

Summary
Type 1 diabetes (T1D) is a chronic condition in which beta cells are destroyed by immune cells. Despite progress in immunotherapies that could delay T1D onset, early detection of autoimmunity remains challenging. Here, we evaluate the utility of machine learning for early prediction of T1D using single-cell analysis of islets. Using gradient-boosting algorithms, we model changes in gene expression of single cells from pancreatic tissues in T1D and non-diabetic organ donors. We assess if mathematical modeling could predict the likelihood of T1D development in non-diabetic autoantibody-positive donors. While most autoantibody-positive donors are predicted to be non-diabetic, select donors with unique gene signatures are classified as T1D. Our strategy also reveals a shared gene signature in distinct T1D-associated models across cell types, suggesting a common effect of the disease on transcriptional outputs of these cells. Our study establishes a precedent for using machine learning in early detection of T1D.
Keywords: single-cell RNA-seq, type 1 diabetes, machine learning, human islets, autoantibody-positive
Graphical abstract
Highlights
-
•
Cells from a subset of autoantibody-positive donors are classified as T1D
-
•
A shared gene signature in distinct T1D-associated models across cell types
-
•
Machine learning and single-cell profiling can be used to model T1D
Despite progress in therapeutic approaches that could delay T1D onset, early detection of this autoimmune disease remains challenging. Patil et al. evaluate the utility of machine learning for early prediction of T1D and demonstrate the feasibility of modeling of T1D based on single-cell profiling of islets.
Introduction
Muscle and adipose tissues respond to insulin to increase glucose uptake. Insulin is a hormone that is made by specialized cells called beta cells positioned in the islets of Langerhans in the pancreas. In the autoimmune disease type 1 diabetes (T1D), which arises from a complicated interplay between genetic and environmental factors, T cells attack and destroy beta cells. During the early stages of the autoimmune process, autoantibodies (AAbs) against pancreatic islets can frequently be detected in the serum, and the presence of multiple AAbs is a strong predictor of T1D progression.1 Although the discovery of insulin was a milestone in T1D research that made the survival of millions of patients possible, insulin therapy fails to provide complete protection against diabetes-associated complications. Recent research has revealed various immune cell types and secreted cytokines responsible for beta cell destruction.2 These findings have led to the development of therapies to slow down or prevent T1D onset. For example, blocking T cells using teplizumab was recently approved by the FDA and has been reported to delay progression to clinical T1D in high-risk participants by 2 years.3 Moreover, multiple clinical trials including tumor necrosis factor-α (TNF-α) inhibition using golimumab4,5 or regulatory T cell-based therapies6 are actively pursued as opportunities to delay T1D onset in at-risk individuals. Despite these breakthroughs and the thrilling prospects presented by ongoing immunotherapy trials for T1D, the unmet clinical need is to reliably identify individuals fated to develop T1D at the earliest possible stages and substantially delay or prevent the disease onset.
In this work, we evaluated the feasibility of modeling early molecular events in tissues relevant to the etiology of T1D using machine learning and single-cell transcriptomic maps of individual cells from pancreatic tissues. The JDRF-supported nPOD7 and the NIDDK-supported HPAP consortia8,9 are ongoing efforts, collecting pancreatic tissues and immune-related organs from hundreds of controls, non-diabetic but islet AAb-positive (AAb+), and T1D organ donors. Among numerous genomics and molecular assays, the revolutionizing single-cell transcriptomics (single-cell RNA sequencing [scRNA-seq]) has become a standard technology to study T1D development. The first series of human donor islets analyzed by our team at HPAP released transcriptional profiles of islets in 24 non-diabetic control (CTL), AAb+, and T1D donors across more than ∼80,000 cells.10 In Fasolino et al., we reported a surprising correlation between the expression level of around 1,000 genes in beta cells, but not any other cell types, with anti-glutamic acid decarboxylase (GAD) AAb levels detected in the serum of AAb+ donors, suggesting that the progression of the autoimmunity process is reflected in the transcriptome of AAb+ beta cells. Despite the new insights gained from scRNA-seq profiling in this study and other reports,11 many questions related to the early molecular events leading to autoimmunity in T1D remain unanswered. For example, it is not clear if there are consensus transcriptional changes associated with T1D in different islet cell populations across the human population. In addition, it remains unknown whether there are any consensus transcriptional changes associated with T1D progression that can be detected at early stages of autoimmunity in AAb+ donors.
Although statistical strategies for differential gene expression analysis have been developed to address such questions, the agreement of differentially expressed genes identified through various approaches is very low,12 and choosing the best approach to select differentially expressed genes is challenging.10,13,14,15,16,17,18 Here, we aimed to model T1D progression using machine learning approaches employing scRNA measurements from 50 organ donors, acquired through the HPAP program. We reasoned that machine learning strategies, which can learn patterns from data, may identify consensus changes in gene expression associated with T1D for cells in pancreatic tissues at prediabetic stages. A machine learning model is trained to perform a task by receiving several examples of input data, such as gene counts as features, with corresponding output labels, e.g., individual cells labeled as T1D, AAb+, or CTL. The model then updates internal parameters to enhance prediction accuracy. We devised a machine learning classifier based on the extreme gradient boosting19 (XGBoost) algorithm and carried out classifications of single cells across the three donor groups. Remarkably, we report that T1D can be modeled by the XGBoost algorithm with high accuracy using solely islet cell transcriptomic data from T1D and CTL donors. Interestingly, our classifier reported T1D-like islet cells in a subset of AAb+ donors, demonstrating that the transcriptional adaptations that occur in islets of patients with T1D are already initiated in some AAb+ donors. Considering the inaccessibility of the pancreatic tissues, our model using single cells from islets cannot be used directly to predict early stages of T1D in living individuals. Nonetheless, our study reports the utility of machine learning algorithms in the early detection of molecular changes in T1D using single-cell transcriptomics.
Results
scRNA-seq data in human pancreatic islets
We took advantage of our most recent release of scRNA-seq experiments in the HPAP program across 50 donors in three groups, namely T1D (n = 9), AAb+ (n = 10), and CTL (n = 31) (Figure 1A). The preprocessing of scRNA-seq data included filtering low-quality cells, doublet removal, and dimensionality reduction, similar to our previously described protocol.20,21 The total number of cells obtained after processing the scRNA-seq data across all conditions was ∼169,000 (Figure 1B). Considering variability in tissue isolation and surgical procedures, different numbers of cells were incorporated from each donor across corresponding conditions (Figures S1A–S1C). We annotated individual cells based on the expression of known marker genes using scSorter22 and reported the frequency of 10 different cell types across conditions (Figure 1C). Overall, acinar, alpha, and beta cells were the largest cell populations with 43,401, 47,988, and 36,837 cells, respectively (Figure 1C). The percentage of acinar and alpha cells was evenly distributed across different donor groups (Figure 1D). Expectedly, beta cells were significantly less abundant in T1D donors compared to other donor groups, reflecting the autoimmune destruction of this cell type (Figures 1D and S1D). Conversely, ductal cells were more abundant in tissues collected from T1D donors than the other two groups, reflecting the difficulty of isolating pure islets from these donors (Figure 1D). The cell-type annotation and composition across different groups were also consistent with previously published studies.10,20,21 The expression of marker genes across major cell types such as acinar (PRSS1), alpha (GCG), beta (INS), delta (SST), ductal (KRT19), endothelial (VWF), epsilon (GHRL), immune (NCF2),pancreatic polypeptide (PP) (PPY), and stellates (COL1A1) further corroborated cell annotations across all the samples combined (Figures 1E and S2). Together, we compiled high-quality transcriptional data generated by HPAP and annotated major cell types in islets of three donor groups.
Figure 1.

scRNA-seq reveals the cell populations of the human pancreatic islets in CTL, AAb+, and T1D donors
(A) The complete workflow depicting the scRNA-seq and machine learning workflow using human pancreatic islet tissue samples.
(B) Pie chart showing the number of cells and donor distribution across different biological conditions.
(C) Uniform manifold approximation and projection (UMAP) plot showing the scSorter cell classification of islet cells.
(D) Stacked bar chart showing the percentage-wise distribution of cell types across AAb+, control, and T1D donors.
(E) Multiple feature plots UMAPs depicting the validation of cell-type-specific expression of marker genes. Acinar cells (PRSS1 high), alpha cells (GCG high), beta cells (INS high), delta cells (SST high), ductal cells (KRT19 high), endothelial cells (VWF high), epsilon cells (GHRL high), immune (NCF2 high), PP cells (PPY high), and stellate cells (COL1A1 high).
Performance of the machine learning model on scRNA-seq islet data
We aimed to devise three binary classifiers using single cells between any pair of donor groups: (1) single cells from T1D vs. single cells from AAb+ donors, (2) single cells from T1D vs. single cells from CTL donors, and (3) single cells from AAb+ vs. single cells from CTL donors. We followed two distinct strategies to build a model across donor groups. In the first strategy, which we refer to as “unannotated” classification, all cells from each donor group were combined and used for training and testing purposes. We reasoned that this approach could take advantage of all cells in our scRNA-seq measurements, improving the performance of our classification. The strategy’s disadvantage is the uneven number of cell types across different donor groups, making the frequency of a particular cell type enriched in a class as the driver of the training process. In the second strategy, which we refer to as “annotated” classification, cells of each donor group with the same annotation, e.g., alpha cells in AAb+ donors, were combined and used for training and testing purposes, leading to the development of one classifier per annotated cell type. The advantage of this approach is that only changes in gene expression of the same cell type would be used to classify cells from different disease groups. However, in this strategy, subsets of cells are utilized for training and testing steps compared to the unannotated approach, potentially influencing the performance of our classification.
For both annotated and unannotated strategies, we divided individual cells into training and testing groups and subjected the training data to hyperparameter optimization (HPO) using the XGBoost algorithm. After performing HPO using a 5-fold cross-validation procedure, we obtained the optimal parameter set, which was used for training and testing the final model. We performed 100 repetitions of the above procedure by randomly shuffling the training data (i.e., random sampling without replacement). In the unannotated XGBoost classifier built for all cells, the T1D vs. AAb+ and T1D vs. CTL binary classifiers performed exceptionally well, averaging ∼99% accuracy, ∼99% sensitivity, and ∼97% specificity. The AAb+ vs. CTL classifier demonstrated an accuracy of ∼96% and a specificity of ∼88%. The relatively small decrease in performance in the AAb+ vs. CTL comparison likely reflects the similarity in transcriptional landscapes of single cells from AAb+ and CTL donors (Figure 2B; Table S1). We also compared the performance of XGBoost with other machine learning models such as a support vector machine (SVM) with linear kernel, a SVM with radial kernel, and naive Bayes methods across all these pairwise comparisons and observed that XGBoost outperformed the other models (Figure S3A; Table S2).
