

Summary
There is a compelling need for approaches to predict the efficacy of immunotherapy drugs. Tumor-on-chip technology exploits microfluidics to generate 3D cell co-cultures embedded in hydrogels that recapitulate simplified tumor ecosystems. Here, we present the development and validation of lung tumor-on-chip platforms to quickly and precisely measure ex vivo the effects of immune checkpoint inhibitors on T cell-mediated cancer cell death by exploiting the power of live imaging and advanced image analysis algorithms. The integration of autologous immunosuppressive FAP+ cancer-associated fibroblasts impaired the response to anti-PD-1, indicating that tumors-on-chips are capable of recapitulating stroma-dependent mechanisms of immunotherapy resistance. For a small cohort of non-small cell lung cancer patients, we generated personalized tumors-on-chips with their autologous primary cells isolated from fresh tumor samples, and we measured the responses to anti-PD-1 treatment. These results support the power of tumor-on-chip technology in immuno-oncology research and open a path to future clinical validations.
Keywords: cancer models, microfluidics, tumor-on-chip, tumor microenvironment, immunotherapy, anti-PD-1, lung cancer, patient-derived, cancer-associated fibroblasts, immuno-oncology
Graphical abstract
Highlights
-
•
Tumors-on-chips (ToCs) measure ex vivo immunotherapy effects on cancer ecosystems
-
•
ToCs are personalized with patient-derived primary autologous cells
-
•
Lung ToC platforms are used for profiling anti-PD-1 response of each patient
-
•
Adding FAP+ cancer-associated fibroblasts to ToC impairs the response to anti-PD-1
Veith and Nurmik et al. present a tumor-on-chip (ToC) approach to quantify ex vivo responses to immunotherapy treatments and to recapitulate stroma-dependent mechanisms of resistance. Personalized responses to anti-PD-1 are evaluated by using tumors-on-chips generated with autologous primary cells from lung cancers, supporting patient-derived ToCs as a powerful technology in immuno-oncology.
Introduction
The lack of adequate model systems is a critical obstacle in the development and deployment of new effective treatments against cancer. It is now well recognized that conventional cell cultures or animal models fail to accurately predict human responses to oncology treatments, as they do not properly mimic human physiopathology, particularly regarding the immune system.1 In the last decade, a novel concept has emerged: the use of micro-physiological systems to achieve a rational simplification of the human body. This led to the creation of new research fields, named organ-on-chip and tumor-on-chip (ToC).2,3,4 More specifically, ToC technology exploits micro-fabrication and microfluidics to generate cell co-cultures embedded in 3D hydrogels that mimic the extracellular matrix, recapitulating the immune and stromal characteristics of the tumor ecosystem. While the ToC field is exponentially growing,5 current efforts have mainly relied on established, mostly immortalized cell lines. It is now time to move toward clinically relevant cell models, such as primary autologous cells, and to prepare the ground for translational applications in personalized medicine.
Clinical oncology is currently undergoing an extraordinary therapeutic revolution driven by the new immunotherapy drugs, in particular immune checkpoint inhibitors (ICI), which can induce impressive long-lasting responses and increase patients’ life expectancy. Melanoma, lung cancer, head and neck cancer, and bladder cancer have most benefitted from these immunotherapies, while many clinical trials are still ongoing to show survival improvements in other common cancers. There is a compelling need for new concepts and methods to develop and test immuno-oncology drugs. By definition, these new experimental models must be immunocompetent; i.e., they must be able to recapitulate drug effects that rely on the immune components of the tumor microenvironment. In this regard, ToC platforms have great potential that is only waiting to be exploited.3,5 For example, we previously showed that a specific immune response, antibody-dependent cell-mediated cytotoxicity, can be recapitulated in a HER2+ breast ToC treated with trastuzumab (Herceptin).6 Another ToC study revealed a cooperative behavior between cytotoxic T cells in tumor killing.7
In this work, we aimed to generate ToC platforms as an ex vivo experimental paradigm for preclinical studies on ICI responses in order to pave a way to address immuno-oncology issues in fully human, controllable, and directly observable tumor 3D ecosystems. We chose to focus on lung cancers, the leading cause of cancer-related death worldwide, among which non-small cell lung cancer (NSCLC) is the most frequent lung cancer type (80% of cases). Despite the major improvements achieved by immunotherapy drugs, only 20% to 40% of NSCLC patients benefit from ICI drugs.8,9 What makes non-responder patients resistant to ICI still remains elusive. Undoubtedly, intrinsic cancer cell features such as PD-L1 expression, tumor mutational burden (TMB), or specific genetic alterations (p53, K-Ras, PTEN, EGFR, STK11, and KEAP11 mutations), impact ICI response.10,11,12 However, at the individual level, such biomarkers cannot be reliably used to exclude a patient from immunotherapy, since major responses are still reported in patients with low TMB or low PD-L1 expression.13,14 Besides cancer cells, stromal cell populations, such as cancer-associated fibroblasts15 and endothelial cells,16 also contribute to immunotherapy response or resistance. Overall, despite intense research, there is still an unmet medical need for predictive tools and biomarkers to identify more accurately which patients will derive long-term benefits from ICI. In addition, a crucial challenge is to more accurately understand the mechanisms of primary or secondary resistance to immunotherapy in order to conceive therapeutic interventions capable of switching non-responders to long-term-responding patients by shattering these protection processes.
We reasoned that the study of the cancer-immune interplay, and of its responses to ICI, requires the use of autologous cytotoxic T lymphocytes (CTLs) to avoid any allogeneic reaction. We first used an already established pair of a NSCLC cell line (IGR-Heu) and autologous CTLs (H5B)17,18 in order to implement robust methods to precisely quantify T-cell-mediated anti-tumor activity upon immunotherapy treatment using ToC platforms. Next, we moved to primary cells, freshly isolated from NSCLC samples, in order to evaluate the possibility to use patient-derived ToCs for personalized immunotherapy response profiling, in a time window of a few days, compatible with the decision-making process in clinics. Importantly, our experimental strategy involves the continuous live imaging for 2 days of the autologous 3D ToC co-cultures, rather than endpoint assays, in order to quantify the dynamics of crucial cellular processes within the tumor ecosystem, such as cancer cell apoptosis19 and cancer-immune interactions.6,20,21
This original combination of ToC 3D co-cultures, patient-derived autologous cell models, and advanced computational methods for image analysis allowed us to develop and validate a procedure in order to measure ex vivo the effects of immunotherapy treatments on T cell-mediated anti-tumor activity, opening avenues for both fundamental and translational research in immuno-oncology.
Results
Lung ToC platforms for personalized immunotherapy response profiling
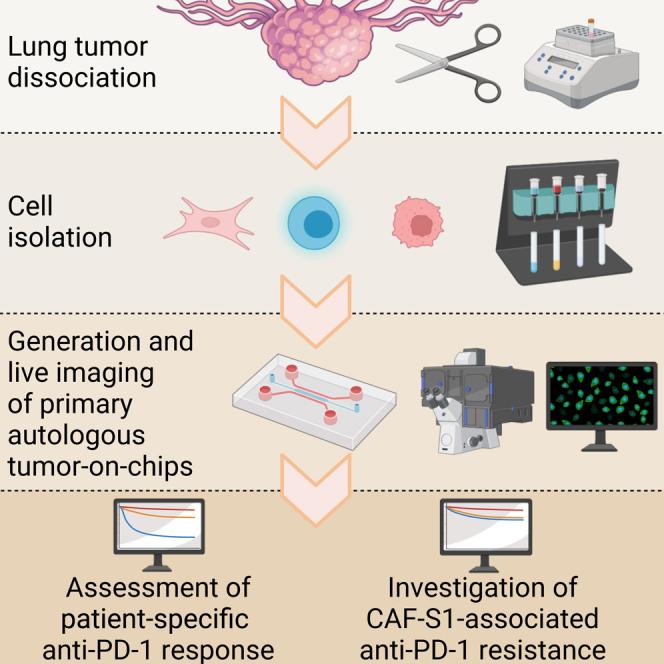
We conceived a strategy to efficiently generate patient-derived lung ToCs, to treat them with immunotherapy drugs, and to precisely measure drug response via computational image analysis (Figure 1A). We used a simple microfluidics design developed a decade ago22 and used in several studies.6,23,24,25 We chose a commercially available version of the device, made of plastic and of a gas-permeable membrane (AIM-Biotech). The central micro-chamber contains a 3D biomimetic collagen I gel (volume of 3.4 μL), while the two lateral chambers contain medium to which drugs were added in static condition or by microfluidic perfusion. Cell populations were embedded in the central collagen gel (2.3 mg/mL), at given densities: cancer cells (2,000 cells/μL), immune cells (usually at 1,000 cells/μL, up to 10.000 cells/μL for some experiments), and cancer-associated fibroblasts (CAFs) (usually at 400 cells/μL). Confocal microscopy confirmed that, within the lung ToC, the various cell populations were well distributed along the z axis and made many cell-cell physical contacts (Figure 1B). The numbers of seeded cells were chosen mainly to offer a proper imaging of the tumor ecosystem and in particular to assess cell-cell interactions; the ToC cell densities were therefore much lower than the in vivo real cell densities, approximatively 100-fold less. To determine the ratios of different cell types in the ToC, approaching at the closest the pathological observations in human tumor samples, we performed precise immunohistochemistry quantifications of cancer cells, FAP+ CAFs (also called CAF-S1), and CD8+ immune cells, in the lung cancer samples from the patient cohort assessed in this study (Figure 1C). Regarding the cancer cell-to-CAF ratio, we chose an experimental ToC condition (5:1) very close to the in vivo situation, 6:1 on average. Regarding the cancer-to-immune cell ratio, as expected,26,27 the tumor pathology sample density of CD8+ cells showed large variations, with an average ratio of 3:1 (Figures 1D and 1E). The necessity to achieve an efficient T cell-mediated killing in few days obliged to use a slight excess of immune cells in ToC (from 2:1 to 1:5) with respect to the pathology sample observations.
Figure 1.

Lung tumor-on-chip (ToC) platforms for personalized immunotherapy response profiling
(A) Workflow for lung ToC generation and analysis. Cancer cells, T cells, and fibroblasts are isolated from the tumor and co-cultured embedded in a biomimetic collagen gel within microfluidics devices. The microfluidics setup allows us to perfuse the immunotherapy drugs into the ToC, which is live imaged by video microscopy. Automated advanced methods of image analysis are used to measure the anti-cancer cytotoxic activity and the kinematics of immune cells.
(B) Representative confocal images of the reconstituted 3D lung tumor microenvironment. Autologous cancer cells (IGR-Heu) and CD8+ CTLs (H5B) are labeled in red and blue (Cell Trace), respectively. CAFs (heterologous) are labeled in green. a: top view. b: lateral view. c: magnified view.
(C) Patients’ clinical data. N/D, not determined.
(D) Representative immunostaining of human lung adenocarcinoma. Top: co-immunostaining of pancytokeratin (brown), highlighting tumor cells, and FAP (red), highlighting CAFs, with a magnified view on the right, used for manual counting. Bottom: CD8 immunostaining before (left) and after (right) supervised automated quantification using QuPath software (red, CD8+ T lymphocytes; blue, CD8−lymphocytes and tumor cells).
(E) Density of tumor cells, FAP+ CAFs, and CD8+ T cells for all patients and cell ratio calculation.
