

Abstract
The E1 and E2 proteins from bovine papillomavirus bind cooperatively to the viral origin of DNA replication (ori), forming a complex which is essential for initiation of DNA replication. Cooperative binding has two components, in which (i) the DNA binding domains (DBDs) of the two proteins interact with each other and (ii) the E2 transactivation domain interacts with the helicase domain of E1. By generating specific point mutations in the DBD of E2, we have defined two patches of amino acids that are involved in the interaction with the E1 DBD. These same mutations, when introduced into the viral genome, result in severely reduced replication of the viral genome, as well as failure to transform mouse cells in tissue culture. Thus, the interaction between the E1 and E2 DBDs is important for the establishment of the viral genome as an episome and most likely contributes to the formation of a preinitiation complex on the viral ori.
Cooperative DNA binding plays a prominent role as a regulatory device for control of gene expression and DNA replication. One of the functions of cooperative DNA binding is to achieve increased specificity or affinity for DNA binding contributed by combinations of DNA binding factors (19). The ability of a DNA binding domain (DBD) to occupy a given site may thus be determined not only by its interaction with DNA but also by protein-protein interactions involving the DBD itself and/or its associated domains and other DNA binding factors. In consequence, in the cell, an elaborate network of protein-protein interactions may influence DNA binding, and DBDs, in addition to being passive tools with a tethering function, may play other roles, such as making contacts with other proteins that alter or augment their DNA binding properties. Interactions between DBDs (for example, homeodomains) have been studied extensively, and the consequences and mechanisms of interaction are well established in several cases (5, 12, 25, 33, 35).
Cooperative DNA binding between E1 and E2 performs a specific and essential function in initiation of papillomavirus DNA replication (22). The initiator E1, on its own, binds with low specificity to the origin of replication (ori), and specific and efficient recognition is accomplished by cooperative binding of E1 and the transcription factor E2 to immediately adjacent sites (15, 18, 22, 24). Once the complex is formed, in an ATP-dependent step, E2 can be displaced and additional E1 molecules can be added to the complex (20). Several observations indicate that formation of this cooperative complex is more elaborate than simply a tethering protein-protein interaction. Several separate interaction domains appear to exist in both the E1 and E2 proteins (1–4, 7, 11, 14, 16, 18, 21, 26, 34), and the mapped interactions appear to be different in nature. The interaction between the E1 and E2 DBDs is weak in the absence of DNA and, in the presence of DNA, shows strong dependence on the precise distance and positioning of the two binding sites, indicating that it corresponds to a short-range interaction (3, 4). In contrast, for example, the interaction between the E2 activation domain and E1 can readily be detected both in the absence and in the presence of DNA and shows little dependence on the relative positions of the respective binding sites (2, 7, 15, 16, 18). Indeed, in vivo replication assays indicate that this interaction can occur over distances of several kilobase pairs (27). Furthermore, whereas the interaction between the E2 activation domain and E1 by itself is sufficient for DNA replication, the interaction between the E2 and E1 DBDs by itself does not allow DNA replication; indeed, E2 lacking the activation domain has no activity for replication in vivo (3, 13, 28).
The relationship between these two interactions is interesting. Previous experiments have indicated that these two interactions are not independent. In experiments with chimeric proteins, replacement of the bovine papillomavirus (BPV) E2 DBD with the human papillomavirus type 11 (HPV-11) E2 DBD, which is unable to interact with BPV E1, also abolished the interaction between the BPV E2 activation domain of the chimeric protein and E1. However, when the distance between the E1 and E2 binding sites was increased from 3 bp, as in the wild-type (wt) ori, to 22 bp, no dependence on the identity of the DBD was observed and both (i) the interaction between the E2 activation domain and E1 and (ii) DNA replication could be detected (3, 4).
To determine the function of the interaction between the E1 and E2 DBDs, we have mapped the region within the 85-amino-acid (aa) minimal E2 DBD required for interaction with E1. We found that mutation of five residues, in two separate patches, affects the ability of the E2 DBD to interact with E1. The interaction between the DBDs is required for DNA replication, as determined by in vivo replication assays, and is also important for transformation by the viral DNA, demonstrating the importance of this interaction for the viral life cycle.
MATERIALS AND METHODS
Mutagenesis and plasmid constructs. (i) Mutant E2 DBDs.
Single alanine substitutions were generated in the pET11CE2DBD (aa 323 to 410) bacterial expression plasmid (3) by site-directed oligonucleotide mutagenesis. Mutations were confirmed by DNA sequencing. Mutations were placed into full-length E2 by digesting mutant pET11CE2DBD with KpnI and BamHI and isolating the fragment between the two sites and ligated into pET11CE2 cut with KpnI and BamHI. For in vivo transient replication assays, full-length E2 mutants were transferred to pCG mammalian expression vectors. The mutations were also placed into the background of the cloned full-length BPV type 1 genome for viral transformation assays.
(ii) ori constructs.
All of the ori constructs used in this study have been described previously (3, 22).
Protein expression. (i) E2 DBD.
Escherichia coli BL21(DE3) was transformed with pET11CE2DBD (3). Liquid cultures were inoculated and grown at 18°C until an optical density at 600 nm of 0.6 to 0.8 was reached. Cultures were induced with 0.4 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and grown for an additional 6 h at 18°C. Bacterial pellets were resuspended in lysis buffer (50 mM Tris [pH 7.5], 0.1 M NaCl, 5 mM EDTA, 5 mM dithiothreitol, 20% sucrose, 1 mM phenylmethylsulfonyl fluoride) and treated with lysozyme (100 μg/ml) for 10 min on ice. A 0.1% concentration of Nonidet P-40 was added, and the lysate was sonicated, cleared by centrifugation, and frozen in liquid nitrogen. The protein was quantitated by Western blot analysis.