Figure 2.

Classification performance of machine learning model on scRNA-seq islet data
(A) A schematic workflow of XGBoost and performance. The machine-learning-based XGBoost model was built for gene selection and classification. The dotted lines show the training and testing procedures, where T denotes the gradient boosting tree models. The double lines show 100 repetitions of the entire workflow.
(B) Boxplots depicting a pairwise comparison of the XGBoost method across all cells (unannotated) in the dataset.
(C) Performance of XGBoost across major cell types for T1D vs. CTL comparison using boxplots.
(D) Performance of XGBoost across major cell types for T1D vs. AAb+ comparison using boxplots.
(E) Performance of XGBoost across major cell types for AAb+ vs. CTL comparison using boxplots.
In the annotated classification between T1D and CTL groups built for each annotated cell type, the XGBoost method performed exceptionally well on our three metrics in the acinar, alpha, beta, and ductal cells. However, binary classification using delta cells or immune cells, which contained fewer cells compared to other cell types, demonstrated a reduced performance (Figure 2C). Similar results were observed in the T1D vs. AAb+ and AAb+ vs. CTL comparisons (Figures 2D and 2E). Additionally, the average standard deviation in the comparisons of T1D vs. CTL, T1D vs. AAb+, and AAb+ vs. CTL across 100 repetitions were found to be very low (<1%), demonstrating the robustness and stability of XGBoost models (Table S1). Of note, the comparison of XGBoost with SVMs or naive Bayes across annotated cells further demonstrated the superiority of XGBoost models (Figure S3B; Table S2). Taking these results altogether, the annotated XGBoost classifier exhibited high performance across all cell-type comparisons.
Top-ranked genes selected from the machine learning model and pathway enrichment analysis
A major reason for our choice of XGBoost over other machine learning algorithms such as convolutional neural networks is the interpretability and transparency in the XGBoost’s decision-making process. In particular, XGBoost produces feature importance rankings, allowing us to understand which features, i.e., genes, drove predictions. In contrast, neural networks are often considered “black boxes,” making it challenging to interpret their predictions.23 To better understand which features drove the high-performance predictions across single cells, we obtained the key gene signatures for each comparison and used two strategies: (1) we ranked the lists of genes based on the robust ranking algorithm (RRA) approach24 (Tables S3, S4, and S5) and (2) we examined the unranked list of genes based on their selection frequency across 100 repetitions (Tables S6, S7, and S8). These top-selected genes were used for downstream pathway or protein-protein interaction (PPI) analysis to further compare gene signatures associated with three clinical conditions. The ranked list of genes with p < 0.05 based on the RRA approach obtained from the unannotated T1D vs. CTL classifier was enriched with genes annotated as “lipid mRNA metabolic process,” “defense to external biotic,” and “antimicrobial” pathways (Figure 3A). The 20 KEGG pathways (false discovery rate [FDR] < 0.05) enriched within the top features (ranked genes with p < 0.05) of unannotated and annotated T1D vs. CTL classifiers were related to HIV and human papillomavirus infections in addition to cytokine signaling (Figures 3B, S4, and S5).
Figure 3.

Top-ranked genes selected from the machine learning model and pathway enrichment analysis
(A) GO cluster analysis showing pathways based on genes obtained from T1D vs. CTL comparison across unannotated all cells.
(B) Top 20 KEGG pathways based on ranked genes obtained from T1D vs. CTL comparison across all cells (unannotated) and different cell types (annotated) (Table S3).
(C) UpSetR chart showing the common and unique count of genes across annotated cells in T1D vs. CTL comparison.
(D) Pathways based on shared RRA_combined gene list in T1D vs. CTL annotated cells.
(E) UpSetR chart showing the common and unique count of genes across annotated cells in T1D vs. AAb+ comparison.
(F) UpSetR chart showing the common and unique count of genes across annotated cells in AAb+ vs. CTL comparison.
We next aimed to assess whether there are consensus transcriptional changes associated with T1D in different islet cell populations across donors and evaluated commonality among top features (i.e., genes) across different cell types. We applied the RRA approach again but now on the ranked list of genes across cell types. This strategy can produce reliable and consistent rankings even in the presence of noisy or incomplete data. In the T1D vs. CTL classification, while more than 100 genes were uniquely detected in classifiers built on each annotated cell type such as acinar, alpha, ductal, and beta cells, 66 genes were common across classifiers of all annotated cell types based on their high RRA scores, suggesting that changes in the transcriptional outputs of these genes occur across T1D islets independent of their cellular ontogeny (Figure 3C). In particular, “neutrophil degranulation,” “ER-phagosome pathway,” and “pancreatic secretion” were enriched within this set of 66 common genes associated with models based on annotated cell types (Figure 3D). Moreover, in AAb+ vs. CTL and T1D vs. AAb+ classifiers, both common and unique genes across different annotated cell types were detected, suggesting the relevance of multiple pathways to changes in cells from AAb+ donors (Figures 3E and 3F). Hence, despite a clear manifestation of autoimmunity associated with beta cells, our modeling strategy reports shared changes in transcriptional landscapes of distinct cell types of islets. Together, the XGBoost classifiers based on training the model using single cells grouped as different cell types revealed the link between multiple genes and pathways associated with T1D and AAb positivity.
One reason for choosing the XGBoost classifier in our modeling strategy was to extract top features associated with T1D across different cell types. Although statistical approaches performing differential expression (DE) analysis also aim to determine genes with different expression between disease groups, there are numerous concerns related to these approaches, which are described in the discussion. Nonetheless, we also followed two DE analysis approaches as complementary strategies: (1) DE gene lists obtained from individual cells in pairwise comparisons of different donor groups using Wilcoxon rank-sum tests (Tables S9, S10, and S11) and (2) DE gene lists obtained by performing individual donor-wise pseudobulk analysis using DESeq2 (Tables S12, S13, and S14). There was no consensus on these two strategies. Single-cell-based DE analysis reports too many genes to be differentially expressed (∼11,000) and pseudobulk-based DE analysis reports too few genes (∼10) to be differentially expressed. Hence, the DE-based approaches fail to reliably detect disease-associated genes.
Expression of HLA class I genes in beta cells across healthy and T1D donors
We next focused on beta cell classifiers within all three groups and focused on genes with selection frequencies higher than 50% across 100 iterations following the unranked gene selection approach. In particular, we examined the enrichment of highly frequent genes as top features within KEGG pathways (Figures 4A and 4B). Among the top 10 significant pathways (FDR < 0.05), “type 1 diabetes mellitus” and “antigen processing” were found among the highly frequent features of T1D vs. CTL and T1D vs. AAb+ classifiers for beta cells (Tables S15 and S16). The specific genes involved in T1D and antigen processing and presentation pathways were predominantly from HLA class I, i.e., HLA-A, HLA-B, HLA-C, and HLA-E. Additionally, these genes were detected across all 100 repetitions of modeling, suggesting that HLA class I genes were reproducible top features across donors.
Figure 4.

Expression of HLA-1 genes in beta cells across healthy and T1D donors
(A) Comparison of significant KEGG pathways (FDR < 0.05) obtained from different pairwise classifiers.
(B) Significant KEGG pathways for T1D vs. CTL (FDR < 0.05).
(C–E) The top 3 modules were obtained from the PPI network using the MCODE algorithm.
(F) Average expression of beta cells in non-diabetic controls and T1D donors.
Next, we created a PPI network map with significant genes as nodes and their connections as edges. We further applied the MCODE algorithm25 on the PPI network map and obtained three clusters or key modules (Figure 4C). All the HLA class I proteins were grouped into one cluster that was significant (p < 0.05). In the beta cell classification, genes encoding the HLA class I proteins were significantly upregulated in T1D donors compared to CTL donors (Figure 4F). Additional significant clusters contained genes important for mitochondrial function and transcriptional elongation. Next, we analyzed the expression of the HLA class I genes in CTL and T1D beta cells individually (Figure 4F). The dots inside the violin plots represent individual donors, where the cell-level gene counts were aggregated into pseudobulk counts. Strikingly, the HLA class I genes were upregulated within the few remaining beta cells from T1D donors (Figure 4F). Modeling differences in beta cells of T1D and CTL donors suggest that small residues of beta cells in T1D donors express high levels of HLA class I genes. These results are in agreement with prior findings of increased HLA class I expression in islets from patients with T1D obtained using antibody staining.26
We followed a comprehensive and robust resampling approach with over 100 iterations and additional cross-validations. To ensure an unbiased modeling strategy, the test datasets from the 100 iterations in the outer loop were independent of the training set. Nevertheless, we generated scRNA-seq data in new sets of T1D and CTL organ donors and assessed the significance of predicted genes in this confirmatory donor cohort that were not utilized for testing or training (Table S17). We first performed gene set enrichment analysis and examined the enrichment of the top features predicted by our model in up- or down-regulated genes in two T1D donors compared to two CTL donors in this confirmatory cohort. Remarkably, we found that the top predicted genes based on our model were significantly enriched across major cell types such as alpha, immune, and ductal cells in T1D donors (Figure S6A). Moreover, HLA class I genes in addition to other top features predicted by our model demonstrated increased expression levels in T1D, but not CTL, organ donors in this confirmatory cohort (Figure S6B).
