

Summary
Population-based genomic screening may help diagnose individuals with disease-risk variants. Here, we perform a genome-first evaluation for nine disorders in 29,039 participants with linked exome sequences and electronic health records (EHRs). We identify 614 individuals with 303 pathogenic/likely pathogenic or predicted loss-of-function (P/LP/LoF) variants, yielding 644 observations; 487 observations (76%) lack a corresponding clinical diagnosis in the EHR. Upon further investigation, 75 clinically undiagnosed observations (15%) have evidence of symptomatic untreated disease, including familial hypercholesterolemia (3 of 6 [50%] undiagnosed observations with disease evidence) and breast cancer (23 of 106 [22%]). These genetic findings enable targeted phenotyping that reveals new diagnoses in previously undiagnosed individuals. Disease yield is greater with variants in penetrant genes for which disease is observed in carriers in an independent cohort. The prevalence of P/LP/LoF variants exceeds that of clinical diagnoses, and some clinically undiagnosed carriers are discovered to have disease. These results highlight the potential of population-based genomic screening.
Keywords: genome-first, underdiagnosis, biobank, cardiomyopathy, breast cancer, precision medicine, exome sequence, American College of Medical Genetics and Genomics, penetrance, electronic health record
Graphical abstract
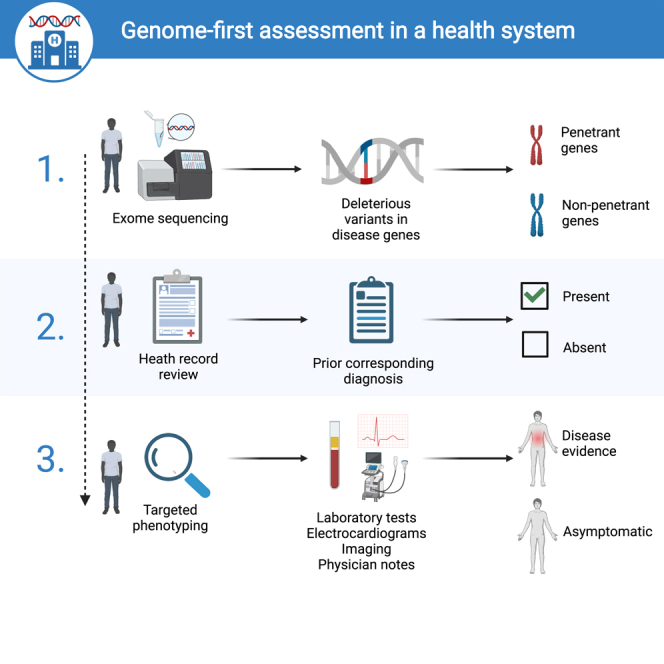
Highlights
-
•
Persons with disease-risk variants and exome and clinical phenotype data are evaluated
-
•
3 in 4 observations of variants lack a corresponding diagnosis, 15% of which show symptoms
-
•
Exome analysis enables targeted phenotyping and new diagnoses missing from clinical care
Forrest et al. pilot a genome-first strategy for assessing individuals genetically predisposed to disease who may be missing a diagnosis in their routine healthcare. Participants carrying disease-risk variants are identified and undergo targeted phenotyping with laboratory measurements, electrophysiology, imaging, and physician notes to uncover new diagnoses in previously undiagnosed individuals.
Introduction
A major endeavor of precision medicine is to leverage genetic data to improve the diagnosis and risk stratification of genetic disorders.1,2 Most genetic tests are applied to Mendelian disorders driven by a single gene mutation, such as familial breast cancer caused by a deleterious mutation in BRCA1 or BRCA2.3,4 The American College of Medical Genetics and Genomics has released guidelines for reporting secondary findings from clinical genomic sequencing.5 These vetted disease-predisposition genes are clinically actionable, but their use in population screening has not yet been determined.6 In addition, ClinVar7 and ClinGen8 advance the clinical interpretation of variants with annotations of pathogenicity, including pathogenic/likely pathogenic (P/LP), which is useful for genomic screening, since P/LP variants can be selected.
The increasing use of exome sequencing has permitted an assessment of the prevalence of P/LP variants in the population for numerous genetic diseases.9,10 Notably, the allelic prevalence of P/LP variants has been shown to exceed the estimated disease prevalence attributed to P/LP variants.11,12 This indicates at least three possibilities: (1) incompletely penetrant variants, in which the presence of a variant does not always result in disease;13 (2) underdiagnosed disease, where an individual with the variant expresses disease but is not clinically diagnosed; or (3) a combination of the two. In familial hypercholesterolemia, causative genetic variants may be highly penetrant, and the disorder is often underdiagnosed and undertreated.14 Monogenic cardiac diseases have also been examined recently for underdiagnosis; a study of Noonan syndrome found that 67% of individuals with P/LP variants in PTPN11 had a probable missed clinical diagnosis, while a study of hereditary transthyretin amyloidosis-induced heart failure showed that just 10 of 67 individuals with TTR V112I and disease had a clinical diagnosis.15,16
Population-based genomic screening can help fill the gaps in the clinical diagnosis of genetic disorders. The identification of individuals carrying clinical variants in known disease predisposition genes leads to targeted phenotyping and can improve diagnostic yield relative to a genetic-agnostic approach.17,18 This genome-first strategy, defined as identifying variants first and then evaluating relevant phenotypes,19 was adopted in recent studies to detect hereditary breast and ovarian cancer syndrome, Lynch syndrome, and familial hypercholesterolemia (tier 1 conditions designated by the Centers for Disease Control and Prevention [CDC]) in individuals with no prior diagnosis.20,21 However, the value of genomic screening and extent of underdiagnosis across most genetic disorders are uncertain. Moreover, prior studies have not addressed the selection of genes and age of individuals carrying variants. Variants conventionally classified as P/LP do not always cause disease (incomplete penetrance), and penetrance estimates depend on individuals’ age (i.e., age-dependent penetrance);13,22 obtaining variants that are observed to occur with disease in real-world populations is important for conducting genome-first assessments but has not been studied.
Here, we asked whether exome sequencing in a large healthcare system has diagnostic utility in individuals lacking clinical diagnoses from routine care. We used a genome-first approach to evaluate individuals with clinical variants on a systematic level for monogenic disorders in a cohort of 29,039 participants from an electronic health record (EHR)-linked clinical care biobank. First, individuals harboring P/LP or predicted loss-of-function (LoF) variants in known disease predisposition genes from a prior study13 were identified, including many who lacked a clinical diagnosis for the disease corresponding to their variant. Second, clinically undiagnosed individuals were evaluated for EHR evidence of disease symptoms and findings informed by their genotype. This permitted the discovery of individuals with P/LP/LoF variants who had EHR evidence of symptomatic disease but were not medicated or clinically diagnosed with the disease and motivated an investigation into the genetic and clinical factors driving this phenomenon.
Results
Summary of the study population and variants
The study design is shown in Figure 1. The study population included 29,039 individuals from the BioMe Biobank (BioMe) with exome sequence and EHR phenotype data who passed quality control (STAR Methods). The mean age was 59 years (standard deviation [SD], 16 years); 17,353 (60%) were female; 7,190 (25%), 1,349 (4.6%), 9,376 (32%), and 8,528 (29%) were of African, Asian, European, and Hispanic ethnicities, respectively; and 614 (2.1%) had at least one P/LP/LoF variant (Tables 1, S1, and S2). The prevalence of clinical diagnoses ranged from >20% for common conditions (e.g., 7,044 [24%] diagnosed with type 2 diabetes) to <1% for rare disorders (e.g., 15 [0.052%] diagnosed with retinitis pigmentosa).
