

Summary
The investigation of the mechanisms behind p53 mutations in acute myeloid leukemia (AML) has been limited by the lack of suitable mouse models, which historically have resulted in lymphoma rather than leukemia. This study introduces two new AML mouse models. One model induces mutant p53 and Mdm2 haploinsufficiency in early development, showing the role of Mdm2 in myeloid-biased hematopoiesis and AML predisposition, independent of p53. The second model mimics clonal hematopoiesis by inducing mutant p53 in adult hematopoietic stem cells, demonstrating that the timing of p53 mutation determines AML vs. lymphoma development. In this context, age-related changes in hematopoietic stem cells (HSCs) collaborate with mutant p53 to predispose toward myeloid transformation rather than lymphoma development. Our study unveils new insights into the cooperative impact of HSC age, Trp53 mutations, and Mdm2 haploinsufficiency on clonal hematopoiesis and the development of myeloid malignancies.
Keywords: TP53 mutations, clonal hematopoiesis, Mdm2 haploinsufficiency, hematopoietic stem cells, cholesterol biosynthesis, acute myeloid leukemia, mouse model, myeloid-biased hematopoiesis, mevalonate pathway
Graphical abstract
Highlights
-
•
Allele-specific loss of MDM2 in TP53-mutant acute myeloid leukemia (AML)
-
•
Mdm2 haploinsufficiency collaborates with mutant Trp53 in AML
-
•
p53-independent mechanism of Mdm2 in controlling the mevalonate pathway
-
•
Targeting the mevalonate pathway enhances the efficacy of MDM2 inhibitors in AML
Pourebrahim et al. develop two mouse models for p53-mutant acute myeloid leukemia, revealing how Mdm2 interaction and the timing of mutations in blood stem cells influence disease onset and type. This work offers new insights into AML pathogenesis and potential treatment strategies.
Introduction
TP53 abnormalities are observed in 5%–10% of de novo myelodysplastic syndrome (MDS) and AML patients, but the frequency increases up to 40% in older patients or those with therapy-related myeloid malignancies.1 The mechanisms by which TP53 mutations lead to the development of AML are not well understood. Previous data from studies with Trp53 and Mdm2 knockout mice suggested that once p53 is inactivated, Mdm2 becomes redundant in cells.2,3 However, recent findings have challenged this established belief, revealing that Mdm2 plays a crucial role in the survival and proliferation of p53-mutated and deleted human cancer cells, such as triple-negative breast cancer.4
The regulation of p53 protein by Mdm2 plays a crucial role in maintaining cellular integrity following DNA damage.5,6 Mdm2 is an E3 ubiquitin ligase that targets p53 for degradation in the 26S proteasome, providing a mechanism for fine-tuning p53 levels after DNA damage.7,8,9 Li-Fraumeni syndrome (LFS), caused by germline mutations in the TP53 gene, is associated with increased susceptibility to developing lymphoid malignancies at a young age, and myeloid malignancies in the setting of therapy-related disease and old age.10 Similarly, mice with germline Trp53 mutations also predominantly develop lymphomas rather than leukemias,11 further emphasizing the role of germinal p53 mutations in lymphoma predisposition. Yet, the role of p53 and Mdm2 in hematopoietic differentiation and their link to the development of specific hematological malignancies have yet to be fully elucidated.
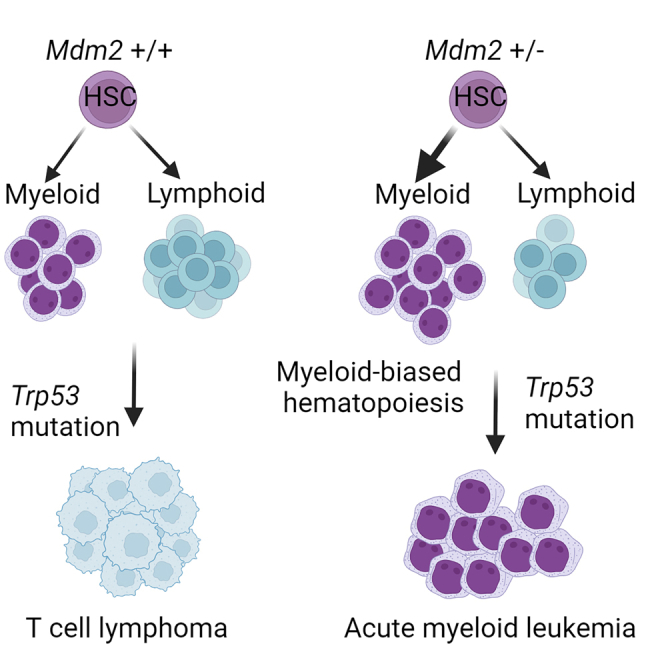
In this study, we explored the impact of Mdm2 haploinsufficiency on the development of lymphoma vs. AML in the presence of Trp53 R172H mutation. Our analysis revealed concurrent heterozygous loss of MDM2 in TP53-mutated AML, along with a significant enrichment of Mdm2SNP309 in TP53-mutant AML. We show that Mdm2 haploinsufficiency leads to myeloid-biased hematopoiesis, which cooperates with somatic Trp53-mutations in the development of AML. Furthermore, we uncovered a novel function of Mdm2 in regulation of the mevalonate pathway and demonstrate that its inhibition promotes myeloid differentiation, independently of its role in p53 regulation. Through a series of complementary genetic models, we show that the age of mice is a critical factor in determining the transformation to myeloid vs. lymphoid malignancies in the context of p53 mutations. Together, our findings present a valuable mouse model for dissecting the role of mutant p53 in AML and offer important insights into the impact of Mdm2 haploinsufficiency on myeloid-biased hematopoiesis, potentially guiding the development of therapeutic strategies for TP53-mutant AML.
Results
Frequency and association of MDM2 alterations and TP53 mutations and allele-specific characteristics of MDM2SNP309 in AML
To assess the significance of MDM2 alterations and the potential implications of MDM2 haploinsufficiency in AML, we conducted an analysis using data from the Pan-Cancer Analysis of Whole Genomics study.12 Our objective was to determine the frequency of MDM2-Hetloss specifically in TP53 mutant tumors. Among the 2,583 patients examined, we observed MDM2-Hetloss in approximately 7% of all cancers. However, TP53-mutant cases exhibited MDM2 Hetloss in more than 17% of instances, indicating a strong association (log2 odds ratio = 2.04, p < 0.001) (Figure 1A). It is worth noting that no homozygous deletions of MDM2 were detected in any cancer types, suggesting the essential nature of MDM2, even in TP53-mutant tumors, which has been previously reported.13 We also assessed the transcript levels of MDM2 with respect to TP53 mutation status and found that MDM2 expression levels are significantly lower in TP53-mutant tumors (t test, p = 0.0001) (Figure 1B). In the TP53-mutant group, MDM2 expression was significantly lower in MDM2-Hetloss cases compared with MDM2-diploid cases (t test, p = 0.001) (Figure 1C). Additionally, we searched for available AML datasets on cBioPortal (400 cases) and found that MDM2-Hetloss was concomitant with TP53 missense mutations (log2 odds ratio >3, p < 0.001) (Figure 1D). This association was not present in lymphomas, where we found MDM2-Hetloss and TP53 mutations to be mutually exclusive, suggesting that the observed MDM2 copy number changes may have a specific link to the myeloid lineage (Figure 1D). Among AML patients with MDM2-Hetloss, TP53 mutations were the most frequently observed mutation (Figure 1E). To validate these genomic findings, we expanded our analysis to a TP53-mutant AML dataset consisting of 187 AML cell lines documented in the Cancer Cell Line Encyclopedia.14 The AML cell lines with TP53 mutations exhibited significantly lower MDM2 expression compared with TP53 wild-type cell lines (p = 0.0001) (Figure 1F).
Figure 1.

Analysis of TP53 mutations, MDM2 alterations, and MDM2 expression patterns in cancer
(A) Frequency of TP53 mutations and MDM2 copy number alterations across various cancer types using whole-genome analysis of ICGC/TCGA Pan-Cancer data.
(B) Expression of MDM2 in TP53–wild-type (WT) and TP53-mutant (Mut) cases in pan-cancer analysis of whole genomes.
(C) Relative expression of MDM2 mRNA based on MDM2 allele status pan-cancer analysis of whole genomes, ∗∗∗p < 0.001.
