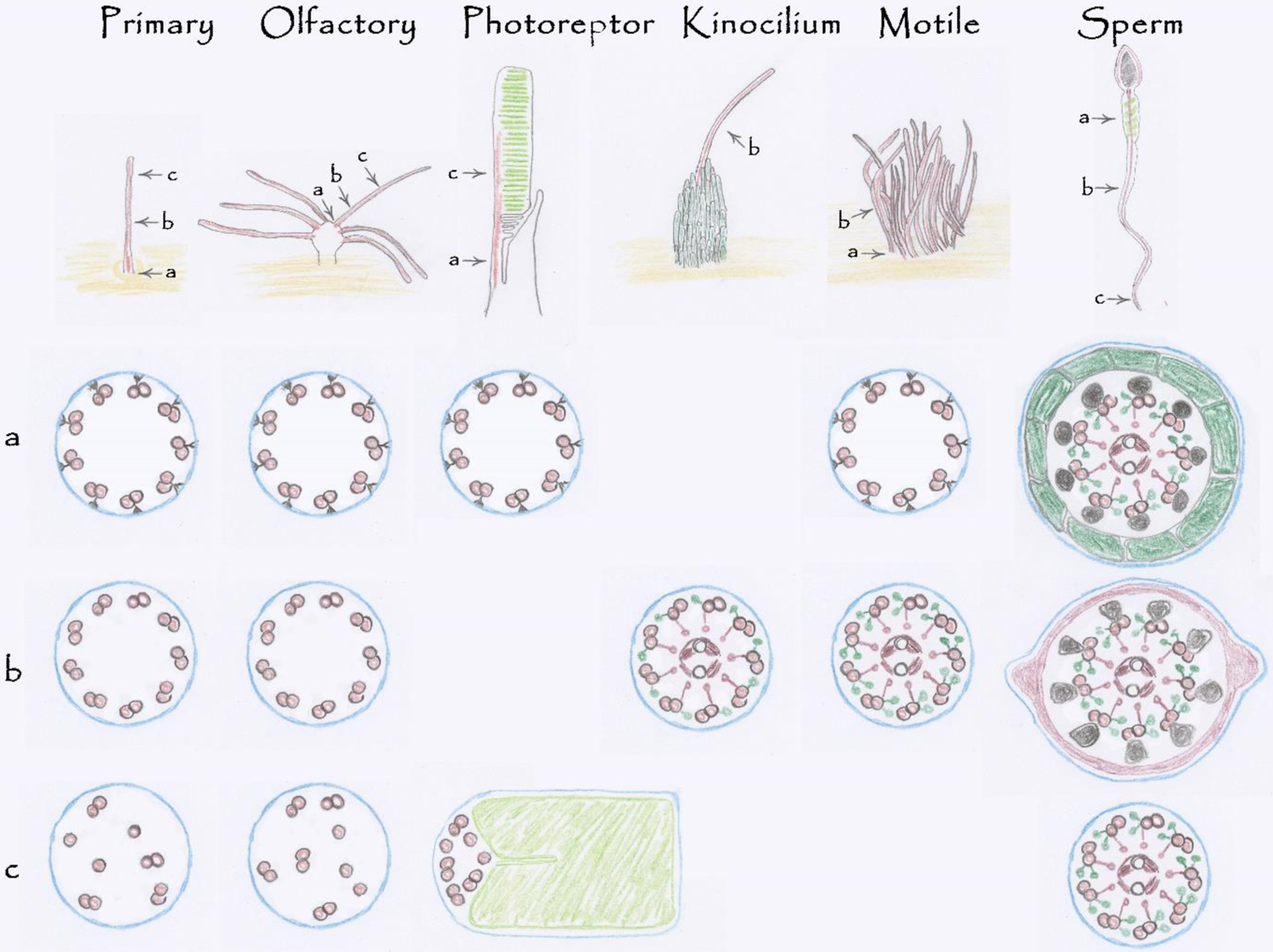
Mammalian Cilia Diversity.
Mammalian cells express a variety of cilia to serve diverse sensory and motile functions. All cilia contain a microtubule-based cytoskeleton or axoneme enclosed by membrane (blue). Primary and olfactory cilia share similar axonemal structure with a transition zone at the base (a) followed by a 9+0 (b) cytoskeleton that gradually loses microtubules as it extends distally (c). Photoreceptor cilia have an extended transition zone called the connecting cilium (a). The 9+0 arrangement is lost as the axoneme extends along the photoreceptor disks (green) in the outer segment (c). Kinocilia extend from the side of an array of microvilli known as stereocilia (green). Structurally, kinocilia contain axonemal structures found in motile cilia including the central apparatus, outer dynein arms and radial spokes. Stereocilia have an actin-based cytoskeleton and are not cilia. Motile cilia have a transition zone (a) and a well-defined 9+2 cytoskeleton with a central apparatus, radial spokes, and dynein arms. Sperm flagella share features with motile cilia but have accessory structures. In the midpiece, each outer doublet is associated with an outer dense fiber (black) that is wrapped with a layer of mitochondria (green). The outer dense fibers extend through the tail to almost the tip (c). The outer dense fibers distal to the midpiece are wrapped with a fibrous sheath (red)
Cilia Assembly
The assembly of most types of eukaryotic cilia and flagella presents a unique challenge to the cell. These complex organelles have no protein synthesis capability and require all components to be made in the cell body and transported into the cilium for assembly. It is thought that intraflagellar transport (IFT) is the major force for moving proteins into the cilium (Kozminski et al., 1993). During IFT, large protein complexes called IFT particles are carried along the ciliary microtubules by kinesin-2 (Cole et al., 1998) and dynein-2 (Pazour et al., 1998). The IFT particles are thought to shuttle proteins into cilia for assembly and maintenance and to remove proteins from cilia as needed (Petriman and Lorentzen, 2020; Rosenbaum and Witman, 2002). IFT is critical to ciliary signaling by delivering receptors to cilia (Mukhopadhyay et al., 2010) and by directly participating in the signaling cascades (Eguether et al., 2018; Wang et al., 2006).