Prediction of AAb+ cells using classification models from annotated cells in T1D and CTL donors
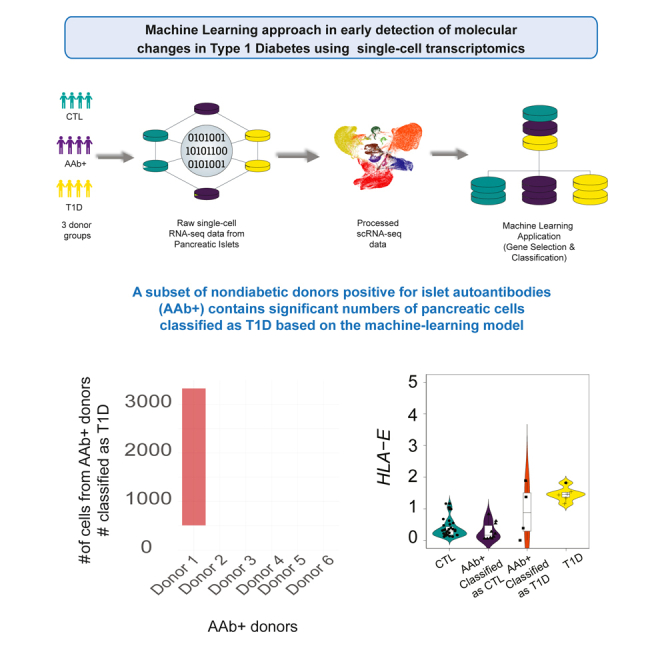
One key goal in this study was to evaluate whether any subset of islet cells from AAb+ organ donors demonstrate transcriptional similarity to those from T1D individuals and, if such a similarity can be modeled, the expression of which genes can classify a single AAb+ cell as a T1D cell. Hence, for each annotated cell type, we used the trained T1D vs. CTL classifier, where cells from T1D donors were labeled as class 1 and cells from CTL donors were labeled as class 0. We tested how this model predicted the class of AAb+ cells. Using the probability scores obtained for each individual cell from the AAb+ donor group, we determined the predicted class of a cell where a probability of >0.5 is classified as T1D and less than 0.5 as CTL. Although 90% of cells from AAb+ donors were predicted to be non-diabetic (class 0), around 10% of cells were classified as T1D across different cell types (class 1) (Figure 5A). Importantly, these “T1D-like” cells were not present at uniform abundance among all organ donors but were highly enriched among specific donors, in particular donors labeled as HPAP092 and HPAP107 (Figure 5A). Strikingly, the beta cells classified as T1D from AAb+ donors had transcriptomic signatures of HLA class I genes extremely similar to the T1D group (Figure 5B). We also compared the expression of alpha cells from AAb+ donors classified as T1D and observed similar results, where HLA class I genes were upregulated in those AAb+ cells that were predicted as T1D (Figure 5C). The pancreatic alpha cells are known to have a key role in the development of diabetes mellitus.27,28 It has been reported that the alpha cells from donors with recent-onset T1D demonstrate reduced glucagon secretion and dysregulated gene expression.29 Another study using immunofluorescence staining showed that the majority of HLA class I genes are expressed on pancreatic alpha cells and are particularly hyperexpressed in the T1D group.30 We further combined all the cells from the two AAb+ donors that were majorly predicted as T1D and compared their HLA class I genes between all groups. We confirmed that the HLA-A, HLA-B, HLA-C, and HLA-E genes were highly upregulated in cells from AAb+ donors predicted as T1D, which might reflect that autoimmunity had already progressed in these AAb+ individuals (Figure 5D). Projection of cells from these donors highlights their distribution across the uniform manifold approximation (UMAP) (Figures S7A and B). In addition, we created a module gene score for HLA class I genes and observed a similar enrichment of this pathway in AAb+ and T1D donor groups compared to CTL donors (Figure S7C). Of note, HPAP107 is an organ donor expressing both GAD and IA-2 AAbs, indicative of further disease progression, while HPAP092 is a single GAD AAb+ donor. Together, our modeling strategy discovered that a subset of cells in specific AAb+ organ donors have transcriptional patterns resembling those typically found in islets of T1D donors.
Figure 5.

Prediction of AAb+ cells using trained T1D-CTL classifier across major cell types
(A) Distribution of cells misclassified as T1D in different cell types.
(B) Comparing the average expression of HLA-I genes among beta cells from AAb+ donors classified as T1D with other conditions.
(C) Comparing the average expression of HLA-I genes among alpha cells from AAb+ donors classified as T1D with other conditions.
(D) Comparing the average expression of HLA-I genes among all cell types from two AAb+ donors classified as T1D with other conditions.
Prediction of AAb+ cells using classification models from unannotated cells in T1D and CTL donors
We next compared transcriptomic differences and similarities between T1D, AAb+, and CTL groups using our pretrained unannotated T1D-CTL classifier. Among ten AAb+ donors, different percentages of cells from six AAb+ donors were classified as T1D, with the majority belonging to the HPAP092 donor (Figure 6A). We next sought to evaluate transcriptional profiles of the AAb+ cells that were predicted as T1D. To provide a reference for these T1D-like cells, we first focused on significant genes obtained in unannotated T1D vs. CTL, T1D vs. AAb+, and AAb+ vs. CTL classifiers (Tables S6, S7, and S8). We checked the gene selection frequency of HLA class I, HLA class II, and non-HLA genes across different unannotated classifiers (Figure 6B). The selection frequency of HLA class I genes was higher compared to some of the HLA class II genes. In addition, we also checked the selection frequency of non-HLA genes including INS, IL32, TNFAIP3, and LMO7 that have been associated with T1D pathology.31,32 The expression of HLA class I and II genes among all the AAb+ cells from HPAP092 classified as T1D had a similar expression pattern to the T1D group (Figures 6D and 6E). Previous studies have shown the inherited risk for T1D to be largely determined by specific HLA-DQA1, HLA-DQB1, HLA-DRA, HLA-DPA1, and HLA-DRB1 alleles.31,33,34 The expression of HLA-DPB1 was found to be higher in endocrine cell types of single-cell islet data from diabetic individuals.10 We also observed a down-regulation of the INS gene in AAb+ cells (HPAP092), which was similar to the T1D group (Figure 6F). Moreover, the IL32, TNFAIP3, and LMO7 genes were found to be upregulated in AAb+ cells predicted to be in the T1D class (HPAP092). Previously, an association to T1D had been reported for these non-HLA genes (INS,31,32 TNFAIP3,32,35 LMO7,32,36 and IL3237). The similarities between the transcript levels of the HLA class I (Figure 6D), HLA class II (Figure 6E), and non-HLA genes (Figure 6F) observed between AAb+ cells predicted as T1D and the actual T1D group suggest that in specific AAb+ organ donors, the pathogenic process toward T1D had progressed further than that in typical AAb+ individuals. Altogether, modeling of the transcriptomic differences between islets cells from T1D and CTL donors using either an annotated or an unannotated classifier revealed individual cells from AAb+ donors with transcriptional similarity to those from T1D donors.
Figure 6.

Prediction of AAb+ cells using trained T1D-CTL classifier across all cells together
(A) Distribution of AAb+ cells predicted as T1D using trained T1D-CTL classifier for all cells.
(B) Selection frequency of genes from HLA-I and HLA-II class.
(C) Selection frequency of genes from non-HLA class relevant to T1D.
(D) Comparing the average expression of HLA-I genes among all cells from HPAP092 AAb+ donor classified as T1D.
(E) Comparing the average expression of HLA-II genes among all cells from HPAP092 AAb+ donor classified as T1D.
(F) Comparing the average expression of non-HLA genes among all cells from HPAP092 AAb+ donor classified as T1D.
Donor-wise classification using the LOOCV strategy
To evaluate the performance accuracy per donor, we applied a unique splitting criterion for testing and training purposes and implemented the leave-one-out cross-validation (LOOCV) strategy. We trained the model based on cells from all donors except one and tested the model’s performance on the one donor left out of the training step. This process was repeated across all donors. Remarkably, in this analysis, the AAb+ donor HPAP092 was classified as T1D across all the annotated cell-type comparisons (Figure S8). For evaluation purposes, we considered LogNormalize data (RNA assay) in addition to the default SCTransform (SCT assay) data. Similar results were observed on the RNA assay where the AAb+ donor HPAP092 was classified as T1D across all the annotated cell types (Figure S9). These results from the LOOCV strategy confirmed of our previous observations (Figure 5A). The mean classification accuracy values for each annotated cell type using the LOOCV strategy were between 80% and 90% for most cell types except ∼72% for the beta cell classifier in both the SCT assay (Figure S10) and RNA assay (Figure S11). This is likely caused by the very few beta cells remaining in several of the donors; for instance, the islets recovered from donors HPAP021 and HPAP022 had only two beta cells each among the total 4,410 and 864 cells analyzed, respectively. This low beta cell count in a subset of T1D donors led to lower overall accuracy by the annotated beta cell classifier of T1D vs. AAb+ using LOOCV. In contrast, when we had employed the annotated beta cell classifier for T1D vs. AAb+ without the LOOCV strategy, all the beta cells were pooled together, leading to high prediction accuracy.
Expression of CXCL8 gene in cell types across healthy, AAb+, and T1D donors
Another key finding based on top predicted features relates to the expression of CXCL8, which is commonly known to be involved in the immune system’s response to inflammation. We observed that among cytokines and chemokines, CXCL8 was the only cytokine other than IL32 that was selected across all training instances for the T1D-CTL comparison (Figure S12A). Next, we checked the selection of CXCL8 across classifiers for annotated cell types and found that among T1D-AAb+ classifiers, CXCL8 was selected majorly in ductal and immune cell types (Figure S12B). We also compared the expression of CXCL8 across individual donors to understand the expression at the donor level. Strikingly, CXCL8 was highly expressed only in one AAb+ donor (HPAP092), who was classified as a T1D donor based on our machine learning approach (Figures S12C and S12F). Evaluating the expression abundance at the cell-type level across conditions demonstrated that ductal, immune, and stellate cells had similar expression patterns for CXCL8 in AAb+ and T1D donors, with the percentage of cells expressing CXCL8 being the highest in T1D (∼55%) and AAb+ donors (∼42%) compared to CTL donors (∼22%) (Figures S12D–S12E). Lastly, we measured the expression of CXCL8 in our independent cohort and observed higher expression in ductal cells of the T1D group (Figure S6C). Together, the results of our machine learning strategy pinpointed changes in gene expression in T1D and AAb cohorts that were reproducibly detected across donors at the single-cell level.
Discussion
Although substantial progress has been made over the past decades in our understanding of the alterations in the pancreas of patients with T1D, the underlying molecular processes of disease progression from healthy to AAb positivity to T1D remain to be elucidated fully. Using scRNA-seq islet data from 50+ human organ donors, we performed a comprehensive analysis by applying a machine learning approach on the large islet gene expression data from T1D, AAb+, and CTL individuals to understand the disease progression at the single-cell level.