(ii) Full-length E1 and the E1 DBD.
The expression and purification of E1 and the E1 DBD have been described previously (4, 23).
Probes.
Probes for electrophoretic gel mobility shift assays (EMSAs) and McKay assays were generated by PCR amplification of ori constructs cloned into pUC19 using the universal primers USP and RSP.
EMSAs.
A probe containing a high-affinity E2 binding site from the HPV-11 ori (ACCGAAAACGGT) was incubated with crude E. coli extracts containing the E2 DBD and 20 ng of nonspecific competitor DNA (pUC119) in 10 μl of binding buffer (20 mM potassium phosphate [pH 7.4], 0.1 M NaCl, 1 mM EDTA, 10% glycerol, 0.1% Nonidet P-40, 3 mM dithiothreitol, 0.7 mg of bovine serum albumin per ml) for 30 min. For all binding reactions, equivalent amounts of E2 DBD protein were used, as determined by Western blot analysis. Binding reactions were directly subjected to 5% polyacrylamide gel electrophoresis (PAGE) in 0.5× Tris-borate-EDTA buffer.
McKay assays.
Purified full-length E1 with an N-terminal glutathione S-transferase (GST) fusion was incubated with crude extracts containing the E2 DBD in binding buffer and 100 ng of nonspecific competitor DNA [poly(dA-dT)]. After 30 min, a 2.5-μl volume of glutathione-agarose beads was added to each 10-μl binding reaction mixture and the total volume was brought to 50 μl by the addition of binding buffer. After 20 min of rotational mixing at room temperature, the beads were washed three times with 200 μl of binding buffer. A 100-μl volume of stop buffer (1% sodium dodecyl sulfate, 50 mM EDTA, 0.1 M NaCl, 25 μg of tRNA per ml, 5 μg of mussel glycogen) was added; this was followed by phenol extraction and ethanol precipitation. Samples were analyzed by denaturing 6% PAGE.
Transient replication assays.
Transient replication assays were performed with C127 cells as previously described (28).
Transformation assay.
C127 cells were transfected with the wt or mutant BPV genome by electroporation. At 2 to 3 weeks posttransfection, plates were stained with methylene blue and the number of transformed foci was recorded.
RESULTS
Mutations in the E2 DBD.
The X-ray crystal structure of the E2 DBD complexed with DNA has been determined (9). To identify residues in the E2 DBD which affect the ability of E2 to interact with E1, we changed individual amino acids on the surface of the E2 DBD to alanines. Based on our previous observation that the HPV-11 E2 DBD failed to stimulate E1 binding to the BPV ori (3), we initially mutated residues in the E2 DBD that were not conserved between the HPV-11 and BPV type 1 E2 DBDs. Furthermore, based on the crystal structure of the E2 DBD, only residues located on the surface of the E2 DBD which are not involved in DNA recognition and dimerization were mutated. After an initial screen to identify mutations that disrupted the ability of the E2 DBD to interact with E1 in a DNA binding assay, we identified three mutant proteins that were defective in stimulating E1 binding to the ori (348, 388, and 401 in Table 1). We then mutated to alanines an additional nine residues flanking the three original mutations. These additional residues were not necessarily on the surface of the protein or not conserved with the HPV-11 E2 DBD. Of the total of 35 mutations, 4 had side chains that were not accessible. The mutant proteins were expressed in E. coli, and expression levels were determined by Western blot analysis with a polyclonal antibody to E2.
TABLE 1.
Summary of the DNA binding activities of 35 point mutant proteinsa
| Residue mutated | DNA binding activity |
|---|---|
| 334T | <0.06 |
| 347K | <0.06 |
| 348N | 0.5 |
| 349H | <0.06 |
| 350R | 0 |
| 352R | 0.5 |
| 355N | No expression |
| 365D | 0.25 |
| 366N | wt |
| 367G | wt |
| 369E | wt |
| 371N | 0.5 |
| 372G | <0.06 |
| 373Q | wt |
| 381G | wt |
| 383P | wt |
| 384S | wt |
| 385Q | wt |
| 386R | <0.06 |
| 387Q | wt |
| 388D | wt |
| 389Fb | No expression |
| 390Lb | wt |
| 391K | wt |
| 392H | 0.25 |
| 394P | wt |
| 395Lb | <0.06 |
| 398G | wt |
| 400N | 0.5 |
| 401Ib | wt |
| 402S | wt |
| 403G | <0.06 |
| 405F | 0.03 |
| 409D | 0.03 |
| 410Fb | wt |
By using mutant protein concentrations normalized with respect to the wt, the DNA binding activities of all of the point mutant proteins were determined by EMSA using a probe containing a high-affinity E2 binding site. The binding activity of each mutant protein is expressed relative to the activity of the wt E2 DBD.
Not on the surface.
Effects of mutations on E2 DNA binding activity.