The IFT particle that transports material along the ciliary axoneme is built from IFT-A, IFT-B, and BBSome subcomplexes with stoichiometry of ~3 IFT-A to 6 IFT-B to 1 BBSome (Lacey et al., 2023; Lechtreck et al., 2009). IFT-A contains 6 subunits (Behal et al., 2012), IFT-B contains 16 (Follit et al., 2009), and BBSome 8 (Nachury et al., 2007) subunits. These proteins are highly conserved across ciliated eukaryotes and incredibly all 30 of these proteins are conserved between humans and Chlamydomonas suggesting that the last common eukaryotic ancestor had an IFT particle similar to what we study today. The remarkable fact that all IFT proteins were maintained across this evolutionary timeframe suggests that each IFT protein has an important function to play in ciliary biology. It is thought that IFT and coated vesicle transport are evolutionarily related, likely evolving from a transport process in cells prior to the last common ancestor (Avidor-Reiss et al., 2004). Across eukaryotes, IFT has been lost from all organisms that lost cilia suggesting that its only purpose is ciliary assembly. Most ciliated organisms retain all three complexes, but exceptions exist. For example, Toxoplasma, Selaginella and Physcomitrella lost the BBSome but retained most of IFT-A and IFT-B. Others like Thalassiosira have lost the BBSome and IFT-A but retained a portion of IFT-B. All three complexes have been lost from the ciliated organism Plasmodium, which assembles cilia in the cytoplasm before the structure becomes covered by the ciliary membrane. This observation suggests IFT is not needed to build cilia per se but is needed to move material into cilia for assembly. The emergent theme from analysis of taxonomic distribution and cell biology is that IFT-B is the most critical for ciliary assembly as no organism with cilia assembled in a membrane projection has lost IFT-B and most IFT-B mutants completely lack cilia. Exceptions exist, including IFT25 and IFT27, which are lost from metazoans that do not use cilia for Hedgehog signaling (Keady et al., 2012). IFT-A appears to be critical in transport of membrane proteins into cilia and IFT-A mutants often assemble short cilia (Mukhopadhyay et al., 2010). IFT-A mutant cilia are typically filled with IFT-B and other proteins suggesting that IFT-A is critical for retrograde transport (Iomini et al., 2009). Reviews often state that IFT-B plays an analogous role in anterograde transport but there is no data to support this idea and all three complexes are transported together in both directions. The BBSome is not required for assembly of cilia but appears to be necessary for removal of non-ciliary proteins from cilia (Lechtreck et al., 2009) and is important for delivery of some GPCRs to cilia (Berbari et al., 2008).
Ciliary Diseases or Ciliopathies
Advances in genome sequencing have allowed the identification of several hundred ciliary diseases in the human population and there are likely hundreds more to be identified. Human cilia genes and associated diseases are cataloged at ChlamyFP.org/human. These diseases evince the main functions of cilia in generating force and sensing the environment. The phenotypes observed in patients range from single organ dysfunction to wide ranging dysfunction in many organs. The phenotypes reflect the function of the encoded protein and the nature of the allele. For example, ciliary genes encoding proteins needed only for motility result in respiratory, reproduction, and left-right phenotypes, while gene products needed more broadly will result in the motility phenotypes along with structural birth defects and sensory perception deficits. In addition, the nature of the allele is critical. For example, individual alleles of IFT140 can result in syndromic disease with multi organ involvement (Schmidts et al., 2013) or they can result in non-syndromic blindness (Xu et al., 2015), male infertility (Wang et al., 2019), or cystic kidney disease (Senum et al., 2022). While there is significant overlap between the ciliary diseases, unique features allow classification into three large groups. The first being the motile ciliopathies. The second and third groups represent defects in non-motile cilia and result in ciliopathies caused by primary cilia dysfunction or sensory diseases. For this discussion, they have been split into two groups to highlight unique phenotypes. However, it should be kept in mind that many patients have pathology that spans all three groups.
Motile Cilia Dysfunction
Defects in ciliary motility cause a syndrome of phenotypes known as primary ciliary dyskinesia (PCD). This disease was historically known as Kartagener’s syndrome and consists of recurrent respiratory infections, male infertility, and disturbances of the left-right organization of the thoracic and abdominal organs (Afzelius, 1976). Recurrent respiratory infections result from ciliary dysfunction failing to properly move mucus over the epithelium of the lungs and trachea allowing bacteria and particles to remain in the airways. This leads to chronic cough and infections, which often lead to lung degeneration and significant loss of quality of life (Shah and Laguna, 2022). Nearly 100 PCD genes have been identified, most of which encode proteins required for ciliary force generation or regulation of motility.