For precise quantifications of cell dynamics and interactions, while we initially seeded isolated cells, tumor cells often had the tendency to form multicellular clusters, the situation being very heterogeneous depending on each individual patient. Live fluorescent dyes were used to selectively pre-stain the different cell populations (CellTrace, red fluorescence) and to monitor apoptotic death (Cell Event Caspase-3/7, green fluorescence). Quantitative image analysis methods were developed to measure parameters that were chosen for their relevance to evaluate immunotherapy responses: death rate of cancer cells and number and time of cancer-immune interactions. The ToC videos were analyzed as pseudo-2D videos since the gel height is relatively small (250 μm). This had the great advantage of avoiding the complexity of 3D tracking algorithms, although the reconstituted lung tumor microenvironments actually have a 3D architecture.
Autologous lung ToC platforms respond to anti-PD-1 treatment
To develop our model, we first used an already established pair of an NSCLC cell line (IGR-Heu) and its autologous CD8+ T cell counterpart (H5B), generated from tumor-infiltrating lymphocytes (TILs).17,18 It has been shown previously that, in mice transplanted with IGR-Heu tumors, adoptive transfer of autologous CTL clones supported their antitumor effect in vivo.18 The autologous ToC co-cultures of cancer and CTLs were imaged for 48 h before and after treatment with an anti-PD-1 immunotherapy drug (nivolumab) (Figure 2A; Video S1). Drug injection was achieved by infusing 10 μg/mL anti-PD-1 drug, at a 1 μL/min flow rate, starting at 16 h of co-culture. An appropriate microfluidics setup allowed for simultaneous and parallel injection of 3 chips per experiment.
Figure 2.

Direct visualization and quantification of ex vivo CTL-mediated anti-tumor cytotoxic activity upon anti-PD-1 treatment
(A) Experimental design. Autologous 3D co-cultures at a 2:1 ratio of lung cancer cells (IGR-Heu) and CTLs (H5B) were generated in the central chamber of microfluidics devices and imaged by video microscopy for 48 h. Anti-PD-1 immunotherapy drug (nivolumab) is perfused in the lateral medium chamber after 16 h of co-culture.
(B) Representative time-lapse images of co-cultures at the indicated time points, before and after the addition of control isotype antibodies (top) or of anti-PD-1 immunotherapy (bottom). Cancer cells are stained in red (Cell Trace). CTLs are not stained. A green apoptosis reporter (Cell Event) is added to the medium; cells dying by apoptosis become green. Scale bar, 50 μm. See also Video S1.
(C) Quantification of the CTL-mediated anti-tumor cytotoxic activity upon anti-PD-1 treatment. The apoptosis rates of cancer cells (i.e., the percentage of cancer cells dying at 4-h time intervals) are computed using the STAMP method.19 The averages are computed every 1 h using a 4-h sliding window. The red arrow indicates the moment of drug injection (16 h). The graph reports means ± SEM from n = 4 independent experiments.
(D) Statistical analysis of apoptosis rates. The areas under the curve from 16 h to 48 h were measured for the control and treated conditions from the 4 experiments. Unpaired Student’s t test was used.
(E) Quantification of the CTL-mediated anti-tumor cytotoxic activity upon anti-PD-1 treatment. The survival curves of cancer cells (i.e., the percentage of surviving cancer cells calculated with respect to the initial number of living cells) are computed using the STAMP method.19 The red arrow indicates the moment of drug injection (16 h). The graph reports means ± SEM from 4 independent experiments.
(F) Statistical analysis of survival curves. The linear regression slopes were measured for the control and treated conditions from the 4 experiments. Unpaired Student’s t test was used.
(G) Temporal analysis of the death signal at the single-cell level. The green signal (Cell Event apoptosis reporter) of one representative dying cell is shown. By automatic signal analysis, characteristic times (tstart, tend, tmax, trise, tmed1, and tmed2) are computed. Then, the rising time and the band pass are measured.
(H) Plot of the rising time and of the band pass from 4 conditions: before and after isotype control injection and before and after anti-PD-1 injection. The distribution for each condition, represented by kernel density, is also shown. n = 121 cells total. Student’s t test showed that differences are statistically significant before versus after anti-PD-1 for rising time (p = 0.01) and band pass (p = 0.026) but not before versus after isotype control.
Time length, 48 h. Drug is added at 16 h. Cells, pre-stained in red, become green when dying by apoptosis (Cell Event apoptosis reporter). Scale bar, 100 μm.
Cancer cells were pre-stained in red (Cell Trace). A green apoptosis reporter (Cell Event) was added to the medium at the beginning of co-culture. Red fluorescent cancer cells dying by apoptosis became green (Figure 2B; Video S1). The ToC videos were analyzed by the open-source computational method STAMP (Spatiotemporal Apoptosis Mapper), that we recently developed.19
By counting the red-to-green transition signals, STAMP analysis allows us to specifically quantify the apoptotic death of only the cancer cells in co-cultures, excluding the death of immune cells or CAFs. This strategy allowed us to achieve direct visualization and evaluation of specific antitumor T cell-mediated cytotoxic activity upon anti-PD-1 treatment. The apoptosis rates of cancer cells (Figures 2C and 2D) and the survival curves of cancer cells (Figures 2E and 2F) started to diverge immediately upon anti-PD-1 addition as compared to the control isotype antibody control, indicating that CTLs very quickly reacted ex vivo to anti-PD-1 blockade.
We analyzed the green fluorescent signal emitted by dying cells at the single-cell level. This green signal is a reporter of intracellular caspase 3/7 activity. Dying cells were manually selected (n = 121), and the time profiles of their emitted signals were automatically characterized (Figure 2G). The distribution analysis showed that, after addition of anti-PD-1, but not the isotype control, there was a significant decrease in the values of two characteristic signal descriptors: the rising time and band pass (Figure 2H) (see STAR Methods for definitions), indicating that immunotherapy-induced death is characterized by a sharper increase and shorter length of caspase 3/7 activity than “natural” death spontaneously occurring in the absence of drug.
Next, we asked the question whether the kinetics of CTL immune cells were modulated by effective anti-PD-1 treatment. For robust tracking of the fast-moving immune cells, phase-contrast ToC videos were generated with high temporal resolution (every 30 s for a total of 6 h) and with z stack acquisitions at each position and each time point (Video S2). Cells were tracked using Cell Hunter software as described previously.6
Time length, 6 h. Drug is added at the start. Dying cells are in magenta (DRQ7 dye). Scale bar, 100 μm.
Cancer-immune interactions were defined as events in which a CTL moved into the circular neighborhood of a cancer cell. This neighborhood was set as 34 μm, twice the sum of the average radius of CTLs (3.4 μm) and cancer cells (13.6 μm) (Figures 3A and 3B). The time of interaction between cancer cells and CTLs significantly increased in the presence of anti-PD-1, from a median value of 24 min ± 8 min under the control condition to 29 min ± 12 min in the presence of immunotherapy treatment (p = 0.012) (Figure 3C). The number of cancer-immune cell interactions during the 6-h observation time, normalized to the number of cancer cells per video field, significantly increased in the presence of anti-PD-1, from average value of 0.07 under the control condition to 0.14 in the presence of immunotherapy treatment (p < 0.0001) (Figure 3D). In addition, the cell distribution analysis indicated that the addition of anti-PD-1 increased the percentage of both CTL and cancer cells that had a high number of interactions (Figures 3E and 3F); the fraction of CTLs with 3–4 interactions increased from 14% to 19%; the fraction of cancer cells with ≥5 interactions increased from 1% to 7.7%. Moreover, the addition of anti-PD-1 triggered the appearance of a cancer cell fraction (1.5%) with an extremely high number of immune interactions (>15).
Figure 3.

Impact of effective anti-PD-1 treatment on the kinetics and plasticity of CTL immune cells
(A) Tracking strategy. ToC videos of lung cancer cells (IGR-Heu) and autologous CTLs (H5B) were acquired with high temporal resolution (every 30 s) for 6 h. A cancer cell and an immune were considered to interact when their distance was closer than the interaction radius (here defined as 34 μm, twice the sum of the average radius of detected CTLs and cancer cells). See also Video S2.
(B) Representative output of the Cell Hunter tracking algorithm.
(C) Quantification of the time of interaction between cancer cells and CTLs. 669 time events with a duration longer than 10 min were counted in total. Statistical significance was assessed using Mann-Whitney test.
(D) Quantification of the number of interactions between cancer cells and CTLs. The number of interactions counted for each cancer cell was normalized by the total number of cancer cell trajectories detected along the video. 2,880 interaction events were counted in total. Mann-Whitney statistical test was used.
(E) Number of interactions per each immune CTL. 625 interactions in total were counted.
(F) Number of interactions per each cancer cell. 382 interactions in total were counted.
(G) Quantification of the speeds of immune CTLs. 1,542 speed values were counted in total. Mann-Whitney test was used.
(H) Quantification of the track curvatures of immune CTLs. 1,542 curvature values were counted in total. Mann-Whitney test was used.
(I) Experimental design for analysis of T cell plasticity in ToC co-cultures. CTLs were co-cultured in microfluidics devices with or without autologous cancer cells and treated with the isotype control or with anti-PD-1 (nivolumab). After 3 days, the cells were retrieved from the gel by collagenase digestion, stained, and analyzed by flow cytometry. The following markers were measured: CD25 and CD69 (activation markers); PD-1, TIM-3, TIGIT, LAG-3, CD244, and CTL-4 (inhibitory immune checkpoints); and OX-40, CD137, and GITR (activatory immune checkpoints). It was not possible to measure the PD-1 marker in the presence of anti-PD-1 treatment because of antibody competition. Three conditions were assessed: CTLs only, with cancer cells without anti-PD-1, and with cancer cells with anti-PD-1. Heatmaps report averages from 2–4 independent experiments depending on the condition.
(J) Fold change of specific MFI for CTL markers of H5B cells. The specific MFI for the condition CTLs only is set as 1.
(K) Percentage of positive H5B cells for CTL markers.
See Figure S1 for full datasets.
Regarding the immune kinematics, while the average speed of CTLs did not substantially change in the presence of anti-PD-1 (Figure 3G), the track curvature of CTLs significantly decreased, from a median value of 0.67 μm−1 and median absolute deviation of 0.19 μm−1 under the control condition to 0.60 μm−1 with a median absolute deviation of 0.16 μm−1 in the presence of immunotherapy treatment (Figure 3H), indicating that the treatment promotes directionality of CTL movements in a 3D setting.
Taken together, these results all indicate that autologous lung ToC platforms can reveal drastic and surprisingly rapid cell behavior changes upon anti-PD-1 treatment, as shown by different quantifications: at the cancer cell level (apoptosis rate and apoptotic signal), at the immune cell level (directionality of motility), and at the cancer-immune interaction level (number and duration of interactions). This demonstrates the strong added value of investigating the response to immunotherapy at a tumor ecosystem level and of incorporating multiparametric analysis. The ToC response was fast and precisely quantifiable, supporting the feasibility of immunotherapy response profiling on patient-derived ToCs.
Analysis of T cell plasticity in ToC co-cultures
We reasoned that ToCs might be useful to study T cell plasticity, in particular their activation and exhaustion status, under controlled co-culture conditions, with or without immunotherapy drugs, and to identify possible alternative targets for ICI treatments. We developed a method to recover cells from ToCs by collagenase digestion and to analyze T cell activation and exhaustion markers using multicolor flow cytometry (Figure 3I). CD25 and CD69 were selected as activation markers. As exhaustion markers, we looked at PD-1, TIM-3, TIGIT, LAG-3, CD244, and CTLA-4 immune checkpoints and at OX-40, CD137, and GITR co-stimulatory receptors.