Figure 1.

Schematic of genome-first assessment of pathogenic/likely pathogenic or predicted loss-of-function (P/LP/LoF) variants in the BioMe Biobank at Mount Sinai and UK Biobank
(A) Study design for genome-first evaluation in the BioMe Biobank at Mount Sinai using the plakophilin 2 (PKP2) W538Ter variant as an example. A total of P/LP/LoF variants in non-recessive monogenic genes for nine genetic disorders were curated: (1) variants reported as P/LP in the ClinVar repository with a minimum review status of two (multiple submitters) and previously unreported LoF variants identified by Variant Effect Predictor were obtained, and (2) non-recessive genes with a monogenic disease predisposition were identified from Online Mendelian Inheritance in Man and corroborated with literature review. A total of 29,039 participants with exome sequence and electronic health record (EHR) data were included in the study. This yielded 644 observations of 303 P/LP/LoF variants in 614 individuals. As an example, PKP2 W538Ter was identified in seven individuals, of whom two had a prior clinical diagnosis of cardiomyopathy in the EHR. The remaining five individuals lacked a clinical diagnosis, of whom two (40%) were discovered to have EHR evidence of cardiomyopathy. This procedure was repeated for all variants to produce a dataset of the percentage of clinically undiagnosed observations that had evidence of disease, which was used to assess factors associated with the presence of disease in clinically undiagnosed individuals. Factors comprised the gene containing the variant, age of individuals, disease, and symptoms.
(B) Evaluation of clinically undiagnosed but symptomatic individuals with P/LP/LoF variants in BioMe Biobank by gene penetrance observed in UK Biobank. A total of 34 target genes were identified in an independent cohort from UK Biobank, for which disease was either observed in individuals with P/LP/LoF variants in the gene (19 penetrant genes) or not observed (15 non-penetrant genes). The proportion of clinically undiagnosed observations with disease evidence in BioMe Biobank was then compared between the penetrant genes and non-penetrant genes.
Table 1.
Baseline demographic traits and clinical diagnoses in a clinical care cohort of 29,039 individuals
| Trait | All participants (n = 29,039) | Individuals with P/LP/LoF variants (n = 614) |
|---|---|---|
| Age, mean (SD) years | 59 (16) | 59 (16) |
| Female, n (%) | 17,353 (60) | 1,143 (62) |
| Ethnicity, n (%) | ||
| African | 7,190 (25) | 114 (19) |
| Asian | 1,349 (4.6) | 28 (4.6) |
| European | 9,376 (32) | 242 (39) |
| Hispanic | 8,528 (29) | 182 (30) |
| Other | 2,596 (8.9) | 48 (7.8) |
| Clinical diagnoses, n (%) | ||
| Amyotrophic lateral sclerosis | 7 (0.024) | 1/21 (4.8) |
| Breast cancer | 1,488 (5.2) | 62/168 (37) |
| Cardiomyopathy | 823 (2.8) | 78/213 (37) |
| Colorectal cancer | 218 (0.75) | 2/19 (11) |
| Endometrial cancer | 115 (0.40) | 1/13 (7.7) |
| Hypercholesterolemia | 4,460 (15) | 5/11 (45) |
| Prostate cancer | 474 (1.6) | 2/2 (100) |
| Retinitis pigmentosa | 15 (0.052) | 3/175 (1.7) |
| Type 2 diabetes | 7,044 (24) | 1/1 (100) |
Demographic traits and prevalence of clinical diagnoses are shown for all study participants and in a subset of individuals carrying variants reported as pathogenic/likely pathogenic in ClinVar with a minimum review status of two or variants of predicted LoF molecular consequence (P/LP/LoF variants). The denominator of the proportion of clinical diagnoses in the second column refers to the number of individuals carrying variants corresponding to the disease for each row (e.g., for cardiomyopathy, 213 individuals carry cardiomyopathy-predisposition variants, of whom 78 have a clinical diagnosis of cardiomyopathy). An overview of baseline traits among individuals clinically diagnosed with each disease in the electronic health record (EHR) is provided (Table S2). Clinical diagnoses were identified by International Classification of Diseases 10 (ICD-10) codes in the EHR. Ethnicity, self-reported ethnicity; other ethnicity, miscellaneous ethnicities other than those listed; n, number; SD, standard deviation.
P/LP/LoF variants from a previous study13 were curated for a set of nine genetic disorders: 303 P/LP/LoF variants in 54 genes corresponding to the nine diseases were identified in the exomes of 614 individuals, yielding 644 observations of variants in individuals (Tables S1 and S3). Among these individuals harboring P/LP/LoF variants, the mean number of unique P/LP/LoF variants per person was 1.0 (SD, 0.16; range, 1–2). The diseases with the largest number of observations of variants in individuals were cardiomyopathy (n = 234 observations [36%]), retinitis pigmentosa (n = 175 [27%]), and familial breast cancer (n = 168 [26%]) (Table 2). Demographic traits for individuals carrying P/LP/LoF variants are summarized in Table 1. The mean age of individuals with P/LP/LoF variants was 59 years (SD, 16 years); 371 (60%) were female; and 114 (19%), 28 (4.6%), 242 (39%), and 182 (30%) were of African, Asian, European, and Hispanic ethnicities, respectively.
Table 2.
Clinical diagnosis and phenotypes for 644 observations of pathogenic/likely pathogenic or loss-of-function (P/LP/LoF) variants in 614 individuals
| Disease | Genes | Variants | Observations, n | +Dx, n (% of observations) | −Dx -Sx, n (% of −Dx observations) | −Dx +Sx, n (% of −Dx observations) |
|---|---|---|---|---|---|---|
| Amyotrophic lateral sclerosis | 4 | 19 | 21 | 1 (4.8) | 15 (80) | 5 (20) |
| Cardiomyopathy | 21 | 151 | 234 | 80 (34) | 124 (81) | 30 (19) |
| Colorectal cancer | 7 | 16 | 19 | 2 (11) | 15 (88) | 2 (12) |
| Endometrial cancer | 1 | 1 | 13 | 1 (7.7) | 8 (67) | 4 (33) |
| Familial breast cancer | 4 | 64 | 168 | 62 (37) | 83 (78) | 23 (22) |
| Familial hypercholesterolemia | 1 | 7 | 11 | 5 (45) | 3 (50) | 3 (50) |
| Monogenic diabetes | 1 | 1 | 1 | 1 (100) | – | – |
| Prostate cancer | 1 | 2 | 2 | 2 (100) | – | – |
| Retinitis pigmentosa | 15 | 42 | 175 | 3 (1.7) | 164 (95) | 8 (4.7) |
A total of 644 observations of 303 pathogenic/likely pathogenic or predicted LoF (P/LP/LoF) variants carried by 614 individuals were identified for nine diseases. Observations were first categorized by whether the disease corresponding to the variant was clinically diagnosed (+Dx) or not clinically diagnosed (−Dx) in the individual with the variant. Electronic health records (EHRs) of clinically undiagnosed individuals were then evaluated and categorized by whether evidence of symptomatic disease was present (+Sx) or absent (−Sx). This resulted in three distinct groups: (1) clinically diagnosed (+Dx), (2) no clinical diagnosis and no EHR evidence of disease (−Dx −Sx), and (3) no clinical diagnosis but EHR evidence of disease (−Dx +Sx). n (% of observations), number and percentage of the observations of individuals with P/LP/LoF variants; n (% of −Dx observations), number and percentage of the observations of individuals with P/LP/LoF variants lacking a clinical diagnosis. Genes, number of disease predisposition genes containing the P/LP/LoF variants; Variants, number of P/LP/LoF variants; –, not applicable, as all observations for monogenic diabetes and prostate cancer were diagnosed with the corresponding disease.