(D) Frequency of TP53 mutations and MDM2 copy number alterations in patients with AML and lymphoma, sourced from cBioPortal for Cancer Genomics.
(E) Frequency of mutations in different genes categorized by MDM2 WT and MDM2 LOH, retrieved from cBioPortal.
(F) Relative expression of MDM2 based on TP53 status using data from the Cancer Cell Line Encyclopedia, ∗∗∗∗p < 0.0001.
(G) MDM2 expression in AML samples, highlighting cases with mutant TP53: green for missense, brown for splice, and black for truncation mutations.
(H and I) Scatter diagram graphs the correlation of CD34 and KIT versus MDM2 at the mRNA levels in patients with AML (TCGA).
(J) Overall survival of p53 mutant AML patients with or without MDM2 LOH.
(K) Bar plot displaying the distribution of the G allele on the major or minor allele in germline T/G cases and in the context of somatic MDM2 loss, depending on TP53 mutational status (altered or wild type) and the presence or absence of the T allele (no LOH; left) or its absence (LOH; right). This demonstrates a significant enrichment for the G allele on the minor allele in altered TP53 cases without LOH (adjusted PFisher = 4.8e−2).
(L) Allele-specific expression analysis of GG (blue), TG (green), and TT (red) in indicated patient samples.
To investigate if the reduced expression of MDM2 in TP53-mutant patients is due to MDM2-Hetloss or impaired transcriptional activity of TP53 resulting from mutations, we compared MDM2 expression in subjects with MDM2-Hetloss with those with a diploid MDM2 status. Significantly lower MDM2 expression was observed in TP53-mutant/MDM2-Hetloss cases compared with TP53-mutant/MDM2-diploid cases (p = 0.001) (Figure 1G). This indicates that in TP53-mutant patients, MDM2 levels are predominantly influenced by MDM2 copy number. Further analysis was conducted to determine whether MDM2-Hetloss is linked to a specific phenotype in TP53-mutant AML. As depicted in Figures 1H and 1I, an inverse relationship was found between MDM2 levels and the expression of CD34 and KIT, suggesting a possible role in the differentiation of leukemia cells. Moreover, a significant correlation was found between MDM2-Hetloss and lower survival rates (p = 0.002) (Figure 1J), suggesting the potential prognostic significance of MDM2-Hetloss in TP53-mutant AML.
Next, we aimed to investigate whether MDM2 loss of heterozygosity (LOH) exhibited allele-specific characteristics or varied between alleles of MDM2 single nucleotide polymorphisms (SNPs), with particular focus on MDM2SNP309 (rs2279744; T>G). MDM2SNP309 is located in the intronic promoter region of MDM2 (Figure S1A) and has been associated with increased MDM2 expression and earlier cancer onset in patients with LFS and sporadic cancers.15 We analyzed the MDM2SNP309 genotype within the pan-cancer analysis of whole genomes (PCAWG) cohort. Due to the limited availability of TP53-mutant AML cases (only eight) in a curated list of 2,658 PCAWG cases, we leveraged whole-genome sequencing data to achieve sufficient statistical power. Our analysis revealed similar patterns across the T/T, T/G, and G/G genotypes, indicating no significant association between the MDM2SNP309 genotype and TP53 inactivation, alteration, or wild-type status (Pchi2 = 0.71; Figure S1B). We also examined the relationship between MDM2 copy number status and TP53 mutational status within the T/T and G/G genotypes. In a wild-type TP53 context, we observed a higher frequency of MDM2 losses in the G/G genotype (13.22%) compared with the T/T genotype (7.66%) (adjusted PFisher test = 3.99e−2; Figure S1C). Subsequently, we assessed MDM2 allelic imbalances in T/G germline cases and found that the G allele was retained on the major allele in 64% of wild-type TP53 cases, whereas only 30% of altered-TP53 cases exhibited this pattern in losses without LOH (Figure 1K). Of note, no significant differences in T/G were observed in losses with LOH.
Additionally, we investigated the MDM2SNP309 T/G genotype in bone marrow samples from AML patients, distinguishing between those with wild-type (n = 10) and mutant TP53 (n = 46). Our findings revealed that the T/T genotype was present in 45% of wild-type TP53 cases, while 89% of mutant TP53 cases exhibited the T/T genotype, indicating selective loss of the G allele in TP53-mutant AML (Figure 1L). These results suggest an association between allele-specific loss of the G allele and TP53 mutations in various cancer types and AML patients. Together, these data reveal a strong association between MDM2-Hetloss and TP53 mutations in AML, indicating functional implications. Additionally, allele-specific loss of the G allele suggests a link between MDM2 expression levels and TP53 mutations across various cancer types and AML.
Mdm2 haploinsufficiency cooperates with mutant p53 to promote the development of AML
Building on the observed correlation between MDM2 expression levels and TP53 mutations in AML, we developed specific genetic models to further elucidate the potential interplay between Mdm2 levels and Trp53 mutations, thereby enhancing our understanding of AML pathogenesis. As a brief introduction, the gain-of-function (GOF) activity of mutant p53 has been largely studied through comparison of mice harboring a conditional mutant Trp53 allele known as LSL-Trp53R172H.16 Hematopoietic-specific induction of LSL-Trp53R172H results in thymic lymphoma in most animals.17 However, the LSL cassette in LSL-Trp53R172H mice leads to Trp53 heterozygosity in the whole mouse, allowing for a heterozygous Trp53 microenvironment that facilitates tumorigenesis.18 To tackle this challenge, we utilized an inducible mutant Trp53R172H allele, which we refer to as “wmR172H” (where “wm” denotes the transition from wild type to mutant). This allele features an R172H mutation, analogous to the R175H mutation in humans, positioned downstream of a wild-type Trp53 cDNA sequence, which is in turn flanked by loxP sites (Figure 2A). This design ensures the maintenance of wild-type Trp53 expression until recombination occurs.19,20 To evaluate the impact of Trp53 LOH and Mdm2 haploinsufficiency in our model, we utilized the Trp53fl 21 and Mdm2fl22 respectively. To monitor recombined cells in mice, we utilized an mTmG allele, a fluorescent reporter with a tdTomato (mT) cassette. Upon Cre-mediated recombination, this cassette is replaced by a green fluorescent protein (mG) cassette (Figure 2A).
Figure 2.

Overview of alleles and pathological findings in a novel p53 mutant AML mouse model
(A) Schematic representation of the genetic modifications in the mouse model. The activation of Cre recombinase under the Vav1 promoter leads to deletion of wild-type Trp53 cDNA flanked by loxP sites (black triangles) resulting in a Trp53-mut allele. The diagram also shows the recombination of the mTmG reporter allele, facilitating GFP expression in hematopoietic cells, with “mT” for membranous tomato, “mG” for membranous GFP, and “A” indicating a polyadenylation sequence. The red star marks the Trp53 R172H mutation on exon 5.
(B) Image of a white thoracic lymphoma mass in a Vav-Cre;Trp53fl/wmR172H mouse.
(C) Immunohistochemistry for p53, showing nuclear accumulation in lymphoma cells. Flow cytometry analysis identifying a distinct CD3+ T lymphocyte population, with absence of CD11b+ myeloid cells.
(D) Kaplan-Meier survival curve with a p value of 0.2 from the log rank (Mantel-Cox) test, showing the overall survival rate of the mice.
(E) Representative image of AML infiltration in liver and spleen of moribund Vav-Cre;Mdm2+/fl;Trp53fl/wmR172H mice.
(F) Hematoxylin and eosin (H&E)-stained bone marrow (BM) section from the AML mouse depicted in (J), showing characteristic features of AML.
(G) Immunostaining for p53 protein, visualized as brown staining, demonstrates the presence of p53 in leukemia cells within bone marrow (BM).
(H) Flow cytometry analysis of CD3, CD11b, and Ly6G markers in the bone marrow of AML mice.
(I) Western blot analysis of p53 protein expression in BM samples comparing p53 protein levels in p53 wild-type control and five AML samples.
(J) Immunofluorescence analysis of Ki-67 in BM. MSCs, mesenchymal stromal cells.
(K) Mutant p53 accumulation in GFP-positive AML cells in BM sections.
(L) Kaplan-Meier analysis of overall survival of the indicated mice. p = 0.01, log rank (Mantel-Cox) test.