At the basal level, i.e., without co-culture or drug treatment, H5B CTLs displayed a high percentage of positivity (>60%) for many of these markers (CD25, CD69, TIM-3, TIGIT, and LAG-3), possibly because these CTLs have been already activated during the ex vivo amplification procedure (Figure S1). After 3 days of co-culture on a chip with cancer cells, the H5B CTLs showed a further increase in the expression of activation markers, such as CD25 and CD69, as well as in exhaustion markers, notably PD-1, TIM-3, CD137 and GITR, as assessed by measuring both specific mean fluorescent intensity (MFI) (Figure 3J) and percentage of positive cells (Figure 3K). However, treatment with anti-PD-1 had very mild effects on T cell expression of both immune checkpoints and co-stimulatory receptors. Only TIM-3 expression appeared to increase, reaching statistical significance for MFI measurements (Figure S1B).
To confirm these observations, we tested another established autologous pair: the NSCLC cell line IGR-Pub and the autologous P62 CTL clone28 (Figures S1 and S2). Again, we observed increased expression in P62 cells of the activation markers CD25 and CD69 upon 3D co-culture with the autologous IGR-Pub cancer cells as well as of several exhaustion markers, such as PD-1, TIM-3, LAG3, CTL4, OX-40, CD137, and GITR, but no significant modulation by the addition of anti-PD-1 treatment.
These results indicate that, when CTLs are already strongly committed into an activation state, by ex vivo amplification and/or by co-culture with their target cells, the addition of anti-PD-1 does not substantially impact the expression of immune checkpoints.
CAFs dampen the response to anti-PD-1 in lung ToCs
We have reported previously that the abundancy of specific types of immunosuppressive FAP+ CAFs is indicative of primary resistance to immunotherapy in lung cancer patients.15 However, the impact of CAFs in immunotherapy response was never experimentally addressed ex vivo with patient cells. Primary lung CAFs can be easily obtained from tumor samples because CAFs survive relatively well on standard culture dishes. As expected, after in vitro amplification, CAF populations are quite homogeneous, positive for both FAP and CD29 markers (Figure S3), corresponding to the so-called CAF-S1 subset, which has immunosuppressive properties.15,29,30,31,32 We added these allogeneic CAF-S1 (isolated from patient #2) into the autologous cancer-immune co-cultures (IGR-Heu and H5B). Then we treated the cancer-immune-CAF lung ToCs with anti-PD-1 immunotherapy (Figures 4A and 4B; Video S3). Strikingly, when CAFs were present at a relative cancer/CAF ratio very similar to the in vivo tumor observed ratio (5:1 cancer to CAF) (Figure 1E), the anti-PD-1 treatment was unable to stimulate apoptotic death (Figures 4C and 4D) or to decrease cancer cell survival (Figures 4E and 4F), indicating that lung FAP+ CAFs actually promote immunotherapy resistance in the ToC devices.
Figure 4.

CAFs promote resistance to anti-PD-1 immunotherapy in lung ToC
(A) Experimental design. Tri-cultures of lung cancer cells (IGR-Heu), autologous CTLs (H5B), and heterologous lung CAFs (CAF#2), at ratios of 5:1 cancer to CAF and 2:1 cancer to immune cells, were generated in the central chamber of microfluidics devices and imaged by video microscopy for 48 h. An anti-PD-1 immunotherapy drug (nivolumab) is perfused in the lateral medium chamber after 16 h of co-culture.
(B) Representative time-lapse images of tri-cultures at the indicated time points, before and after the addition of control isotype antibodies (top) or of anti-PD-1 immunotherapy (bottom). Cancer cells are stained in red (Cell Trace). CAFs and CTLs are not stained. A green apoptosis reporter (Cell Event) is added to the medium; cells dying by apoptosis become green. Scale bar, 50 μm. See also Video S3.
(C) Quantification of the CTL-mediated anti-tumor cytotoxic activity upon anti-PD-1 treatment. The apoptosis rates of cancer cells (i.e., the percentage of cancer cells dying at 4-h time intervals) are computed using the STAMP method.19 The averages are computed every 1 h using a 4-h sliding window. The red arrow indicates the moment of drug injection (16 h). The graph reports means ± SEM from 4–5 independent experiments.
(D) Statistical analysis of apoptosis rates. The areas under the curve from 16 h to 48 h were measured for the control and treated conditions from 4–5 experiments. Unpaired Student’s t test was used.
(E) Quantification of the CTL-mediated anti-tumor cytotoxic activity upon anti-PD-1 treatment. The survival curves of cancer cells (i.e., the percentage of surviving cancer cells calculated with respect to the initial number of living cells) are computed using the STAMP method.19 The red arrow indicates the moment of drug injection (16 h). The graph reports means ± SEM from 4–5 independent experiments.
(F) Statistical analysis of survival curves. The linear regression slopes were measured for the control and treated conditions from 4–5 experiments. Unpaired Student’s t test was used.
Time length, 48 h. Drug is added at 16 h. Cells, pre-stained in red, become green when dying by apoptosis (Cell Event apoptosis reporter). Scale bar, 100 μm.
Personalization of ToCs using fresh lung cancer samples
To exclude an artifact due to allogenic reaction, we aimed to confirm this finding in fully autologous ToC co-cultures. We developed a strategy to freshly isolate the three cell types (cancer cells, CD8+ CTLs, and CAFs) from surgical NSCLC tumor samples by sequential magnetic cell sorting (MACS)33 and to shortly amplify them ex vivo before ToC generation (Figure 5A). Each cell type was expanded for 3 weeks in its appropriate medium and growth support: CD8+ cells in suspension in V-bottom multiwell plates, cancer cells as spheroids in ultra-low attachment flasks, and adherent CAFs in Petri dishes. Primary autologous cells from patient #13 were used to generate ToCs without and with CAF-S1, without and with anti-PD-1 treatment (added at time 0 [t0]) (Video S4). Infiltrating CD8+ T cells were able to efficiently kill the cancer cells, and their killing capacity was stimulated by anti-PD-1. The addition of autologous CAF-S1 (at an experimental 1:1 cancer to CAF ratio) did impair both the action of CD8+ T cells and the response to immunotherapy (Figures 5B and 5C). When using patient-derived cells, we simplified the experimental setting by adding the drug in the medium of the lateral chambers at the beginning of the on-chip co-cultures without the need of microfluidics setups to inject the drug. Of note, in agreement with the literature,34 our previous analysis35,36 showed that NSCLC CTLs are essentially effector memory (CD45RO+) or terminally differentiated effector memory cells (CD45RA+), negative for CD62L, CD28, CCR7, and TCF1, with an exhausted T cell profile (PD1+, TIM3+, CTLA-4+).
Figure 5.

Personalization of ToCs using patient-derived cells isolated from fresh lung cancer samples
(A) Strategy to generate primary autologous ToCs. Right after the surgery, the tumor sample is transferred from the hospital to the research facility and mechanically and enzymatically dissociated. Cancer cells, tumor-infiltrating CD8+ CTLs, and CAFs are isolated by MACS and amplified ex vivo before ToC generation.
(B) Apoptosis rates for patient #13 post amplification. Autologous cancer cells, CD8+ CTLs, and CAF-S1 were co-cultured at ratios of 1:1 cancer to CAF and 1:1 cancer to immune cells. The anti-PD-1 drug was added at the beginning of the on-chip co-culture (t0). The graph reports means ± SEM of >200 cells from 2–3 view fields. For this specific experiment, the counting of apoptosis was done manually to precisely distinguish cancer cells from CAFs.
(C) Cancer cell survival curves for patient #13 post amplification.
Time length, 19 h. Drug is added at the start. Cells, pre-stained in red, become green when dying by apoptosis (Cell Event apoptosis reporter). Scale bar, 100 μm.
Next, we implemented a fast pipeline from patient to ToC to achieve personalized immunotherapy response profiling. Autologous ToC co-culture of cancer and CD8+ CTLs (without CAFs) was generated the same day or the day after of surgery by MACS (Figure 6A; Video S5). Cancer and immune cells were directly transferred from the in vivo environment to ex vivo ToC 3D collagen gel without any culture step on standard dishes. We processed lung tumor samples (30–1,125 mm3 range size; median, 130 mm3) from a cohort of 12 patients, representative of the major NSCLC subtypes: adenocarcinoma, large cell carcinoma, and squamous cell carcinoma (see Figure 1C for clinical data). The cell yields were variable, as was cell viability (Figure 6B). Big tumors usually have necrotic cores, leading to low cell viability (see patient #5) and less efficiency in ToC generation. The viability of cancer cells after isolation was the critical parameter to achieve good-quality ToCs. We considered ToC generation as a failure when the viability of cancer cells was less than 65%–70% before seeding, since, under this condition, the co-cultures appeared very heavily influenced by large quantities of initial cell death. In 9 of 12 patients, cell numbers (0.18–5.75 × 106 CD8+ cells, 0.17–4.05 × 106 cancer cells) and survival percentages (68%–96%) were perfectly suitable for ToC generation (up to 12 conditions per patient), resulting in an overall 75% success rate.
Figure 6.

Efficiency of patient-derived ToC generation
(A) Workflow from surgery to ToCs and live-imaging microscopy. Right after the surgery, the tumor sample is transferred from the hospital to the research facility. Autologous cancer cells and tumor-infiltrating CD8+ CTLs are isolated by MACS and immediately used to generate ToC platforms.
(B) Efficiency of patient-derived cell isolation. For each patient, the tumor sample size, the number and viability of CD8+ CTLs and cancer living cells retrieved, the number of generated ToC, and the drug treatment are indicated.
Patient #8. Time length, 48 h. Drug is added at the start. Cells dying by apoptosis are in green (Cell Event apoptosis reporter). Scale bar, 100 μm.
Quantification of cancer death induced by autologous CD8+ TILs and by anti-PD-1 immunotherapy in patient-derived lung ToCs
Fully automated quantification of cancer death with the original STAMP method was not possible due to the heterogeneity of cancer cell morphology in 3D collagen gels, which is very likely a consequence of the expected patient heterogeneity. Moreover, freshly isolated primary lung cancer cells very often constituted multicellular aggregates, quickly proliferating, with consequent rapid loss of red Cell Trace dye. To overcome these obstacles, we conceived the transmission STAMP (TM-STAMP) method, in which the segmentation of cancer cell regions was automatically performed in the transmission channel (STAR Methods). In order to confirm the reliability of the modified algorithm, we compared original STAMP and TM-STAMP using ToC videos from co-cultures of the IGR-Heu cell line and autologous CD8+ H5B T cells. The output results were very similar both in terms of time-lapse profiles and of impact of anti-PD-1 drug averaged over the experimental time length (Figure S4), validating the precision of the TM-STAMP method. It is worth mentioning that, even though this TM-STAMP version is more adaptable to different ToC morphologies, it is more time consuming for the experimenter and less massive (leading to less data generation).
We analyzed results generated by TM-STAMP from ToC experiments from 6 patients (Figures 7 and S5). For all of these patients, the autologous infiltrating CD8+ CTLs had a robust, precisely quantifiable, cytotoxic activity. Interestingly, the cytotoxic activities of the isolated CTLs greatly varied among the patients, reflecting patient heterogeneity. For patient #1, we compared the killing effects of different densities of autologous CTLs, at ratios of 1:1 and 1:5, highlighting a dose-response effect, as expected (Figures 7A and 7B). ToCs from the other 5 patients (#2, #3, #7, #8, and #9) were generated at a 1:3 cancer-to-immune cell ratio and treated with anti-PD-1. The addition of anti-PD-1 treatment did not stimulate CTL activity at all in 3 of 5 patients (#2, #3, and #9) but appeared to accelerate cancer cell death in the other 2 patients (#7 and #8), as shown by the steeper slops of survival curves (Figure 7) and by the increase in apoptosis rates (Figure S5), suggesting that these 2 patients may be potential responders. The percentage of on-chip responders (2 out of 5; i.e., 40%) is close to the 20%–40% observed in clinical practice for NSCLC.8,9 So far, only patient #9 received immunotherapy, two cycles of anti-PD-1/anti-LAG3 immunotherapy after a relapse, with rapid metastatic progression, indicating that he was ICI refractory, as suspected by the lack of any effect of anti-PD-1 in his personalized ToC (Figure 7G). In conclusion, these results demonstrate the feasibility to efficiently generate immunocompetent patient-derived ToC, to treat them with immunotherapy, and to semi-automatically analyze their responses to treatment, at a timescale compatible with clinical needs.