There are several tools useful for a broad range of concepts in the single-cell field; however, some of the key measurements, such as DE analysis, remain challenging. Single-cell data are often sparse, heterogeneous, and multidimensional in nature, and it becomes challenging to perform differential state analysis. There is a high level of noise and dropouts (zero values),38 and the data also encompass a large amount of biological variability.39 At present, there are three approaches to performing DE analysis in scRNA-seq data: (1) the individual cell approach where cell-level DE measurements are performed using a negative binomial generalized linear model or Wilcoxon rank-sum tests, (2) the pseudobulk approach where the cell-level counts are aggregated into pseudobulk counts for DE analysis using bulk RNA-seq tools such as edgeR and DESeq2, and (3) mixed modeling with random effects where sample-level inferences are considered. Overall, the individual cell approach developed for scRNA-seq was outperformed by pseudobulk and mixed-modeling approaches40 through a well-controlled FDR. However, these two approaches reveal a lack of consensus for DE analysis.10 While the computational time required for analyzing data through the mixed-modeling approach is understood to be extremely high compared to pseudobulk even in a down-sampled dataset,17,18,40 Zimmerman et al.41 described the pseudobulk approach as conservative, where many DE genes were not detected. Additionally, the agreement of DE genes identified through various approaches was very low.12 Hence, choosing the best approach to select DE genes remains challenging.10
In this study, we focused on the XGBoost method to achieve insights into the prediabetic and diabetic disease states of pancreatic islets. Sparse read counts are a main characteristic of scRNA-seq data, which is important to consider when performing differential gene expression analysis. The superior performance and robustness of the XGBoost method on high-throughput gene expression studies42,43,44,45 led to its increased popularity in single-cell biology,46,47,48,49,50 and this approach remains more powerful compared to other machine learning approaches, including neural networks.50 To the best of our knowledge, there is no literature using the machine learning approach to identify gene signatures and classification of cells into relevant disease states on single-cell data from T1D donors. We demonstrate excellent performance of XGBoost classifiers built for T1D vs. CTL, T1D vs. AAb+, and AAb+ vs. CTL comparisons across all cells and individual cell types compared to other classifiers such as linear or radial SVM and naive Bayes methods. The average accuracy, sensitivity, and specificity obtained from 100 repetitions were found to be higher in the T1D vs. CTL comparison compared to the AAb+ vs. CTL one, likely because of the high similarity of the latter two states. In addition, the top-selected gene lists obtained for each of these comparisons were found to be upregulated in several key pathways, such as the T1D and antigen processing and presentation pathways, with special enrichment seen in beta cells. The genes involved in these pathways belonged to HLA class I; therefore, we measured their individual expression in beta cells and found that they were more highly expressed in T1D donors than in CTL donors. We also evaluated the expression of these genes in an independent cohort and found that the expression levels were indeed consistently upregulated in T1D donors compared to CTL donors.
Increased expression of HLA class I genes in T1D has been reported in the past. Benkahla et al.30 reported hyperexpression of HLA class I genes in T1D donors through immunofluorescence staining and microscopic image analysis. Hamilton-Williams et al.51 used the non-obese diabetic mice model and showed that hyperglycemia was observed in those mice that exhibited higher major histocompatibility complex class I expression in beta cells. Richardson et al.52 performed enteroviral capsid protein vp1 staining on a large cohort of neonatal, pediatric control, and T1D groups and observed hyperexpression only in T1D donors. Nejentsev et al.53 reported the contribution of HLA-A, HLA-B, and HLA-C toward T1D. In contrast to these studies, Skog et al.54 performed staining through immunohistochemical staining to measure protein expression and RNA-seq to measure mRNA expression in non-diabetic controls and patients with T1D; however, they reported no changes in the HLA class I genes across these groups. Using imaging mass cytometry, Wang et al. found HLA-A, -B, and -C expression to be upregulated in islets from short-, but not long-, duration T1D and also overexpressed in beta cells in very recent onset disease.55 Detecting this gene signature in T1D-like AAb+ cells in our study further demonstrates the upregulation of this pathway at the early stages of autoimmunity.
A surprising discovery from this study is the observation that a subset of non-diabetic donors positive for islet AAbs contain significant numbers of pancreatic cells with gene expression profiles that do not resemble AAb−, non-diabetic controls as expected but rather those present in the pancreas of T1D individuals. It is tempting to speculate that those AAb+ individuals with a large proportion of T1D-like pancreatic cells would have been the ones to progress to the diabetic state more rapidly than those in whom these cells are not present. Another surprising finding from our study was the consistent upregulation of CXCL8 in these AAb+ and T1D donors, especially in ductal cells as predicted by our machine learning models. Only few studies discussed the role of CXCL8 in T1D,56,57 with the primary factors of T1D disease being immune dysregulation and inflammation, where several cytokines and chemokines contribute to the inflammatory process; we speculate that CXCL8 would be a potential biomarker in the AAb+ and T1D conditions. Unfortunately, it is impossible to test this hypothesis due to the cross-sectional nature of our study and the fact that the human pancreas cannot be biopsied safely. Nevertheless, this finding provides strong evidence that the transcriptomic changes that occur during the pathogenesis of T1D are not simply a consequence of the hyperglycemic state; rather, they appear to be an integral part of disease progression. Our future studies will focus on examining the utility of machine learning approaches in peripheral blood collected from T1D individuals.
Limitations of the study
In this study, we used scRNA-seq data and applied machine learning approaches to train our models. The machine learning models required large computational resources to train because of the high-dimensional nature of single-cell transcriptomic data. Another limitation is that the biomarkers identified could not be tested due to the cross-sectional nature of our study since human pancreas cannot be biopsied safely.
STAR★Methods
Key resources table
Resource availability
Lead contact
Correspondence and requests for materials should be addressed to Lead Contact, Golnaz Vahedi (vahedi@pennmedicine.upenn.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The pancreatic islet sequencing data and processed scRNA-seq Seurat object can be found in PANCDB (https://hpap.pmacs.upenn.edu/analysis). The scripts used for scRNA-seq data processing and machine learning modeling are available on GitHub (https://github.com/AbhijeetRPatil/ML_Islets). Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
Experimental model and subject details
Our study does not include animals, plants, microbe strains, cell lines, primary cell cultures. Pancreatic islets were procured by the HPAP consortium (RRID:SCR_016202; https://hpap.pmacs.upenn.edu), part of the Human Islet Research Network (https://hirnetwork.org/), with approval from the University of Florida Institutional ReviewBoard (IRB # 201600029) and the United Network for Organ Sharing (UNOS).
Method details
We present an overview of the ML-based XGBoost approach for the classification of pancreatic scRNA-seq islet data from different conditions (Figure 1A). The complete process included the procurement of human islet tissues, the preparation of a single-cell suspension and 10x Genomics sample processing.
We used R programming language62 to perform all the computations, including data pre-processing, ML model building, and downstream calculations and visualizations. The caret,63 dplyr,64 and matrix65 packages were used for data wrangling tasks and Seurat58 for working with the scRNA-seq object. The plots were generated using ggplot2,66 ggpubr,67 and cowplot.68 The ML model was built using XGBoost19 for classification purposes, and the gene selection was performed using Ckmeans.1d.dp.69,70 All ML computations, including hyperparameter optimization tasks, were performed through parallel computing using #cores between 30 to 100.62
scRNA-seq data description and analysis
The single-cell sequencing (scRNA-seq) experiments were performed using the Single-cell 3′ Reagent v2 and v3 kits from 10X Genomics. The detailed clinical information of donors showing their medical history, BMI, age, auto-islet antibody test, HbA1c, and C-peptide levels is provided (Table S17). The libraries were processed using 10X Genomics Cell Ranger v6.1 software which aligns the reads and generates feature-barcode matrices (3' gene expression data) using a collection of several pipelines.71 The experimental details and pre-processing steps for all the samples from pancreatic-islet data were followed as described previously.21,72 We first obtained HPAP samples from different biological conditions such as auto-antibody positive (AAb+; n = 10; cells = 36,244), type 1 diabetes (T1D; n = 9; cells = 34,524), and healthy controls (CTL; n = 31; cells = 123,102). The AAb+ samples were determined based on the AAb+ screening test to measure the levels of antibodies against glutamic acid decarboxylase (GAD) to determine positivity.21,72 The combined feature-barcode matrix, including cells from all HPAP samples, added to a total of 193,870 cells. We pre-processed the raw data following the protocol described previously,21,72 excluding type-2 diabetes (T2D) samples. We also performed an additional quality control step by removing the mitochondrial and ribosomal reads from the raw data. We then used the exact pipeline described in21,72 for downstream analysis using Seurat v4.1.058 for creating the single-cell object, scDblFinder v1.8.059 for removing doublets from the data, SingleCellExperiment v1.16.060 for data wrangling, sctransform v0.3.361 for data transformation through normalization and scaling, and finally scSorter v0.0.222 for cell-type annotations. The final processed and annotated scRNA-seq data object contained a total of 169,027 cells and 30,002 genes for which transcripts could be detected in at least on cell.
Machine learning classification network architecture and training protocol
The annotated scRNA-seq data includes 50 HPAP donor samples with 169,027 cells and 30,002 genes from three groups of CTL (n = 31), AAb+ (n = 10), and T1D (n = 9). The XGBoost machine learning framework is constituted of inner and outer loops. In the outer loop, we first randomly split the pre-processed SCT normalized single-cell data into training and testing sets with 70% and 30% of HPAP samples, respectively. In the inner loop, the training was subjected to hyperparameter tuning, and a 5-fold cross-validation was performed across 200 sub-training model iterations to select the best model with a minimum error rate. We repeated this hyperparameter optimization procedure ten times to select the best weights and parameters. The final best weights and parameters obtained were applied to the complete 70% of the training data matrix to obtain the final best training model. Lastly, we applied 30% of the test data matrix on the best-trained model to evaluate predictions. We used the evaluation metrics of accuracy, sensitivity, and specificity on the test data to measure the performance of the model. Along with these metrics, we also obtained the ranked list of genes, also called the best features, from the best-trained model.