We initially assessed the impact of Trp53 mutation/loss in early hematopoietic stem cells (HSCs) using the Vav-Cre system. Two mouse cohorts were generated: one with a conditional Trp53 mutant allele exhibitingTrp53 LOH (Vav-Cre;Trp53wmR172 H/fl), and another with homozygous Trp53 floxed alleles (Vav-Cre;Trp53 fl/fl), allowing us to compare the effects of p53 mutation vs. complete loss of p53 in HSCs. Some mice also carried the mTmG allele for tracking. To validate our model, we analyzed E10.5 Vav-Cre-mTmG embryos and confirmed successful recombination of the mTmG allele in the aortogonadal mesenchyme and fetal liver by fluorescence microscopy (Figure S2). Mice with Trp53 mutations and Trp53 deletion introduced early in life developed lymphoma with complete penetrance, suggesting that the mutation or deletion of Trp53 at an early age is more likely to result in the development of lymphoma (Figure 2B). Immunohistochemical analysis of lymphoma tissue showed nuclear p53 staining and flow cytometry confirmed CD3 expression and absence of CD11b in lymphoma cells (Figure 2C). The Kaplan-Meier survival analysis revealed comparable survival of Vav-Cre;Trp53wmR172 H/fl mice to that of Vav-Cre;Trp53 fl/fl mice, indicating that there was no GOF of mutant p53 in this model (Figure 2D). Both models had full penetrance of thymic lymphoma, suggesting that the loss of p53 function alone is sufficient to drive lymphoma development in these mice. This observation is consistent with previous reports comparing Trp53null with Trp53R172H mice in which Trp53 was deleted or mutated in the whole body.23,24,25 The absence of AML in both cohorts indicates that additional genetic aberrations may be necessary in this context for the development of AML.
Next, we aimed to explore the phenotype of Mdm2 haploinsufficiency and Trp53 mutations in early HSCs. We crossed mice carrying the Mdm2fl alleles,22 with progeny from Trp53wmR172 H/fl to generate Vav-Cre;Mdm2+/fl; Trp53wmR172 H/fl mice (referred to as Vav-Mdmfl/+;p53mut/fl), some of which also carried the mTmG allele for tracing. Vav-Mdmfl/+;p53mut/fl mice developed AML with full penetrance, characterized by myelomonocytic leukemia cells infiltrating both bone marrow (BM) and liver (Figure 2E; Table S1). Three out of 12 mice succumbed to lymphoma. However, analysis of DNA isolated from tumor tissue revealed a lack of recombination of the Mdm2 allele in the tumor. Histopathological analysis of BM, spleen, and liver samples revealed myeloid-derived leukemias (Figure 2F). Some mice showed multiclonality with the presence of more than one type of morphologic nonlymphoid hematopoietic neoplasm. Immunostaining analysis showed strong nuclear expression of mutant p53 in the BM (Figure 2G). Flow cytometry analysis revealed a CD3–,CD11b+, Ly6G+ phenotype, consistent with our histopathology findings (Figure 2H). Western blot analysis of a handful of spleen samples revealed high levels of mutant p53 (Figure 2I). Myelomonocytic AML features were observed in BM samples from Vav-Mdmfl/+;p53mut/fl mice, with GFP+ leukemic cells invading periosteum and muscles (Figure S3A). Immunostaining displayed a highly proliferative disease, with p53 accumulation in most malignant BM cells (Figures 2J and 2K). We observed a few clusters of CD3+ cells in the BM indicating suppressed lymphoid lineage (Figure S3B). Clonal expansion of p53-mutant AML cells was heterogeneously distributed throughout the BM (Figures S3C and S3D). Mdm2 haploinsufficiency significantly extended survival of p53-mutant mice compared with Mdm2-diploid p53-mutant mice (p = 0.01)(Figure 2L). Notably, mice with the deletion of both Mdm2 alleles (Vav-Mdmfl/fl;p53mut/fl) developed lymphoma, a phenotype comparable to that of Vav-cre;p53mut/fl mice, suggesting that the complete loss of Mdm2 does not directly contribute to the development of AML. Together, these findings confirm that Mdm2 haploinsufficiency and Trp53 mutations synergistically induce aggressive AML with full penetrance.
Trp53 somatic mutation in adult HSCs leads to pre-leukemic clonal expansion
To explore the clonal evolution of p53 mutant subclones and their progression into myeloid malignancies, we created an inducible mouse model using the Mx1-cre driver and polyinosinic:polycytidylic acid (pIpC) induction. The inducible conditional mouse model replicates clonal hematopoiesis by inducing somatic mutations in a fraction of adult HSCs at about 4 months of age, which is equivalent to young adult humans, and track the recombined cells using the mTmG allele (Figure 3A). Initially, we confirmed that Mx1-cre effectively targets HSCs in the BM of Mx1-cre;mTmG mice. Analysis of BM cells from these mice revealed that the GFP+ population was present within the CD150+/Lin−/Sca1+/c-Kit+ (LSK) cell population, indicating the successful targeting of the hematopoietic stem cell compartment by the Mx1-cre driver (Figure S4A). This finding is consistent with previous reports demonstrating the effectiveness of the Mx1-cre system in vivo.26 Fluorescence microscopy analysis of the BM displayed clear evidence of efficient recombination of the mTmG allele, as indicated by the presence of clonal clusters of GFP+ cells (Figure 3B). The GFP+ cells were detectable throughout the BM and formed distinct colonies. The presence of GFP+ cells was also detected in the peripheral blood (PB), and spleen (Figure 3C).
Figure 3.

Clonal evolution and generation of a p53 mutant clonal hematopoiesis mouse model
(A) The schematic outlines the creation of the Trp53R172H clonal hematopoiesis model in adult HSCs. Following pIpC injection, Cre recombinase expressed under the Mx1-Cre promoter induces recombination in hematopoietic cells, leading to a fluorescence switch in the mTmG allele from red (tdTomato) to green (GFP), simultaneously converting wild-type p53 to its mutant form.
(B and C) (B) Direct fluorescence image of the bone marrow of Mx1-Cre;Trp53wmR172 H/fl;mTmG mouse showing the distribution of GFP+ cells in the bone marrow (BM), peripheral blood (PB), and (C) spleen (SP). High magnification displaying a colony of GFP+ cells marked by white arrow.
(D) Flow cytometry plots showing the expression of GFP and Tomato in the peripheral blood of Mx1-Cre;Trp53wmR172 H/fl;mTmG mice at different time points.
(E) Flow cytometry plots of CD3 and CD11b expression gated on GFP or RFP in (D).
(F) tSNE plots representing a CyTOF analysis of the indicated marker distribution in BM.
(G) Graph showing the percentage of GFP+ cells in the peripheral blood of indicated mice over 3 months after pIpC injection. The data are presented as mean ± standard deviation.
(H) UMAP plot showing the cell subsets detected in pooled BM samples isolated from Mx1-Cre;mTmG (p53 wild-type), Mx1-Cre;Trp53fl/fl;mTmG (p53 null), and Mx1-Cre;Trp53wmR172 H/fl;mTmG (p53 mutant) mice subjected to UMAP dimension reduction. Each linage is marked by colors indicating cell type. The stacked bar plots summarize the subset frequencies in indicated genotypes.
Next, we investigated the clonal evolution of p53 mutant clonal hematopoiesis by generating cohorts of Mx1-Cre;mTmG and Mx1-Cre;Trp53wmR172 H/fl;mTmG mice, and analyzing PB samples on a monthly basis, starting at 4 months of age. The population of p53-mutant (GFP+) cells increased over time (Figure 3D). Flow cytometry analysis of PB after 3 months revealed an increased myeloid population defined as CD11b+ (Figure 3E).
To identify specific lineages that were expanded in p53 mutant clonal hematopoiesis, we isolated BM samples from Mx1-Cre;mTmG (p53 wild-type), Mx1-Cre;Trp53wmR172 H/fl;mTmG (p53 mutant) and Mx1-Cre;Trp53 fl/fl;mTmG (p53 null) mice and performed CyTOF analysis using a panel of 31 antibodies (Table S2). Antibodies specific to RFP and GFP were employed to distinguish the p53-mutant (GFP+) and p53 wild-type (RFP+) populations (Figure 3F). The p53 wild-type group showed relatively stable population levels over time, whereas the p53 mutant population displayed a significant expansion with increasing duration following polyinosinic:polycytidylic acid (pIpC) injection (Figure 3G). The CyTOF analysis revealed a slight increase in the myeloid lineage in p53 mutant and null mice compared with p53 wild-type (Figures 3H and S4B). We did not observe a significant expansion of a specific stem/progenitor cell population in p53 mutant GFP+ cells. These findings indicate that loss of p53 function may contribute to alterations in hematopoietic lineage development, without causing a dramatic shift in the stem cell population. The overall increase in the myeloid population may indicate a transformation occurring in the myeloid progenitors. Together, these data highlight the significant impact of Trp53 mutation on the clonal expansion and evolution of HSCs into myeloid lineage within an adult mouse model.