Figure 7.

Quantification of CTL-mediated anti-tumor cytotoxic activity in autologous patient-derived lung ToCs
(A) Apoptotic death induced by autologous CD8+ CTLs as function of CTL density. ToC data shown correspond to patient #1 using primary cancer cells alone or co-cultured with autologous CD8+ TILs at a 1:1 or 1:5 ratio. The apoptosis rates of cancer cells were computed over 24 h using the TM-STAMP method and averaged. The graph reports mean ± SEM from 4 view fields.
(B) Cancer cell survival curves for patient #1. The percentage of surviving cancer cells, calculated with respect to the initial number of living cells, was computed using the TM-STAMP method. The anti-PD-1 drug was added at the beginning of the on-chip co-culture (t0). The graph reports means ± SEM from 4 view fields.
(C) Cancer cell survival curves for patient #2 (1:3 cancer to immune cell ratio).
(D) Cancer cell survival curves for patient #3.
(E) Cancer cell survival curves for patient #7.
(F) Cancer cell survival curves for patient #8.
(G) Cancer cell survival curves for patient #9.
(H) Summary of observed responses in all patients.
Discussion
Resistance to immunotherapy has dramatic consequences on the outcomes of cancer patients, but currently the only valuable investigation approaches are based on the trial-and-error strategies used in clinical trials and in management of each patient. These empiric approaches are clearly not satisfactory. The success rates in oncology therapeutic clinical trials is extremely low, being recently evaluated to be only at 3.4%.37 Moreover, only 20%–40% of lung cancer patients treated by immunotherapy derive long-term benefits, meaning that 60%–80% of patients are receiving ineffective treatments that are potentially toxic, not to mention their elevated cost, leading to the so called “financial toxicity” for health systems.
In this work, we show that ToC platforms can innovatively improve our understanding of patient-dependent immunotherapy response as well as resistance mechanisms. Our ToC procedure for the quantification immunotherapy efficacy has three strengths: it can be personalized by using patient-derived autologous primary cells directly isolated from fresh tumors, it is precise because it exploits the power of live imaging and advanced image analysis algorithms, and it is relatively fast, in that results can be obtained 3–4 days after patient surgery, a timing compatible with clinical decisions.
Strikingly, we found that the on-chip response to anti-PD-1 injection was already detected 4 h after drug injection (Figures 2C and 2E), indicating that reactivation of CTL toxicity develop very fast, which was not known previously. We also showed the feasibility of quantifying CTL toxicity using patient-derived ToCs composed of autologous cancer and immune cells freshly isolated from tumor samples. We generated two kinds of patient-derived ToC: “post-dissociation” ToCs immediately after surgery (Figure 6A) and “post-amplification” ToCs after ex vivo cell expansion for a few weeks (Figure 5A). Post-dissociation ToCs will be more suitable for drug response prediction in personalized medicine, while post-amplification ToCs may be used as 3D cancer models for mechanistic studies in basic research.
Among the five patients we tested in ToCs with anti-PD-1, three were clearly non-responders, as determined by apoptosis quantification, and two showed a noticeable ex vivo response. Interestingly, ToCs from patients #7 and #8 responded ex vivo to anti-PD-1 even though cancer cells had very low PD-L1 expression (≤1%), and, conversely, ToC from patient #9 did not respond even though cancer cells had very high PD-L1 expression (80%), indicating that PD-L1 expression is not a reliable marker for patient stratification, as suggested by clinical observations.13,14 Even more interestingly, patient #9 was clearly refractory to ICI when treated at disease progression, providing first evidence of a plausible clinical correlation between ToC and in vivo responses, but such correlation needs further confirmation in a larger series of patients. We could not validate our predictive power with such a small patient cohort, as most of the patients never received an anti-PD-1 drug, since their tumors were surgically removed, and they did not receive adjuvant post-operative immunotherapy, which was not yet reimbursed in France for such early-stage disease. Despite this limitation, our results provided a proof of concept and of feasibility, building a discovery basis to support the subsequent clinical development steps. A robust in vivo validation can be obtained only with clinical trials in humans, since mouse models are not suitable for studying ICIs targeting human T cells. Future phase 0 (pre-phase I) trials with patient-derived ToCs will be required to evaluate their precision for in vivo drug response predictions. Then, as for validation of any biomarker in medical oncology, only a prospective randomized phase 3 trial could definitively validate the ToC approach to select the right patients for the right treatment.
These clinical developments will require further miniaturization of the chips to make them compatible with biopsy samples. This will allow testing ToC-based predictions in a large prospective cohort of metastatic patients receiving immunotherapy by using relative fast endpoints (progression-free survival) as a clinical correlate. Biopsy endoscopy samples are usually 1–2 mm3 and provide a much lower number of cells (a few thousand) than surgical samples (a few million). Based on our previously published experience,38,39 we estimate that we can easily obtain 10,000 cells by combining 3 biopsies. We are currently working to design chips with very small gel volumes (3–5 μL) to achieve such biopsy-derived ToCs.
Other complementary approaches have been proposed for drug screening in personalized therapy design, the main ones being organoids and organotypic cultures (such as tissue slices or tumor fragments). Regarding organoids, despite encouraging experimental evidence supporting the notion that they can recapitulate patient responses in the clinic to chemotherapies or targeted therapies for some cancers,40,41,42 there are doubts regarding their utility for immunotherapy profiling for lung cancer patients, as they require long culture times (weeks) that are incompatible with immune cell survival and because the overall establishment rate of pure NSCLC organoids is only 17%.43 The success rate of our lung ToC was actually much higher, 75%, although the low number of samples requires caution. Regarding organotypic cultures, a very recent study showed that tumor fragment platforms that preserve the tumor microenvironment (TME) architecture, can respond to anti-PD-1 treatment, as assessed by a combination of selected cytokines and T cell activation markers. Remarkably, in such models, the capacity of tumor-resident T cells to be reactivated ex vivo was predictive of clinical response.44 Similarly, profiles of secreted cytokines from organotypic tumor spheroids were used to screen for the response of patient tumors to anti-PD-1 therapy.23,45 As compared to these organotypic approaches, ToCs provide a different type of information since they rely on direct visualization by video microscopy as a potent readout. Indeed, ToC videos produce a wealth of imaging data at single-cell resolution about the dynamics of the TME (e.g., motility, cell-cell interactions, death mitosis, etc.), which can be exploited to deeply investigate the spatiotemporal features of the tumor ecosystem. For example, we showed that a deep learning approach is able to correctly identify responding breast ToC cancer-immune co-cultures treated by an anti-HER2 antibody (trastuzumab), using an atlas of immune cell trajectories.46 Moreover, ToCs allow fine TME control of cell parameters (cell types, density, ratio, compartmentalization, etc.), matrix properties (composition, stiffness, porosity, etc.), and physicochemical environment (dissolved oxygen,47 nutrient gradients, etc.), which is very advantageous for deciphering the resistance mechanisms to immunotherapy.
To this regard, we found that the presence of lung CAFs, specifically the FAP+ CAF-S1, both allogeneic and autologous, dampen the response to anti-PD-1 in lung ToCs, experimentally supporting the clinically relevant notion that CAFs play an important role in immunotherapy resistance. Indeed, several in vivo studies showed that CAFs, by acting on immune components, can induce an immunosuppressive microenvironment that is likely involved in resistance to immunotherapy and in cancer progression. It has been reported that, in immunocompetent mouse models, FAP+ CAFs are associated with immunosuppression48,49,50,51 and induce resistance to common ICIs, such as anti-PD-1 and anti-CTLA4, through the action of the chemokine CXCL12.52 In breast and ovarian human cancers, the CAF-S1 subset displays immunosuppressive functions through a multistep mechanism.29,31 Notably, in metastatic melanoma and NSCLC, specific CAF-S1 populations (namely the extracellular matrix [ECM]-myCAF, wound-myCAF, and transforming growth factor β-myCAF clusters) are enriched at diagnosis in non-responders, indicating that they might be predictive of immunotherapy responses.15 Moreover, we recently found, by functional assays, that the ANTXR1+ ECM-myCAF cluster reduces CD8+ T cell cytotoxicity through a YAP-dependent mechanism.32 In this context, our ToC platforms offer an ideal experimental setting to investigate strategies to overcome CAF-dependent immunotherapy resistance, possibly using YAP/TEAD interaction inhibitors.53
In conclusion, the last 15 years have witnessed the emerging of ToC platforms as 3D models in oncology, as we recently reviewed.5 In our hands, ToC are clearly capable to recapitulate the expected cancer death responses upon a variety of anti-cancer treatments, such chemotherapies,19 targeted therapies,6 oncolytic viruses,21 anti-PD-1 (this work), and, even more important, the drug resistance promoted by FAP+ CAF-S16,19 (this work), which has been largely documented by in vivo studies. This evidence supports the biological relevance of the ToC approach and the interest in extending these efforts to the immuno-oncology field54 (1) to investigate the efficacy of immunotherapy molecules eventually in combination with other drugs, (2) to study the resistance mechanisms, and (3) to potentially provide future predictive tools for personalized medicine in the field of immuno-oncology.