To increase the robustness of our approach and attain reliable results, we repeated this entire process which consists of randomly splitting the single-cell data into training (70%) and testing (30%) by using random sampling without replacement in the outer loop for 100 iterations. The mean and SD classification accuracy, sensitivity, and specificity across 100 XGBoost iterations were calculated for the final average performance evaluation. For the final significant gene selection, we built a matrix of genes X iterations. The ranked list of genes obtained in each outer loop iteration was added to the matrix. We counted the gene occurrences across all 100 iterations and ranked them accordingly. The final significant gene list consisted of the list of genes with the count showing how many times they were selected in the best-trained model in 100 iterations. The complete workflow of the XGBoost scRNA-seq ML framework is shown in Figure 2A. Lastly, we also evaluated several other machine learning models such as support vector machines (SVMs) with linear and radial kernels,73 and naive bayes74 methods for comparing the performance with XGBoost method.
XGBoost method
eXtreme Gradient Boosting (XGBoost)19 is an extension of gradient-boosted decision trees (GBDT) and is specifically optimized to provide faster computations through parallel and distributed computing. This ensemble learning method uses a boosting approach to improve prediction accuracy by building many aggregated trees to form a single consensus prediction model.75 The least squares loss function is used to reduce the loss.76 The XGBoost method creates trees, and the residuals obtained from previous trees are given as input to the subsequent tree, which improves the overall prediction by modeling the errors. For each tree sequence, a subsample of training data is randomly drawn without replacement from the entire training data and is used to fit the tree and compute the model update. Additional trees are not allowed to be added after reaching the pre-specified maximum threshold or if the models converges and the performance does not improve which helps to avoid over parametrization. Overall, this highly effective scalable tree boosting system was originally proposed for sparse data and weighted quantile sketch for approximate tree learning.19 XGBoost method is applied in a wide range of applications such as regression, classification, ranking, and user-defined prediction problems.
Feature importance score
The trained XGBoost model automatically provides a feature importance score.77 The score indicates how important the feature (gene in this study) was used for the model’s prediction. The feature importance score was obtained for each trained model. We selected the genes with non-zero importance scores for all models. We obtained the ranked lists of genes across 100 repetitions of train-test splits.
Hyperparameter optimization (HPO)
The XGBoost method uses several parameters to control the bias-variance tradeoffs. The tree-based boosting models could suffer from overfitting; however, the XGBoost method provides several parameters, such as maximum tree depth, minimum leaf weight, and minimum split gain, which helps to avoid overfitting. Additionally, it adds randomness to the model during the training phase, making it more robust to noise. Following are the list of parameters set during cross validation on training, max_depth-the maximum depth of the tree between 3 and 7, gamma-represents the minimum loss reduction required to make a further partition on a leaf node of the tree and it was set between 0 and 0.2, eta-the step size of each boosting step was set using random uniform distribution between 0.01 and 0.3, the min_child_weight-was set to 30 as large number is usually conservative, subsample-the subsample ratio will randomly sample the training data prior to growing trees which will avoid overfitting was set between 0.5 and 0.8 using random uniform distribution, colsample_bytree-was also set between 0.5 and 0.8, cv.nrounds-200 represents the maximum number of rounds for cross validation, and the cv.nfold = 5 shows the 5-fold cross validation, early_stopping_rounds = 100 which helps the model to stop training further if the best performance was achieved, eval_metric=logloss was used to minimize the loss, Binary=logistic was used for performing logistic regression for binary classification and output the probabilities, nthreads = 30, for parallelization of all the tasks. In each inner loop, we iterated ten times over the list of above-described parameters to identify the best parameters and used them to train the XGBoost model and test the predictions through the test dataset. Lastly, this procedure was repeated 100 times in the outer loop, and the average performance metrics were calculated.
Leave one out cross-validation strategy (LOOCV)
We used a special case of k-fold cross-validation called LOOCV where for each sample in the dataset, removing that sample (testing), training was performed on all the remaining samples. We used the LOOCV instead of previous criteria of training (70%) and testing (30%), where random sampling without replacement was considered in the outer loop for 100 iterations. The same parameters previously mentioned in HPO pipeline were used here for optimizing the model during training. The LOOCV helps to classify disease vs. normal group at subject level where the subject being tested is classified as either disease or normal.
Evaluating performance
We measure the performance of XGBoost models using averages of accuracy, sensitivity, and specificity obtained across 100 iterations. The equations for these metrics with respect to true positive (TP), true negative (TN), false negative (FN), and false positive (FP) are as follows:
The classification accuracy of the model is defined as the ratio of correctly predicted instances (TP + TN) to the total number of instances in the dataset (TP + TN + FP + FN)
Sensitivity measures the model’s ability to correctly identify positive instances (TP) out of all the instances that are actually positive (TP + FN).
And finally, Specificity shows the model’s ability to correctly identify negative instances (TN) out of all the instances that are actually negative (TN + FP).
Gene selection and pathway enrichment analysis
We obtained the ranked lists across 100 repetitions of train-test splits (Methods, section- Feature Importance score) and followed two strategies with different criteria to obtain list of ranked gene lists:
Ranked gene selection
We aggregated the ranked lists across 100 repetitions by preserving the ranks by applying the robust rank aggregation (RRA)24 method to obtain the final ranked list of genes in a given comparison. RRA is a statistical technique used to combine rankings from multiple sources into a single, aggregated ranking that is robust to noise, inconsistencies, and outliers in the individual rankings. The RRA method assigns scores in terms of P-values for each gene to determine the significance level. We used genes with P-value <0.05 for the final ranked list of genes for all comparisons (Table S3. Ranked significant gene lists obtained through RRA approach (p < 0.05) in unannotated (all cells) and annotated cells (acinar, alpha, beta, delta, ductal, immune, endothelial, and stellates) for T1D vs. CTL pairwise comparison related to Figure 3, Table S4. Ranked significant gene obtained through RRA approach (p < 0.05) in unannotated (all cells) and annotated cells (acinar, alpha, beta, delta, ductal, immune, endothelial, and stellates) for T1D vs. AAb+ pairwise comparison, related to Figure 3, Table S5. Ranked significant gene lists obtained through RRA approach (p < 0.05) in unannotated (all cells) and annotated cells (acinar, alpha, beta, delta, ductal, immune, endothelial, and stellates) for AAb+ vs. CTL pairwise comparison, related to Figure 3, Table S18. GO-BP and KEGG pathways for unannotated (all cells) and annotated cells (acinar, alpha, beta, delta, ductal, immune, endothelial, and stellates) using clusterProfiler in T1D vs. CTL pairwise comparison based on ranked gene lists based on RRA (p < 0.05) from Table S3, related to Figure 3, Table S19. GO-BP and KEGG pathways for unannotated (all cells) and annotated cells (acinar, alpha, beta, delta, ductal, immune, endothelial, and stellates) using clusterProfiler in T1D vs. AAb+ pairwise comparison based on ranked gene lists based on RRA (p < 0.05) from Table S4, related to Figure 3, Table S20. GO-BP and KEGG pathways for unannotated (all cells) and annotated cells (acinar, alpha, beta, delta, ductal, immune, endothelial, and stellates) using clusterProfiler in AAb+ vs. CTL pairwise comparison based on ranked gene lists based on RRA (p < 0.05) from Table S5, related to Figure 3, Table S21. Shared list of ranked genes called RRA_combined obtained by combining gene lists from Table S3 using RRA approach in T1D vs. CTL, related to Figure 3, Table S21. Shared list of ranked genes called RRA_combined obtained by combining gene lists from Table S3 using RRA approach in T1D vs. CTL, related to Figure 3, Table S22. Shared list of ranked genes called RRA_combined obtained by combining gene lists from Table S4 using RRA approach in T1D vs. AAb+, related to Figure 3, Table S22. Shared list of ranked genes called RRA_combined obtained by combining gene lists from Table S4 using RRA approach in T1D vs. AAb+, related to Figure 3, Table S23. Shared list of ranked genes called RRA_combined obtained by combining gene lists from Table S5 using RRA approach in AAb+ vs. CTL, related to Figure 3, Table S23. Shared list of ranked genes called RRA_combined obtained by combining gene lists from Table S5 using RRA approach in AAb+ vs. CTL, related to Figure 3).
Unranked gene selection
We aggregated the ranked lists across 100 repetitions by ignoring the ranks and focusing on the number of times a gene was selected across 100 repetitions. For example, in beta cells of T1D vs. CTL classifier results, if gene INS is selected 80 times among 100 repetitions, then a selection frequency score of 80 was assigned to INS. The final ordered list of genes with score as gene selection frequency among 100 repetitions for all comparisons (Table S6. Unranked significant gene lists with gene selection frequency in unannotated (all cells) and annotated cells (acinar, alpha, beta, delta, ductal, immune, endothelial, and stellates) for T1D vs. CTL pairwise comparison, related to Figure 3, Table S7. Unranked significant gene lists with selection frequency in unannotated (all cells) and annotated cells (acinar, alpha, beta, delta, ductal, immune, endothelial, and stellates) for T1D vs. AAb+ pairwise comparison, related to Figure 3, Table S8. Unranked significant gene lists with selection frequency in unannotated (all cells) and annotated cells (acinar, alpha, beta, delta, ductal, immune, endothelial, and stellates) for AAb+ vs. CTL pairwise comparison, related to Figure 3, Table S15. GO (GO-BP, GO-MF, GO-CC) and KEGG pathways for unannotated (all cells) and annotated cells (acinar, alpha, beta, delta, ductal, immune, endothelial, and stellates) using DAVID in T1D vs. CTL pairwise comparison based on unranked gene lists based on gene selection frequency >50 from Table S6, related to Figure 3, Table S16. GO (GO-BP, GO-MF, GO-CC) and KEGG pathways for unannotated (all cells) and annotated cells (acinar, alpha, beta, delta, ductal, immune, endothelial, and stellates) using DAVID in T1D vs. AAb+ pairwise comparison based on unranked gene lists based on gene selection frequency >50 from Table S7, related to Figure 3, Table S24. GO (GO-BP, GO-MF, GO-CC) and KEGG pathways for unannotated (all cells) and annotated cells (acinar, alpha, beta, delta, ductal, immune, endothelial, and stellates) using DAVID in AAb+ vs. CTL pairwise comparison based on unranked gene lists based on gene selection frequency >50 from Table S8, related to Figure 3).