Clonal expansion of p53 mutant cells and development of myeloid malignancies in adult HSCs
We expanded the cohorts of Mx1-Cre;Trp53wmR172H/+;mTmG mice (referred to as Mx-p53H/+), Mx1-Cre;Trp53wmR172 H/fl;mTmG mice (referred to as Mx-p53mut/fl), and Mx1-Cre;Mdm2fl/+:Trp53wmR172 H/fl;mTmG mice (referred to as Mx-Mdm-p53mut/fl) and injected them with pIpC to induce the Mx1-Cre. Then, we closely monitored them to observe the development of leukemia in these mice cohorts. After a follow-up period of over a year, the Mx-p53mut/fl and Mx-Mdm-p53mut/fl mice developed various myeloid malignancies (Figure 4A). In contrast, the Mx-p53H/+ mice did not develop similar transformations during the observation period. These findings suggest that the loss of the wild-type Trp53 allele was a crucial factor in the transformation process. Mx-p53mut/fl mice displayed a significant expansion of myeloid cells in the BM, categorized as myeloid leukemia with or without maturation, aligning with the Bethesda proposals for classifying nonlymphoid hematopoietic neoplasms in mice27 (Figures 4B and S5A). Immunostaining of the BM confirmed the accumulation of the mutant p53 in leukemic clones (Figure 4C), which were scattered throughout the bone marrow (Figure S5B), while normal stromal and hematopoietic cells (Tomato-positive) did not show p53 positivity (Figure 4E). GFP+ leukemia blast cells were observed in the PB, indicating the progression and systemic spread of the disease (Figures 4D and 4F). The Mx-p53mut/fl mice exhibited a significant incidence of mixed leukemia/lymphoma phenotype (50%). In contrast, all Mx-Mdm-p53mut/fl mice presented with myeloid malignancies, showing no expansion of the lymphoid lineage (Figure 4G). The Mx-Mdm-p53 fl/fl mice (n = 3) developed leukemia with the same penetrance as Mx-Mdm-p53mut/fl, suggesting that the role of Mdm2 haploinsufficiency in this context was p53-independent.
Figure 4.

Characterization of AML development in Mx1-Cre;Trp53wmR172 H/fl;mTmG mice
(A) Infiltration of AML cells into the liver (LV) and spleen (SP), indicative of AML development, is shown.
(B) Hematoxylin and eosin (H&E)-stained sections of bone marrow (BM) showing extensive leukemia cell infiltration.
(C) p53 protein immunostaining in leukemia cells in BM.
(D) Direct fluorescence image of BM depicting colonies of GFP+ cells. The stromal cells and normal hematopoietic cells are labeled with tomato fluorescence.
(E) Fluorescent imaging of p53 immunostaining in BM shows GFP+ hematopoietic cells expressing mutant p53 protein (magenta) among Tomato+ stromal cells.
(F) Peripheral blood smear and fluorescence imaging of GFP+ blast cells.
(G) Table representing the phenotype of different leukemia subtypes in p53 mutant AML mice with indicated Mdm2 genotypes.
(H) Kaplan-Meier analysis of overall survival of the indicated mice.
(I) Flow cytometry analysis of BM for indicated markers showing the expansion of myeloid and erythroid markers within the GFP+ population.
(J) DNA content analysis in selected BM cell populations showing higher DNA content in p53 mutant AML cells (GFP+) compared with normal BM cells (RFP+).
(K) Heat map of copy number variation in bone marrow cells derived from Mx1-Cre;Trp53wmR172H/fl;mTmG mice with AML. Each row represents a separate bone marrow sample, while each column represents specific genomic regions. The color scale indicates the relative copy number, with shades ranging from blue (deletion) to red (amplification).
(L) Colony forming unit (CFU) assay shows granulocyte macrophage (GM) colonies under brightfield and fluorescence, highlighting the clonogenic ability of GFP+ cells, with Tomato+ cells showing no colony formation.
(M) Erythroid colony formation in AEL mice is shown through brightfield and direct fluorescence images, showing expansion of GFP+ colonies.
To investigate the gain of function of mutant p53 in our AML model, we compared the survival rates of Mx-p53mut/fl mice with those of Mx-p53 fl/fl mice. The findings suggest that the mutant p53 does not exhibit a pronounced GOF effect beyond the loss of wild-type p53 tumor-suppressing functions (Figure 4H). The survival rates of Mx-Mdm-p53mut/fl mice were also similar to those of Mx-p53mut/fl mice, suggesting that in this AML model, Mdm2 haploinsufficiency did not alter survival (Figure 4H). Further characterization of each BM sample by flow cytometry showed that the myeloid leukemia cells were mainly CD11b+Ly6G+CD3−, while the erythroid leukemia cells were CD34+CD71+ (Figure 4I). We observed that p53 mutant cells (GFP+) were highly enriched in CD71 population whereas the population of GFPnegRFPneg cells was predominantly Ter119 positive, indicating the expansion of mature erythroblasts (Figure 4I). To evaluate aneuploidy, which is a common feature of p53 mutant AML, we initially performed DNA content analysis. The population of GFP+ AML cells had higher DNA content compared to Tomato-positive p53 wild-type cells (Figure 4J). Further, whole exome sequencing analysis conducted on 16 AML samples with p53 mutations, and five control samples identified significant copy number alterations across various chromosomes (Figure 4K). Interestingly, we observed a recurrent microdeletion at 11q, and a deletion at 6q in 12 out of 16 (75%) AML samples, resembling the 7q deletion commonly found in human TP53 mutant AML. The Integrative Genomics Viewer analysis of the sequencing data verified the presence of the Trp53 R172H mutation in all 16 samples (Figure S5C).
Colony-forming analysis of BM cells from the Mx-p53mut/fl mice revealed granulocyte-macrophage (GM) colonies of GFP+ cells (Figure 4L), while erythroid leukemia cells showed BFU colonies (Figure 4M). In subsequent secondary transplant studies conducted in the syngeneic background or NSG mice, no leukemia developed even after a period of 6 months, despite the presence of detectable GFP and RFP cells in the recipients, suggesting that the p53 mutant leukemia cells may possess a more mature phenotype.
In conclusion, these data demonstrate the role of p53 mutations in the development of myeloid malignancies in adult HSCs, and that Mdm2 haploinsufficiency in adult HSCs leads to the development of myeloid, but not lymphoid malignancies.
Mdm2 haploinsufficiency leads to myeloid-biased hematopoiesis
Both Mdm2 and p53 play crucial roles in the regulation of HSC differentiation and hematopoiesis.28 To explore the role of Mdm2 in hematopoietic differentiation and myeloid-biased hematopoiesis, we focused on studying the hematopoietic phenotype of Mdm2 haploinsufficient hematopoietic cells using Vav-Cre;Mdm2fl/+;mTmG which will hereafter be referred to as Vav-Mdmfl/+ mice. Evaluation of BM cells of Vav-Mdmfl/+ mice at 10 days post-birth showed a reduction in cellularity compared to the control mice (Figure 5A). Furthermore, BM cells from Vav-Mdmfl/+mice showed a significant reduction in LSK cells (Figure 5B), and a significant reduction in PB counts, coupled with an increase in the myeloid lineage and a decrease in the erythroid lineage (Figures S6A and S6B). Flow cytometry analysis of BM-derived cells indicated a significant increase in CD11b+ and Ly6G + cell populations in Vav-Mdmfl/+mice, suggesting myeloid-biased hematopoiesis (Figure 5C).
Figure 5.

Myeloid-biased hematopoiesis resulting from Mdm2 haploinsufficiency
(A) Direct fluorescence images of longitudinal sections of femurs derived from the indicated mice on postnatal day 10. GFP marks hematopoietic cells. A high-magnification view of the boxed region in the left panel is shown in the right panel.