Limitations of the study
A major limitation of our study is that, at this stage, we could not provide evidence that ToC tools may correctly predict immunotherapy efficacy in patients. Therefore, the potential predictive power of such a lung cancer-on-chip approach could not be validated. Larger patient cohorts and biopsy-compatible chips will be necessary. A second major limitation is that the question of whether or not ToC is a good preclinical platform remains open. More work will be required to achieve a complete and direct comparison between ToC and in vivo; for example, in terms of cancer cell proliferation, cell heterogeneity (including other CAFs and immune cell subtypes), gene expression patterns, or cell morphodynamics. ToC cultures are still obviously far from fully recapitulating the complexity of the in vivo situation.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD29 AF700 | Biolegend | #303020 RRID:AB_2130079 |
| CD29 AF700 Isotype | Biolegend | #400144; RRID:AB_2923250 |
| FAP | R&D Systems | #MAB3715-500; RRID:AB_2102368 |
| FAP Isotype | R&D Systems | #MAB002; RRID:AB_357344 |
| CD3 AF700 | BD Biosciences | #561805; RRID:AB_396938 |
| CD4 BV510 | BD Biosciences | #566804; RRID:AB_2869877 |
| CD8 BV510 | BD Biosciences | #563919; RRID:AB_2722546 |
| CD69 BB700 | BD Biosciences | #747520; RRID:AB_2744097 |
| CD25 BUV563 | BD Biosciences | #741365; RRID:AB_2870865 |
| CD279 BV421 | BD Biosciences | #562516; RRID:AB_11153482 |
| TIM-3 BV711 | BD Biosciences | #565567; RRID:AB_2744370 |
| LAG-3 PE | BD Biosciences | #565616; RRID:AB_2571727 |
| TIGIT BV605 | BD Biosciences | #747841; RRID:AB_2872304 |
| CD152 PE-CF594 | BD Biosciences | #562742; RRID:AB_2737761 |
| CD134 PE-Cy7 | Biolegend | #350012; RRID:AB_10901161 |
| CD137 BUV395 | BD Biosciences | #745737; RRID:AB_2743209 |
| CD357 APC | Thermo-Fisher Scientific | #17-5875-41; RRID:AB_10714991 |
| CD224 FITC | BD Biosciences | #550815; RRID:AB_393900 |
| BTLA APC | BD Biosciences | #564800; RRID:AB_2738959 |
| CompBeads Anti-Mouse Ig; κ/Negative Control | BD Biosciences | #552843; RRID:AB_10051478 |
| Zenon™ Mouse IgG1 Labeling Kit | Thermo-Fisher Scientific | #Z25051; RRID:AB_2736969 |
| anti-PD-1 (Nivolumab) | Selleck Chemicals | #A2002; RRID:AB_2810223 |
| Ultra-LEAF™ Purified Human IgG4 Isotype | Biolegend | #403701; RRID: N/A |
| Cytokeratin Pan Plus (AE1 & AE3 & 5D3) | Zytomed Systems | #MSK098-05; RID:AB_2864454 |
| Recombinant Anti-Fibroblast activation protein, alpha antibody | Abcam | #ab207178; RRID:AB_2864720 |
| anti-CD8 | Agilent | #M710301-2 |
| Biological samples | ||
| Patient histological blocks (NSCLC) | Bichat-Claude Bernard Hospital (AP-HP) | N/A |
| Patient histological blocks (NSCLC) | Institut Mutualiste Montsouris (IMM) | N/A |
| Tumor samples (NSCLC) | Bichat-Claude Bernard Hospital (AP-HP) | N/A |
| Tumor samples (NSCLC) | Institut Mutualiste Montsouris (IMM) | N/A |
| Chemicals, peptide, and recombinant proteins | ||
| Human IL-2 Recombinant Protein | Thermo-Fisher Scientific | #PHC0021 |
| rIL-2 | Gibco | #PHC0021 |
| DNAse I | Sigma-Aldrich | #D5025-15KU |
| Collagen type I, rat tail | Thermo-Fisher Scientific | #A1048301 |
| Collagenase type I | Merck Millipore | #SCR103 |
| Human EGF Recombinant Protein | Thermo-Fisher Scientific | #PHG0311 |
| Human FGF-basic (FGF-2/bFGF) Recombinant Protein | Thermo-Fisher Scientific | #13256-029 |
| Hydrocortisone | Sigma-Aldrich | #H0888-1G |
| B-27™ Supplement (50X); serum free | Thermo-Fisher Scientific | #17504044 |
| Heparin | Sigma-Aldrich | #H3149-10KU |
| Insulin (Human) | Sigma-Aldrich | #I9278-5ML |
| UltraPure™ 0.5M EDTA; pH 8.0 | Thermo-Fisher Scientific | #15575020 |
| Sodium Pyruvate | Thermo-Fisher Scientific | #11-360-070 |
| Sodium Hydroxide Solution | Sigma-Aldrich | #S2770 |
| Penicillin/Streptomycin | Thermo-Fisher Scientific | #15140122 |
| Critical commercial assays | ||
| Tumor Dissociation Kit; human | Miltenyi Biotec | #130-095-929 |
| CD8 Microbeads; human | Miltenyi Biotec | #130-045-201 |
| Tumor Cell Isolation Kit; human | Miltenyi Biotec | #130-108-339 |
| Red Blood Cell Lysis Solution (10×) | Miltenyi Biotec | #130-094-183 |
| Dead Cell Removal Kit | Miltenyi Biotec | #130-090-101 |
| CellTrace™ Yellow Cell Proliferation Kit | Thermo-Fisher Scientific | #C34567 |
| CellEvent™ Caspase-3/7 Detection Reagent | Thermo-Fisher Scientific | #C10423 |
| Zombie NIR™ Fixable Viability Kit | BioLegend | #423105 |
| Venor®GeM Classic | Minerva Biolabs | #11-1250 |
| Experimental models: Cell lines | ||
| IGR-Heu/H5B autologous cells | Dorothée et al., 200228 | Dorothée et al., 200228 |
| IGR-Pub/P62 autologous cells | Echchakir et al., 2000 and Asselin-Paturel et al., 200117,18 | Echchakir et al., 2000 and Asselin-Paturel et al., 200117,18 |
| Software and algorithms | ||
| ImageJ | Schneider et al., 2012, https://doi.org/10.1038/nmeth.2089 | https://imagej.nih.gov/ij/ |
| MetaMorph | Molecular Devices | https://www.moleculardevices.com/products/cellular-imaging-systems/high-content-analysis/metamorph-microscopy |
| Adobe Illustrator | Adobe Inc. | https://www.adobe.com/products/illustrator.html |
| Biorender | Biorender | https://www.biorender.com/ |
| FlowJo | BD Biosciences | https://www.flowjo.com/ |
| Spatiotemporal Apoptosis Mapper (STAMP) | Veith et al., 201819 | Veith et al. 19 |
| TM-STAMP | University of Rome TorVergata | https://zenodo.org/records/10944892 |
| Cell Hunter | Biselli et al., 2017. 55, Parlato et al., 201756 | Biselli et al., 2017. 55, Parlato et al., 201756 |
| GraphPad Prism 10 | Dotmatics | https://www.graphpad.com |
| MATLAB R2022b | Mathworks | https://www.mathworks.com/products/matlab.html |
| Other | ||
| DMEM F12 medium | Gibco | #11330-032 |
| RPMI-1640 medium | Cytiva | #SH30027.01 |
| DMEM | Sigma-Aldrich | #D0822 |
| Fetal Bovine Serum | Biosera | #FB-1001 |
| Human AB serum | Institut Jacques Boy, Reims, France | #201021334 |
| Ultroser G | Sartorius | #15950-017 |
| Penicillin/Streptomycin | Gibco | #P4333 |
| Mycoplasms detection kit, VenorGem Classic | BioValley | #11–1250 |
| LS Columns | Miltenyi Biotec | #130-042-401 |
| idenTx 3 microfluidic chips | aimbiotech | #DAX-1 |
| QuadroMACS Seperator | Miltenyi Biotec | #130-090-976 |
| ZE5 flow cytometer | Bio-Rad | #12004279 |
| LSRFortessa™ Cell Analyzer | BD Biosciences | https://www.bdbiosciences.com/en-fr/products/instruments/flow-cytometers/research-cell-analyzers/bd-lsrfortessa |
| Ultra-Low Attachment 75cm2 U-Flask | Corning | #3814 |
| 96-well V-bottom plate | Greiner | #651261 |
| Petri dish; PS; 94/16 mm | Greiner | #353003 |
| Falcon® 100 μm Cell Strainer | Corning | #352360 |
| Masterflex Transfer Tubing, Microbore PTFE | Cole-Parmer | #EW-06417-11 |
| Leica BOND III | Leica Biosystems | #21.2201 |
| Bond Polymer Refine Detection | Leica Biosystems | #DS9800 |
| BOND Polymer Refine Red Detection | Leica Biosystems | #DS9390 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed and will be fulfilled by the lead contact Maria Carla Parrini (maria-carla.parrini@curie.fr)
Materials availability
This study did not generate unique reagents.
Data and code availability
-
•
All source data reported in this paper will be shared by the lead contact upon request.
-
•
The original code TM-STAMP has been deposited at Nextcloud and is publicly available as of the date of publication. The link is listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Cell cultures
The IGR-Heu large cell carcinoma cells, the IGR-Pub lung adenocarcinoma cells, and the autologous H5B T cell line and P62 T cell clone, respectively, were generated in one of our laboratories.17,18,28 IGR-Heu and H5B cells were obtained from a 61-year old male patient (Heu). Our previous analysis showed that H5B CD8+ T cells display a TEMRA (terminally differentiated effector memory) phenotype characterized by the expression of CD45RA+ in the absence of CCR7, while P62 CD4+ T cells display an effector memory phenotype characterized by CD45RO expression in the absence of CD45RA.
The IGR-Heu and IGR-Pub cancer cells were cultured in DMEM F12 (Gibco) supplemented with 10% fetal bovine serum (FBS) (Biosera), 1% of Sodium Pyruvate (Thermo-Fisher Scientific), 1% of Ultroser G (Pall), and 1% Penicillin/Streptomycin (Gibco). H5B, P62, and patient-derived CD8+ T cells were cultured in 96-well V-bottom plate (Greiner, #651261) in T cell medium: RPMI-1640 (GE Healthcare) supplemented with 10% human AB serum (Institut Jacques Boy, Reims, France), rIL-2 (20 U/ml, Gibco), 1% of Sodium Pyruvate and 0.1% Penicillin/Streptomycin. H5B and P62 were stimulated with irradiated feeder cells as described.17,28 Primary CAFs were cultured on Petri dish in DMEM (Sigma-Aldrich, #D0822) supplemented with 10% FBS, 1% of Sodium Pyruvate and 0.1% Penicillin/Streptomycin. For amplification as spheroids, isolated tumor cells were transferred into Ultra-Low-Attachment (ULA) flasks (Corning, #3814) and cultured in serum-free growth media composed of DMEM-F12 supplemented with 0.1X/mL B27 Supplement (Gibco, #17504044), 5 ng/ml Insulin (Sigma-Aldrich, #I9278), 1 μg/ml Hydrocortisone (Sigma-Aldrich, #H0888-1G), 4 μg/ml Heparin (Sigma-Aldrich, #H3149), 1% Penicillin/Streptomycin, 20 ng/mL basic fibroblast growth factor (Gibco, #13256-029), and 20 ng/mL epidermal growth factor (Gibco, #PHG0311). Cells were periodically tested to exclude mycoplasma contamination using a qPCR-based method (VenorGem Classic, BioValley, #11–1250) or by a plasmid degradation test. All ToC co-cultures were performed using T cell medium.
Patients' samples
Fresh tumor samples were provided by the Pathology department of the University Bichat Hospital (AP-HP) and by the Institut Mutualiste Montsouris (IMM) in Paris, France, from patients with NSCLC having undergone standard-of-care surgical resection. Tissue samples were taken from surgical residues available after histopathological analysis and not required for diagnosis. Patients were informed of the potential use for research of their samples/data and signed an informed non-opposition form according the French regulation on non-interventional studies using leftover biological specimens for research. The human experimental procedures follow the Declaration of Helsinki guidelines. Ethical approval was obtained from the institutional review board of the French Society of Respiratory Medicine (Société de Pneumologie de Langue Française, SPLF) (number CEPRO 2020-051) and from the Institutional Review Board and Ethics Committee of Institut Curie Hospital group (CRI-DATA190154 and CRI-DATA230112). Specimens at IMM were collected under a dedicated protocol approved by the French Ethics and Informatics Commission (EUdract 2017-A03081-52).
Fresh NSCLC samples were preferentially processed immediately after surgery, but in some cases cell suspensions after tumor digestion were stored over night at 4°C in RPMI (10% FBS, 1% Penicillin/Streptomycin). Patients #1, #3, #5, #6, #9, and #11 were male, while patients #2, #7, #8, #10, #12, and #13 were female. Tumors were cut in small pieces of approximately 2 mm3 then incubated with Human tumor dissociation kit (Miltenyi Biotec, #130-095-929) and placed at 37°C in agitation (500 rpm) for 45 min. After enzymatic digestion, the samples were filtered through a 100μm cell strainer (Falcon, Cat#352360) and the remaining pieces smashed with a 1 mm syringe plunger and washed abundantly with Medium A (RPMI 1640, 10% human serum, 10X sodium pyruvate and 0.1% Penicillin/Streptomycin). Necrotic tumors were additionally treated with DNAse I (Sigma, #D5025-15KU, 240 U/mL) for 5 min at 37°C to avoid column clogging during magnetic isolations. After centrifugation the single cell suspension was incubated with RBC lysis buffer (Miltenyi Biotec, #130-094-183) and incubated 2–3 min at room temperature, then washed with Medium A. For CD8+ T cells magnetic isolation we incubated the cell suspension 15 min incubation at 4°C with CD8 microbeads (Miltenyi Biotec, #130-045-201), then cells were passed through LS magnetic columns (Miltenyi Biotec, #130-042-401) installed on a MACS Quadro magnetic stand (Miltenyi Biotec, #130-090-976). To isolate cancer cells, the negative fraction of CD8+ T cells selection was collected and incubated with the Tumor Cell Isolation Kit (Miltenyi Biotec, #130-108-339) for 15 min incubation at 4°C. Cancer cells and CD8+ cells were either immediately used to prepare ToC (post-dissocation ToC), or amplified ex vivo, as spheroids in ULA flasks for cancer cells, to prepare ToC few weeks later (post-amplification ToC). In order to retrieve CAFs, the cells remaining from the sequential isolations were put in culture in Petri dishes and amplified.