Assessment of the clinical diagnosis and phenotype of individuals with P/LP/LoF variants
We systematically evaluated the clinical diagnosis and phenotype of individuals with P/LP/LoF variants by examining their EHR for diagnoses and traits of diseases expected with each of the variants they carried. These observations of variants in individuals were categorized into three distinct groups based on the presence or absence of a clinical diagnosis and symptoms of disease corresponding to the variant (STAR Methods; Tables S4, S5, and S6): (1) 157 of 644 observations (24%) had a clinical diagnosis of the disease corresponding to the variant recorded in the EHR. Of the 487 observations without a clinical diagnosis, (2) 412 (85%) did not have EHR evidence of disease, and (3) 75 (15%) had EHR evidence of disease (Figure 2). Of the 75 individuals in the last group, 40 (53%) had P variants, 3 (4%) had LP variants, and 32 (43%) had LoF variants. None of the individuals in the last group had received a medication specifically indicated for treatment of the disease (Table S5), and the findings were consistent across different ancestries (Table S7).
Figure 2.

Assessment of the clinical diagnosis and phenotypes of 644 observations of 303 pathogenic/likely pathogenic or predicted LoF (P/LP/LoF) variants in 614 individuals
Observations were categorized into three distinct groups: (1) clinically diagnosed (+Dx), where the individual with a P/LP/LoF variant had a prior clinical diagnosis for the genetic disorder corresponding to the variant; (2) no clinical diagnosis and no evidence of disease (−Dx −Sx), where the individual with a P/LP/LoF variant lacked a prior clinical diagnosis for the corresponding genetic disorder and had no electronic health record (EHR) evidence of disease symptoms or findings; and (3) no clinical diagnosis but evidence of disease (−Dx +Sx), where the individual with a P/LP/LoF variant lacked a clinical diagnosis for the corresponding genetic disorder but had EHR evidence of disease symptoms or findings. The latter group was the primary outcome of interest for downstream analyses, comprising 75 of 487 (15%) clinically undiagnosed observations that had EHR evidence of symptomatic untreated disease.
We focused on the third group—individuals with P/LP/LoF variants who had EHR evidence of symptomatic disease but were not clinically diagnosed or medicated—as the primary outcome of interest to assess underdiagnosis for most of the subsequent analyses. We investigated factors contributing to smaller or larger proportions of these underdiagnosed observations, including the gene containing the variant, age of individuals, and disease and symptoms corresponding to the variant.
Clinically undiagnosed individuals with evidence of disease stratified by genes
Population genomic screening would ideally use variants in penetrant genes with disease observed in real-world populations to increase clinical yield. We tested this hypothesis by comparing the proportion of clinically undiagnosed observations with disease evidence in BioMe for 19 penetrant genes and 15 non-penetrant genes observed in UK Biobank (STAR Methods). The median proportion of clinically undiagnosed observations that had disease evidence was 25% (interquartile range [IQR], 33) for penetrant genes compared to 0% (IQR, 29) for non-penetrant genes (p = 9.7 × 10−5). Using linear regression adjusted for age, we observed a 10-percentage point increase (SE = 4.3) in clinically undiagnosed observations that had disease evidence for penetrant genes compared to non-penetrant genes (p = 0.02). A larger proportion of penetrant genes had ≥20% observations with disease evidence compared to non-penetrant genes (odds ratio = 1.09 adjusted for age; 95% confidence interval [CI], 1.03–1.17; p = 0.004). These trends were consistent across three age groups of individuals who were at least 20, 40, and 60 years old (Figure 3).
Figure 3.

Rates of disease evidence found in clinically undiagnosed individuals with pathogenic/likely pathogenic or predicted LoF (P/LP/LoF) variants in BioMe Biobank (BioMe) for genes with versus without disease occurrence in UK Biobank (UKB)
A subset of 34 genes and six diseases that had clinically undiagnosed observations in BioMe were identified in UKB. Disease was observed in individuals from UKB with P/LP/LoF variants in 19 genes (penetrant genes) and not observed for 15 genes (non-penetrant genes) (STAR Methods). Yield of disease evidence in BioMe was then compared for P/LP/LoF variants found in penetrant genes versus non-penetrant genes. For each gene, clinically undiagnosed observations were evaluated for electronic health record evidence of symptomatic disease, and the proportion of observations with disease evidence was categorized as 0% (blue), between 0% and 20% (tan), or ≥20% (red). This analysis was completed three times using individuals who were ≥20, ≥40, and ≥60 years of age. For example, for individuals ≥40 years of age, 14 of 18 (78%) genes with disease observed in UKB had ≥20% observations with disease evidence compared to 4 of 11 (36%) genes with no disease observed in UKB.
Of medical importance, the American College of Medical Genetics and Genomics secondary findings (ACMG SF) v.3.1 genes are vetted disease predisposition genes with clinical actionability,5 but the clinical utility of population screening for individuals with variants in these genes has not been established.6 We therefore assessed the diagnoses and phenotypes of individuals with P/LP/LoF variants in ACMG SF v.3.1 genes comprising 22 of 54 genes (41%) (Figure S1). A greater proportion of observations of ACMG SF v.3.1 gene variants in individuals had a corresponding clinical diagnosis for the expected disease than non-ACMG SF v.3.1 gene variants (134 of 396 [34%] versus 23 of 248 [9.3%], p = 1.6 × 10−13). There was also a greater proportion of clinically undiagnosed observations with evidence of disease for ACMG SF v.3.1 gene variants (57 of 262 [22%]) compared to variants in non-ACMG SF v.3.1 genes (18 of 225 [8.0%]) (p = 2.6 × 10−5).
Underdiagnosis stratified by disease and symptoms
Next, we evaluated the phenotype of individuals without a clinical diagnosis carrying P/LP/LoF variants for each of the nine genetic disorders (Table 2; Figure 4). Genetic disorders are diverse, with different affected systems and clinical manifestations, which may be differentially detected and reported in a healthcare setting. The proportion of clinically undiagnosed observations with evidence of disease varied by genetic disorder, with a median of 22% (IQR, 14) across all nine disorders. The greatest proportion of clinically undiagnosed observations with evidence of disease was noted for familial hypercholesterolemia (3 of 6 [50%]), endometrial cancer (4 of 12 [33%]), amyotrophic lateral sclerosis (5 of 20 [25%]), breast cancer (23 of 106 [22%]), and cardiomyopathy (30 of 154 [19%]). All observations of monogenic diabetes and prostate cancer were diagnosed. In terms of absolute numbers, cardiomyopathy had the most clinically undiagnosed observations, with evidence of disease with 30 such observations.
Figure 4.

Evidence of symptomatic disease in clinically undiagnosed observations of variants in individuals from BioMe
The proportion of clinically undiagnosed observations that had electronic health record evidence of disease is depicted along with the 95% CIs (error bars) for three age groups and each of the seven disorders: amyotrophic lateral sclerosis (ALS), cardiomyopathy (CM), colorectal cancer (CRC), endometrial cancer (EC), familial breast cancer (FBC), familial hypercholesterolemia (FH), and retinitis pigmentosa (RP). The mean age of diagnosis is listed below each disease on the x axis. Monogenic diabetes (MD) and prostate cancer (PC) are not shown, as they both had zero observations lacking a clinical diagnosis (i.e., all individuals with MD or PC variants were diagnosed with the disease).