For the ranked list of genes, we performed pathway analysis using the ClusterProfiler78 tool. The clusters were created for the gene ontology (GO) biological process (BP) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways based on the ranked list of genes across each comparison. Our analysis determined the GO biological processes BP and KEGG pathways with an adjusted P-value <0.05 as statistically significant (Table S18. GO-BP and KEGG pathways for unannotated (all cells) and annotated cells (acinar, alpha, beta, delta, ductal, immune, endothelial, and stellates) using clusterProfiler in T1D vs. CTL pairwise comparison based on ranked gene lists based on RRA (p < 0.05) from Table S3, related to Figure 3, Table S19. GO-BP and KEGG pathways for unannotated (all cells) and annotated cells (acinar, alpha, beta, delta, ductal, immune, endothelial, and stellates) using clusterProfiler in T1D vs. AAb+ pairwise comparison based on ranked gene lists based on RRA (p < 0.05) from Table S4, related to Figure 3, Table S20. GO-BP and KEGG pathways for unannotated (all cells) and annotated cells (acinar, alpha, beta, delta, ductal, immune, endothelial, and stellates) using clusterProfiler in AAb+ vs. CTL pairwise comparison based on ranked gene lists based on RRA (p < 0.05) from Table S5, related to Figure 3). Next, we created a shared list of ranked genes using the RRA method and performed GO and KEGG pathway analysis using clusterProfiler78 and Metascape,79 based on the ranked gene lists obtained for different cell types. The ranked gene lists across different cell types in each comparison (Table S3. Ranked significant gene lists obtained through RRA approach (p < 0.05) in unannotated (all cells) and annotated cells (acinar, alpha, beta, delta, ductal, immune, endothelial, and stellates) for T1D vs. CTL pairwise comparison related to Figure 3, Table S4. Ranked significant gene obtained through RRA approach (p < 0.05) in unannotated (all cells) and annotated cells (acinar, alpha, beta, delta, ductal, immune, endothelial, and stellates) for T1D vs. AAb+ pairwise comparison, related to Figure 3, Table S5. Ranked significant gene lists obtained through RRA approach (p < 0.05) in unannotated (all cells) and annotated cells (acinar, alpha, beta, delta, ductal, immune, endothelial, and stellates) for AAb+ vs. CTL pairwise comparison, related to Figure 3, Table S18. GO-BP and KEGG pathways for unannotated (all cells) and annotated cells (acinar, alpha, beta, delta, ductal, immune, endothelial, and stellates) using clusterProfiler in T1D vs. CTL pairwise comparison based on ranked gene lists based on RRA (p < 0.05) from Table S3, related to Figure 3, Table S19. GO-BP and KEGG pathways for unannotated (all cells) and annotated cells (acinar, alpha, beta, delta, ductal, immune, endothelial, and stellates) using clusterProfiler in T1D vs. AAb+ pairwise comparison based on ranked gene lists based on RRA (p < 0.05) from Table S4, related to Figure 3, Table S20. GO-BP and KEGG pathways for unannotated (all cells) and annotated cells (acinar, alpha, beta, delta, ductal, immune, endothelial, and stellates) using clusterProfiler in AAb+ vs. CTL pairwise comparison based on ranked gene lists based on RRA (p < 0.05) from Table S5, related to Figure 3, Table S21. Shared list of ranked genes called RRA_combined obtained by combining gene lists from Table S3 using RRA approach in T1D vs. CTL, related to Figure 3, Table S21. Shared list of ranked genes called RRA_combined obtained by combining gene lists from Table S3 using RRA approach in T1D vs. CTL, related to Figure 3, Table S22. Shared list of ranked genes called RRA_combined obtained by combining gene lists from Table S4 using RRA approach in T1D vs. AAb+, related to Figure 3, Table S22. Shared list of ranked genes called RRA_combined obtained by combining gene lists from Table S4 using RRA approach in T1D vs. AAb+, related to Figure 3, Table S23. Shared list of ranked genes called RRA_combined obtained by combining gene lists from Table S5 using RRA approach in AAb+ vs. CTL, related to Figure 3, Table S23. Shared list of ranked genes called RRA_combined obtained by combining gene lists from Table S5 using RRA approach in AAb+ vs. CTL, related to Figure 3) were given as input to the RRA method. The obtained shared list of ranked genes was called “RRA_combined” and reported along with the corresponding GO and KEGG pathways (Table S21. Shared list of ranked genes called RRA_combined obtained by combining gene lists from Table S3 using RRA approach in T1D vs. CTL, related to Figure 3, Table S21. Shared list of ranked genes called RRA_combined obtained by combining gene lists from Table S3 using RRA approach in T1D vs. CTL, related to Figure 3, Table S22. Shared list of ranked genes called RRA_combined obtained by combining gene lists from Table S4 using RRA approach in T1D vs. AAb+, related to Figure 3, Table S22. Shared list of ranked genes called RRA_combined obtained by combining gene lists from Table S4 using RRA approach in T1D vs. AAb+, related to Figure 3, Table S23. Shared list of ranked genes called RRA_combined obtained by combining gene lists from Table S5 using RRA approach in AAb+ vs. CTL, related to Figure 3, Table S23. Shared list of ranked genes called RRA_combined obtained by combining gene lists from Table S5 using RRA approach in AAb+ vs. CTL, related to Figure 3).
For the unranked list of genes, the publicly available databases such as the ‘database for annotation, visualization, and integrated discovery’ (DAVID, version 6.8) bioinformatics web server80 was used to identify various biological pathways of significant genes through a set of functional annotation tools. We first filtered high confidence genes by selection genes with a selection frequency of greater than 50 and used the GO81,82 and the KEGG83,84,85 databases for pathway enrichment analysis in the DAVID database. The GO classifies the gene functionalities into three categories: biological processes (BP), cellular component (CC), and molecular function (MF), and the KEGG database provides an overview of high-level gene functions and biological signaling pathways. Our analysis determined the GO and KEGG terms with false discovery rate (FDR) < 0.05 as statistically significant (Tables S15, S16, and S24).
Protein-protein interaction networks and gene modules selection
The Search Tool for Retrieval of Interacting proteins database (STRINGdb v11)86 was used to identify the various protein-protein interaction networks (PPI) of significant gene lists based on the medium confidence score of 0.7. The loaded PPI network from STRINGdb86 was analyzed using the open-source Cytoscape87,88 tool. Cytoscape is a bioinformatics software used for visualizing and integrating highly complex PPI networks through several supported plugins. The StringApp87 within Cytoscape is used to load the raw PPI network, and the network analyzer89 plugin is used to measure the degree of interaction between nodes and display the up/down-regulated genes. Finally, we applied the molecular complex detection (MCODE)25 clustering-based algorithm for further splitting the network into modules/clusters that helped identify the densely connected regions. We used the default parameters (degree cutoff = 2, node score cut-off = 0.2, k-core = 2, and max.depth = 100) to filter and identify key clusters in the network. We selected the top modules from each analysis group to show the degree of interactions.
Differential expression analysis
The differential expression (DE) analysis was performed on the LogNormalized data using Seurat’s ‘FindMarkers’ function58 where the DE test uses non-parametric Wilcoxon rank-sum test as default to test for the DE genes between two groups of cells within same cell type across different conditions. Here, each cell is treated as an independent replicate. We also performed pseudobulk analysis where the gene counts of all cells within donors were aggregated using Seurat’s ‘AggregateExpression’ and the sample level DE analysis was performed using DESeq2.90 In both individual single-cell and pseudobulk strategies, the significant DE genes were filtered based on threshold of adjusted p-value <0.05.
Quantification and statistical analysis
The differentially expressed genes were determined based on adjusted p-value <0.05 in both individual cells and Pseudobulk strategies using non-parametric Wilcoxon rank-sum test and DESeq2 approaches respectively.
Acknowledgments
We thank the Vahedi, Faryabi, Naji, and Kaestner lab members for discussions. This work was supported by National Institutes of Health grants UC4 DK112217, U01DK112217 (A.N., K.H.K., R.B.F., and G.V.), R01HL145754, and U01DK127768 and awards from the Burroughs Wellcome Fund, the Chan Zuckerberg Initiative, the W.W. Smith Charitable Trust, and the Sloan Foundation (G.V.).
Author contributions
A.N. and C.L. performed organ procurement; D.L. and H.C.D. generated scRNA-seq libraries in the K.H.K. lab; all computational analyses in this work were performed by A.R.P. with some help from R.B.F., J.S., and G.V.; the original draft was prepared by A.R.P.; and G.V. and A.R.P. edited and revised drafts. All authors have read and agreed to the published version of the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: April 26, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101535.
Supplemental information
The corresponding GO and KEGG pathways for shared ranked genes are also shown.
The corresponding GO and KEGG pathways for shared ranked genes are also shown.
The corresponding GO and KEGG pathways for shared ranked genes are also shown.