Defects in sperm motility are a major cause of male infertility. Sperm flagella and respiratory cilia share many components and have structural similarities. In most cases, mutations disrupting ciliary motility affect both respiratory cilia and sperm flagella. However, sperm flagella and respiratory cilia each have unique structural adaptations that are critical to function and some mutations affect only sperm motility (Whitfield et al., 2019) while others affect only respiratory cilia (Jonsson et al., 1982). Human sperm flagella are about 50 microns long with a typical 9+2 axoneme covered with membrane and are divided into three distinct domains, the midpiece which is next to the head, the principal piece in the middle, and the end piece. In sperm, the nine outer doublet microtubules are flanked by nine outer dense fibers that extend along the axoneme. These fibers originate from the segmented column surrounding the centriole and extend to the tip of the principal piece. In the midpiece, the outer dense fibers are surrounded by a layer of mitochondria. In the principal piece, the outer dense fibers are surrounded by a fibrous sheath (Inaba, 2011). Like other cilia, sperm axonemes are templated from a basal body, which in sperm is called the distal centriole. Curiously, in sperm the basal body may be derived from the daughter centriole rather than the mother centriole as is typically the case (Alieva et al., 2018). Once assembly is complete, the basal body can be modified or lost. Sperm competition is thought to be a major driving force for centriole evolution with more extensive modifications found in species where females may have more than one partner. In humans, the proximal centriole maintains a typical structure, but in the distal centriole (basal body) the triplets become doublets and are splayed out, and the centriole loses its typical barrel structure (Fishman et al., 2018). In most mammals, the proximal and distal centrioles form a dynamic link between the head and tail (Khanal et al., 2021). However, in mice the centrioles appear to be completely lost or so highly modified as to not be recognizable (Avidor-Reiss et al., 2020).
In addition to defects in sperm motility, defects in efferent duct cilia also contribute to male infertility (Hoque et al., 2022). The efferent ductules connect the rete testis to the epididymis. The epithelium lining the ductules is composed of two cell types. One type is similar to the kidney proximal tubule epithelium in that the cells are covered with an extensive brush border and resorb ~90% of the lumenal fluid that passes into the duct. The second type is multiciliated and shares features with respiratory epithelium. Motility mutations that affect respiratory cilia but not sperm flagella disrupt the motility of the efferent duct cilia and defective motility of these cilia is likely the cause of infertility in males carrying these mutations (Aprea et al., 2021; Terre et al., 2019; Terre et al., 2016). Initial reports suggested that ciliary motility moved the sperm along the tubule. Further analysis indicated that the cilia did not produce a metachronal wave like that seen in respiratory epithelium but instead have a whip-like chaotic motility that generates turbulence. This motility is thought to keep the immotile sperm in suspension as they move through the duct by peristaltic contractions (Yuan et al., 2019).
The abnormal development of the left-right axis is thought to result from nodal cilia dysfunction failing to properly define the left and right sides of the embryo. Mechanisms driving the initial determination of left and right in the embryo are hotly debated but it is clear that left-ward flow generated by ciliary motility in the embryonic node is critical. Normal left-ward flow triggers nodal signaling on the left side of the embryo producing the normal situs solitus structure of the heart and asymmetric placement of spleen, stomach, and other internal organs (Nakamura and Hamada, 2012). When nodal cilia are dysfunctional, the side of nodal activation is randomized. This can lead to normal situs or a complete reversal of internal organ placement, which is known as situs inversus. In some cases, the nodal activation is not fully left nor fully right leading to heterotaxy where the internal organs have incomplete determination and are neither normal nor completely reversed. Situs inversus is a relatively benign condition, but heterotaxy is thought to be responsible for much of the structural cardiac disease seen in the human population (Gabriel et al., 2021).
In addition to the major roles of cilia in respiratory health, male fertility, and left-right determination, motile cilia are found in the brain, in the female reproductive tract, and the efferent ducts of the testis. In the brain, the cilia are found on ependymal cells lining the ventricles and canals connecting the ventricles and are thought to move cerebrospinal fluid over the cell surfaces. In dogs and rodents, ciliary motility defects lead to a high incidence of hydrocephaly. In humans, PCD patients have an increased risk of hydrocephaly, but it is a relatively rare comorbidity in the population (Ibanez-Tallon et al., 2004; Wallmeier et al., 2022). In the female reproductive tract, the cilia are abundant in the oviduct and endometrium. It is thought that motile cilia in the infundibulum are essential for oocyte pickup whereas other cilia assist smooth muscle in the movement of the egg from the ovary to the uterus (Halbert et al., 1976; McGlade et al., 2023; Yuan et al., 2021). Female patients with PCD are reported to be sub fertile, but many are able to procreate without assistance (Newman et al., 2023; Raidt et al., 2015).
Primary Cilia Dysfunction
Most vertebrate cells project a solitary non motile primary cilium from their cell surface (Pazour and Witman, 2003). These organelles are enriched in receptors allowing the cell to monitor the extracellular environment and coordinate their behavior and physiology with the cells around them. Dysfunction in these organelles leads to a large group of rare genetic diseases (Pazour et al., 2020). The complete loss of primary cilia in mice causes severe cardiac malformations and embryo demise at the time of heartbeat initiation (Li et al., 2015; Marszalek et al., 1999; Murcia et al., 2000). This has not been studied in humans, but the equivalent developmental landmark occurs in the first trimester. Less severe defects in cilia cause structural or degenerative defects in every organ that has been examined. Predominant phenotypes in the human population include brain malformations like holoprosencephaly, craniofacial abnormalities, skeletal malformations including polydactyly, shortened ribs, and long bone malformations, along with hair and skin abnormalities, structural cardiac defects, genital malformations, obesity, and cystic/fibrotic diseases of the kidney, liver, and pancreas (Gabriel et al., 2021; Schmidts, 2014; Thomas et al., 2019). Obesity and the cystic/fibrotic diseases will be discussed below but many if not all of the other phenotypes result from defective Hedgehog signaling (Huangfu et al., 2003). In vertebrates the Hedgehog pathway is organized around cilia. Cilia defects dampen responses to Hedgehog ligands as the Hedgehog receptors (Ptch1, GPR161, Smo) localize to cilia (Corbit et al., 2005; Mukhopadhyay et al., 2013; Rohatgi et al., 2007). Furthermore, the Gli transcription factors are transiently localized to the cilium where they are processed in response to Hedgehog signaling to become activators or repressors (Haycraft et al., 2005).