We reasoned that the symptoms associated with greater risk of hospitalization may be better detected and recorded in a healthcare setting and, therefore, more prevalent in our analysis of disease evidence. Hence, we investigated the association of symptom hospitalization risk and prevalence of symptoms detected as evidence of disease in clinically undiagnosed observations of variants in individuals, using a score23 for adverse phenotypes ranging from 0 (lowest risk of hospitalization and death) to 1 (highest risk of hospitalization and death) (Table S8). There was a positive association between symptom score and prevalence of symptoms detected in observations lacking a clinical diagnosis with a 4.6-percentage point increase (95% CI, 2.6–6.6) in the prevalence of symptoms per 0.1 increase in symptom score (p = 6.7 × 10−5) (Figure S2). The symptom with the lowest score (breast enlargement) was found in just 1.9% of observations without a clinical diagnosis for breast cancer, while the symptom with the highest score (acute cardiac failure) was found in 58% of observations without a clinical diagnosis for cardiomyopathy.
Genome-first identification of new diagnoses in clinically undiagnosed individuals
We undertook a detailed case series analysis of clinically undiagnosed individuals carrying P/LP/LoF variants in plakophilin 2 (PKP2) and low-density lipoprotein receptor (LDLR) genes who had EHR evidence of symptomatic disease as examples for demonstrating how a genome-first evaluation can guide a targeted phenotypic evaluation and reveal new diagnoses. Seven individuals harbored the P/LoF PKP2 W538Ter variant (NM_004572.3:c.1613G>A), of whom five (71%) lacked a clinical diagnosis of cardiomyopathy (Figure 1). Of these five individuals, two (40%) were found to have EHR evidence of cardiomyopathy. Physician notes, including for electrocardiograms and echocardiography, for these two individuals were reviewed and summarized. Both individuals presented with multiple syncopal episodes, palpitations, and chest pain along with marked tachycardia and prolonged QRS duration on the electrocardiogram. These findings in two individuals with PKP2 W538Ter suggested a diagnosis of arrhythmogenic right ventricular cardiomyopathy according to the 2010 Task Force Criteria.24
Of 11 individuals with P/LP/LoF variants in LDLR, six (55%) were missing a corresponding clinical diagnosis, of whom three (50%) had EHR evidence of familial hypercholesterolemia and carried three different P/LP/LoF variants (NM_000527.4:c.772G>T, p.E258Ter; NM_000527.4:c.1860G>T, p.W620C; and NM_000527.4:c.590G>A, p.C197Y). Physician notes and lipid panel results for the three individuals were obtained and reviewed. All individuals presented on at least three occasions with chest pain, at least one occasion with claudication, and in one individual, one occasion with ischemic stroke due to an occluded carotid artery. Elevated total cholesterol and low-density lipoprotein cholesterol levels were noted in all individuals on multiple occasions, and one individual had a family history of myocardial infarction and coronary artery bypass grafting. These findings in three individuals with P/LP/LoF variants in LDLR indicated a diagnosis of familial hypercholesterolemia based on the Simon-Broome criteria.25
Discussion
Here, we performed a population-based genome-first evaluation of individuals carrying P/LP/LoF variants for nine genetic disorders in a large clinical care cohort, using a rich set of phenotype data to characterize individuals lacking a clinical diagnosis. The diagnostic utility of exome sequencing was assessed comprehensively for a wide array of diseases and revealed a preponderance of individuals with P/LP/LoF variants missing a relevant diagnosis in the EHR: 487 of 644 observations (76%) of variants in individuals lacked a corresponding clinical diagnosis. Individuals with evidence of symptomatic but clinically undiagnosed disease were identified by targeted phenotype assessment tailored to each person’s genetic risk profile, such as a cardiovascular-specific evaluation for those with variants in cardiomyopathy-predisposition genes, demonstrating how precision medicine can be achieved with the integration of genomic and clinical data. We also interrogated key factors driving the detection of underdiagnosed disease. For example, not all clinically undiagnosed individuals had manifestations of disease, attributed in part to some P variants not associated with disease;13 we found a greater prevalence of disease evidence in individuals with variants in genes observed to be penetrant in an independent cohort, emphasizing the importance of prioritizing genetic variation with demonstrable disease occurrence (e.g., penetrance) in the population rather than presumed pathogenicity.
Using a combination of phenotypes captured in the EHR, we observed that most individuals carrying P/LP/LoF variants lacked a relevant clinical diagnosis and, notably, some also had evidence of untreated symptomatic disease. This suggests a need to improve diagnostic approaches for individuals carrying P/LP/LoF variants. One possibility explored in the current study is to use a genome-first approach, screening for deleterious variants in well-known and empirically supported disease predisposition genes, the presence of which triggers a targeted disease evaluation in individuals with the variant. Such information gained from exome sequencing facilitated new genome-informed diagnoses for several individuals who were missing a clinical diagnosis from routine care. For example, the finding of a P/LP/LoF PKP2 variant in two undiagnosed individuals informed a targeted evaluation of cardiac phenotypes in the EHR that supported a diagnosis of cardiomyopathy, while the identification of P/LP/LoF LDLR variants in three undiagnosed individuals guided an assessment of their cardiovascular and lipidic EHR profile that indicated a diagnosis of familial hypercholesterolemia. Guidelines for genetic testing of cardiomyopathy, familial hypercholesterolemia, and other inherited cardiovascular diseases hinge on a clinical diagnosis of the condition in the individual and/or the individual’s family to trigger genetic testing10; similarly, genetic testing for hereditary cancers is recommended for individuals with a personal and/or family history of clinically diagnosed disease.26,27,28 Instead, we implemented a genome-first testing29 strategy to diagnose genetic disorders in the population that may otherwise have been missed with the standard of care.
Calls have been growing to use genomic sequencing to obtain a genetic diagnosis,9,10,11 and nascent efforts have increased the diagnostic yield of several genetic disorders.14,15,30 These have predominantly targeted three conditions designated tier 1 by the CDC.31 The present study adds to this literature a systematic evaluation of nine disorders beyond solely tier 1 conditions. Whereas previous studies have focused on small clinical cohorts of individuals with a family or personal history of disease, we used a large sample of unrelated individuals from a non-disease-ascertained cohort. Furthermore, we examined the clinical yield of variants in many ACMG SF v.3.1 genes; the ACMG has called for population screening studies of these clinically actionable genes,5,6 and we found that 1 in 4 clinically undiagnosed observations of variants in ACMG SF v.3.1 genes had disease evidence in the population. We also accounted for variable disease risk of P/LP/LoF variants and the age of individuals.13 It was previously unclear whether the lack of clinical diagnosis in genetically predisposed individuals is due to absence of disease, as in the case of incomplete penetrance and/or younger age, or presence of disease that is underdiagnosed. We assessed both of these factors; we evaluated the prevalence of disease evidence in clinically undiagnosed individuals who had variants in penetrant genes in an independent cohort from UK Biobank and in three age groups of individuals. Individuals with variants in penetrant genes had a greater prevalence of disease evidence than those with variants in non-penetrant genes, and age-stratified analyses showed consistent trends across the three age groups. Together, these results indicate that clinically undiagnosed individuals who have positive disease expression in the EHR are underdiagnosed, rather than simply lacking disease as a product of incompletely penetrant variants or younger age.