References
- 1.Ziegler A.-G., Kick K., Bonifacio E., Haupt F., Hippich M., Dunstheimer D., Lang M., Laub O., Warncke K., Lange K., et al. Yield of a Public Health Screening of Children for Islet Autoantibodies in Bavaria, Germany. JAMA. 2020;323:339–351. doi: 10.1001/jama.2019.21565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bluestone J.A., Buckner J.H., Herold K.C. Immunotherapy: Building a bridge to a cure for type 1 diabetes. Science. 2021;373:510–516. doi: 10.1126/science.abh1654. [DOI] [PubMed] [Google Scholar]
- 3.Herold K.C., Bundy B.N., Long S.A., Bluestone J.A., DiMeglio L.A., Dufort M.J., Gitelman S.E., Gottlieb P.A., Krischer J.P., Linsley P.S., et al. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N. Engl. J. Med. 2019;381:603–613. doi: 10.1056/NEJMoa1902226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quattrin T., Haller M.J., Steck A.K., Felner E.I., Li Y., Xia Y., Leu J.H., Zoka R., Hedrick J.A., Rigby M.R., et al. Golimumab and Beta-Cell Function in Youth with New-Onset Type 1 Diabetes. N. Engl. J. Med. 2020;383:2007–2017. doi: 10.1056/NEJMoa2006136. [DOI] [PubMed] [Google Scholar]
- 5.Rigby M.R., Hayes B., Li Y., Vercruysse F., Hedrick J.A., Quattrin T. Two-Year Follow-up From the T1GER Study: Continued Off-Therapy Metabolic Improvements in Children and Young Adults With New-Onset T1D Treated With Golimumab and Characterization of Responders. Diabetes Care. 2023;46:561–569. doi: 10.2337/dc22-0908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bettini M., Bettini M.L. Function, Failure, and the Future Potential of Tregs in Type 1 Diabetes. Diabetes. 2021;70:1211–1219. doi: 10.2337/dbi18-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perry D.J., Shapiro M.R., Chamberlain S.W., Kusmartseva I., Chamala S., Balzano-Nogueira L., Yang M., Brant J.O., Brusko M., Williams M.D., et al. A genomic data archive from the Network for Pancreatic Organ donors with Diabetes. Sci. Data. 2023;10:323. doi: 10.1038/s41597-023-02244-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaestner K.H., Powers A.C., Naji A., Atkinson M.A., HPAP Consortium NIH Initiative to Improve Understanding of the Pancreas, Islet, and Autoimmunity in Type 1 Diabetes: The Human Pancreas Analysis Program (HPAP) Diabetes. 2019;68:1394–1402. doi: 10.2337/db19-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shapira S.N., Naji A., Atkinson M.A., Powers A.C., Kaestner K.H. Understanding islet dysfunction in type 2 diabetes through multidimensional pancreatic phenotyping: The Human Pancreas Analysis Program. Cell Metab. 2022;34:1906–1913. doi: 10.1016/j.cmet.2022.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fasolino M., Schwartz G.W., Patil A.R., Mongia A., Golson M.L., Wang Y.J., Morgan A., Liu C., Schug J., Liu J., et al. Single-cell multi-omics analysis of human pancreatic islets reveals novel cellular states in type 1 diabetes. Nat. Metab. 2022;4:284–299. doi: 10.1038/s42255-022-00531-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chiou J., Geusz R.J., Okino M.L., Han J.Y., Miller M., Melton R., Beebe E., Benaglio P., Huang S., Korgaonkar K., et al. Interpreting type 1 diabetes risk with genetics and single-cell epigenomics. Nature. 2021;594:398–402. doi: 10.1038/s41586-021-03552-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang T., Li B., Nelson C.E., Nabavi S. Comparative analysis of differential gene expression analysis tools for single-cell RNA sequencing data. BMC Bioinf. 2019;20:40. doi: 10.1186/s12859-019-2599-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He L., Davila-Velderrain J., Sumida T.S., Hafler D.A., Kellis M., Kulminski A.M. NEBULA is a fast negative binomial mixed model for differential or co-expression analysis of large-scale multi-subject single-cell data. Commun. Biol. 2021;4:629. doi: 10.1038/s42003-021-02146-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kharchenko P.V., Silberstein L., Scadden D.T. Bayesian approach to single-cell differential expression analysis. Nat. Methods. 2014;11:740–742. doi: 10.1038/nmeth.2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miao Z., Deng K., Wang X., Zhang X. DEsingle for detecting three types of differential expression in single-cell RNA-seq data. Bioinformatics. 2018;34:3223–3224. doi: 10.1093/bioinformatics/bty332. [DOI] [PubMed] [Google Scholar]
- 16.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Squair J.W., Gautier M., Kathe C., Anderson M.A., James N.D., Hutson T.H., Hudelle R., Qaiser T., Matson K.J.E., Barraud Q., et al. Confronting false discoveries in single-cell differential expression. Nat. Commun. 2021;12:5692. doi: 10.1038/s41467-021-25960-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thurman A.L., Ratcliff J.A., Chimenti M.S., Pezzulo A.A. Differential gene expression analysis for multi-subject single cell RNA sequencing studies with aggregateBioVar. Bioinformatics. 2021;37:3243–3251. doi: 10.1093/bioinformatics/btab337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen, T. & Guestrin, C. in Proceedings of the 22nd acm sigkdd international conference on knowledge discovery and data mining. 785-794.
- 20.Patil A.R., Schug J., Naji A., Kaestner K.H., Faryabi R.B., Vahedi G. Single-cell expression profiling of islets generated by the Human Pancreas Analysis Program. Nat. Metab. 2023;5:713–715. doi: 10.1038/s42255-023-00806-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patil A.R., Schug J., Naji A., Kaestner K.H., Faryabi R.B., Vahedi G., HPAP Consortium Computational workflow and interactive analysis of single-cell expression profiling of islets generated by the Human Pancreas Analysis Program. bioRxiv. 2023 doi: 10.1101/2023.01.03.522578. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo H., Li J. scSorter: assigning cells to known cell types according to marker genes. Genome Biol. 2021;22 doi: 10.1186/s13059-021-02281-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swanson K., Wu E., Zhang A., Alizadeh A.A., Zou J. From patterns to patients: Advances in clinical machine learning for cancer diagnosis, prognosis, and treatment. Cell. 2023;186:1772–1791. doi: 10.1016/j.cell.2023.01.035. [DOI] [PubMed] [Google Scholar]
- 24.Kolde R., Laur S., Adler P., Vilo J. Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics. 2012;28:573–580. doi: 10.1093/bioinformatics/btr709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bader G.D., Hogue C.W.V. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinf. 2003;4:2. doi: 10.1186/1471-2105-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richardson S.J., Rodriguez-Calvo T., Gerling I.C., Mathews C.E., Kaddis J.S., Russell M.A., Zeissler M., Leete P., Krogvold L., Dahl-Jørgensen K., et al. Islet cell hyperexpression of HLA class I antigens: a defining feature in type 1 diabetes. Diabetologia. 2016;59:2448–2458. doi: 10.1007/s00125-016-4067-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gromada J., Chabosseau P., Rutter G.A. The alpha-cell in diabetes mellitus. Nat. Rev. Endocrinol. 2018;14:694–704. doi: 10.1038/s41574-018-0097-y. [DOI] [PubMed] [Google Scholar]
- 28.Doliba N.M., Rozo A.V., Roman J., Qin W., Traum D., Gao L., Liu J., Manduchi E., Liu C., Golson M.L., et al. alpha Cell dysfunction in islets from nondiabetic, glutamic acid decarboxylase autoantibody-positive individuals. J. Clin. Invest. 2022;132 doi: 10.1172/JCI156243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brissova M., Haliyur R., Saunders D., Shrestha S., Dai C., Blodgett D.M., Bottino R., Campbell-Thompson M., Aramandla R., Poffenberger G., et al. α Cell Function and Gene Expression Are Compromised in Type 1 Diabetes. Cell Rep. 2018;22:2667–2676. doi: 10.1016/j.celrep.2018.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Benkahla M.A., Sabouri S., Kiosses W.B., Rajendran S., Quesada-Masachs E., von Herrath M.G. HLA class I hyper-expression unmasks beta cells but not alpha cells to the immune system in pre-diabetes. J. Autoimmun. 2021;119 doi: 10.1016/j.jaut.2021.102628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Redondo M.J., Steck A.K., Pugliese A. Genetics of type 1 diabetes. Pediatr. Diabetes. 2018;19:346–353. doi: 10.1111/pedi.12597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klak M., Gomółka M., Kowalska P., Cichoń J., Ambrożkiewicz F., Serwańska-Świętek M., Berman A., Wszoła M., et al. Type 1 diabetes: genes associated with disease development. Cent. Eur. J. Immunol. 2020;45:439–453. doi: 10.5114/ceji.2020.103386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pociot F., McDermott M.F. Genetics of type 1 diabetes mellitus. Genes Immun. 2002;3:235–249. doi: 10.1038/sj.gene.6363875. [DOI] [PubMed] [Google Scholar]
- 34.Russell M.A., Redick S.D., Blodgett D.M., Richardson S.J., Leete P., Krogvold L., Dahl-Jørgensen K., Bottino R., Brissova M., Spaeth J.M., et al. HLA Class II Antigen Processing and Presentation Pathway Components Demonstrated by Transcriptome and Protein Analyses of Islet β-Cells From Donors With Type 1 Diabetes. Diabetes. 2019;68:988–1001. doi: 10.2337/db18-0686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fung E.Y.M.G., Smyth D.J., Howson J.M.M., Cooper J.D., Walker N.M., Stevens H., Wicker L.S., Todd J.A. Analysis of 17 autoimmune disease-associated variants in type 1 diabetes identifies 6q23/TNFAIP3 as a susceptibility locus. Genes Immun. 2009;10:188–191. doi: 10.1038/gene.2008.99. [DOI] [PubMed] [Google Scholar]
- 36.Bradfield J.P., Qu H.Q., Wang K., Zhang H., Sleiman P.M., Kim C.E., Mentch F.D., Qiu H., Glessner J.T., Thomas K.A., et al. A Genome-Wide Meta-Analysis of Six Type 1 Diabetes Cohorts Identifies Multiple Associated Loci. PLoS Genet. 2011;7 doi: 10.1371/journal.pgen.1002293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Albuquerque R., Komsi E., Starskaia I., Ullah U., Lahesmaa R. The role of Interleukin-32 in autoimmunity. Scand. J. Immunol. 2021;93 doi: 10.1111/sji.13012. [DOI] [PubMed] [Google Scholar]
- 38.Kiselev V.Y., Andrews T.S., Hemberg M. Challenges in unsupervised clustering of single-cell RNA-seq data. Nat. Rev. Genet. 2019;20:273–282. doi: 10.1038/s41576-018-0088-9. [DOI] [PubMed] [Google Scholar]