Obesity is often seen in patients with ciliary dysfunction and many cilia-defective mice become obese. Obesity is thought to result from hyperphagia due to a failure of the pons neurons in the brain to properly regulate feeding behavior (Davenport et al., 2007). Numerous GPCRs including somatostatin receptor 3 (Sstr3), serotonin receptor 6 (5HT6), dopamine receptor 1 (Drd1), neuropeptide Y receptor 2 (Npy2r), and kisspeptin receptor 1 (Kiss1r) have been localized to neuronal cilia and may be involved in sensing satiety (Vaisse et al., 2017).
Cells lining the uriniferous tubules of the kidney, the biliary ducts of the liver, and the intra-acinar and intercalated ducts of the exocrine pancreas display prominent primary cilia extending from the apical surface into the lumen of the duct. The function of these cilia is unknown. However, defects lead to tubule expansion and cyst formation in these organs (Pazour et al., 2000). Cyst formation is often accompanied by fibrosis. In humans, autosomal dominant polycystic kidney disease (PKD) is mainly caused by mutations in the genes encoding polycystin-1 and polycystin-2. These channel-like proteins localize to primary cilia and by unknown mechanisms monitor the tubule diameter. When the polycystins or the cilia are defective, the cell perceives this as damage and responds by proliferating. The excess proliferation converts the normal tubule into a cyst. Cyst formation activates immune cells and causes tissue damage and fibrosis. While autosomal dominant PKD predominately affects the kidney, patients also can develop cysts in the liver and pancreas. In addition to autosomal dominant PKD, ciliary dysfunction is thought to drive kidney, liver, and pancreas cyst formation in autosomal recessive PKD and in the numerous syndromic diseases caused by ciliary dysfunction (Smith et al., 2022).
Sensory Dystrophies: Blindness, Anosmia, Deafness
Throughout the eukaryotic kingdom, cilia are widely used to detect light, odorants, and sound for the senses of sight, smell, and hearing. In mammals, cilia are used for detection of light in the eye and detection of odorants in the nose. In the eye, light is detected by the outer segments of photoreceptor rods and cones. During development, photoreceptor cells become post mitotic and assemble a primary cilium. The outer segment is formed from the primary cilium by elaborating the ciliary membrane to become the disks of the outer segment. The disks are highly enriched in rhodopsin and opsin GPCRs and the initial steps of photodetection occur in the cilium. The elaboration of the ciliary membrane into the disks of the outer segment requires extensive protein and lipid transport from the inner segment. To initially build the structure and to replace disks lost through normal shedding, ~4000 molecules of opsin must be transported through the connecting cilium each minute (Williams, 2002). IFT appears to be the major driver of transport in photoreceptor outer segments (Gupta and Pazour, 2023; Pazour et al., 2002).
In the nose, odorants are detected by the olfactory sensory neurons. These cells project cilia enriched in olfactory GPCRs into the nasal cavity for the detection of odorants. Similar to photoreceptors, the first steps in odorant detection occur in the cilia. Unlike most cells with sensory cilia, the olfactory sensory neurons are multiciliated. During development, the centrioles are amplified and then migrate into an apical protrusion called the dendritic knob. The centrioles dock on the membrane of the dendritic knob and extend cilia (Jenkins et al., 2009).
Hearing in mammals depends on an array of actin-based microvilli on hair cells to detect sound. Unfortunately, the hair cell microvilli are named stereocilia despite not being true microtubule-based cilia structures. Development of the stereocilia array starts with the assembly of a true cilium called the kinocilium. The kinocilium marks the center of the tallest row of microvilli and is thought to provide planar cell polarity cues for organization of the array (Jones et al., 2008). In some species the kinocilium is disassembled after development of the microvilli array is complete (Wang and Zhou, 2021). However, in species able to regenerate microvilli after damage, the cilium reappears at the initiation of regeneration, indicating that it is critical to the formation of the array (Sobkowicz et al., 1995). Curiously, the kinocilium has a 9+2 arrangement of microtubules with outer arm dynein and radial spokes suggesting that it may be motile. However, the role of motility is unknown and PCD patients do not have associated deafness.
Concluding Remarks
The following manuscripts in this special issue will highlight our understanding of the role of cilia in endocrinology and reproductive biology. Equally important, they will highlight areas for further work as our understanding of cilia function is far from complete and there remain hundreds of cilia genes with little ascribed function. This is an exciting time for cilia research with the focus moving from the identification of cilia genes to directly ascribing function to individual proteins and uncovering new roles for cilia in endocrinology and reproductive biology.
Acknowledgments
Work in the laboratory is supported by a National Institutes of Health grant GM060992. I thank Tomer Avidor-Reiss (University of Toledo) and Helen May-Simera (Johannes Gutenberg-University Mainz) for helpful comments on this work.
Abbreviations:
- GPCRs
G-protein coupled receptors
- IFT
intraflagellar transport
- PCD
primary ciliary dyskinesia
- PKD
polycystic kidney disease
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: none.
References
- Afzelius BA 1976. A human syndrome caused by immotile cilia. Science. 193:317–319. 10.1126/science.1084576. [DOI] [PubMed] [Google Scholar]; ** Electron microscopy identified defects in respiratory cilia from patients with Kartagener’s syndrome, now known as Primary Ciliary Dyskinesia. This was the first ciliopathy identified.