Clinical actionability of genetic findings is crucial to consider. Many genes in the study are in the CDC tier 1 genomic applications and the ACMG SF v.3.1, both of which comprise conditions for which early detection and intervention can reduce morbidity and mortality. Among the 75 individuals with clinical manifestations but no diagnosis, 57 (76%) had variants in ACMG SF v.3.1 genes, and 25 (33%) had variants in CDC tier 1 genes. This includes 3 individuals with LDLR variants who would be candidates for targeted lipid-lowering therapy and 22 individuals with BRCA1/BRCA2 variants who would be candidates for earlier mammography and breast cancer screening. Furthermore, a higher prevalence of clinical manifestations was observed among individuals carrying variants in penetrant genes; this suggests that penetrant genetic variation resulting in clinical manifestations that can be targeted by clinical interventions and treatment should be prioritized for genomic screening. Notably, most individuals carrying P/LP/LoF variants were of European ancestry, owing in part to the predominance of clinically interpreted variants identified in Europeans and underrepresentation of non-European ancestries in genetic databases, populations, and biobanks.32,33 Further inclusion of diverse ancestries in clinical and population genetic studies will help identify and characterize more clinically relevant variants in non-European ancestries.
In conclusion, the findings of this population-based genome-first study demonstrate the untapped potential of using exome sequencing in healthcare systems to assess individuals carrying P/LP/LoF variants. We used exome data not yet part of routine clinical care in an EHR setting to inform a targeted strategy of phenotyping individuals at genetic risk for nine diseases. These data motivate the development and implementation of population-based genomic screening programs, which should be tested prospectively to diagnose individuals in the population who carry demonstrably deleterious genetic variation in well-known disease predisposition genes.
Limitations of the study
There were several study limitations. First, disease symptoms were identified in part by International Classification of Diseases 10 (ICD-10) diagnosis codes. ICD-10 codes are commonly used to ascertain phenotypes in biobank studies;34,35 however, we cannot exclude the possibility of some misclassification. Importantly, we supplemented ICD-10-based phenotypes with a thorough review of physician notes in the EHR (Table S6). This provided a check for accuracy of ICD-10 diagnosis codes and captured additional symptoms that may have otherwise been missed with an exclusively ICD-10-based phenotyping strategy. Nonetheless, not all phenotypes will be captured in the EHR, as individuals may have received care elsewhere, provider ICD-10 coding may not be standardized, and known biases exist when using ICD-10 diagnosis codes. Second, subgroups stratified by age had smaller sample sizes than in the primary analysis, particularly for the older age groups, which may account for more variability in the proportions of disease evidence in these subgroups. Furthermore, variants have heterogeneous effects on disease onset, including some instances where large-effect size variants are associated with earlier onset of disease; hence, age should be considered as a risk factor for disease. Third, a few of the disorders were associated with little to no detection of symptoms in the EHR, such as retinitis pigmentosa. It is possible that these individuals were seen by specialists in ophthalmology. Specialized tests and evaluations documented outside the standard physician note and diagnosis code system in the EHR would not have been detected in our analysis, possibly decreasing the number of clinically undiagnosed individuals with disease evidence. Another possibility is that some diseases and their manifestations are more evident to patients; symptoms associated with greater risk of hospitalization were more prevalent among observations of disease evidence. Fourth, while this study illuminated several important factors contributing to missing clinical diagnoses for genetic disorders, other reasons remain unknown. These include socioeconomic determinants, insurance status, access to healthcare systems, health literacy, environmental and lifestyle factors, demographics, and biological factors. Future studies are needed to further scrutinize the differences between individuals with genetic predisposition for disease who have and have not received a clinical diagnosis.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| Summary table of clinical variants analyzed in BioMe Biobank | Mendeley | Mendeley: https://doi.org/10.17632/c2g66gycvx.1 |
| Penetrance of clinical variants in UK Biobank | Publication and Mendeley |
https://doi.org/10.1001/jama.2021.23686 https://data.mendeley.com/datasets/v37dmjkbfj/draft?a=a2f58e92-da19-461d-991a46dc70128860 |
| ClinVar database | NCBI | https://www.ncbi.nlm.nih.gov/clinvar/ |
| Institute for Personalized Medicine summary and access to BioMe Biobank | NCBI | https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000925.v1.p1 |
| Human reference genome NCBI build 38, GRCh38 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| Software and algorithms | ||
| R 3.5.3 | R | https://cran.r-project.org/ |
| PLINK 2.0 | PLINK | https://www.cog-genomics.org/plink2/ |
| Variant Effect Predictor | Github | https://github.com/Ensembl/ensembl-vep |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Ron Do (ron.do@mssm.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
All data associated with this study are present in the paper or Supplementary Material and use existing data from a previous study.13 A tabulated summary of all variants analyzed in the study is found in Table S3. This comprehensive list is annotated with information regarding genomic location, gene, associated disease, variant effect, amino acid change, ClinVar clinical significance and review status, minor allele frequencies in gnomAD, and minor allele frequencies in BioMe. In addition, this complete list has been deposited in Mendeley and its DOI is listed in the key resources table. Individual-level data, including sequencing, EHR phenotypes, and physician notes analyzed in this study are not publicly available due to Institutional Review Board (IRB) restrictions and research participant privacy concerns; however, requests from accredited researchers for access to data relevant to this manuscript can be made by contacting the lead contact.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
-
•
No custom code was generated in this study.
Experimental model and study participant details
Study design
A genome-first approach was applied to a population-based cohort whereby individuals carrying P/LP/LoF variants were identified and then phenotyped for the relevant genetic disorder. A schematic of the study’s assessment of clinically undiagnosed individuals with P/LP/LoF variants is shown (Figure 1A). A diverse set of nine genetic disorders was analyzed: amyotrophic lateral sclerosis, cardiomyopathy, colorectal cancer, endometrial cancer, familial breast cancer, familial hypercholesterolemia, monogenic diabetes, prostate cancer, and retinitis pigmentosa. These were selected from a previous study13 to ensure representation of a range of different systems (cardiac, neoplastic, metabolic, ocular, etc.) and prevalence (rare and common), and diseases that are analyzable in the EHR. Clinical diagnosis of disease was defined by the presence of a corresponding ICD-10 diagnosis code used in a previous study13 (Table S4).
Individuals with P/LP/LoF variants were categorized phenotypically on the basis of their clinical diagnosis and disease symptoms extracted from the EHR. Evidence of symptomatic disease comprised ICD-10 codes and physician notes for symptoms captured by a set of curated clinical criteria (Table S6). This genome-first approach of first identifying individuals at high genetic risk for disease and then examining their ICD-10 codes, physician notes, and medications has been used previously.15,16,36 This produced three distinct groups of individuals: clinically diagnosed (with a clinical diagnosis of the relevant disease), no clinical diagnosis and no evidence of disease (no clinical diagnosis and no EHR evidence of symptomatic disease), or no clinical diagnosis but has evidence of disease (EHR evidence of symptomatic disease but no clinical diagnosis). This latter group was the outcome of interest for most analyses unless otherwise stated. We reviewed the EHR pharmacy record and confirmed that all clinically undiagnosed individuals with evidence of disease were not treated with a disease-specific medication, defined as a medication with an indication specific to the disease of interest (Table S5). Medication indications were obtained from Drugbank.37
The study protocols were approved by the IRB at Mount Sinai (GCO#07–0529; STUDY-11-01139) and written informed consent was obtained for all participants. Use of data from the UK Biobank was approved under application number 16218 in the UK Biobank Resource. The study used de-identified genetic and EHR data for research purposes only.