- 39.Chen G., Ning B., Shi T. Single-Cell RNA-Seq Technologies and Related Computational Data Analysis. Front. Genet. 2019;10:317. doi: 10.3389/fgene.2019.00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crowell H.L., Soneson C., Germain P.L., Calini D., Collin L., Raposo C., Malhotra D., Robinson M.D. muscat detects subpopulation-specific state transitions from multi-sample multi-condition single-cell transcriptomics data. Nat. Commun. 2020;11:6077. doi: 10.1038/s41467-020-19894-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zimmerman K.D., Espeland M.A., Langefeld C.D. A practical solution to pseudoreplication bias in single-cell studies. Nat. Commun. 2021;12:738. doi: 10.1038/s41467-021-21038-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li W., Yin Y., Quan X., Zhang H. Gene Expression Value Prediction Based on XGBoost Algorithm. Front. Genet. 2019;10:1077. doi: 10.3389/fgene.2019.01077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Q., Yang H., Wang P., Liu X., Lv K., Ye M. XGBoost-based and tumor-immune characterized gene signature for the prediction of metastatic status in breast cancer. J. Transl. Med. 2022;20:177. doi: 10.1186/s12967-022-03369-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shen C., Li H., Li M., Niu Y., Liu J., Zhu L., Gui H., Han W., Wang H., Zhang W., et al. DLRAPom: a hybrid pipeline of Optimized XGBoost-guided integrative multiomics analysis for identifying targetable disease-related lncRNA-miRNA-mRNA regulatory axes. Brief. Bioinform. 2022;23 doi: 10.1093/bib/bbac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Y., Kang K., Krahn J.M., Croutwater N., Lee K., Umbach D.M., Li L. A comprehensive genomic pan-cancer classification using The Cancer Genome Atlas gene expression data. BMC Genom. 2017;18:508. doi: 10.1186/s12864-017-3906-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Galdos F.X., Xu S., Goodyer W.R., Duan L., Huang Y.V., Lee S., Zhu H., Lee C., Wei N., Lee D., Wu S.M. devCellPy is a machine learning-enabled pipeline for automated annotation of complex multilayered single-cell transcriptomic data. Nat. Commun. 2022;13:5271. doi: 10.1038/s41467-022-33045-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lieberman Y., Rokach L., Shay T. CaSTLe – Classification of single cells by transfer learning: Harnessing the power of publicly available single cell RNA sequencing experiments to annotate new experiments. PLoS One. 2018;13 doi: 10.1371/journal.pone.0205499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Le H., Peng B., Uy J., Carrillo D., Zhang Y., Aevermann B.D., Scheuermann R.H. Machine learning for cell type classification from single nucleus RNA sequencing data. PLoS One. 2022;17 doi: 10.1371/journal.pone.0275070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen Y., Zhang S. Automatic Cell Type Annotation Using Marker Genes for Single-Cell RNA Sequencing Data. Biomolecules. 2022;12 doi: 10.3390/biom12101539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Köhler N.D., Büttner M., Andriamanga N., Theis F.J. Deep learning does not outperform classical machine learning for cell-type annotation. bioRxiv. 2021 doi: 10.1101/653907. Preprint at. [DOI] [Google Scholar]
- 51.Hamilton-Williams E.E., Palmer S.E., Charlton B., Slattery R.M. Beta cell MHC class I is a late requirement for diabetes. Proc. Natl. Acad. Sci. USA. 2003;100:6688–6693. doi: 10.1073/pnas.1131954100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Richardson S.J., Willcox A., Bone A.J., Foulis A.K., Morgan N.G. The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia. 2009;52:1143–1151. doi: 10.1007/s00125-009-1276-0. [DOI] [PubMed] [Google Scholar]
- 53.Nejentsev S., Howson J.M.M., Walker N.M., Szeszko J., Field S.F., Stevens H.E., Reynolds P., Hardy M., King E., Masters J., et al. Localization of type 1 diabetes susceptibility to the MHC class I genes HLA-B and HLA-A. Nature. 2007;450:887–892. doi: 10.1038/nature06406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Skog O., Korsgren S., Wiberg A., Danielsson A., Edwin B., Buanes T., Krogvold L., Korsgren O., Dahl-Jørgensen K. Expression of Human Leukocyte Antigen Class I in Endocrine and Exocrine Pancreatic Tissue at Onset of Type 1 Diabetes. Am. J. Pathol. 2015;185:129–138. doi: 10.1016/j.ajpath.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 55.Wang Y.J., Traum D., Schug J., Gao L., Liu C., Atkinson M.A., Powers A.C., Feldman M.D., Naji A., et al. HPAP Consortium Multiplexed In Situ Imaging Mass Cytometry Analysis of the Human Endocrine Pancreas and Immune System in Type 1 Diabetes. Cell Metab. 2019;29:769–783.e4. doi: 10.1016/j.cmet.2019.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.ALHAMAR G., FALLUCCA S., PIERALICE S., VALENTE L., POZZILLI P. 1492-P: IL-8/CXCL8 May Identify a New Type 1 Diabetes Endotype. Diabetes. 2023;72 doi: 10.2337/db23-1492-P. [DOI] [Google Scholar]
- 57.Cimini F.A., Barchetta I., Porzia A., Mainiero F., Costantino C., Bertoccini L., Ceccarelli V., Morini S., Baroni M.G., Lenzi A., Cavallo M.G. Circulating IL-8 levels are increased in patients with type 2 diabetes and associated with worse inflammatory and cardiometabolic profile. Acta Diabetol. 2017;54:961–967. doi: 10.1007/s00592-017-1039-1. [DOI] [PubMed] [Google Scholar]
- 58.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., 3rd, Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Germain P.-L., Lun A., Garcia Meixide C., Macnair W., Robinson M.D. Doublet identification in single-cell sequencing data using scDblFinder. F1000Res. 2021;10:979. doi: 10.12688/f1000research.73600.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Amezquita R.A., Lun A.T.L., Becht E., Carey V.J., Carpp L.N., Geistlinger L., Marini F., Rue-Albrecht K., Risso D., Soneson C., et al. Orchestrating single-cell analysis with Bioconductor. Nat. Methods. 2020;17:137–145. doi: 10.1038/s41592-019-0654-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hafemeister C., Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20 doi: 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.R Core Team . 2021. R: A Language and Environment for Statistical Computing.https://www.R-project.org/> [Google Scholar]
- 63.Kuhn M. Building Predictive Models in R Using the caret Package. J. Stat. Softw. 2008;28:1–26. doi: 10.18637/jss.v028.i05. [DOI] [Google Scholar]
- 64.Wickham H., Francois R., Henry L., Muller K. 2022. A Grammar of Data Manipulation.https://github.com/tidyverse/dplyr> [Google Scholar]
- 65.Bates D., Maechler M. 2021. Matrix: Sparse and Dense Matrix Classes and Methods.https://CRAN.R-project.org/package=Matrix> [Google Scholar]
- 66.Valero-Mora P.M. ggplot2: Elegant Graphics for Data Analysis. J. Stat. Softw. 2010;35:1–3. doi: 10.18637/jss.v035.b01. [DOI] [Google Scholar]
- 67.Kassambara A. 2020. ggpubr: 'ggplot2' Based Publication Ready Plots.https://CRAN.R-project.org/package=ggpubr> [Google Scholar]
- 68.Wilke C.O. 2020. cowplot: Streamlined Plot Theme and Plot Annotations for 'ggplot2'.https://CRAN.R-project.org/package=cowplot> [Google Scholar]
- 69.Song M., Zhong H. Efficient weighted univariate clustering maps outstanding dysregulated genomic zones in human cancers. Bioinformatics. 2020;36:5027–5036. doi: 10.1093/bioinformatics/btaa613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang H., Song M. Optimal k-means Clustering in One Dimension by Dynamic Programming. R J. 2011;3:29–33. doi: 10.32614/RJ-2011-015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zheng G.X.Y., Terry J.M., Belgrader P., Ryvkin P., Bent Z.W., Wilson R., Ziraldo S.B., Wheeler T.D., McDermott G.P., Zhu J., et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017;8 doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Patil A.R. 2022. HPAP scRNA-seq workflow.https://github.com/faryabiLab/HPAP-scRNA-seq-Workflow-2022 [Google Scholar]
- 73.Hearst M.A., Dumais S.T., Osuna E., Platt J., Scholkopf B. Support vector machines. IEEE Intell. Syst. Their Appl. 1998;13:18–28. doi: 10.1109/5254.708428. [DOI] [Google Scholar]
- 74.Rish I. An Empirical Study of the Naïve Bayes Classifier. IJCAI 2001 Work Empir Methods Artif Intell. 2001;3 [Google Scholar]
- 75.Schapire R.E. In: Lecture Notes in Statistics. Denison D.D., Hansen M.H., Holmes C.C., Mallick B., Yu B., editors. New York: Springer; 2003. Nonlinear Estimation and Classification; pp. 149–171. [Google Scholar]
- 76.Elith J., Leathwick J.R., Hastie T. A working guide to boosted regression trees. J. Anim. Ecol. 2008;77:802–813. doi: 10.1111/j.1365-2656.2008.01390.x. [DOI] [PubMed] [Google Scholar]
- 77.Li Y., Umbach D.M., Bingham A., Li Q.J., Zhuang Y., Li L. Putative biomarkers for predicting tumor sample purity based on gene expression data. BMC Genom. 2019;20:1021. doi: 10.1186/s12864-019-6412-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., Feng T., Zhou L., Tang W., Zhan L., et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation. 2021;2 doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhou Y., Zhou B., Pache L., Chang M., Khodabakhshi A.H., Tanaseichuk O., Benner C., Chanda S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019;10:1523. doi: 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sherman B.T., Hao M., Qiu J., Jiao X., Baseler M.W., Lane H.C., Imamichi T., Chang W. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update) Nucleic Acids Res. 2022;50:W216–W221. doi: 10.1093/nar/gkac194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gene Ontology Consortium The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res. 2021;49:D325–D334. doi: 10.1093/nar/gkaa1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kanehisa M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019;28:1947–1951. doi: 10.1002/pro.3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kanehisa M., Furumichi M., Sato Y., Kawashima M., Ishiguro-Watanabe M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023;51:D587–D592. doi: 10.1093/nar/gkac963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kanehisa M., Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Szklarczyk D., Gable A.L., Lyon D., Junge A., Wyder S., Huerta-Cepas J., Simonovic M., Doncheva N.T., Morris J.H., Bork P., et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–D613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Doncheva N.T., Morris J.H., Gorodkin J., Jensen L.J. Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data. J. Proteome Res. 2019;18:623–632. doi: 10.1021/acs.jproteome.8b00702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Doncheva N.T., Assenov Y., Domingues F.S., Albrecht M. Topological analysis and interactive visualization of biological networks and protein structures. Nat. Protoc. 2012;7:670–685. doi: 10.1038/nprot.2012.004. [DOI] [PubMed] [Google Scholar]
- 90.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The corresponding GO and KEGG pathways for shared ranked genes are also shown.
The corresponding GO and KEGG pathways for shared ranked genes are also shown.
The corresponding GO and KEGG pathways for shared ranked genes are also shown.
Data Availability Statement
The pancreatic islet sequencing data and processed scRNA-seq Seurat object can be found in PANCDB (https://hpap.pmacs.upenn.edu/analysis). The scripts used for scRNA-seq data processing and machine learning modeling are available on GitHub (https://github.com/AbhijeetRPatil/ML_Islets). Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.