Of the 35 mutants, 33 were successfully expressed in E. coli (Table 1). The level of expression of the mutant proteins was determined by Western blot analysis and amounted to approximately 5% of the total bacterial protein in crude extracts. Most of the mutant proteins were expressed at similar levels (i.e., within twofold of the wt protein level), with the exception of mutant proteins 355 and 389, which were not expressed at detectable levels (Table 1). As a measure of activity and an assurance of proper protein folding and stability, we performed DNA binding assays. We assayed equivalent amounts of protein, based on Western blot analysis, for the ability to bind a probe containing a high-affinity E2 binding site in EMSAs using twofold dilutions of crude E. coli extracts. Figure 1 shows the expression levels of five different mutant proteins as determined by Western blot analysis (bottom panel) and their DNA binding activities after normalization of the protein concentrations. Of the five mutant proteins tested as shown in Fig. 1, 366, 373, 381, and 401 showed levels of binding virtually identical to that of the wt for each titration used (compare lanes 1 to 3 with lanes 11 to 13, 16 to 20, 21 to 23, and 26 to 28). An approximately twofold lower level of binding was observed for mutant protein 348 (compare lanes 1 to 3 with lanes 6 to 8).
FIG. 1.

DNA binding activities of E2 DBD point mutant proteins. Levels of mutant E2 DBDs were determined by Western blot analysis using a polyclonal antibody to E2 and quantitated by using an IS 1000 Digital Imaging system. Three different dilutions of crude E. coli extracts containing the wt E2 DBD (lanes 1 to 3, bottom panel) were used as standards for levels of expression of five different mutant proteins (lanes 4 to 8, bottom panel), and equivalent amounts of protein were used to test for DNA binding activity by EMSA. Twofold titrations of the wt protein (lanes 1 to 5, top panel) or mutant proteins (lanes 6 to 30, top panel) were analyzed on a 5% polyacrylamide gel. Lane 31 contained probe alone.
As summarized in Table 1, the majority of the other mutant proteins also showed nearly wt levels of DNA binding activity (within twofold). Nine mutant proteins exhibited significantly lower-than-wt DNA binding (<25% of the wt). In three of these, the substitutions, at residues 334, 347, and 349, are located within well-conserved α helix 1 of the protein, which has been termed the recognition helix because it contains residues involved in important contacts with DNA. Therefore, these three residues may have affected the ability of the E2 DBD to recognize DNA. The remaining six mutations that had severe effects on DNA binding are located in the four β strands which contribute to formation of the β barrel. It is possible that these residues interfered with the formation of half of the β-barrel subunit in one E2 DBD monomer or with the dimerization interface between two E2 monomers. Since the structural integrity of these nine mutant proteins was in question, they were not evaluated for cooperative DNA binding with E1.
Residues required for interaction with E1 map to two discrete patches on the surface of the E2 DBD.
To measure the abilities of the mutant proteins to stimulate E1 binding to the ori, we used a quantitative DNA binding assay known as the McKay assay. In the McKay assay, GST-E1 is incubated together with two probes of different lengths, one longer probe containing the minimal ori with a high-affinity E2 binding site (I) and a shorter probe lacking the E2 binding site (II). We chose to use a high-affinity E2 binding site rather than the naturally occurring low-affinity E2 binding site because DNA binding by the E2 DBD mutant proteins was measured by using the high-affinity E2 site and the use of the high-affinity site increased the sensitivity of the assay. Probes bound by GST-E1 were recovered with glutathione agarose beads and analyzed by PAGE. The longer probe, which contains the E2 binding site, allows the formation of the complex containing both E1 and the E2 DBD and provides a measure of the level of stimulation of GST-E1 binding by the E2 DBD to the ori. The shorter probe, which lacks the E2 binding site, serves as an internal control for binding by GST-E1 in the absence of cooperative binding with E2. We can determine the stimulation of GST-E1 binding by the E2 DBD by comparing the amounts of the two probes recovered. The 24 mutant proteins that showed DNA binding activity within fourfold of that of the wt were tested for cooperative binding with E1. For mutant proteins with a two- to fourfold reduction in DNA binding activity compared to the wt (i.e., mutant proteins 348, 352, 365, 371, 392, and 400), the titration of crude extract used in the McKay assay was increased two- to fourfold, respectively, in order to compensate for their lower DNA binding activity.
Figure 2A shows the results of McKay assays with seven different mutant proteins. In the presence of the wt E2 DBD, we observed approximately fivefold stimulation of binding of GST-E1 to probe I relative to probe II, which lacks the E2 binding site (top panel, lanes 1 to 4, and bottom panel, lanes 1 to 4). This stimulation of GST-E1 binding was not observed in the absence of E2 (bottom panel, lane 21). For mutant protein 387, the level of cooperative binding with GST-E1 was nearly identical to that of the wt (bottom panel, lanes 9 to 12). However, in the presence of either mutant protein 401, 348, 385, 390, or 410, virtually no stimulation of binding by GST-E1 was observed (top panel, lanes 5 to 8 and 9 to 12, bottom panel, lanes 5 to 8, 13 to 16, and 17 to 20), with a ratio of approximately 1 for binding of GST-E1 to probes I and II. Mutant protein 388 was capable of cooperative binding with GST-E1, but at a level intermediate between those of the wt and mutant proteins 348 and 401 (I/II ratio, approximately 2). For the remaining 18 mutant proteins, McKay assays demonstrated wt or nearly wt levels of cooperative binding between E1 and the mutant E2 DBDs (data not shown).
FIG. 2.