Method details
Immunohistochemical analysis
For each tumor, one representative formalin-fixed and paraffin-embedded (FFPE) block was selected. Immunohistochemical stainings were performed on 3 μm serial sections using an automated immunohistochemical stainer (Leica BOND III). Antigen retrieval was conducted by heat, at pH9 in TRIS Buffer. PBS was substituted for the primary antibody and used as a negative control. Used antibodies were: anti-CD8 (C8/144B, Agilent technologies, Singapore, dilution 1:300), anti-pancytokeratin (AE1/AE3/5D3, Zytomed, #MSK098-05, dilution: 1/200), and anti-FAP (Fibroblast activation protein, alpha antibody [EPR20021], Abcam, #ab207178, dilution: 1/200). Detection of CD8 was performed by simple immunohistochemistry revealed by DAB (Bond Polymer Refine Detection, Leica Biosystem, #DS9800). Detection of FAP and pancytokeratin was performed by sequential co-immunolabeling. The anti-pancytokeratin antibody, applied first, was revealed by DAB. The anti-FAP antibody was secondly applied and revealed by Fast Red (Bond Polymer Refine Red Detection, Leica Biosystem, #DS9390).
Manual quantification of CAFs cells (FAP+) and tumor cells (pancytokeratin+) was performed by two pathologists (NP, EG) on 15 representative randomly selected areas of 1200 μm2 using Qpath software. Automated quantification of CD8+ lymphocytes was performed on the whole tumor area under supervision using the "positive cell detection" function (Qpath).
Tumor-on-chip preparation
The microfluidic devices were purchased from AIM-Biotech (#DAX-1). Cells were seeded at the final density of 2x106 cells/ml in the central chamber of the DAX-1 chips embedded in a matrix composed of type I rat tail collagen at the final concentration of 2.3 mg/mL (Thermofisher, #A1048301), prepared according to manufacturer protocol with 10X PBS (Gibco, #12579099), distilled H2O (Gibco, #15230162). The pH of the resulting collagen mixture was neutralized using 1M NaOH solution (Sigma-Aldrich, #S2770) to a final pH = 7. All pipettes, tips, chips, and reagents were kept chilled to preempt premature polymerization. Each chip was loaded with 10μL of gel (with a concentration of 2000 cancer cells/μL), equivalent to 20000 cancer cells/chip. The targeted cell to cell ratios were from 2:1 to 1:5 cancer to immune cells and 5:1 cancer to CAF. After addition of the gel in the microfluidic device, in order to allow gel polymerization, chip was incubated in a humidified chamber for 25 min in the incubator (37°C, 5%CO2). After gel polymerization, 120 μL of T cell medium were added into each lateral chamber, supplemented with 6μM CellEvent Caspase-3/7 Green Detection Reagent (Thermofisher, #C10423, green fluorescence). Cancer cell lines (IGR-Heu and IGR-Pub) were pre-stained with 5μM CellTrace Yellow (Thermofisher, #C34567, red fluorescence) in PBS for 25 min at 37°C then washed with warm medium and seeded in the gel. Anti-PD-1 antibody (Selleck, #SE-A2002-5MG) or IgG4 isotype control (Biolegend, #403702) were added at a concentration of 10 μg/ml.
Microfluidic setup
An adapted microfluidic setup was developed in order to inject drugs in the microfluidic devices in real-time while imaging the cells. The microfluidic setup includes, in order: a pressure controller (Fluigent, Flow-EZ), 15mL-tube reservoirs for culture medium, flow control detectors (Fluigent, Flow Unit-M) and the microfluidic device with cells in the video-microscope incubator. The constant flowrate was set at 0.5-2 μL/min. All the parts of the microfluidic system are connected through PTFE tubing (Cole Parmer, #06417-11). The tubing was connected to the microfluidic device through custom 3D-printed fluidic adaptors, printed on a DWS 028J + HR 3D SLA printer with DS3000 clear resin (DWS Systems) and a 100 μm layer height. After printing, the adaptors were rinsed and sonicated in isopropanol for 2 min before UV curing for 25 min in a UV curing unit (DWS Systems).
Live cell imaging
The tumor-on-chips were mounted on the motorized stage of an inverted wide-field fluorescence video-microscope (DMi8, Leica), enclosed in an incubation chamber to provide a humid atmosphere with 5% CO2 at 37°C, equipped with a Retiga R6 camera and Lumencor SOLA SE 365 light engine, and piloted by MetaMorph software. 5X objective was used. For each gel, 2–4 positions were acquired. For cancer death analysis, acquisition intervals were 1 frame every 30–60 min for 48 h. Three channels were acquired at each position and each time point: phase-contrast, green and red fluorescence. For green fluorescence, the filters were: excitation 470/40 nm, emission 525/50 nm, dichroic mirror 495 nm. For red fluorescence, the filters were: excitation 560/40 nm, emission 630/75 nm, dichroic mirror 585 nm. For immune cell kinematics, acquisition intervals were 1 frame every 30 s for 6 h, only in the phase-contrast channel to avoid photo-toxicity. To increase number and length of immune cell tracks, z stack acquisition mode (6 planes every 5 μm) and the plugin Stack Focuser of ImageJ software were used.
Flow cytometry
T cell markers were assessed by flow cytometry after 72 h of co-culture on-chip with cancer cells, with or without anti-PD-1 treatment. In order to digest the gel and harvest the cells, after a wash with PBS, collagenase I (Millipore, # SCR103, resuspended at 4 mg/ml in DMEM F12 and passed through 0.22μm filter) was added to chip lateral chambers for 30 min at 37°C. After collagen digestion, cells were transferred in a 96-well V-bottom plate (Greiner, Kremsmünster, Austria, #651101) for flow cytometry staining. First cells were stained with Zombie NIR viability staining (Biolegend, #423105, diluted 1:500 in PBS) for 25 min at room temperature. After washing with PBS+ (PBS, 1% FBS, 2mM EDTA) cells were stained with antibody or isotype control mixes for 30 min at room temperature. The antibody mix was composed of anti-CD3 (BD, #561805, diluted 1:60), anti-CD4 (BD, #566804, diluted 1:40), anti-CD8 (BD, #563919, diluted 1:80), anti-CD69 (BD, #747520, diluted 1:40), anti-CD25 (BD, #741365, diluted 1:20), anti-CD279 (BD, #562516, diluted 1:40), anti-TIM-3 (BD, #565567, diluted 1:40), anti-LAG-3 (BD, #565616, diluted 1:40), anti-TIGIT (BD, #747841, diluted 1:40), anti-CTLA-4 (BD, #562742, diluted 1:50), anti-OX-40 (Biolegend, #350012, diluted 1:20), anti-CD137 (BD, #745737, diluted 1:60), anti-GITR (eBiosciences, #15588146, diluted 1:40), anti-CD244 (BD, #550815, diluted 1:20), anti-BTLA (BD, #564800, diluted 1:60). After staining, cells were washed in PBS+ and were analyzed at the ZE5 flow cytometer (Biorad). Compensations were performed using single stained beads (BD biosciences, #552843) for each antibody. For analysis of CAF markers, CAFs were cultured in standard dishes, detached by trypsin digestion, washed, counted and plated in a 96-well V-bottom plate for staining in a 50 μL mix containing antibody in PBS+ (PBS, 1% human serum (Institut de Biotechnologies Jacques Boy, #201021334), 2 mM EDTA (Invitrogen, #15575020)). The mixture was incubated for 30 min at room temperature, protected from light. After centrifugation, the pellet was resuspended in 50 μL PBS+, and the samples were then directly analyzed using a LSRFortessa Cell Analyzer (BD). The used antibodies were: CD29 AF700 (Biolegend, #303020) and FAP (R&D Systems, #MAB3715-500), conjugated to APC using the Zenon Mouse IgG1 Labeling Kit (Invitrogen, #Z25051). Data analysis was performed on FlowJo (v 10.5.2).
Automatic video analysis
Cancer death
The quantifications of cancer apoptosis rates and survival were done using the original STAMP (SpatioTemporal Apoptosis Mapper) method, as previously reported,19 for ToC experiments using the cancer cell line (IGR-Heu). For patient-derived ToC experiments, using freshly isolated primary cancer cells, a modification of this method was implemented, named TM-STAMP (TransMission STAMP). In TM-STAMP, the regions occupied by cancer cells, isolated or as aggregates, were automatically segmented at each time frame by using the Circular Hough transform (CHT),57 in the transmission channel instead of red channel. Then, images were merged to create a unique segmentation mask of only the time-lapse regions that were selected in a number of time frames higher than an automatic threshold value estimated through Otsu algorithm.58 Since the cancer regions do not move much during the observation time, this unique segmentation mask was applied along the entire video analysis by the STAMP algorithm (Figure S4). One only change was introduced in STAMP code. Since tumor cell aggregates often had heterogeneous shapes, we could not assume cancer cell regions to have circular shapes and we could not anymore define cell background by simply expanding the cell circle in an annulus. Therefore, we applied scaling to the shape manually extracted by the experimenter through morphological operators of dilation.58 The resulting background can have diversified shapes according to original shapes of cell aggregates.
Single-cell death signal
Video crops of dying cancer cells were manually selected. By applying automatic cell localization by Cell-Hunter software,6 the region belonging to the cell was discriminated from the background in order to automatically analyze the average apoptotic green signal (CellEvent Caspase-3/7 reporter) emitted from each cell region. First, a smoothing spline approximation59 was applied over the period 0–48 h. In this way, by smoothing small fluctuations due to errors in cell localization and segmentation, peaks and local minima of green signals were more accurately detected. Then, after detection of maximum intensity value (gmax) and the corresponding time (tmax), we applied a local minima finding procedure and retained the two minima, gstart and gend, whose times are the closest times to tmax, one before (tstart) and one after (tend) respectively. We computed a baseline value g0 as the average values between gstart and gend. Then, we computed the time, trise, in the range [tstart, tmax], as the time at which the green signal reaches the 25th percentile in the range [g0,gmax]. We defined the Rising Time (DTrise) the quantity tmax-trise. We then computed the times tmed1 and tmed2 at which the green signal reaches the median value in the range [g0,gmax]. We defined the Band Pass (BP) the quantity tmed2-tmed1.
Cell kinematics
The quantifications of average speeds and track curvatures of T cells, as well as of the interaction times between cancer and immune cells, were done as previously reported.21
Briefly, the instantaneous speed of T cells along the track was computed as follows:
| (Equation 1) |
The instantaneous curvature value along the track was computed as follows:
| (Equation 2) |
where and and indicate the first and the second-order derivatives along t of the and coordinates, respectively. Subsequently, the average values along the track were computed for each trajectory.
The total number of detected cell tracks was 1600 for CTL and 645 for cancer cells. CTL tracks had an average duration of about 81 min, whereas cancer cells average duration was 137 min. CTL tracks with maximum displacement less than 23 μm and with duration less than 50 min were eliminated and similarly for cancer cell tracks displacement less than 9 μm and duration less than 25 min. Such selection was applied to avoid false cell detection (e.g., phase ring) and to eliminate tracks too short for cell-cell interaction computation.