Study participants
A cohort of participants from an EHR-linked population-based biobank (BioMe) was included in the study. BioMe consists of over 60,000 individuals of African, Hispanic, European, Asian, and other self-reported ethnicities who were recruited from outpatient centers in the Mount Sinai Health System from 2007 onwards. All individuals in BioMe consented to providing biological and DNA samples linked to de-identified EHRs. Exome sequencing and quality control were performed for the first 31,250 participants. Samples with discordance between genetic sex and recorded sex, low coverage, contamination, low call rate, or duplications were excluded, leaving 30,813 samples. Those without complete demographic data (n = 345), younger than 20 years of age (n = 610), or without ICD-10 diagnosis data (n = 819) were excluded, leaving a set of 29,039 samples for analysis.
Method details
Variant sequencing and selection
Exome sequence data and variant call files (VCFs) were generated by the Regeneron Genetics Center (preparation and quality control described extensively elsewhere13). The average 20X coverage was 95% and greater than 99% of the samples had more than 85% of the targeted bases covered at 20X or more. Briefly, 9,202,884 variants were called in the samples and the Goldilocks Filter (GF) was applied to the VCFs.38 For single nucleotide polymorphisms (SNPs), cells with depth-normalized quality scores <3 or depth of coverage <7 were set to missing. For insertions and deletions (indels), cells with depth-normalized quality scores <5 or depth of coverage <10 were set to missing. Variant sites were then filtered, whereby sites of heterozygous variation that failed the Allele Balance (AB) cutoff were removed. SNP sites required ≥1 sample to carry an alternate AB ≥15% and indel sites required ≥1 sample to carry an alternate AB ≥20%. These site filters left 8,761,478 variants after GF. Next, sites with missing genotypes for >2% of individuals in the dataset (267,955 sites) were removed. AB was calculated for biallelic SNPs and 320,877 sites with AB <0.3 or >0.8 were removed, leaving 8,172,646 sites. Lastly, the dataset was filtered to regions within the target regions of the exome capture platform (IDT xGen capture platform; 4,256,827 sites) and separated into two file sets for biallelic and multiallelic sites (3,948,623 and 308,204, respectively). All variants were ascertained from VCFs using PLINK (version 2.0).39
Variants in the analysis were derived from a set of P/LP/LoF exonic variants (LoF variants in genes mediating disease via LoF mechanism) without recessive inheritance (excluded due to insufficient sample size) that were previously characterized in BioMe.13 The previous study assessed population-based penetrance of a wide array of variants, while the present study investigated clinical underdiagnosis of individuals carrying rare P/LP/LoF variants, factors associated with underdiagnosis, and optimization of genome-first approaches for clinical utility. P/LP variants reported in ClinVar and previously unreported variants with a LoF molecular consequence (splice acceptor/donor, stop gained/lost, frameshift, or start lost) annotated by Variant Effect Predictor40 were identified. Variants in the last exon or last 50 base pairs of the penultimate exon were considered not LoF due to a predicted lack of efficient nonsense-mediated decay, with exception for variants predicted to delete over 20% of the gene. Only LoF variants present in MANE Select or MANE Plus Clinical transcripts were retained.41 LoF variants in a gene were then mapped to disease based on prior P/LP variant submissions in ClinVar linking genes to diseases (e.g., BRCA1 LoF variants mapped to breast cancer). Variants with benign, uncertain, or conflicting clinical significance in ClinVar and predicted synonymous consequence were removed, as were variants in genes with recessive inheritance reported in Online Mendelian Inheritance in Man.42 ClinVar variants were further restricted to those reported by multiple clinical testing labs or reviewed by an expert panel such as ClinGen.8 Rare LoF variants with ancestry-specific allele frequency <0.001 in BioMe and ancestry-specific allele frequency <0.001 in or absent from gnomAD v3.1.243 were included. This yielded 303 P/LP/LoF variants in 54 disease-predisposition genes corresponding to nine genetic disorders (Table S1).
Analysis of genes, age, diseases, and symptoms in clinically undiagnosed individuals
Importantly, the evaluation of disease in clinically undiagnosed individuals with P/LP/LoF variants accounted for different genes, ages, diseases, and symptoms. First, we hypothesized that a genome-first approach using variants in genes for which disease is observed in real-world non-disease ascertained populations would result in higher diagnostic yield. Although all variants in the target genes had evidence of pathogenicity, expected pathogenicity does not always translate to occurrence of disease in the population.13 Population genomic screening would ideally use variants in genes with disease observed in real-world populations to increase clinical yield. We tested this hypothesis by first identifying 19 target genes for which disease was observed in individuals with any P/LP/LoF variants in the gene in an independent cohort from UK Biobank13 (penetrant) and 15 target genes for which disease was not observed (non-penetrant) in UK Biobank, and then comparing the proportion of clinically undiagnosed observations with disease evidence in BioMe for penetrant genes versus non-penetrant genes (Figure 1B). The exome sequence and EHR data used to characterize disease-associated genes from the UK Biobank have been previously described.13 The proportion of observations of individuals with variants who had symptomatic evidence of disease in BioMe was then compared for variants in the penetrant genes versus non-penetrant genes.
Second, the presence or absence of disease evidence may be explained by age; for example, younger individuals may not have manifested the disease yet. To address this, we separately evaluated the proportion of clinically undiagnosed observations that had evidence of disease in three age groups of individuals ≥20 years, ≥40 years, and ≥60 years.
Third, genetic disorders are heterogeneous in terms of affected systems and clinical presentation, such that certain diseases and symptoms may be better detected in the healthcare setting. We therefore examined the proportion of clinically undiagnosed observations of variants in individuals with evidence of disease for each genetic disorder and the prevalence of each symptom detected. We tested the hypothesis that symptoms associated with hospitalization would be better detected and have a higher prevalence in our analysis using a previously published score23 on an ordinal scale ranging from 0 (smallest hospitalization and mortality risk) to 1 (greatest hospitalization and mortality risk) assigned to each of the 39 disease symptoms (Table S8).
Quantification and statistical analysis
Statistical analysis
Differences in categorical and continuous variables were assessed with a two-sided unpaired Fisher’s exact test and t-test, respectively. Individuals with at least one allele for a P/LP/LoF variant were identified and the proportions of observations of variants in individuals with 1) a clinical diagnosis; 2) no clinical diagnosis or evidence of disease; and 3) no clinical diagnosis but have evidence of disease were determined. Analyses of disease in clinically undiagnosed observations were stratified by gene, age groups (≥20 years, ≥40 years, ≥60 years), and disease. Multivariable linear regression adjusted for age was used to model the proportion of clinically undiagnosed observations with evidence of disease as a function of gene category (penetrant gene versus gene without penetrant in UK Biobank) and to model the prevalence of symptoms in clinically undiagnosed observations as a function of symptom score. All statistical tests and plots were generated with R (version 3.5.3).
Acknowledgments
Bruce D. Gelb, MD; Sander Houten, PhD; Paz Polak, PhD; Stuart Scott, PhD; and Ethylin Jabs, MD, all of whom were on the thesis advisory committee of ISF, provided critical feedback and expertise. I.S.F. is supported by the National Institute of General Medical Sciences of the National Institutes of Health (NIH) (T32-GM007280). R.D. is supported by the National Institute of General Medical Sciences of the NIH (R35-GM124836) and the National Heart, Lung, and Blood Institute of the NIH (R01-HL139865 and R01-HL155915). L.R.P. is supported by the National Eye Institute of the NIH (R01EY015473 and R01EY032559) and an unrestricted grant from Research to Prevent Blindness, NYC. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author contributions
I.S.F., A.D., and R.D. conceived and designed the study. All authors aided in the acquisition, analysis, and interpretation of data. I.S.F. and R.D. drafted the manuscript, and all authors provided critical revisions of the manuscript. I.S.F., A.D., J.K.P., and H.M.T.V. performed statistical analyses. G.N.N., J.H.C., and R.D. provided administrative, technical, and material support. R.D. supervised the study. I.S.F. and R.D. had access to and verified all of the data in the study.