(B) Quantification of the expression of the LSK population expression in the indicated mice. Each data point represents a measurement from an individual mouse; bars indicate means, and whiskers indicate SDs. ∗∗∗∗p < 0.0001, n = 5.
(C) Expression of CD19, CD11b, Ly6G, and CD3 presented as percentages of total white blood cells (means ± SDs). ∗∗p < 0.01.
(D) Schematic depiction illustrating the experimental setup wherein BM cells from indicated donor mice were transplanted into recipient mice.
(E) Dot plot showing the percent of chimerism in indicated time points (n = 5).
(F) Quantification of ROS levels (means ± SDs) in indicated populations of BM isolated from indicated mice. ∗p < 0.01, n = 3.
(G) Kaplan-Meier analysis of the overall survival of indicated mice after irradiation.
(H) Representative histologic images of hematoxylin and eosin (H&E)-stained bone marrow sections from the indicated mice, taken 1 week after irradiation (IR).
(I) Schematic view of the transplant strategy. Mice were transplanted with a mixture of bone marrow cells-GFP (Trp53wt), Tomato (p53 mut), and GFP+Tomato (p53 mut, Mdm2 het) in equal proportions.
(J) Fluorescence images of peripheral blood (PB) and spleen show clone distribution.
(K) The bar plot shows cell population frequencies in peripheral blood 4 weeks after transplant, identified by flow cytometry. Each data point represents a measurement from an individual mouse; bars indicate means.∗p < 0.01.
(L) UMAP visualization depicting distinct cell subsets identified in pooled BM samples cells-GFP (Trp53wt), Tomato (p53 mut), and GFP+Tomato (p53 mut, Mdm2 het). Different cell lineages are color-coded for easy identification. The stacked bar plots provide a summary of subset frequencies across the specified genotypes.
To investigate the effects of Mdm2 haploinsufficiency on hematopoietic stem/progenitor cells, we conducted a comprehensive analysis using both in vitro (colony formation and serial replating assays) and in vivo (competitive repopulation) functional assays. LSK cells were isolated from Vav;Mdmfl/+ and control mice to evaluate their colony-forming capabilities. Notably, the Vav;Mdmfl/+ mice demonstrated small colonies and increase in colony-forming unit-granulocyte-macrophage (CFU-GM) progenitors, and a significant reduction in colony formation in serial replating assays (Figures S6C–S6E). These findings suggest that Mdm2 haploinsufficiency specifically impacts the formation of GM progenitors, potentially leading to alterations in myeloid lineage development.
Long-term repopulation assay in irradiated recipient mice over 3 months was performed to evaluate the HSC self-renewal and differentiation potential. Equal numbers of BM cells from Vav;Mdmfl/+ (GFP+) and control mTmG mice (Tomato+) were transplanted into lethally irradiated recipients and monitored to assess engraftment and reconstitution ability (Figure 5D). A significant decrease in engraftment and multilineage reconstitution ability was observed in Vav;Mdmfl/+ compared with control mice (Figure 5E). Flow cytometry analysis of BM cells derived from Vav-Mdm2fl/+ mice further revealed higher levels of reactive oxygen species (ROS) in the LSK population (Figure 5F), suggesting that Mdm2 haploinsufficiency may contribute to increased oxidative stress and impaired HSC function. In addition, Vav;Mdmfl/+ mice displayed heightened radiosensitivity and lethal hematopoietic failure following irradiation (Figure 5G). Histological examination of the BM in Vav;Mdmfl/+ mice after irradiation revealed a broad loss of hematopoietic lineage, indicating the essential role of Mdm2 levels in maintaining hematopoietic integrity after DNA damage (Figure 5H). These data indicate that Mdm2 haploinsufficiency in Vav-Mdm2fl/+ mice leads to impaired HSC function and maintenance in response to DNA damage stress.
Next, we examined if the observed phenotype in Vav;Mdmfl/+ mice was p53 dependent. Deletion of p53 in Vav;Mdmfl/+ mice rescued the LSK population and improved colony formation, showing that p53 loss can counteract some defects in Mdm2+/− HSCs (Figures S6F and S6G). To evaluate the phenotype in vivo, we conducted a competitive transplant experiment using BM cells from Vav;mTmG (p53WT-green), Vav;RosaLsl-TdTomato;p53mut/fl (p53-mutant, red) and Vav;RosaLsl-TdTomato;mTmG;Mdm-p53mut/fl (p53-mutant-Mdm2+/−, yellow) cells (Figure 5I). Six weeks post-transplant, these transplanted populations were detectable in the PB and spleen (Figure 5J). Notably, the population of p53 mutant cells was significantly higher than the other groups (Figure 5K). To further investigate the impact of Mdm2 haploinsufficiency on the hematopoietic hierarchies, we performed multiplexed CyTOF analysis of the BM in indicated mice. We employed unsupervised analysis to identify distinct lineages in the BM and assessed the impact of each genetic modification on cell composition. Notably, our analysis revealed the preferential expansion of granulocytic lineages in p53-mutant-Mdm2+/− BM cells (Figure 5L). This expansion occurred at the expense of lymphoid lineage and other lineages, indicating a significant skewing of BM differentiation trajectories toward myeloid differentiation. This result further supports our findings that the Mdm2 haploinsufficiency promotes differentiation toward the myeloid lineage, and that this effect is p53-independent.
Our collective findings suggest that Mdm2 haploinsufficiency leads to impaired HSC function, including myeloid-biased differentiation, engraftment defect, and increased oxidative stress. Moreover, these effects on differentiation appear to be independent of p53 function.
Mechanisms of Mdm2 haploinsufficiency in myeloid-biased hematopoiesis
To investigate how Mdm2 haploinsufficiency results in myeloid-biased hematopoiesis, we conducted RNA sequencing (RNA-seq) on Lin-ckit+ (LK) cells isolated from the BM of adult Vav;MdmWt, Vav;Mdmfl/+, and Vav;Mdmfl/+;p53 fl/fl mice (Figure 6A). We observed 2,961 upregulated and 1,648 downregulated genes (±1.5-fold, false discovery rate [FDR] = 0.05) in Vav;Mdmfl/+ compared with Vav;MdmWt mice (Figure 6B). Surprisingly, the gene expression signature of LK cells isolated from Vav;Mdmfl/+;p53 fl/fl mice was very similar to that of Vav;Mdmfl/+ mice (Figure 6C), indicating that the gene signature associated with Mdm2 haploinsufficiency was independent of p53. Deletion of Trp53 in Vav;Mdmfl/+ mice only led to upregulation of 20 genes and downregulation of 62 genes (Figure 6C). The transcript level of Mdm2 was reduced by 50% in Vav;Mdm2fl/+ and Vav;Mdmfl/+;p53 fl/fl mice, implying that Mdm2 copy numbers, and not p53 levels, determine its expression levels (Figure 6D).
Figure 6.

Mdm2 haploinsufficiency represses the mevalonate pathway independently of p53
(A) Schematic representation of experimental design. The total RNA extracted from Lin− cKit+ cells sorted from the indicated mice were analyzed by RNA sequencing.
(B and C) Mean-average plots of Log2 mean expression vs. Log2 fold change in mice with the indicated genotypes. Red dots indicate genes that were upregulated (Log2FC > 1 and p < 0.05). Blue dots indicate genes that were downregulated (Log2FC < −1 and p < 0.05).
(D) Bar graphs representing the average read counts of genes in the indicated mice (mean ± SD).
(E) Ingenuity pathway analysis (IPA) of differentially expressed genes (DEGs) representing the percentage of significant DEGs out of the total genes in each pathway that were downregulated (green) or upregulated (red) in Mdm2-haploinsufficient hematopoietic progenitors, ranked by p values (pVal).
(F) Heatmap representing changes in gene expression of the cholesterol biosynthesis pathway in Vav-Cre;Mdm2fl/+ mice compared with Vav-Cre;Mdm2+/+mice and Vav-Cre;Mdm2fl/+;Trp53fl/fl.
(G) Heatmap representing changes in gene expression of the oxidative phosphorylation (OxPhos) pathway.
(H) IPA analysis of upregulated genes in Mdm2-haploinsufficient mice.
(I) IPA upstream regulator analysis of DEGs in Vav-Cre;Mdm2+/fl vs. Vav-Cre;Mdm2+/+ mice showing inhibition of indicated upstream regulators and activation of Myc (red).