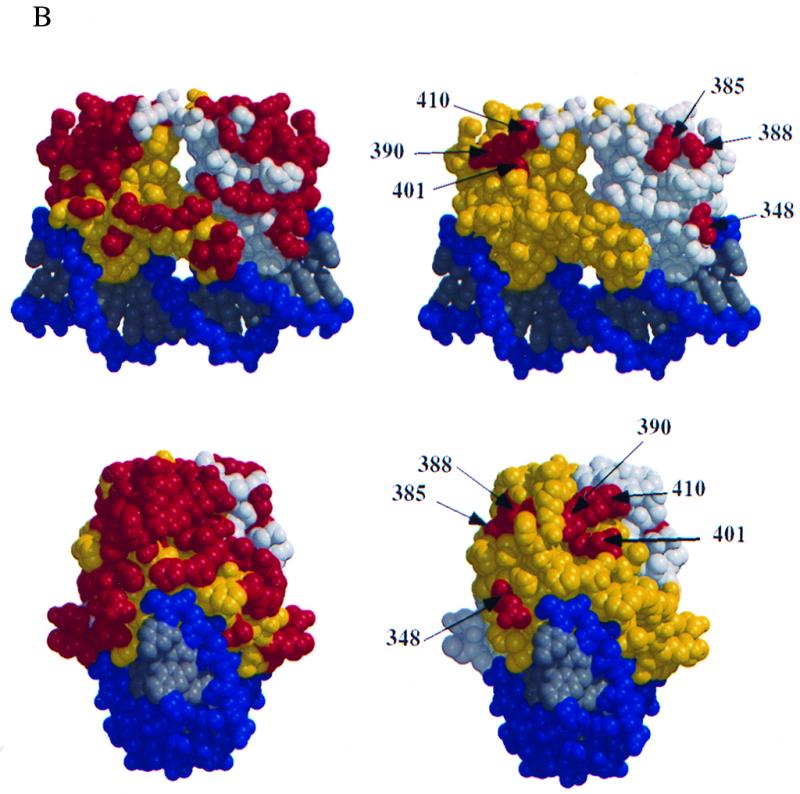
(A) Six mutant proteins are defective for cooperative binding with E1. Two different probes, one containing the BPV minimal ori with a high-affinity E2 binding site (I) and one containing an E1 binding site alone (II), were incubated with 6 ng of GST-E1 alone (lane 21, bottom) or with either the wt E2 DBD (lanes 1 to 4, top and bottom), or seven mutant E2 DBDs (lanes 5 to 16, top, and lanes 5 to 20, bottom). Equal quantities of wt and mutant proteins, based on Western blot analysis, were used in four twofold titrations. Probes bound by GST-E1 were recovered with glutathione-agarose beads and analyzed on a 6% urea gel. E2-dependent stimulation of binding was measured by comparing the amounts of probes II and I recovered. (B) Five mutations which do not affect DNA binding form two distinct patches on the E2 DBD. Shown are two different views of a space-filling model of the E2 DBD bound to its cognate binding site, created by the BOBSCRIPT program and Raster3D (6, 10, 17). In the images on the right, all mutations which were made on the surface of the E2 DBD are in red. The images on the left show the same two views with mutations that affect the ability of the E2 DBD to interact with E1 in red.
Thus, the McKay assays identified six residues that either disrupted or reduced the ability of the E2 DBD to stimulate E1 binding to the ori. The locations of these residues are illustrated in Fig. 2B. Five of the six residues form two discrete patches on the surface of the protein. Residue 348 is located outside of these two defined patches and is, instead, close to the DNA. Proteins with mutations on either side of 348, that is, at residues 347 and 349, were both severely defective for DNA binding. Mutation of residue 348 itself resulted in slightly impaired DNA binding, as shown in Fig. 1. Thus, the phenotype caused by mutation of residue 348 in the McKay assay may be the result of decreased DNA binding stability or affinity, and we therefore focused our subsequent studies on the residues located within the two defined patches.
Combination of mutations in the E2 DBD abolishes cooperative binding with E1.
We constructed two double mutant proteins, namely, 390/385 and 390/388, by combining one mutation from each patch. The double mutant proteins were expressed in E. coli, and the levels of their expression were determined by Western blot analysis (Fig. 3A, bottom panel) and their ability to bind to a high-affinity E2 binding site was determined by EMSA as shown in Fig. 3A. The quantities of mutant proteins used in the assay were normalized with respect to the wt E2 DBD. Mutant protein 390/388 exhibited wt levels of DNA binding activity (compare lanes 2 to 5 with lanes 10 to 13), whereas 390/385 showed a decreased level of DNA binding activity compared to the wt. The level of binding by 390/385 in lanes 6 and 7 approximates that of the wt in lanes 3 and 4, respectively. Since each lane represents a twofold titration, mutant protein 390/385 is approximately fourfold less active in DNA binding than the wt.
FIG. 3.

(A) DNA binding activities of double mutant E2 DBDs. The double mutant proteins 390/385 (lanes 4 and 5, bottom) and 390/388 (lanes 6 and 7, bottom) were quantitated with wt E2 DBD standards (lanes 1 to 3, bottom) as described in the legend to Fig. 1. Equal quantities of mutant and wt E2 DBDs were then tested for DNA binding activity by EMSA using four twofold dilutions of wt E2 DBD (lanes 2 to 5, top), 390/385 (lanes 6 to 9, top), or 390/388 (lanes 10 to 13, top). (B) The double mutant proteins 390/385 and 390/388 failed to interact with E1. To determine the abilities of the mutant proteins to interact with the E1 DBD, an EMSA was performed with a probe containing the BPV minimal ori with a high-affinity E2 binding site. A 0.4-ng sample of the E1 DBD (aa 142 to 308) was incubated with four different twofold titrations of E. coli extracts containing the wt E2 DBD (lanes 3 to 6) or the mutant protein 390 (lanes 7 to 10), 401 (lanes 11 to 14), 390/385 (lanes 15 to 18), or 390/388 (lanes 19 to 22). Lane 1 contained 0.4 ng of the E1 DBD (aa 142 to 308) alone, lane 2 contained the wt E2 DBD alone, and lane 23 contained probe alone.