Declaration of interests
R.D. reported receiving grants from AstraZeneca and grants and nonfinancial support from Goldfinch Bio and being a scientific co-founder, consultant, and equity holder for Pensieve Health (pending) and a consultant for Variant Bio, all not related to this work. G.N.N. reported being a scientific co-founder, consultant, advisory board member, and equity owner of Renalytix AI; a scientific co-founder and equity holder for Pensieve Health (pending); and a consultant for Variant Bio and receiving grants from Goldfinch Bio and personal fees from Renalytix AI, BioVie, Reata, AstraZeneca, and GLG Consulting. L.R.P. is a consultant for Eyenovia, Twenty Twenty, and Skye Bioscience.
Published: May 21, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101518.
Supplemental information
References
- 1.Verdonschot J.A.J., Hazebroek M.R., Krapels I.P.C., Henkens M.T.H.M., Raafs A., Wang P., Merken J.J., Claes G.R.F., Vanhoutte E.K., van den Wijngaard A., et al. Implications of genetic testing in dilated cardiomyopathy. Circ. Genomic Precis. Med. 2020;13:476–487. doi: 10.1161/CIRCGEN.120.003031. [DOI] [PubMed] [Google Scholar]
- 2.Pal T., Agnese D., Daly M., La Spada A., Litton J., Wick M., Klugman S., Esplin E.D., Jarvik G.P., Professional Practice and Guidelines Committee Points to consider: is there evidence to support BRCA1/2 and other inherited breast cancer genetic testing for all breast cancer patients? A statement of the American College of Medical Genetics and Genomics (ACMG) Genet. Med. 2020;22:681–685. doi: 10.1038/s41436-019-0712-x. [DOI] [PubMed] [Google Scholar]
- 3.US Preventive Services Task Force. Owens D.K., Davidson K.W., Krist A.H., Barry M.J., Cabana M., Caughey A.B., Doubeni C.A., Epling J.W., Jr., Kubik M., et al. Risk Assessment, Genetic Counseling, and Genetic Testing for BRCA -Related Cancer: US Preventive Services Task Force Recommendation Statement. JAMA, J. Am. Med. Assoc. 2019;322:652–665. doi: 10.1001/jama.2019.10987. [DOI] [PubMed] [Google Scholar]
- 4.Abul-Husn N.S., Soper E.R., Odgis J.A., Cullina S., Bobo D., Moscati A., Rodriguez J.E., CBIPM Genomics Team. Regeneron Genetics Center. Loos R.J.F., et al. Exome sequencing reveals a high prevalence of BRCA1 and BRCA2 founder variants in a diverse population-based biobank. Genome Med. 2019;12:2. doi: 10.1186/s13073-019-0691-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McGurk K.A., Zheng S.L., Henry A., Josephs K., Edwards M., de Marvao A., Whiffin N., Roberts A., Lumbers T.R., O’Regan D.P., Ware J.S. ACMG SF v3.1 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG) Genet. Med. 2022;24:744–746. doi: 10.1016/j.gim.2021.10.020. [DOI] [PubMed] [Google Scholar]
- 6.ACMG Board of Directors The use of ACMG secondary findings recommendations for general population screening: a policy statement of the American College of Medical Genetics and Genomics (ACMG) Genet. Med. 2019;21:1467–1468. doi: 10.1038/s41436-019-0502-5. [DOI] [PubMed] [Google Scholar]
- 7.Landrum M.J., Lee J.M., Riley G.R., Jang W., Rubinstein W.S., Church D.M., Maglott D.R. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42:D980–D985. doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rehm H.L., Berg J.S., Brooks L.D., Bustamante C.D., Evans J.P., Landrum M.J., Ledbetter D.H., Maglott D.R., Martin C.L., Nussbaum R.L., et al. ClinGen — The Clinical Genome Resource. N. Engl. J. Med. 2015;372:2235–2242. doi: 10.1056/nejmsr1406261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O’Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B., et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Musunuru K., Hershberger R.E., Day S.M., Klinedinst N.J., Landstrom A.P., Parikh V.N., Prakash S., Semsarian C., Sturm A.C. Genetic testing for inherited cardiovascular diseases: A scientific statement from the american heart association. Circ. Genomic Precis. Med. 2020;13:373–385. doi: 10.1161/HCG.0000000000000067. [DOI] [PubMed] [Google Scholar]
- 11.Bick A.G., Flannick J., Ito K., Cheng S., Vasan R.S., Parfenov M.G., Herman D.S., Depalma S.R., Gupta N., Gabriel S.B., et al. Burden of rare sarcomere gene variants in the framingham and jackson heart study cohorts. Am. J. Hum. Genet. 2012;91:513–519. doi: 10.1016/j.ajhg.2012.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Semsarian C., Ingles J., Maron M.S., Maron B.J. New perspectives on the prevalence of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2015;65:1249–1254. doi: 10.1016/j.jacc.2015.01.019. [DOI] [PubMed] [Google Scholar]
- 13.Forrest I.S., Chaudhary K., Vy H.M.T., Petrazzini B.O., Bafna S., Jordan D.M., Rocheleau G., Loos R.J.F., Nadkarni G.N., Cho J.H., Do R. Population-Based Penetrance of Deleterious Clinical Variants. JAMA. 2022;327:350–359. doi: 10.1001/JAMA.2021.23686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nordestgaard B.G., Chapman M.J., Humphries S.E., Ginsberg H.N., Masana L., Descamps O.S., Wiklund O., Hegele R.A., Raal F.J., Defesche J.C., et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: Guidance for clinicians to prevent coronary heart disease. Eur. Heart J. 2013;34:3478–3490. doi: 10.1093/eurheartj/eht273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abdulrahim J.W., Kwee L.C., Alenezi F., Sun A.Y., Baras A., Ajayi T.A., Henao R., Holley C.L., McGarrah R.W., Daubert J.P., et al. Identification of Undetected Monogenic Cardiovascular Disorders. J. Am. Coll. Cardiol. 2020;76:797–808. doi: 10.1016/j.jacc.2020.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Damrauer S.M., Chaudhary K., Cho J.H., Liang L.W., Argulian E., Chan L., Dobbyn A., Guerraty M.A., Judy R., Kay J., et al. Association of the V122I Hereditary Transthyretin Amyloidosis Genetic Variant with Heart Failure among Individuals of African or Hispanic/Latino Ancestry. JAMA. 2019;322:2191–2202. doi: 10.1001/jama.2019.17935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adams D.R., Eng C.M. Next-Generation Sequencing to Diagnose Suspected Genetic Disorders. N. Engl. J. Med. 2018;379:1353–1362. doi: 10.1056/NEJMra1711801. [DOI] [PubMed] [Google Scholar]
- 18.Turro E., Astle W.J., Megy K., Gräf S., Greene D., Shamardina O., Allen H.L., Sanchis-Juan A., Frontini M., Thys C., et al. Whole-genome sequencing of patients with rare diseases in a national health system. Nature. 2020;583:96–102. doi: 10.1038/s41586-020-2434-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park J., Levin M.G., Haggerty C.M., Hartzel D.N., Judy R., Kember R.L., Reza N., Regeneron Genetics Center. Ritchie M.D., Owens A.T., et al. A genome-first approach to aggregating rare genetic variants in LMNA for association with electronic health record phenotypes. Genet. Med. 2020;22:102–111. doi: 10.1038/s41436-019-0625-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buchanan A.H., Lester Kirchner H., Schwartz M.L.B., Kelly M.A., Schmidlen T., Jones L.K., Hallquist M.L.G., Rocha H., Betts M., Schwiter R., et al. Clinical outcomes of a genomic screening program for actionable genetic conditions. Genet. Med. 2020;22:1874–1882. doi: 10.1038/s41436-020-0876-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grzymski J.J., Elhanan G., Morales Rosado J.A., Smith E., Schlauch K.A., Read R., Rowan C., Slotnick N., Dabe S., Metcalf W.J., et al. Population genetic screening efficiently identifies carriers of autosomal dominant diseases. Nat. Med. 2020;26:1235–1239. doi: 10.1038/s41591-020-0982-5. [DOI] [PubMed] [Google Scholar]
- 22.Hu C., Hart S.N., Gnanaolivu R., Huang H., Lee K.Y., Na J., Gao C., Lilyquist J., Yadav S., Boddicker N.J., et al. A Population-Based Study of Genes Previously Implicated in Breast Cancer. N. Engl. J. Med. 2021;384:440–451. doi: 10.1056/nejmoa2005936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gottlieb A., Hoehndorf R., Dumontier M., Altman R.B. Ranking adverse drug reactions with crowdsourcing. J. Med. Internet Res. 2015;17 doi: 10.2196/jmir.3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marcus F.I., McKenna W.J., Sherrill D., Basso C., Bauce B., Bluemke D.A., Calkins H., Corrado D., Cox M.G.P.J., Daubert J.P., et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Eur. Heart J. 2010;31:806–814. doi: 10.1093/eurheartj/ehq025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neil H.A.W., Betteridge D.J., Broome K., Durrington P.N., Hawkins M.M., Humphries S.E., Mann J.I., Miller J.P., Thompson G.R., Thorogood M., et al. Mortality in treated heterozygous familial hypercholesterolaemia: Implications for clinical management. Atherosclerosis. 1999;142:105–112. doi: 10.1016/S0021-9150(98)00200-7. [DOI] [Google Scholar]
- 26.CDC (2019). Genetic Testing for Hereditary Breast and Ovarian Cancer. Off. Genomics Precis. Public Heal. https://www.cdc.gov/genomics/disease/breast_ovarian_cancer/testing.