(J) IPA disease or functions annotation of DEGs between Vav-Cre;Mdm2+/fl vs. Vav-Cre;Mdm2+/+ mice.
Ingenuity pathway analysis (IPA) of differential gene expression revealed significant downregulation of cholesterol biosynthesis and the mevalonate pathway (−log2 p = 20) in Vav;Mdmfl/+ mice compared with Vav;MdmWt mice (Figure 6E). In addition, TREM1 signaling, Th1 and Th2 activation, and IL8 signaling, were significantly enriched in the downregulated gene set (Figure 6E). Strikingly, 85% of genes involved in cholesterol biosynthesis, including seven encoding enzymes within the mevalonate pathway (Hmgcr, Mvk, Mvd, Fdps, Sqle, Lss, Dhcr7), were downregulated in Vav;Mdmfl/+ mice (Figure 6F). Importantly, deletion of Trp53 in Vav;Mdmfl/+ mice (Vav;Mdmfl/+;p53 fl/fl) did not change this signature, indicating that the mevalonate pathway downregulation was p53 independent. Furthermore, the gene signature associated with oxidative phosphorylation (OxPhos) was significantly upregulated in Vav;Mdmfl/+ mice independent of p53 (Figures 6G and 6H), suggesting a potential compensatory mitochondrial stress response in HSCs with Mdm2 haploinsufficiency.
The key regulatory factors influencing the observed gene expression in Vav;Mdmfl/+ mice (lipopolysaccharide, IFNG, TP53, and IL2) were all downregulated (Figure 6I). Notably, the expression of Myc and its associated signature was upregulated in Vav;Mdmfl/+ mice (Figure 6I). IPA also showed a strong correlation between the Mdm2 signature with the gene signature observed in non-hematological malignancies (Figure 6J), suggesting that the Mdm2 haploinsufficiency-associated gene signature is highly enriched in cancers. In summary, these data demonstrate that Mdm2 haploinsufficiency leads to significant changes in gene expression in HSCs, particularly in the downregulation of cholesterol biosynthesis and the upregulation of oxidative phosphorylation. These changes appear to be independent of p53, as deletion of Trp53 did not alter the gene signature associated with Mdm2 haploinsufficiency.
Disruption of the mevalonate pathway by Mdm2 haploinsufficiency, and synergistic induction of apoptosis through combined inhibition of mevalonate pathway and Mdm2
The mevalonate pathway, regulated by Srebp, is responsible for synthesizing cholesterol and isoprenoids. Statin drugs inhibit the mevalonate pathway, affecting CoQ production and blocking squalene monooxygenase (Sqle)-mediated cholesterol synthesis (Figure 7A). To elucidate the effects of Mdm2 haploinsufficiency on the mevalonate pathway and its impact on hematopoietic differentiation, we compared the levels of p21, known to regulated HSC differentiation,29 p53, and Sqle in Vav-Mdmfl/+ mice and control mice. The Vav-Mdm2fl/+ mice exhibited a notable reduction in Sqle protein (Figure 7B). In Mdm2 haploinsufficient mice, p53 levels were comparable to those in wild-type mice, yet p21 levels were elevated compared with the control mice (Figures 7B and 7C). Analysis of BM cells derived from irradiated mice revealed an upregulation of p21 and downregulation of Srebp2 precursor levels in Vav-Mdmfl/+ mice (Figure 7D). The observed decrease in Srebp2 expression further supports the idea that Mdm2 haploinsufficiency may downregulate the mevalonate pathway via Srebp2 levels.
Figure 7.

Inhibition of mevalonate metabolism induces myeloid differentiation and synergizes with MDM2i to induce apoptosis in AML cells
(A) Schematic representation of the mevalonate pathway. The mevalonate pathway, regulated by SREBP, is responsible for synthesizing cholesterol and isoprenoids. Statin drugs inhibit SREBP activation, affecting CoQ production and blocking Sqle-mediated cholesterol synthesis.
(B) Western blot analysis of Sqle, p53, p21, and b-actin levels in spleen cells derived from Vav-Cre;Mdm2+/+ and Vav-Cre;Mdm2+/fl, before and after irradiation (6 Gy). The figure displays bands corresponding to the molecular weights of the respective proteins.
(C) Quantification of p21 protein levels in (B), normalized to beta-actin. Bar graphs represent the mean protein expression level relative to the control, with error bars indicating the standard error of the mean (SEM) based on two separate blots.
(D) Western blot analysis of SREBP, Mdm2, p21, and β-actin protein levels in bone marrow cells derived from Vav-Cre;Mdm2+/+ and Vav-Cre;Mdm2+/fl, after irradiation (6 Gy). The bands at the corresponding molecular weights are displayed.
(E) Western blot analysis of K562 cells treated with indicated concentrations of XY018. The figure displays bands corresponding to the molecular weights of the respective proteins. The normalized levels of phospho-eIF2α are shown on the right.
(F) Quantification of myeloid cells (CD11b+) in CFU culture treated with atorvastatin, displaying means ± SDs. Each data point represents a measurement from an individual mouse. ∗∗∗∗p < 0.0001.
(G) Representative flow cytometry plot and quantification of the frequency of CD11b+ in HL60 cells after 48 h treatment with atorvastatin. ∗∗∗∗p < 0.0001.
(H) Percentage of live cells in indicated cells treated with different concentration of atorvastatin. Error bars indicate the standard error of the mean (SEM), representing the variability within the experimental replicates.
(I–K) Percentage of dead cells defined as Annexin V/DAPI(+) cells in indicated cell lines treated with indicated compounds and concentrations.
(L) Percentage of cells with high peroxidation index treated with indicated compounds.
To assess the RNA-seq findings that suggested Eif2 signaling activation in Mdm2 haploinsufficient HSC/progenitor cells (Figure 6H), we conducted western blot analysis on spleen cells from Vav-Mdm2fl/+ and control mice. This analysis revealed higher levels of p-Eif2a in the Vav-Mdm2fl/+ mice, suggesting that Mdm2 haploinsufficiency may activate the integrated stress response (ISR) pathway (Figures S7A and S7B). To evaluate if inhibition of the mevalonate pathway may play a role, K562 cells were treated with the RORγ inhibitor XY018, which is known to decrease cholesterol biosynthesis rate without affecting host cholesterol homeostasis.30 This treatment led to significant decreases in Hmgcs and Sqle levels and an increase in p-Eif2a levels, indicating that cholesterol metabolism inhibition might trigger ISR activation (Figure 7E). However, ISR activation through Thapsigargin resulted in increased Hmgcs and Sqle levels, pointing to a complex regulatory interaction between these pathways (Figure S7C).
Next, we investigated the impact of statin-mediated inhibition of cholesterol biosynthesis on myeloid differentiation. LSK cells isolated from wild-type mice were treated with atorvastatin in colony-forming cultures, and showed a significant increase in myeloid differentiation indicated by CD11b+ cells (t test, p = 0.0001) (Figure 7F), suggesting the involvement of the mevalonate pathway in myeloid differentiation. Furthermore, treating p53-null HL60 cells with atorvastatin promoted myeloid differentiation (Figure 7G), accompanied by a dose-dependent inhibition of cell proliferation (Figure 7H). These data further support the contribution of the mevalonate pathway in myeloid differentiation.
To determine whether atorvastatin could synergize with the MDM2 inhibitor (MDM2i) nutlin3a (N3a) to promote cell apoptosis in AML, we treated three AML cell lines with varying concentrations of atorvastatin as previously described,31 nutlin3a, or a combination of both. We also examined the effects of atorvastatin and nutlin3a in the presence and absence of CoQ. All three cell lines were relatively sensitive to both nutlin3a and atorvastatin, but the combination was synergistic in all three cell lines (Figures 7I–7K and S7D–S7F). Notably, CoQ treatment reduced apoptotic cell death, especially in the atorvastatin/nutlin3a combination group, suggesting that inhibiting the mevalonate pathway may enhance MDM2i-mediated oxidative stress by depleting CoQ. Additionally, we measured lipid peroxidation and found that the atorvastatin/nutlin3a combination increased lipid peroxidation (Figure 7L). Together, these results reveal a role for the mevalonate pathway in myeloid differentiation and suggest that a combination of statins and MDM2 inhibitors is potentially effective in the treatment of AML.