To measure the abilities of these double mutant proteins to stimulate E1 binding to the ori, we used an EMSA rather than the McKay assay. We have previously demonstrated that the cooperativity between the E1 and E2 DBDs can be readily observed by EMSA (4). The results are shown in Fig. 3B. In the absence of the E2 DBD, binding of the E1 DBD gives rise to a faint band at the low concentration used in this experiment (lane 1). In the presence of wt E2 DBD, there is an approximately 70-fold stimulation of E1 binding through the formation of the E1 DBD-E2 DBD-ori complex (compare lane 3 to lane 1). This assay is more sensitive than the McKay assay in detecting cooperative interaction between the E1 and E2 DBDs, since the stimulation of binding by the E2 DBD observed by EMSA is greater than that measured by McKay assays. Consequently, mutant proteins that showed no cooperative interaction with E1 in the McKay assay, such as 390 or 401, stimulated E1 DBD binding slightly in the EMSA (twofold and fivefold, respectively; compare lanes 7 and 11 to lane 1).
For double mutant protein 390/388, which exhibited wt levels of DNA binding activity, no stimulation of E1 binding to the ori was observed (compare lanes 19 to 22 with lanes 3 to 6). Double mutant protein 390/385 showed a slightly reduced level of DNA binding activity compared to the wt; however, even with an equivalent amount of DNA binding by 390/385 compared to the wt, there was no cooperative binding between 390/385 and the E1 DBD (compare lanes 15 and 16 with lanes 5 and 6). The low level of the complex formed likely represents co-occupation of the probe by the E1 and E2 DBDs and suggests that the ability of the two proteins to bind to the same probe is not affected by the mutations. The difference in mobility of some of the complexes may reflect differences in the overall structure of the protein-DNA complex in the presence or in the absence of cooperative binding between the E1 and E2 DBDs. The disruption of cooperative binding by the double mutant proteins was also observed when a probe with the low-affinity E2 binding site naturally present in the BPV ori was used (data not shown).
E2 DBD mutations affect the ability of the BPV genome to replicate and transform cells.
To determine the effects of the interaction between the E1 and E2 DBDs on viral DNA replication and transformation, the mutations in the E2 DBD were also placed into the context of the entire BPV genome and the mutated genomes were transfected into mouse C127 cells. The results of a transient replication assay for three different time points (36, 60, and 84 h posttransfection) are shown in Fig. 4A. BPV DNA recovered from cells was digested with DpnI and HindIII. Because mutant 390/388 contains an additional HindIII site created by the mutation, digestion of this mutant viral DNA with HindIII and XbaI resulted a shorter DNA fragment. At the last time point, 390/388 showed at least a 10-fold decrease in levels of replication of the viral genome compared to the wt (compare lane 6 with lane 3). Mutants 401 and 390 showed two- and threefold decreases in DNA replication, respectively. The effects of these mutations were not as severe in the context of the entire viral genome as in the context of a plasmid carrying only the minimal ori, indicating that other E2 binding sites in the viral genome can be utilized. Nonetheless, disruption of the E1-E2 DBD interaction significantly affected the ability of the viral genome to replicate in transient replication assays.
FIG. 4.

(A) The mutations in the E2 DBD affect replication of the viral genome. The BPV genome was mutated at position(s) 390, 401, or 390 and 388. Transient replication assays were performed with mouse C127 cells, and low-molecular-weight DNA was digested with the restriction enzymes DpnI and HindIII. Replication of the wt (lanes 1 to 3) or the 390/388 (lanes 4 to 6), 401 (lanes 7 to 9), or 390 (lanes 10 to 12) mutant genome was measured at 24, 48, and 72 h posttransfection. In lanes 4 to 6, the viral DNA containing mutations at positions 390 and 388 shows increased mobility due to the presence of an additional HindIII site generated by the mutation. (B) Mutations which affect the E2 DBD-E1 interaction affect viral transformation. The wt genome and the genome mutated at position(s) 390, 401, or 390 and 388 were transfected into C127 cells. After 2 weeks, plates were stained with methylene blue and transformed foci were counted.
To determine whether the E2 DBD mutations affect long-term replication and the ability of BPV to transform cells, the wt viral genome and the genome containing a mutation(s) at 390, 401, or 390 and 388 were transfected into C127 cells. After 2 weeks, the cells were stained with methylene blue to identify foci (Fig. 4B). The wt genome produced a large number of foci (approximately 100). The viral genomes containing mutations at 401 and 390 showed approximately two- and fourfold decreases in the number of foci. Viral DNA containing the 390/388 double mutation in the E2 DBD showed a greater-than-20-fold reduction in the number of foci compared to the wt. The reduction in transformation efficiency is similar to the reduction in levels of DNA replication for the different mutants (Fig. 4A), suggesting that the defect in transformation reflects a failure of the viral DNA to replicate efficiently.
Due to the failure of the 390/388 mutant to transform and replicate efficiently, we were unable to establish stable cell lines. In transient transfection assays using the viral genome, E2 is expressed at very low levels. Therefore, to determine whether the wt and mutant proteins were significantly different in stability, we transiently expressed wt E2 and the 390/388 mutant protein from pCG expression vectors in COS cells. At 20 h after transfection, 25 μg of cycloheximide per ml was added and measurements were made 1 and 2 h after inhibition of protein synthesis. The measurements made at these time points were analyzed by Western blotting using a polyclonal antiserum to E2 as shown in Fig. 5. Comparison of the levels of E2 protein before and after inhibition of protein synthesis indicated that under these conditions, the half-lives of both the wt and mutant E2 proteins were very similar, i.e., approximately 50 min (compare lanes 3 to 5 and 6 to 8), indicating that the defect in DNA replication and transformation is not due to a significant reduction in the half-lives of the mutant E2 proteins.