- Alieva I, Staub C, Uzbekova S, and Uzbekov R. 2018. A question of flagella origin for spermatids; mother or daughter centriole? Uzbekov RE, editor. Nova Science Publishers, New York. 109–121. [Google Scholar]
- Aprea I, Nothe-Menchen T, Dougherty GW, Raidt J, Loges NT, Kaiser T, Wallmeier J, Olbrich H, Strunker T, Kliesch S, Pennekamp P, and Omran H. 2021. Motility of efferent duct cilia aids passage of sperm cells through the male reproductive system. Mol Hum Reprod. 27. 10.1093/molehr/gaab009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avidor-Reiss T, Carr A, and Fishman EL. 2020. The sperm centrioles. Mol Cell Endocrinol. 518:110987. 10.1016/j.mce.2020.110987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avidor-Reiss T, Maer AM, Koundakjian E, Polyanovsky A, Keil T, Subramaniam S, and Zuker CS. 2004. Decoding cilia function: defining specialized genes required for compartmentalized cilia biogenesis. Cell. 117:527–539. 10.1016/s0092-8674(04)00412-x. [DOI] [PubMed] [Google Scholar]
- Behal RH, Miller MS, Qin H, Lucker BF, Jones A, and Cole DG. 2012. Subunit interactions and organization of the Chlamydomonas reinhardtii intraflagellar transport complex A proteins. J Biol Chem. 287:11689–11703. 10.1074/jbc.M111.287102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berbari NF, Lewis JS, Bishop GA, Askwith CC, and Mykytyn K. 2008. Bardet-Biedl syndrome proteins are required for the localization of G protein-coupled receptors to primary cilia. Proc Natl Acad Sci U S A. 105:4242–4246. 10.1073/pnas.0711027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloodgood RA 2010. Sensory reception is an attribute of both primary cilia and motile cilia. J Cell Sci. 123:505–509. 10.1242/jcs.066308. [DOI] [PubMed] [Google Scholar]
- Carvalho-Santos Z, Azimzadeh J, Pereira-Leal JB, and Bettencourt-Dias M. 2011. Evolution: Tracing the origins of centrioles, cilia, and flagella. J Cell Biol. 194:165–175. 10.1083/jcb.201011152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole DG, Diener DR, Himelblau AL, Beech PL, Fuster JC, and Rosenbaum JL. 1998. Chlamydomonas kinesin-II-dependent intraflagellar transport (IFT): IFT particles contain proteins required for ciliary assembly in Caenorhabditis elegans sensory neurons. J Cell Biol. 141:993–1008. 10.1083/jcb.141.4.993. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** Purification of the Chlamydomonas intraflagellar transport particle established the organization of the IFT-A and IFT-B sub particles and demonstrated that the IFT subunits are conserved in ciliated eukaryotes.
- Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, and Reiter JF. 2005. Vertebrate Smoothened functions at the primary cilium. Nature. 437:1018–1021. 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]; * This paper along with papers by Rohatgi et al. and Haycraft et al. established that the major components of the Hedgehog pathway are dynamically localized to cilia and that proper Hedgehog signaling in mammals requires primary cilia.
- Davenport JR, Watts AJ, Roper VC, Croyle MJ, van Groen T, Wyss JM, Nagy TR, Kesterson RA, and Yoder BK. 2007. Disruption of intraflagellar transport in adult mice leads to obesity and slow-onset cystic kidney disease. Curr Biol. 17:1586–1594. 10.1016/j.cub.2007.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This work demonstrated for the first time that neuronal cilia regulate behavior in mammals.
- Delling M, DeCaen PG, Doerner JF, Febvay S, and Clapham DE. 2013. Primary cilia are specialized calcium signalling organelles. Nature. 504:311–314. 10.1038/nature12833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguether T, Cordelieres FP, and Pazour GJ. 2018. Intraflagellar transport is deeply integrated in hedgehog signaling. Mol Biol Cell. 29:1178–1189. 10.1091/mbc.E17-10-0600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman EL, Jo K, Nguyen QPH, Kong D, Royfman R, Cekic AR, Khanal S, Miller AL, Simerly C, Schatten G, Loncarek J, Mennella V, and Avidor-Reiss T. 2018. A novel atypical sperm centriole is functional during human fertilization. Nat Commun. 9:2210. 10.1038/s41467-018-04678-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follit JA, Xu F, Keady BT, and Pazour GJ. 2009. Characterization of mouse IFT complex B. Cell Motil Cytoskeleton. 66:457–468. 10.1002/cm.20346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel GC, Young CB, and Lo CW. 2021. Role of cilia in the pathogenesis of congenital heart disease. Semin Cell Dev Biol. 110:2–10. 10.1016/j.semcdb.2020.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilula NB, and Satir P. 1972. The ciliary necklace. A ciliary membrane specialization. J Cell Biol. 53:494–509. 10.1083/jcb.53.2.494. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This seminal work defined the transition zone, which is critically important to maintaining the cilium as a unique cellular compartment and is the site where many ciliopathy gene products localize.
- Gupta M, and Pazour GJ. 2023. Intraflagellar transport: A critical player in photoreceptor development and the pathogenesis of retinal degenerative diseases. Cytoskeleton (Hoboken). 10.1002/cm.21823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbert SA, Tam PY, and Blandau RJ. 1976. Egg transport in the rabbit oviduct: the roles of cilia and muscle. Science. 191:1052–1053. 10.1126/science.1251215. [DOI] [PubMed] [Google Scholar]
- Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, and Yoder BK. 2005. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 1:e53. 10.1371/journal.pgen.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This paper along with papers by Corbit et al. and Rohatgi et al. established that the major components of the Hedgehog pathway are dynamically localized to cilia and that proper Hedgehog signaling in mammals requires primary cilia.
- Hilgendorf KI, Johnson CT, and Jackson PK. 2016. The primary cilium as a cellular receiver: organizing ciliary GPCR signaling. Curr Opin Cell Biol. 39:84–92. 10.1016/j.ceb.2016.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoque M, Kim EN, Chen D, Li FQ, and Takemaru KI. 2022. Essential Roles of Efferent Duct Multicilia in Male Fertility. Cells. 11. 10.3390/cells11030341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q, Milenkovic L, Jin H, Scott MP, Nachury MV, Spiliotis ET, and Nelson WJ. 2010. A septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution. Science. 329:436–439. 10.1126/science.1191054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, and Anderson KV. 2003. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 426:83–87. 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]; ** A genetic screen in mice established that IFT proteins and cilia are required for Hedgehog signaling in mouse. This work laid the foundation for our understanding of much of the developmental pathology that occurs when cilia are defective.