Discussion
AML remains a challenging disease with poor outcomes, particularly in cases involving TP53 mutations, yet the precise molecular mechanisms underlying the disease remain elusive, primarily due to the lack of suitable mouse models. The Trp53R172H mutation in mice and its human equivalent, the R175H allele, contribute to abnormal self-renewal and enhanced cell proliferation in AML.17 Our study, utilizing an inducible Trp53R172H mouse model and Mdm2 heterozygous deletion models, reveals new insights into the cooperative effects of Trp53 mutations and Mdm2 haploinsufficiency on clonal hematopoiesis and myeloid lineage development. While Mdm2 has previously been associated with cell-cycle progression independently of p53,32 our findings uncover an additional p53-independent role for Mdm2 in regulating the mevalonate pathway and cellular stress response, which influence myeloid differentiation in hematopoietic cells.
Mdm2 plays a critical role in regulating cell fate decisions within the hematopoietic system by modulating p53 levels, thereby balancing self-renewal and differentiation in HSCs.33,34 Our investigation into the hematopoietic defects observed in conditional Mdm2 haploinsufficient mice revealed that, contrary to our expectation, p53 levels did not increase in BM cells of these mice, even after irradiation-induced DNA damage, suggesting that the phenotype of Mdm2 haploinsufficiency in BM cells is not solely attributed to increased p53 activity. However, the upregulation of p21, a p53 target gene, suggests a potential p53-independent mechanism, consistent with previous reports of MDM2 inhibition leading to p53-independent increases in p21 levels.35 Furthermore, Mdm2 haploinsufficiency still promoted granulocytic differentiation even in the absence of p53, aligning with previous reports of its p53-independent role in differentiation,36 such as the induction of granulocytic differentiation in p53 null HL-60 cells by nulin-3a.37 In the context of hematologic malignancies, the presence of MDM2(SNP309) was found as a biomarker associated with worse clinical outcomes in AML patients.38
Recent studies underscore the significance of HSC age in determining susceptibility to transformation and the type of malignancy that arises.39,40 Aging HSCs are associated with myeloid-biased hematopoiesis, linked to myeloid malignancies like AML.41,42 The observed activation of ISR pathway in Mdm2 haploinsufficient BM cells suggest an increased state of cellular stress, potentially accelerating the aging process in HSCs. Furthermore, Mdm2 haploinsufficiency compromises the self-renewal and differentiation potential of HSCs, ultimately leading to a skewing toward myeloid lineage. Therefore, further investigations into how ISR activation influences HSC aging in the context of Mdm2 haploinsufficiency are essential. These studies provide invaluable insights into the mechanisms underlying age-related hematopoietic disorders.
The genetic mouse model of Mdm2 haploinsufficiency offers insights into the p53-independent role of Mdm2 in the maintenance and differentiation of HSCs. Pharmacological inhibition of MDM2, unleashing the pro-apoptotic effects of p53, holds promise as a treatment approach for malignancies with intact TP53.43,44 However, concerns arise regarding the possible selection of TP53-mutant subclones with GOF activities.45,46 Previous research, including our own, has indicated that MDM2 inhibition may inadvertently promote the expansion of TP53-mutant clones.47,48,49 Our findings, demonstrating no significant difference between p53 mutant and p53 null groups, suggest that mutant p53 in our model lacks pronounced GOF effects. These results align with recent studies in the field,50,51 further supporting the notion that the mutant p53 does not exhibit enhanced oncogenic properties beyond loss of wild-type p53 function.
Mdm2 haploinsufficiency was previously shown to alter the tumor spectrum in p53-null mice, showing lower incidence of lymphoma.52,53 Importantly, our data clarify the role of Mdm2 haploinsufficiency in leukemogenesis, demonstrating that Mdm2 haploinsufficiency can promote granulocytic differentiation of p53 null BM cells partly through inhibition of the mevalonate pathway. While our findings highlight the significance of Mdm2 haploinsufficiency in myeloid-biased hematopoiesis, its specific role in the transition from clonal hematopoiesis to AML warrants further investigation. We propose that dysregulation of Mdm2 and p53 pathways may contribute to clonal expansion and genomic instability, facilitating the progression from pre-leukemic states to overt AML. However, elucidating the precise mechanisms underlying this transition requires additional research.
Limitations of the study
Our study sheds light on the interaction between Mdm2 haploinsufficiency and mutant p53 in AML development. However, given the heterogeneity of AML in humans, our findings may primarily apply to specific AML subsets, especially those with p53 mutations and MDM2 LOH, hence our results might not extend to all AML patient scenarios. Moreover, while we provide valuable insights into the roles of Mdm2 and mutant p53 in the context of animal models, especially regarding myeloid-biased hematopoiesis, we recognize that our study does not fully elucidate the mechanisms of leukemogenesis. Alternative mechanisms and the direct translation of these findings to human disease requires further research.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| 4% paraformaldehyde | Invitrogen | Cat# FB002 |
| Optimal Cutting Temperature (OCT) | Tissue-Tek | Cat# 4583 |
| 5-Iodo-2′-deoxyuridine (IdU) | Acros Organics | Cat# AC122350000 |
| MitoSOX Red | Molecular Probes | Cat# M36008 |
| C11 BODIPY 581/591 | Molecular Probes | Cat# D-3861 |
| Methylcellulose-based medium (MethoCult) | StemCell Technologies | Cat# M3534 or M3434 |
| RBC lysis buffer | BD Biosciences | Cat# 555899 |
| Protease/Phosphatase Inhibitor Cocktail | Cell Signaling | Cat# 5872 |
| Xylene | Fisher Scientific | Cat# X5-1 |
| Ethanol, Absolute | Fisher Scientific | Cat# E7023 |
| Hydrogen Peroxide, 3% Solution | Sigma-Aldrich | Cat# H1009 |
| Hematoxylin | Sigma-Aldrich | Cat# MHS16 |
| Antibodies | ||
| Rabbit monoclonal anti-Ki67 | Abcam | Cat# ab15580 |
| Rabbit polyclonal anti-p53 | Leica | Cat# CM5P-L |
| Rat monoclonal anti-CD3 | Biolegend | Cat# 100202 |
| Chicken anti-Goat IgG Alexa Fluor 647 | Invitrogen | Cat# A-21469 |
| Donkey anti-Rabbit IgG Alexa Fluor 647 | Invitrogen | Cat# A-31573 |
| DAPI | Life Technologies | Cat# D3571 |
| VECTASHIELD Mounting Medium | Fisher | Cat# H-1000 |
| CD117 (c-Kit) PE/Cy7 | BioLegend | Cat# 105814 |
| Ly-6A/E (Sca-1) Alexa Fluor 700 | BD Biosciences | Cat# 565981-82 |
| Lineage Cocktail Pacific Blue | BioLegend | Cat# 133306 |
| CD150 APC (SLAM) | BioLegend | Cat# 115910 |
| CD19 FITC | BD Biosciences | Cat# 557398 |
| CD11 b PE | BD Biosciences | Cat# 557397 |
| Ly6G APC Cy7 | BD Biosciences | Cat# 560600 |
| CD3 PE | BD Biosciences | Cat# 555275 |
| SREBP2 antibody | Invitrogen | Cat# PA1-338 |
| MDM2 antibody | Sigma-Aldrich | Cat# M4308 |
| Sqle antibody | Cell Signaling | Cat# 40659 |
| p21 antibody | Cell Signaling | Cat# 556430 |
| eIF2a antibody | Cell Signaling | Cat# 2103 |
| Phospho-eIF2a (Ser51) antibody | Cell Signaling | Cat# 53085 |
| ATF-4 antibody | Cell Signaling | Cat# 11815 |
| b-actin antibody | Cell Signaling | Cat# A5316 |
| Alpha-tubulin antibody | Cell Signaling | Cat# 2125 |
| Histone H3 antibody | Cell Signaling | Cat# 3638 |
| Software and algorithms | ||
| PhenoChart | PerkinElmer | Available from PerkinElmer |
| Vectra Polaris | PerkinElmer | PerkinElmer |
| Experimental models: Organisms/strains | ||
| Mdm2-floxed mice | Jackson Laboratory | Mdm2tm2.1Glo/J |
| mTmG mice | Jackson Laboratory | B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J |
| Ai14 reporter | Jackson Laboratory | RosaLsl-TdTomato |
| Trp53-floxed mice | Jackson Laboratory | B6.129P2-Trp53tm1Brn/J |
| Vav-Cre mice | Jackson Laboratory | B6.Cg-Commd10Tg(Vav1-icre)A2Kio/J |
| Mx1-cre mice | Jackson Laboratory | B6.Cg-Tg1Cgn/J |
| Trp53 wmR172H mice | Previously described | Pourebrahim et al.19 and Zhang et al.20 |
| Deposited data | ||
| Differential gene expression data of murine LK cells in Mdm2 haploinsufficient mice | This paper | Dryad: https://doi.org/10.5061/dryad.s4mw6m9dr |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Michael Andreeff (mandreef@mdanderson.org).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
-
•
All primary microscopy and western blot data generated in this study are available from the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze or reproduce data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Animal studies
The following mouse models were utilized in this study: Mdm2-floxed mice (Mdm2tm2.1Glo/J), mTmG mice (B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J), Ai14 reporter (RosaLsl-TdTomato), Trp53-floxed mice (B6.129P2-Trp53tm1Brn/J), Vav-Cre mice (B6.Cg-Commd10Tg(Vav1-icre)A2Kio/J), and Mx1-cre (B6.Cg-Tg1Cgn/J), which were obtained from the Jackson Laboratory. Trp53 wmR172H mice were previously described.19,20 Male and female mice aged between 2 and 6 months were used for the transplantation experiments, ensuring age-matching within each experimental group. Activation of the Mx1-Cre recombinase was achieved by intraperitoneal injections of pIpC. Genotyping was conducted by PCR analysis using tail DNA samples. All animals were housed in compliance with the approved protocols of the Institutional Animal Care and Use Committee (IACUC) at MD Anderson.