FIG. 5.

Stability of wt and mutant E2 proteins. COS cells were transfected with a pCG expression vector encoding wt E2 or the 390/388 mutant protein. At 20 h after transfection, protein synthesis was blocked by the addition of 25 μg cycloheximide per ml. Cells were harvested, and the levels of E2 protein after 1 h (lanes 4 and 7) and 2 h (lanes 5 and 8) in the presence of cycloheximide were compared to E2 levels in the absence of cycloheximide (lanes 3 and 6) by Western blotting using a polyclonal antiserum to E2. Lane 1 contained purified E2 protein; lane 2 contained extract from untransfected cells. The calculated half-life of both the wt and the 390/388 mutant protein, based on quantitation of the Western blot, was approximately 50 min.
DISCUSSION
We undertook these studies to gain an understanding of the nature of the interaction between E1 and E2. We were interested in the role played by the E2 DBD in the interaction between the two proteins. Previous studies have demonstrated that cooperative binding of E1 and E2 to the BPV minimal ori involves an interaction between the E1 and E2 DBDs, in addition to interactions between the E2 activation domain and E1 (3, 4). Here we show that mutations that specifically disrupt the E2 DBD-E1 interaction but have little or no effect on E2 DNA binding, in the context of the viral genome, substantially reduced viral DNA replication and transformation. This indicates that this interaction plays a critical role in the viral life cycle. These results are completely consistent with the in vivo experiments using chimeric E2 proteins (3).
Nature of the interaction between E1 and E2.
The five residues that are important for the interaction between the E2 DBD and E1 are not contiguous residues in the protein sequence but map to two distinct patches. Residues 385 and 388 define one patch, while 390, 401, and 410 define another patch on the E2 DBD. Both patches are on a surface that would face E1 bound to an adjacent site. Residues 385 and 388 reside on the surface of the protein in α helix 2 (9). Curiously, the three residues defining the second patch, 390 (leucine), 401 (isoleucine), and 410 (phenylalanine), have side chains that are not surface accessible and, instead, form a hydrophobic pocket. It is possible that this hydrophobic pocket is engaged in a direct protein-protein interaction with a similar hydrophobic surface in E1 that becomes exposed upon cooperative binding of both proteins to the ori. However, an interesting possibility is that the residues forming the hydrophobic core interact with the DNA upon cooperative binding with E1. Some well-characterized DNA binding factors, such as the TATA binding protein and the Ets-1, have hydrophobic residues that interact specifically with the minor groove of the DNA (30–32). This interaction generates a substantial bend in the DNA through the intercalation of the hydrophobic residues (31). OH-radical and DNase footprinting with the wt E1 and E2 DBDs gave rise to protections virtually identical to those observed with the full-length proteins, including protection of sequences between the E1 and E2 binding sites (22, 23). More importantly, interference studies indicate that sequences between the E1 and E2 binding sites are involved in formation of the combined complex (4, 20, 23). These results are consistent with involvement of the DNA in the interaction; indeed, strong interaction between the E1 and E2 DBDs can only be observed in the presence of DNA. The alteration in complex mobility that we observed when comparing the wt and mutant E2 DBDs (Fig. 3B) is also interesting, since this indicates that the structure of the DNA is altered as a consequence of interaction between the E1 and E2 DBDs. An interesting question is whether the identity of the residues involved in the interaction between E1 and E2 is in any way dependent on the identity of the E2 binding site. Our screen was performed with a high-affinity E2 binding site, and it is possible that use of a different E2 binding site would have resulted in subtle differences. However, the mutations that we have identified, for example, 390/388, disrupt cooperative binding using several high- and low-affinity E2 binding sites, indicating that the effects of these mutations are not dependent on the identity of the E2 binding site (data not shown).
Why is the interaction between the E1 and E2 DBDs important for viral DNA replication and transformation?
Our previous studies have demonstrated that the identity of the E2 DBD is important for DNA replication and for cooperative DNA binding when the binding sites for the E1 and E2 proteins are positioned immediately adjacent to each other (3). When the binding sites for E1 and E2 are placed at a distance from one another, both DNA replication and cooperative DNA binding can be detected with a heterologous DNA binding domain fused to E2, demonstrating that the interaction between the E2 activation domain and E1 is sufficient for cooperative interaction and initiation of DNA replication. In light of our current results, this indicates that the interaction between the two DBDs may be primarily to facilitate the interaction between the E2 activation domain and the E1 helicase domain (Fig. 6). As mentioned above, an interesting possibility is that the interaction between the E1 and E2 DBDs causes alterations in the DNA structure that, in turn, facilitate the interaction between the E2 activation domain and the E1 helicase domain. Although it is generally believed that transcriptional activation domains are intrinsically very flexible, the E2 activation domain may not be typical in this respect, as indicated by the recently reported X-ray crystal structure of the E2 activation domain from HPV-18 (8). Also, the very close juxtaposition of the binding sites is likely to impose constraints due to the inherent stiffness of short sequences of DNA (29). The lack of dependence on the DBD when the E1 and E2 sites are placed at a distance from one another is consistent with this idea. A structural change in DNA, such as a bend or kink produced by the interaction between the E1 and E2 DBDs, could also play a more direct role in initiation of DNA replication.
FIG. 6.