Quantification and statistical analysis
Statistical analysis applied, exact n values, and precision measures are indicated in each figure legend. Statistical analyses were performed using the GraphPad Prism or MATLAB software. When the conditions passed the normality Shapiro-Wilk test, a parametric Student’s t test was applied. If not, the non-parametric Mann-Whitney or Wilcoxon tests were used. Statistical threshold for significance was set for p-values inferior to 0.05.
Acknowledgments
We thank the Biological Resources Center (CRB) of the Institut Mutualiste Montsouris (IMM) for providing patient samples, Jordan Denizeau for technical assistance with tumor sample handling and collection, Rana Mhaidly for help with flow cytometry, as well as Lea Guyonnet from Institut Curie Cytometry Platform. The project was supported by Fondation ARC pour la Recherche sur le Cancer, PGA1 RF20180206991 and PGA12021010002992_3578; INSERM, ITMO N°19CR046-00, ITMO Equipment 2016, and ITMO Equipment N°22CQ036-01; Institut Roche; a CIFRE fellowship to I.V., funded in part by the Association Nationale de la Recherche et de la Technologie (ANRT) on the behalf of the French Ministry of Higher Education and Research and in part by Institut Roche. This work had financial support from ITMO Cancer of Aviesan within the framework of the 2021–2030 Cancer Control Strategy with funds administered by INSERM.
Author contributions
Conceptualization, I.V., G.Z., and M.C.P.; methodology, G.G. and S.C.; investigation, I.V., A.M., M.N., I.D., C.L., S.B., J.F., N.P., E.G., A.C., P.M., J.T., C.S., and N.G.; visualization, I.V., A.M., M.N., C.L., N.P., E.G., and A.C.; supervision, E.P., J.C., H.S., F.M.-C., F.M.-G., S.D., E.M., G.Z., and M.C.P.; writing – original draft, I.V., A.M., M.N., G.Z., and M.C.P.; writing – review & editing, I.V., A.M., M.N., E.P., J.C., F.M.-C., F.M.-G., S.D., E.M., G.Z., and M.C.P.
Declaration of interests
H.S. and I.V. are Roche employees. The STAMP method used in this work has been patented by I.V., A.M., E.M., and M.C.P.
Published: May 3, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101549.
Contributor Information
Gérard Zalcman, Email: gerard.zalcman@aphp.fr.
Maria Carla Parrini, Email: maria-carla.parrini@curie.fr.
Supplemental information
References
- 1.Mestas J., Hughes C.C.W. Of mice and not men: differences between mouse and human immunology. J. Immunol. 2004;172:2731–2738. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 2.Sung K.E., Beebe D.J. Microfluidic 3D models of cancer. Adv. Drug Deliv. Rev. 2014;79–80:68–78. doi: 10.1016/j.addr.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boussommier-Calleja A., Li R., Chen M.B., Wong S.C., Kamm R.D. Microfluidics: A new tool for modeling cancer-immune interactions. Trends Cancer. 2016;2:6–19. doi: 10.1016/j.trecan.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sontheimer-Phelps A., Hassell B.A., Ingber D.E. Modelling cancer in microfluidic human organs-on-chips. Nat. Rev. Cancer. 2019;19:65–81. doi: 10.1038/s41568-018-0104-6. [DOI] [PubMed] [Google Scholar]
- 5.Bouquerel C., Dubrova A., Hofer I., Phan D.T.T., Bernheim M., Ladaigue S., Cavaniol C., Maddalo D., Cabel L., Mechta-Grigoriou F., et al. Bridging the gap between tumor-on-chip and clinics: a systematic review of 15 years of studies. Lab Chip. 2023;23:3906–3935. doi: 10.1039/d3lc00531c. [DOI] [PubMed] [Google Scholar]
- 6.Nguyen M., De Ninno A., Mencattini A., Mermet-Meillon F., Fornabaio G., Evans S.S., Cossutta M., Khira Y., Han W., Sirven P., et al. Dissecting Effects of Anti-cancer Drugs and Cancer-Associated Fibroblasts by On-Chip Reconstitution of Immunocompetent Tumor Microenvironments. Cell Rep. 2018;25:3884–3893.e3. doi: 10.1016/j.celrep.2018.12.015. [DOI] [PubMed] [Google Scholar]
- 7.Ronteix G., Jain S., Angely C., Cazaux M., Khazen R., Bousso P., Baroud C.N. High resolution microfluidic assay and probabilistic modeling reveal cooperation between T cells in tumor killing. Nat. Commun. 2022;13:3111. doi: 10.1038/s41467-022-30575-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paz-Ares L., Luft A., Vicente D., Tafreshi A., Gümüş M., Mazières J., Hermes B., Çay Şenler F., Csőszi T., Fülöp A., et al. Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018;379:2040–2051. doi: 10.1056/NEJMoa1810865. [DOI] [PubMed] [Google Scholar]
- 9.Gadgeel S., Rodríguez-Abreu D., Speranza G., Esteban E., Felip E., Dómine M., Hui R., Hochmair M.J., Clingan P., Powell S.F., et al. Updated Analysis From KEYNOTE-189: Pembrolizumab or Placebo Plus Pemetrexed and Platinum for Previously Untreated Metastatic Nonsquamous Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2020;38:1505–1517. doi: 10.1200/JCO.19.03136. [DOI] [PubMed] [Google Scholar]
- 10.Choucair K., Morand S., Stanbery L., Edelman G., Dworkin L., Nemunaitis J. TMB: a promising immune-response biomarker, and potential spearhead in advancing targeted therapy trials. Cancer Gene Ther. 2020;27:841–853. doi: 10.1038/s41417-020-0174-y. [DOI] [PubMed] [Google Scholar]
- 11.Cyriac G., Gandhi L. Emerging biomarkers for immune checkpoint inhibition in lung cancer. Semin. Cancer Biol. 2018;52:269–277. doi: 10.1016/j.semcancer.2018.05.006. [DOI] [PubMed] [Google Scholar]
- 12.Guaitoli G., Tiseo M., Di Maio M., Friboulet L., Facchinetti F. Immune checkpoint inhibitors in oncogene-addicted non-small cell lung cancer: a systematic review and meta-analysis. Transl. Lung Cancer Res. 2021;10:2890–2916. doi: 10.21037/tlcr-20-941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jardim D.L., Goodman A., de Melo Gagliato D., Kurzrock R. The Challenges of Tumor Mutational Burden as an Immunotherapy Biomarker. Cancer Cell. 2021;39:154–173. doi: 10.1016/j.ccell.2020.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Camidge D.R., Doebele R.C., Kerr K.M. Comparing and contrasting predictive biomarkers for immunotherapy and targeted therapy of NSCLC. Nat. Rev. Clin. Oncol. 2019;16:341–355. doi: 10.1038/s41571-019-0173-9. [DOI] [PubMed] [Google Scholar]
- 15.Kieffer Y., Hocine H.R., Gentric G., Pelon F., Bernard C., Bourachot B., Lameiras S., Albergante L., Bonneau C., Guyard A., et al. Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov. 2020;10:1330–1351. doi: 10.1158/2159-8290.CD-19-1384. [DOI] [PubMed] [Google Scholar]
- 16.Fukumura D., Kloepper J., Amoozgar Z., Duda D.G., Jain R.K. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat. Rev. Clin. Oncol. 2018;15:325–340. doi: 10.1038/nrclinonc.2018.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Echchakir H., Vergnon I., Dorothée G., Grunenwald D., Chouaib S., Mami-Chouaib F. Evidence for in situ expansion of diverse antitumor-specific cytotoxic T lymphocyte clones in a human large cell carcinoma of the lung. Int. Immunol. 2000;12:537–546. doi: 10.1093/intimm/12.4.537. [DOI] [PubMed] [Google Scholar]
- 18.Asselin-Paturel C., Megherat S., Vergnon I., Echchakir H., Dorothée G., Blesson S., Gay F., Mami-Chouaib F., Chouaib S. Differential effect of high doses versus low doses of interleukin-12 on the adoptive transfer of human specific cytotoxic T lymphocyte in autologous lung tumors engrafted into severe combined immunodeficiency disease-nonobese diabetic mice: relation with interleukin-10 induction. Cancer. 2001;91:113–122. [PubMed] [Google Scholar]
- 19.Veith I., Mencattini A., Picant V., Serra M., Leclerc M., Comes M.C., Mami-Chouaib F., Camonis J., Descroix S., Shirvani H., et al. Apoptosis mapping in space and time of 3D tumor ecosystems reveals transmissibility of cytotoxic cancer death. PLoS Comput. Biol. 2021;17 doi: 10.1371/journal.pcbi.1008870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Comes M.C., Casti P., Mencattini A., Di Giuseppe D., Mermet-Meillon F., De Ninno A., Parrini M.C., Businaro L., Di Natale C., Martinelli E. The influence of spatial and temporal resolutions on the analysis of cell-cell interaction: a systematic study for time-lapse microscopy applications. Sci. Rep. 2019;9:6789. doi: 10.1038/s41598-019-42475-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mencattini A., Lansche C., Veith I., Erbs P., Balloul J.-M., Quemeneur E., Descroix S., Mechta-Grigoriou F., Zalcman G., Zaupa C., et al. Direct imaging and automatic analysis in tumor-on-chip reveal cooperative antitumoral activity of immune cells and oncolytic vaccinia virus. Biosens. Bioelectron. 2022;215 doi: 10.1016/j.bios.2022.114571. [DOI] [PubMed] [Google Scholar]
- 22.Zervantonakis I.K., Hughes-Alford S.K., Charest J.L., Condeelis J.S., Gertler F.B., Kamm R.D. Three-dimensional microfluidic model for tumor cell intravasation and endothelial barrier function. Proc. Natl. Acad. Sci. USA. 2012;109:13515–13520. doi: 10.1073/pnas.1210182109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aref A.R., Campisi M., Ivanova E., Portell A., Larios D., Piel B.P., Mathur N., Zhou C., Coakley R.V., Bartels A., et al. 3D microfluidic ex vivo culture of organotypic tumor spheroids to model immune checkpoint blockade. Lab Chip. 2018;18:3129–3143. doi: 10.1039/c8lc00322j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee S.W.L., Adriani G., Ceccarello E., Pavesi A., Tan A.T., Bertoletti A., Kamm R.D., Wong S.C. Characterizing the Role of Monocytes in T Cell Cancer Immunotherapy Using a 3D Microfluidic Model. Front. Immunol. 2018;9:416. doi: 10.3389/fimmu.2018.00416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pavesi A., Tan A.T., Koh S., Chia A., Colombo M., Antonecchia E., Miccolis C., Ceccarello E., Adriani G., Raimondi M.T., et al. A 3D microfluidic model for preclinical evaluation of TCR-engineered T cells against solid tumors. JCI Insight. 2017;2 doi: 10.1172/jci.insight.89762. 89762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lizotte P.H., Ivanova E.V., Awad M.M., Jones R.E., Keogh L., Liu H., Dries R., Almonte C., Herter-Sprie G.S., Santos A., et al. Multiparametric profiling of non-small-cell lung cancers reveals distinct immunophenotypes. JCI Insight. 2016;1 doi: 10.1172/jci.insight.89014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rakaee M., Adib E., Ricciuti B., Sholl L.M., Shi W., Alessi J.V., Cortellini A., Fulgenzi C.A.M., Viola P., Pinato D.J., et al. Association of Machine Learning-Based Assessment of Tumor-Infiltrating Lymphocytes on Standard Histologic Images With Outcomes of Immunotherapy in Patients With NSCLC. JAMA Oncol. 2023;9:51–60. doi: 10.1001/jamaoncol.2022.4933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dorothée G., Vergnon I., Menez J., Echchakir H., Grunenwald D., Kubin M., Chouaib S., Mami-Chouaib F. Tumor-infiltrating CD4+ T lymphocytes express APO2 ligand (APO2L)/TRAIL upon specific stimulation with autologous lung carcinoma cells: role of IFN-alpha on APO2L/TRAIL expression and -mediated cytotoxicity. J. Immunol. 2002;169:809–817. doi: 10.4049/jimmunol.169.2.809. [DOI] [PubMed] [Google Scholar]