- 27.Manahan E.R., Kuerer H.M., Sebastian M., Hughes K.S., Boughey J.C., Euhus D.M., Boolbol S.K., Taylor W.A. Consensus Guidelines on Genetic Testing for Hereditary Breast Cancer from the American Society of Breast Surgeons. Ann. Surg Oncol. 2019;26:3025–3031. doi: 10.1245/s10434-019-07549-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giri V.N., Hyatt C., Gomella L.G. Germline testing for men with prostate cancer: Navigating an expanding new world of genetic evaluation for precision therapy and precision management. J. Clin. Oncol. 2019;37:1455–1459. doi: 10.1200/JCO.18.02181. [DOI] [PubMed] [Google Scholar]
- 29.Samadder N.J., Riegert-Johnson D., Boardman L., Rhodes D., Wick M., Okuno S., Kunze K.L., Golafshar M., Uson P.L.S., Mountjoy L., et al. Comparison of Universal Genetic Testing vs Guideline-Directed Targeted Testing for Patients with Hereditary Cancer Syndrome. JAMA Oncol. 2021;7:230–237. doi: 10.1001/jamaoncol.2020.6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiman O.A., Taylor R.L., Lenassi E., Smith J.C., Douzgou S., Ellingford J.M., Barton S., Hardcastle C., Fletcher T., Campbell C., et al. Diagnostic yield of panel-based genetic testing in syndromic inherited retinal disease. Eur. J. Hum. Genet. 2020;28:576–586. doi: 10.1038/s41431-019-0548-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.CDC (2014). Tier 1 Genomics Applications and their Importance to Public Health. Off. Genomics Precis. Public Heal. https://www.cdc.gov/genomics/implementation/toolkit/tier1.htm.
- 32.Appelbaum P.S., Burke W., Parens E., Zeevi D.A., Arbour L., Garrison N.A., Bonham V.L., Chung W.K. Is there a way to reduce the inequity in variant interpretation on the basis of ancestry? Am. J. Hum. Genet. 2022;109:981–988. doi: 10.1016/J.AJHG.2022.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hindorff L.A., Bonham V.L., Brody L.C., Ginoza M.E.C., Hutter C.M., Manolio T.A., Green E.D. Prioritizing diversity in human genomics research. Nat. Rev. Genet. 2018;19:175–185. doi: 10.1038/NRG.2017.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xue A., Wu Y., Zhu Z., Zhang F., Kemper K.E., Zheng Z., Yengo L., Lloyd-Jones L.R., Sidorenko J., Wu Y., et al. Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nat. Commun. 2018;9:2941–3014. doi: 10.1038/s41467-018-04951-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choquet H., Paylakhi S., Kneeland S.C., Thai K.K., Hoffmann T.J., Yin J., Kvale M.N., Banda Y., Tolman N.G., Williams P.A., et al. A multiethnic genome-wide association study of primary open-angle glaucoma identifies novel risk loci. Nat. Commun. 2018;9:2278–2314. doi: 10.1038/s41467-018-04555-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Forrest I.S., Chaudhary K., Paranjpe I., Vy H.M.T., Marquez-Luna C., Rocheleau G., Saha A., Chan L., Van Vleck T., Loos R.J.F., et al. Genome-wide polygenic risk score for retinopathy of type 2 diabetes. Hum. Mol. Genet. 2021;30:952–960. doi: 10.1093/hmg/ddab067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wishart D.S., Knox C., Guo A.C., Shrivastava S., Hassanali M., Stothard P., Chang Z., Woolsey J. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34:D668–D672. doi: 10.1093/NAR/GKJ067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van Hout C.V., Tachmazidou I., Backman J.D., Hoffman J.D., Liu D., Pandey A.K., Gonzaga-Jauregui C., Khalid S., Ye B., Banerjee N., et al. Exome sequencing and characterization of 49,960 individuals in the UK Biobank. Nature. 2020;586:749–756. doi: 10.1038/s41586-020-2853-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang C.C., Chow C.C., Tellier L.C., Vattikuti S., Purcell S.M., Lee J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R.S., Thormann A., Flicek P., Cunningham F. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morales J., Pujar S., Loveland J.E., Astashyn A., Bennett R., Berry A., Cox E., Davidson C., Ermolaeva O., Farrell C.M., et al. A joint NCBI and EMBL-EBI transcript set for clinical genomics and research. Nature. 2022;604:310–315. doi: 10.1038/s41586-022-04558-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Amberger J.S., Bocchini C.A., Schiettecatte F., Scott A.F., Hamosh A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015;43:D789–D798. doi: 10.1093/nar/gku1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P., et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data associated with this study are present in the paper or Supplementary Material and use existing data from a previous study.13 A tabulated summary of all variants analyzed in the study is found in Table S3. This comprehensive list is annotated with information regarding genomic location, gene, associated disease, variant effect, amino acid change, ClinVar clinical significance and review status, minor allele frequencies in gnomAD, and minor allele frequencies in BioMe. In addition, this complete list has been deposited in Mendeley and its DOI is listed in the key resources table. Individual-level data, including sequencing, EHR phenotypes, and physician notes analyzed in this study are not publicly available due to Institutional Review Board (IRB) restrictions and research participant privacy concerns; however, requests from accredited researchers for access to data relevant to this manuscript can be made by contacting the lead contact.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
-
•
No custom code was generated in this study.