- Ibanez-Tallon I, Pagenstecher A, Fliegauf M, Olbrich H, Kispert A, Ketelsen UP, North A, Heintz N, and Omran H. 2004. Dysfunction of axonemal dynein heavy chain Mdnah5 inhibits ependymal flow and reveals a novel mechanism for hydrocephalus formation. Hum Mol Genet. 13:2133–2141. 10.1093/hmg/ddh219. [DOI] [PubMed] [Google Scholar]
- Inaba K 2011. Sperm flagella: comparative and phylogenetic perspectives of protein components. Mol Hum Reprod. 17:524–538. 10.1093/molehr/gar034. [DOI] [PubMed] [Google Scholar]
- Iomini C, Li L, Esparza JM, and Dutcher SK. 2009. Retrograde intraflagellar transport mutants identify complex A proteins with multiple genetic interactions in Chlamydomonas reinhardtii. Genetics. 183:885–896. 10.1534/genetics.109.101915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins PM, McEwen DP, and Martens JR. 2009. Olfactory cilia: linking sensory cilia function and human disease. Chem Senses. 34:451–464. 10.1093/chemse/bjp020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C, Roper VC, Foucher I, Qian D, Banizs B, Petit C, Yoder BK, and Chen P. 2008. Ciliary proteins link basal body polarization to planar cell polarity regulation. Nat Genet. 40:69–77. 10.1038/ng.2007.54. [DOI] [PubMed] [Google Scholar]
- Jonsson MS, McCormick JR, Gillies CG, and Gondos B. 1982. Kartagener’s syndrome with motile spermatozoa. N Engl J Med. 307:1131–1133. 10.1056/NEJM198210283071807. [DOI] [PubMed] [Google Scholar]
- Kamiya R, and Witman GB. 1984. Submicromolar levels of calcium control the balance of beating between the two flagella in demembranated models of Chlamydomonas. J Cell Biol. 98:97–107. 10.1083/jcb.98.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keady BT, Samtani R, Tobita K, Tsuchya M, San Agustin JT, Follit JA, Jonassen JA, Subramanian R, Lo CW, and Pazour GJ. 2012. IFT25 links the signal-dependent movement of Hedgehog components to intraflagellar transport. Dev Cell. 22:940–951. 10.1016/j.devcel.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanal S, Leung MR, Royfman A, Fishman EL, Saltzman B, Bloomfield-Gadelha H, Zeev-Ben-Mordehai T, and Avidor-Reiss T. 2021. A dynamic basal complex modulates mammalian sperm movement. Nat Commun. 12:3808. 10.1038/s41467-021-24011-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiesel P, Alvarez Viar G, Tsoy N, Maraspini R, Gorilak P, Varga V, Honigmann A, and Pigino G. 2020. The molecular structure of mammalian primary cilia revealed by cryo-electron tomography. Nat Struct Mol Biol. 27:1115–1124. 10.1038/s41594-020-0507-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozminski KG, Johnson KA, Forscher P, and Rosenbaum JL. 1993. A motility in the eukaryotic flagellum unrelated to flagellar beating. Proc Natl Acad Sci U S A. 90:5519–5523. 10.1073/pnas.90.12.5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey SE, Foster HE, and Pigino G. 2023. The molecular structure of IFT-A and IFT-B in anterograde intraflagellar transport trains. Nat Struct Mol Biol. 10.1038/s41594-022-00905-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechtreck KF, Johnson EC, Sakai T, Cochran D, Ballif BA, Rush J, Pazour GJ, Ikebe M, and Witman GB. 2009. The Chlamydomonas reinhardtii BBSome is an IFT cargo required for export of specific signaling proteins from flagella. J Cell Biol. 187:1117–1132. 10.1083/jcb.200909183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis TR, Phan S, Castillo CM, Kim KY, Coppenrath K, Thomas W, Hao Y, Skiba NP, Horb ME, Ellisman MH, and Arshavsky VY. 2023. Photoreceptor disc incisures form as an adaptive mechanism ensuring the completion of disc enclosure. Elife. 12. 10.7554/eLife.89160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Klena NT, Gabriel GC, Liu X, Kim AJ, Lemke K, Chen Y, Chatterjee B, Devine W, Damerla RR, Chang C, Yagi H, San Agustin JT, Thahir M, Anderton S, Lawhead C, Vescovi A, Pratt H, Morgan J, Haynes L, Smith CL, Eppig JT, Reinholdt L, Francis R, Leatherbury L, Ganapathiraju MK, Tobita K, Pazour GJ, and Lo CW. 2015. Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature. 521:520–524. 10.1038/nature14269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marszalek JR, Ruiz-Lozano P, Roberts E, Chien KR, and Goldstein LS. 1999. Situs inversus and embryonic ciliary morphogenesis defects in mouse mutants lacking the KIF3A subunit of kinesin-II. Proc Natl Acad Sci U S A. 96:5043–5048. 10.1073/pnas.96.9.5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlade EA, Stephens KK, Winuthayanon S, Anamthathmakula P, Holtzman MJ, and Winuthayanon W. 2023. Classical Estrogen Signaling in Ciliated Epithelial Cells of the Oviduct Is Nonessential for Fertility in Female Mice. Endocrinology. 165. 10.1210/endocr/bqad163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Wen X, Chih B, Nelson CD, Lane WS, Scales SJ, and Jackson PK. 2010. TULP3 bridges the IFT-A complex and membrane phosphoinositides to promote trafficking of G protein-coupled receptors into primary cilia. Genes Dev. 24:2180–2193. 10.1101/gad.1966210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Wen X, Ratti N, Loktev A, Rangell L, Scales SJ, and Jackson PK. 2013. The ciliary G-protein-coupled receptor Gpr161 negatively regulates the Sonic hedgehog pathway via cAMP signaling. Cell. 152:210–223. 10.1016/j.cell.2012.12.026. [DOI] [PubMed] [Google Scholar]