- 29.Costa A., Kieffer Y., Scholer-Dahirel A., Pelon F., Bourachot B., Cardon M., Sirven P., Magagna I., Fuhrmann L., Bernard C., et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell. 2018;33:463–479.e10. doi: 10.1016/j.ccell.2018.01.011. [DOI] [PubMed] [Google Scholar]
- 30.Pelon F., Bourachot B., Kieffer Y., Magagna I., Mermet-Meillon F., Bonnet I., Costa A., Givel A.-M., Attieh Y., Barbazan J., et al. Cancer-associated fibroblast heterogeneity in axillary lymph nodes drives metastases in breast cancer through complementary mechanisms. Nat. Commun. 2020;11:404. doi: 10.1038/s41467-019-14134-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Givel A.-M., Kieffer Y., Scholer-Dahirel A., Sirven P., Cardon M., Pelon F., Magagna I., Gentric G., Costa A., Bonneau C., et al. miR200-regulated CXCL12β promotes fibroblast heterogeneity and immunosuppression in ovarian cancers. Nat. Commun. 2018;9:1056. doi: 10.1038/s41467-018-03348-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Licaj M., Mhaidly R., Kieffer Y., Croizer H., Bonneau C., Meng A., Djerroudi L., Mujangi-Ebeka K., Hocine H.R., Bourachot B., et al. Residual ANTXR1+ myofibroblasts after chemotherapy inhibit anti-tumor immunity via YAP1 signaling pathway. Nat. Commun. 2024;15:1312. doi: 10.1038/s41467-024-45595-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corgnac S., Lecluse Y., Mami-Chouaib F. Isolation of tumor-resident CD8+ T cells from human lung tumors. STAR Protoc. 2021;2 doi: 10.1016/j.xpro.2020.100267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sheng S.Y., Gu Y., Lu C.G., Tang Y.Y., Zou J.Y., Zhang Y.Q., Wang R.F., Hong H. The Characteristics of Naive-like T Cells in Tumor-infiltrating Lymphocytes From Human Lung Cancer. J. Immunother. 2017;40:1–10. doi: 10.1097/CJI.0000000000000147. [DOI] [PubMed] [Google Scholar]
- 35.Djenidi F., Adam J., Goubar A., Durgeau A., Meurice G., de Montpréville V., Validire P., Besse B., Mami-Chouaib F. CD8+CD103+ tumor-infiltrating lymphocytes are tumor-specific tissue-resident memory T cells and a prognostic factor for survival in lung cancer patients. J. Immunol. 2015;194:3475–3486. doi: 10.4049/jimmunol.1402711. [DOI] [PubMed] [Google Scholar]
- 36.Corgnac S., Malenica I., Mezquita L., Auclin E., Voilin E., Kacher J., Halse H., Grynszpan L., Signolle N., Dayris T., et al. CD103+CD8+ TRM Cells Accumulate in Tumors of Anti-PD-1-Responder Lung Cancer Patients and Are Tumor-Reactive Lymphocytes Enriched with Tc17. Cell Rep. Med. 2020;1 doi: 10.1016/j.xcrm.2020.100127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wong C.H., Siah K.W., Lo A.W. Estimation of clinical trial success rates and related parameters. Biostatistics. 2019;20:273–286. doi: 10.1093/biostatistics/kxx069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fouquet C., Antoine M., Tisserand P., Favis R., Wislez M., Commo F., Rabbe N., Carette M.F., Milleron B., Barany F., et al. Rapid and sensitive p53 alteration analysis in biopsies from lung cancer patients using a functional assay and a universal oligonucleotide array: a prospective study. Clin. Cancer Res. 2004;10:3479–3489. doi: 10.1158/1078-0432.CCR-0994-03. [DOI] [PubMed] [Google Scholar]
- 39.Barlesi F., Mazieres J., Merlio J.-P., Debieuvre D., Mosser J., Lena H., Ouafik L., Besse B., Rouquette I., Westeel V., et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT) Lancet. 2016;387:1415–1426. doi: 10.1016/S0140-6736(16)00004-0. [DOI] [PubMed] [Google Scholar]
- 40.Vlachogiannis G., Hedayat S., Vatsiou A., Jamin Y., Fernández-Mateos J., Khan K., Lampis A., Eason K., Huntingford I., Burke R., et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science. 2018;359:920–926. doi: 10.1126/science.aao2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sachs N., de Ligt J., Kopper O., Gogola E., Bounova G., Weeber F., Balgobind A.V., Wind K., Gracanin A., Begthel H., et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell. 2018;172:373–386.e10. doi: 10.1016/j.cell.2017.11.010. [DOI] [PubMed] [Google Scholar]
- 42.Tiriac H., Belleau P., Engle D.D., Plenker D., Deschênes A., Somerville T.D.D., Froeling F.E.M., Burkhart R.A., Denroche R.E., Jang G.-H., et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov. 2018;8:1112–1129. doi: 10.1158/2159-8290.CD-18-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dijkstra K.K., Monkhorst K., Schipper L.J., Hartemink K.J., Smit E.F., Kaing S., de Groot R., Wolkers M.C., Clevers H., Cuppen E., Voest E.E. Challenges in Establishing Pure Lung Cancer Organoids Limit Their Utility for Personalized Medicine. Cell Rep. 2020;31 doi: 10.1016/j.celrep.2020.107588. [DOI] [PubMed] [Google Scholar]
- 44.Voabil P., de Bruijn M., Roelofsen L.M., Hendriks S.H., Brokamp S., van den Braber M., Broeks A., Sanders J., Herzig P., Zippelius A., et al. An ex vivo tumor fragment platform to dissect response to PD-1 blockade in cancer. Nat. Med. 2021;27:1250–1261. doi: 10.1038/s41591-021-01398-3. [DOI] [PubMed] [Google Scholar]
- 45.Jenkins R.W., Aref A.R., Lizotte P.H., Ivanova E., Stinson S., Zhou C.W., Bowden M., Deng J., Liu H., Miao D., et al. Ex Vivo Profiling of PD-1 Blockade Using Organotypic Tumor Spheroids. Cancer Discov. 2018;8:196–215. doi: 10.1158/2159-8290.CD-17-0833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mencattini A., Di Giuseppe D., Comes M.C., Casti P., Corsi F., Bertani F.R., Ghibelli L., Businaro L., Di Natale C., Parrini M.C., Martinelli E. Discovering the hidden messages within cell trajectories using a deep learning approach for in vitro evaluation of cancer drug treatments. Sci. Rep. 2020;10:7653. doi: 10.1038/s41598-020-64246-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bouquerel C., César W., Barthod L., Arrak S., Battistella A., Gropplero G., Mechta-Grigoriou F., Zalcman G., Parrini M.C., Verhulsel M., Descroix S. Precise and fast control of the dissolved oxygen level for tumor-on-chip. Lab Chip. 2022;22:4443–4455. doi: 10.1039/d2lc00696k. [DOI] [PubMed] [Google Scholar]
- 48.Denton A.E., Roberts E.W., Linterman M.A., Fearon D.T. Fibroblastic reticular cells of the lymph node are required for retention of resting but not activated CD8+ T cells. Proc. Natl. Acad. Sci. USA. 2014;111:12139–12144. doi: 10.1073/pnas.1412910111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ruhland M.K., Loza A.J., Capietto A.-H., Luo X., Knolhoff B.L., Flanagan K.C., Belt B.A., Alspach E., Leahy K., Luo J., et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat. Commun. 2016;7 doi: 10.1038/ncomms11762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang X., Lin Y., Shi Y., Li B., Liu W., Yin W., Dang Y., Chu Y., Fan J., He R. FAP Promotes Immunosuppression by Cancer-Associated Fibroblasts in the Tumor Microenvironment via STAT3-CCL2 Signaling. Cancer Res. 2016;76:4124–4135. doi: 10.1158/0008-5472.CAN-15-2973. [DOI] [PubMed] [Google Scholar]
- 51.Zhang Y., Ertl H.C.J. Depletion of FAP+ cells reduces immunosuppressive cells and improves metabolism and functions CD8+T cells within tumors. Oncotarget. 2016;7:23282–23299. doi: 10.18632/oncotarget.7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Feig C., Jones J.O., Kraman M., Wells R.J.B., Deonarine A., Chan D.S., Connell C.M., Roberts E.W., Zhao Q., Caballero O.L., et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA. 2013;110:20212–20217. doi: 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brosseau S., Abreu P., Bouchez C., Charon L., Kieffer Y., Gentric G., Picant V., Veith I., Camonis J., Descroix S., et al. YAP/TEAD involvement in resistance to paclitaxel chemotherapy in lung cancer. Mol. Cell. Biochem. 2024 doi: 10.1007/s11010-024-04949-7. [DOI] [PubMed] [Google Scholar]
- 54.Ao Z., Cai H., Wu Z., Hu L., Li X., Kaurich C., Gu M., Cheng L., Lu X., Guo F. Evaluation of cancer immunotherapy using mini-tumor chips. Theranostics. 2022;12:3628–3636. doi: 10.7150/thno.71761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Biselli E., Agliari E., Barra A., Bertani F.R., Gerardino A., De Ninno A., Mencattini A., Di Giuseppe D., Mattei F., Schiavoni G., Lucarini V. Organs on chip approach: a tool to evaluate cancer-immune cells interactions. Scientific reports. 2017;7:12737. doi: 10.1038/s41598-017-13070-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Parlato S., De Ninno A., Molfetta R., Toschi E., Salerno D., Mencattini A., Romagnoli G., Fragale A., Roccazzello L., Buoncervello M., Canini I. 3D Microfluidic model for evaluating immunotherapy efficacy by tracking dendritic cell behaviour toward tumor cells. Scientific reports. 2017;7:1093. doi: 10.1038/s41598-017-01013-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Davies E.R. 3rd Edition. Morgan Kauffman Publishers; 2005. Machine Vision: Theory, Algorithms, Practicalities. [Google Scholar]
- 58.Gonzalez R., Woods R. global edition. Vol. 19. 2018. Digital image processing. (Digital Image Processing, Global Edition). [Google Scholar]
- 59.Craven P., Wahba G. Smoothing noisy data with spline functions. Numer. Math. 1978;31:377–403. doi: 10.1007/BF01404567. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Time length, 48 h. Drug is added at 16 h. Cells, pre-stained in red, become green when dying by apoptosis (Cell Event apoptosis reporter). Scale bar, 100 μm.
Time length, 6 h. Drug is added at the start. Dying cells are in magenta (DRQ7 dye). Scale bar, 100 μm.
Time length, 48 h. Drug is added at 16 h. Cells, pre-stained in red, become green when dying by apoptosis (Cell Event apoptosis reporter). Scale bar, 100 μm.
Time length, 19 h. Drug is added at the start. Cells, pre-stained in red, become green when dying by apoptosis (Cell Event apoptosis reporter). Scale bar, 100 μm.
Patient #8. Time length, 48 h. Drug is added at the start. Cells dying by apoptosis are in green (Cell Event apoptosis reporter). Scale bar, 100 μm.
Data Availability Statement
-
•
All source data reported in this paper will be shared by the lead contact upon request.
-
•
The original code TM-STAMP has been deposited at Nextcloud and is publicly available as of the date of publication. The link is listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.