Model of cooperative binding of E1 and E2 to the BPV minimal ori. (A) Cooperative binding between E1 and E2 involves two separate interactions: interaction 1 between the E1 and E2 DBDs and interaction 2 between the E1 helicase domain and the E2 activation domain (AD). As the first required step in the cooperative binding of E1 and E2 on the BPV minimal ori, the E2 DBD interacts with the E1 DBD (interaction 1). This interaction results in bending or kinking of the DNA. As a consequence of the induced DNA bend, the E2 activation domain is placed in a position where it can effectively interact with the E1 helicase domain. The productive interaction between the E2 activation domain and E1 completes the second step in the cooperative binding of E1 and E2 on the ori. (B) Mutations in the E2 DBD which result in failure to interact with the E1 DBD result in loss of the interaction between E1 and E2 despite a wt and functional E2 activation domain.
An interesting question is whether the interaction between the BPV E1 and E2 DBDs that we have analyzed here also occurs between other papillomavirus E1 and E2 proteins. So far, no direct experiments have been carried out to determine whether this is the case. However, if such an interaction exists, it is not sufficiently conserved to allow interaction between heterologous proteins, in contrast to the interaction between E1 and the E2 activation domain (3). Our previous results indicate that the interaction between the DBDs only plays a role when the E1 and E2 binding sites are adjacent. In contrast to the ori from BPV and some other animal papillomaviruses, HPVs all have an E2 binding site in a more distal position, making it unlikely that a DBD interaction is important for binding of E1 and E2 to the ori in these viruses. However, since recognition sequences for E1 are poorly defined, the presence of E1 binding sites adjacent to E2 binding sites outside the ori is a distinct possibility. Indeed, the effects of the 390/388 mutant protein that we observed in BPV could very well be caused by effects on viral gene expression. One of the dramatic consequences of cooperative binding between E1 and E2 to proximal sites is that binding can be achieved even with very low-affinity E2 binding sites, as exemplified by ori-proximal E2 BS12. The BPV genome contains a significant number of very low-affinity E2 binding sites in positions consistent with a role in viral gene expression. Stimulation of binding of E2 to these sites by cooperative binding with E1 could be a means to link control of DNA replication and viral gene expression.
The interaction between the E1 and E2 proteins is a composite interaction of surprising complexity. The contact between the E1 and E2 DBDs may play a role by altering the structure of the DNA component of the complex and thus may have a largely architectural function. The contact between the activation domain of E2 and the E1 helicase domain is clearly the productive interaction that results in initiation of DNA replication, but the exact consequence of this contact remains to be determined.
ACKNOWLEDGMENTS
We thank Nouria Hernandez for critical reading of the manuscript and Leemor Joshua-Tor for assistance with molecular graphics.
This work was supported by National Institutes of Health grant CA13106 to A.S.
REFERENCES
- 1.Abroi A, Kurt R, Ustav M. Transcriptional and replicational activation functions in the bovine papillomavirus type 1 E2 protein are encoded by different structural determinants. J Virol. 1996;70:6169–6179. doi: 10.1128/jvi.70.9.6169-6179.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benson J D, Howley P M. Amino-terminal domains of the bovine papillomavirus type 1 E1 and E2 proteins participate in complex formation. J Virol. 1995;69:4364–4372. doi: 10.1128/jvi.69.7.4364-4372.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berg M, Stenlund A. Functional interactions between papillomavirus E1 and E2 proteins. J Virol. 1997;71:3853–3863. doi: 10.1128/jvi.71.5.3853-3863.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen G, Stenlund A. Characterization of the DNA-binding domain of the bovine papillomavirus replication initiator E1. J Virol. 1998;72:2567–2576. doi: 10.1128/jvi.72.4.2567-2576.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen L, Glover J N M, Hogan P G, Rao A, Harrison S C. Structure of the DNA-binding domains from NFAT, Fos and Jun bound specifically to DNA. Nature. 1998;392:42–48. doi: 10.1038/32100. [DOI] [PubMed] [Google Scholar]
- 6.Esnouf R M. An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J Mol Graphics. 1997;15:132–134. doi: 10.1016/S1093-3263(97)00021-1. [DOI] [PubMed] [Google Scholar]
- 7.Ferguson M K, Botchan M R. Genetic analysis of the activation domain of bovine papillomavirus protein E2: its role in transcription and replication. J Virol. 1996;70:4193–4199. doi: 10.1128/jvi.70.7.4193-4199.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harris S F, Botchan M R. Crystal structure of the human papillomavirus type 18 E2 activation domain. Science. 1999;284:1673–1677. doi: 10.1126/science.284.5420.1673. [DOI] [PubMed] [Google Scholar]
- 9.Hegde R S, Grossman S R, Laimins L A, Sigler P B. Crystal structure at 1.7A of the bovine papillomavirus-1 E2 DNA-binding domain bound to its DNA target. Nature. 1992;359:505–512. doi: 10.1038/359505a0. [DOI] [PubMed] [Google Scholar]