- Murcia NS, Richards WG, Yoder BK, Mucenski ML, Dunlap JR, and Woychik RP. 2000. The Oak Ridge Polycystic Kidney (orpk) disease gene is required for left-right axis determination. Development. 127:2347–2355. [DOI] [PubMed] [Google Scholar]
- Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peranen J, Merdes A, Slusarski DC, Scheller RH, Bazan JF, Sheffield VC, and Jackson PK. 2007. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 129:1201–1213. 10.1016/j.cell.2007.03.053. [DOI] [PubMed] [Google Scholar]
- Nakamura T, and Hamada H. 2012. Left-right patterning: conserved and divergent mechanisms. Development. 139:3257–3262. 10.1242/dev.061606. [DOI] [PubMed] [Google Scholar]
- Newman L, Chopra J, Dossett C, Shepherd E, Bercusson A, Carroll M, Walker W, Lucas JS, and Cheong Y. 2023. The impact of primary ciliary dyskinesia on female and male fertility: a narrative review. Hum Reprod Update. 29:347–367. 10.1093/humupd/dmad003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazour GJ, Agrin N, Leszyk J, and Witman GB. 2005. Proteomic analysis of a eukaryotic cilium. J Cell Biol. 170:103–113. 10.1083/jcb.200504008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazour GJ, Baker SA, Deane JA, Cole DG, Dickert BL, Rosenbaum JL, Witman GB, and Besharse JC. 2002. The intraflagellar transport protein, IFT88, is essential for vertebrate photoreceptor assembly and maintenance. J Cell Biol. 157:103–113. 10.1083/jcb.200107108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, Witman GB, and Cole DG. 2000. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J Cell Biol. 151:709–718. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** This work showed for the first time that primary cilia were critical for maintaining the structural integrity of the kidney tubule. This was the first demonstration that non motile primary cilia are important to mammalian health and are not vestigial organelles.
- Pazour GJ, Quarmby L, Smith AO, Desai PB, and Schmidts M. 2020. Cilia in cystic kidney and other diseases. Cell Signal. 69:109519. 10.1016/j.cellsig.2019.109519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazour GJ, Wilkerson CG, and Witman GB. 1998. A dynein light chain is essential for the retrograde particle movement of intraflagellar transport (IFT). J Cell Biol. 141:979–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazour GJ, and Witman GB. 2003. The vertebrate primary cilium is a sensory organelle. Curr Opin Cell Biol. 15:105–110. [DOI] [PubMed] [Google Scholar]
- Petriman NA, and Lorentzen E. 2020. Structural insights into the architecture and assembly of eukaryotic flagella. Microb Cell. 7:289–299. 10.15698/mic2020.11.734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raidt J, Werner C, Menchen T, Dougherty GW, Olbrich H, Loges NT, Schmitz R, Pennekamp P, and Omran H. 2015. Ciliary function and motor protein composition of human fallopian tubes. Hum Reprod. 30:2871–2880. 10.1093/humrep/dev227. [DOI] [PubMed] [Google Scholar]
- Rohatgi R, Milenkovic L, and Scott MP. 2007. Patched1 regulates hedgehog signaling at the primary cilium. Science. 317:372–376. 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]; * This paper along with papers by Corbit et al. and Haycraft et al. established that the major components of the Hedgehog pathway are dynamically localized to cilia and that proper Hedgehog signaling in mammals requires primary cilia.
- Rosenbaum JL, and Witman GB. 2002. Intraflagellar transport. Nat Rev Mol Cell Biol. 3:813–825. 10.1038/nrm952. [DOI] [PubMed] [Google Scholar]
- Schmidts M 2014. Clinical genetics and pathobiology of ciliary chondrodysplasias. J Pediatr Genet. 3:46–94. 10.3233/PGE-14089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidts M, Frank V, Eisenberger T, Al Turki S, Bizet AA, Antony D, Rix S, Decker C, Bachmann N, Bald M, Vinke T, Toenshoff B, Di Donato N, Neuhann T, Hartley JL, Maher ER, Bogdanovic R, Peco-Antic A, Mache C, Hurles ME, Joksic I, Guc-Scekic M, Dobricic J, Brankovic-Magic M, Bolz HJ, Pazour GJ, Beales PL, Scambler PJ, Saunier S, Mitchison HM, and Bergmann C. 2013. Combined NGS approaches identify mutations in the intraflagellar transport gene IFT140 in skeletal ciliopathies with early progressive kidney Disease. Hum Mutat. 34:714–724. 10.1002/humu.22294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senum SR, Li YSM, Benson KA, Joli G, Olinger E, Lavu S, Madsen CD, Gregory AV, Neatu R, Kline TL, Audrezet MP, Outeda P, Nau CB, Meijer E, Ali H, Steinman TI, Mrug M, Phelan PJ, Watnick TJ, Peters DJM, Ong ACM, Conlon PJ, Perrone RD, Cornec-Le Gall E, Hogan MC, Torres VE, Sayer JA, t.H.P.K.D.C.D.A.M. Genomics England Research Consortium, T.P. studies, and Harris PC. 2022. Monoallelic IFT140 pathogenic variants are an important cause of the autosomal dominant polycystic kidney-spectrum phenotype. American journal of human genetics. 109:136–156. 10.1016/j.ajhg.2021.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah A, and Laguna TA. 2022. Primary Ciliary Dyskinesia: A Rare and Often Underdiagnosed Disease. Pediatr Ann. 51:e82–e85. 10.3928/19382359-20220119-01. [DOI] [PubMed] [Google Scholar]