- 10.Kraulis P J. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 11.Leng X, Ludes-Meyers J H, Wilson V G. Isolation of an amino-terminal region of bovine papillomavirus type 1 E1 protein that retains origin binding and E2 interaction capacity. J Virol. 1997;71:848–852. doi: 10.1128/jvi.71.1.848-852.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li T, Stark M R, Johnson A D, Wolberger C. Crystal structure of the MATa1/MATa2 homeodomain heterodimer bound to DNA. Science. 1995;270:262–269. doi: 10.1126/science.270.5234.262. [DOI] [PubMed] [Google Scholar]
- 13.Lim D A, Gossen M, Lehman C W, Botchan M R. Competition for DNA binding sites between the short and long forms of E2 dimers underlies repression in bovine papillomavirus type 1 DNA replication control. J Virol. 1998;72:1931–1940. doi: 10.1128/jvi.72.3.1931-1940.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lusky M, Fontane E. Formation of the complex of bovine papillomavirus E1 and E2 proteins is modulated by E2 phosphorylation and depends upon sequences within the carboxyl terminus of E1. Proc Natl Acad Sci USA. 1991;88:6363–6367. doi: 10.1073/pnas.88.14.6363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lusky M, Hurwitz J, Seo Y S. Cooperative assembly of the bovine papilloma virus E1 and E2 proteins on the replication origin requires an intact E2 binding site. J Biol Chem. 1993;268:15795–15803. [PubMed] [Google Scholar]
- 16.Masterson P J, Stanley M A, Lewis A P, Romanos M A. A C-terminal helicase domain of the human papillomavirus E1 protein binds E2 and the DNA polymerase alpha-primase p68 subunit. J Virol. 1998;72:7407–7419. doi: 10.1128/jvi.72.9.7407-7419.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Merritt E A, Murphy M E P. Raster3D version 2.0—a program for photorealistic molecular graphics. Acta Crystallogr. 1994;D50:869–873. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]
- 18.Mohr I J, Clark R, Sun S, Androphy E J, MacPherson P, Botchan M R. Targeting the E1 replication protein to the papillomavirus origin of replication by complex formation with the E2 transactivator. Science. 1990;250:1694–1699. doi: 10.1126/science.2176744. [DOI] [PubMed] [Google Scholar]
- 19.Ptashne M. A genetic switch. Cambridge, Mass: Cell Press; 1992. [Google Scholar]
- 20.Sanders C M, Stenlund A. Recruitment and loading of the E1 initiator protein: an ATP-dependent process catalysed by a transcription factor. EMBO J. 1998;17:7044–7055. doi: 10.1093/emboj/17.23.7044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sarafi T R, McBride A A. Domains of the BPV-1 E1 replication protein required for origin-specific DNA binding and interaction with the E2 transactivator. Virology. 1995;211:385–396. doi: 10.1006/viro.1995.1421. [DOI] [PubMed] [Google Scholar]
- 22.Sedman J, Stenlund A. Co-operative interaction between the initiator E1 and the transcriptional activator E2 is required for replicator specific DNA replication of bovine papillomavirus in vivo and in vitro. EMBO J. 1995;14:6218–6228. doi: 10.1002/j.1460-2075.1995.tb00312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sedman T, Sedman J, Stenlund A. Binding of the E1 and E2 proteins to the origin of replication of bovine papillomavirus. J Virol. 1997;71:2887–2896. doi: 10.1128/jvi.71.4.2887-2896.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seo Y S, Muller F, Lusky M, Hurwitz J. Bovine papilloma virus (BPV)-encoded E1 protein contains multiple activities required for BPV DNA replication. Proc Natl Acad Sci USA. 1993;90:702–706. doi: 10.1073/pnas.90.2.702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tan S, Richmond T J. Crystal structure of the yeast MATalpha2/MCM1/DNA ternary complex. Nature. 1998;391:660–666. doi: 10.1038/35563. [DOI] [PubMed] [Google Scholar]
- 26.Thorner L K, Lim D A, Botchan M R. DNA-binding domain of bovine papillomavirus type 1 E1 helicase: structural and functional aspects. J Virol. 1993;67:6000–6014. doi: 10.1128/jvi.67.10.6000-6014.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ustav E, Ustav M, Szymanski P, Stenlund A. The bovine papillomavirus origin of replication requires a binding site for the E2 transcriptional activator. Proc Natl Acad Sci USA. 1993;90:898–902. doi: 10.1073/pnas.90.3.898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ustav M, Stenlund A. Transient replication of BPV-1 requires two viral polypeptides encoded by the E1 and E2 open reading frames. EMBO J. 1991;10:449–457. doi: 10.1002/j.1460-2075.1991.tb07967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J C, Giaever G N. Action at a distance along a DNA. Science. 1988;240:300–304. doi: 10.1126/science.3281259. [DOI] [PubMed] [Google Scholar]
- 30.Werner M H, Burley S K. Architectural transcription factors: proteins that remodel DNA. Cell. 1997;88:733–736. doi: 10.1016/s0092-8674(00)81917-0. [DOI] [PubMed] [Google Scholar]
- 31.Werner M H, Gronenborn A M, Clore G M. Intercalation, DNA kinking, and the control of transcription. Science. 1996;271:778–784. doi: 10.1126/science.271.5250.778. [DOI] [PubMed] [Google Scholar]
- 32.Werner M H, Clore M, Fisher C L, Fisher R J, Trinh L, Shiloach J, Gronenborn A M. The solution structure of the human ETS1-DNA complex reveals a novel mode of binding and true side chain intercalation. Cell. 1995;83:761–771. doi: 10.1016/0092-8674(95)90189-2. [DOI] [PubMed] [Google Scholar]
- 33.Wilson D S, Desplan C. Homeodomain proteins. Cooperating to be different. Curr Biol. 1995;5:32–34. doi: 10.1016/s0960-9822(95)00010-8. [DOI] [PubMed] [Google Scholar]
- 34.Winokur P L, McBride A A. The transactivation and DNA binding domains of the BPV-1 E2 protein have different roles in cooperative origin binding with the E1 protein. Virology. 1996;221:44–53. doi: 10.1006/viro.1996.0351. [DOI] [PubMed] [Google Scholar]
- 35.Wolberger C. Homeodomain interactions. Curr Opin Struct Biol. 1996;6:62–68. doi: 10.1016/s0959-440x(96)80096-0. [DOI] [PubMed] [Google Scholar]