- Smith AO, Jonassen JA, Preval KM, Davis RJ, and Pazour GJ. 2022. c-JUN n-Terminal Kinase (JNK) Signaling in Autosomal Dominant Polycystic Kidney Disease. J Cell Signal. 3:62–78. 10.33696/Signaling.3.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobkowicz HM, Slapnick SM, and August BK. 1995. The kinocilium of auditory hair cells and evidence for its morphogenetic role during the regeneration of stereocilia and cuticular plates. J Neurocytol. 24:633–653. 10.1007/BF01179815. [DOI] [PubMed] [Google Scholar]
- Terre B, Lewis M, Gil-Gomez G, Han Z, Lu H, Aguilera M, Prats N, Roy S, Zhao H, and Stracker TH. 2019. Defects in efferent duct multiciliogenesis underlie male infertility in GEMC1-, MCIDAS- or CCNO-deficient mice. Development. 146. 10.1242/dev.162628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terre B, Piergiovanni G, Segura-Bayona S, Gil-Gomez G, Youssef SA, Attolini CS, Wilsch-Brauninger M, Jung C, Rojas AM, Marjanovic M, Knobel PA, Palenzuela L, Lopez-Rovira T, Forrow S, Huttner WB, Valverde MA, de Bruin A, Costanzo V, and Stracker TH. 2016. GEMC1 is a critical regulator of multiciliated cell differentiation. EMBO J. 35:942–960. 10.15252/embj.201592821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S, Boutaud L, Reilly ML, and Benmerah A. 2019. Cilia in hereditary cerebral anomalies. Biol Cell. 111:217–231. 10.1111/boc.201900012. [DOI] [PubMed] [Google Scholar]
- Vaisse C, Reiter JF, and Berbari NF. 2017. Cilia and Obesity. Cold Spring Harb Perspect Biol. 9. 10.1101/cshperspect.a028217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira OV, Gaus K, Verkade P, Fullekrug J, Vaz WL, and Simons K. 2006. FAPP2, cilium formation, and compartmentalization of the apical membrane in polarized Madin-Darby canine kidney (MDCK) cells. Proc Natl Acad Sci U S A. 103:18556–18561. 10.1073/pnas.0608291103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallmeier J, Dallmayer M, and Omran H. 2022. The role of cilia for hydrocephalus formation. Am J Med Genet C Semin Med Genet. 190:47–56. 10.1002/ajmg.c.31972. [DOI] [PubMed] [Google Scholar]
- Wang D, and Zhou J. 2021. The Kinocilia of Cochlear Hair Cells: Structures, Functions, and Diseases. Front Cell Dev Biol. 9:715037. 10.3389/fcell.2021.715037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Pan J, and Snell WJ. 2006. Intraflagellar transport particles participate directly in cilium-generated signaling in Chlamydomonas. Cell. 125:549–562. 10.1016/j.cell.2006.02.044. [DOI] [PubMed] [Google Scholar]
- Wang X, Sha YW, Wang WT, Cui YQ, Chen J, Yan W, Hou XT, Mei LB, Yu CC, and Wang J. 2019. Novel IFT140 variants cause spermatogenic dysfunction in humans. Mol Genet Genomic Med. 7:e920. 10.1002/mgg3.920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield M, Thomas L, Bequignon E, Schmitt A, Stouvenel L, Montantin G, Tissier S, Duquesnoy P, Copin B, Chantot S, Dastot F, Faucon C, Barbotin AL, Loyens A, Siffroi JP, Papon JF, Escudier E, Amselem S, Mitchell V, Toure A, and Legendre M. 2019. Mutations in DNAH17, Encoding a Sperm-Specific Axonemal Outer Dynein Arm Heavy Chain, Cause Isolated Male Infertility Due to Asthenozoospermia. American journal of human genetics. 105:198–212. 10.1016/j.ajhg.2019.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DS 2002. Transport to the photoreceptor outer segment by myosin VIIa and kinesin II. Vision Res. 42:455–462. 10.1016/s0042-6989(01)00228-0. [DOI] [PubMed] [Google Scholar]
- Witman GB, Carlson K, Berliner J, and Rosenbaum JL. 1972. Chlamydomonas flagella. I. Isolation and electrophoretic analysis of microtubules, matrix, membranes, and mastigonemes. J Cell Biol. 54:507–539. 10.1083/jcb.54.3.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Yang L, Wang F, Li H, Wang X, Wang W, Ge Z, Wang K, Zhao L, Li H, Li Y, Sui R, and Chen R. 2015. Mutations in human IFT140 cause non-syndromic retinal degeneration. Hum Genet. 134:1069–1078. 10.1007/s00439-015-1586-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S, Liu Y, Peng H, Tang C, Hennig GW, Wang Z, Wang L, Yu T, Klukovich R, Zhang Y, Zheng H, Xu C, Wu J, Hess RA, and Yan W. 2019. Motile cilia of the male reproductive system require miR-34/miR-449 for development and function to generate luminal turbulence. Proc Natl Acad Sci U S A. 116:3584–3593. 10.1073/pnas.1817018116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S, Wang Z, Peng H, Ward SM, Hennig GW, Zheng H, and Yan W. 2021. Oviductal motile cilia are essential for oocyte pickup but dispensable for sperm and embryo transport. Proc Natl Acad Sci U S A. 118. 10.1073/pnas.2102940118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Hou Y, McNeill NA, and Witman GB. 2020. The unity and diversity of the ciliary central apparatus. Philos Trans R Soc Lond B Biol Sci. 375:20190164. 10.1098/rstb.2019.0164. [DOI] [PMC free article] [PubMed] [Google Scholar]