Method details
Fluorescence microscopy
Isolated tissues were fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) (Invitrogen, FB002) overnight. Bones were washed in PBS and decalcified in 14% EDTA solution for 10 days at 4°C. Bone samples were subsequently washed in PBS and immersed in 30% sucrose PBS solution overnight. Bones and soft tissues were transferred and submerged in optimal cutting temperature (OCT) compound (Tissue-Tek, 4583). The embedded tissue was cut into 5-μm sections using a cryostat. The sections were then stored as frozen slides at −80°C or further processed. For further processing, tissue slides were washed with PBS and then incubated in 3% BSA for 1 h at room temperature. After three subsequent 5-min washes in PBS, primary antibody incubation against anti-Ki67 antibody (Abcam, ab15580), p53 (CM5, Leica P53-CM5P-L), and CD3 (BioLegend, 100202) was performed at antibody-specific dilutions in a light-protected hydration chamber at 4°C. Secondary antibody staining against the primary antibody host species (Chicken anti-Goat IgG Alexa Fluor 647, Invitrogen, A-21469; and Donkey anti-Rabbit IgG Alexa Fluor 647, Invitrogen, A-31573) was done at dilutions of 1:500 for 1 h at room temperature. Tissue slides were washed thrice in PBS for 5 min, stained with DAPI (Life Technologies, D3571) at 1:750 for 5 min and rinsed with PBS. Slides were mounted with VECTASHIELD Mounting Medium (Fisher, H-1000) and sealed with nail polish.
CyTOF analysis
Bone marrow samples were collected using aseptic techniques by flushing the long bones with cold PBS. After removing red blood cells (RBC lysis), the bone marrow suspension was transferred to sterile tubes. To eliminate contaminants, the cell pellet was washed multiple times with PBS. A panel of 31 antibodies, targeting specific cellular markers of interest in the bone marrow, was designed and selected (Table S2). Antibody cocktails were prepared by combining the individual antibodies in appropriate dilution buffers. The bone marrow cells obtained from the pellet were resuspended in staining buffer, and the antibody cocktails were added to the cell suspension. The mixture was then incubated in the dark at room temperature to allow the antibodies to bind to the target markers. Next, the stained cell suspension was loaded onto the CyTOF instrument. To mark cells in the S-phase of the cell cycle, the cells were labeled with 5-Iodo-2′-deoxyuridine (IdU) (Acros Organics) at a final concentration of 10 μM IdU for 30 min at 37°C. Following intracellular staining, the cells were washed twice and resuspended in an intercalator solution (1.6% paraformaldehyde in PBS with 125 nM iridium nucleic acid intercalator). The suspension was then incubated at 4°C overnight and acquired at a rate of 300 events per second on the Helios instrument, using CyTOF Software version 6.7.1016 (Standard Bio Tools, San Francisco, CA). Subsequently, data processing of samples was performed using FlowJo version 10.8.1.
Imaging and spectral unmixing
Frozen tissue sections and immunofluorescence-stained slides were imaged using a Vectra Multispectral Imaging System version 2 (Akoya Biosciences) at a magnification of 40×. The images were then visualized using Phenochart slide viewer software (Akoya Biosciences). Spectral unmixing was performed using inForm Analysis software (Akoya Biosciences), as previously described. Briefly, the software identifies the spectral signature of each fluorescent probe in the sample and separates overlapping signals into distinct channels based on their unique spectral properties. This process allows for accurate identification and quantification of individual markers in complex samples.
Flow cytometry and ROS analysis
Bone marrow (BM) single-cell suspensions were obtained by intrafemoral flushing and crushing of bone, followed by filtration through 40-μm strainers (Fisher, 08-771-1). Red blood cells (RBCs) were lysed in 1x RBC lysis buffer (BD Biosciences, 555899) for 15 min with slight agitation at room temperature. After two washes in PBS, cells were stained with the following antibodies: CD117 (c-Kit) PE/Cy7 (Biolegend, 105814), Ly-6A/E (Sca-1) Alexa Fluor 700 (BD Bioscience, 565981-82), Lineage Cocktail Pacific Blue (BioLegend, 133306), CD150 APC (SLAM) (BioLegend, 115910), CD19 FITC (BD Biosciences, 557398), CD11 b PE (BD Biosciences, 557397), Ly6G APC Cy7 (BD Biosciences, 560600), and CD3 PE (BD Biosciences, 555275).
To assess oxidative stress upon irradiation, cells were stained with MitoSOX Red (Molecular Probes, M36008) or C11 BODIPY 581/591 (Molecular Probes, D-3861), together with Ghost Dye Violet 540 (Tonbo Biosciences, 13–0879) and the aforementioned surface marker antibodies. Mitochondrial oxidative stress was determined by the percentage of cells exhibiting a high MitoSOX signal, while lipid peroxidation was assessed by the fluorescence ratio of oxidized signal (510 nm) to reduced signal (590 nm) on flow cytometry. The gating strategy is described in the Figure S8. Data were collected and analyzed using the Beckman Coulter Gallios Flow Cytometer and Kaluza Analysis Software.
Colony assays
Isolated total bone marrow and/or progenitor cells from mice were plated at a concentration of 50,000 cells in 2 mL of methylcellulose methylcellulose-based medium (MethoCult M3534 or M3434; StemCell Technologies, Canada), containing IL-3, IL-6, mSCF, EPO. After 5–7 days, colonies were scored microscopically, harvested, and replated up to 5 times. For liquid culture assays, we washed 30,000 cells in PBS and resuspended them in either RPMI 1640 with FBS, Pen/Strep, and various growth factors, or Xvivo Medium supplemented with Flt3-ligand, mSCF, TPO.
Immunoblot analysis
Mouse BM cells were collected, and RBCs were lysed in 1x RBC lysis buffer. After ex vivo irradiation of 400 cGy, cells were lysed at a density of 1 × 106/50 μL in protein lysis buffer (0.25 M Tris-HCl, 2% SDS, 4% β-mercaptoethanol, 10% glycerol, 0.02% bromophenol blue) supplemented with Protease/Phosphatase Inhibitor Cocktail (Cell Signaling) and incubated at 95°C for 5 min for denaturing. Immunoblot analysis was performed as previously reported with some modifications. Briefly, an equal amount of protein lysate was resolved using 4–12% Bir-Tris Mini Protein Gels (Invitrogen) and transferred to polyvinylidene fluoride (PVDF) membranes. Membranes were divided, probed for target proteins, and imaged (Odyssey imaging system, LI-COR). Primary antibodies used were as follows: SREBP2 (Invitrogen, PA1-338), MDM2 (Sigma-Aldrich, M4308), Sqle (Cell Signaling #40659), p21 (BD #556430), eIF2α (cell signaling #2103), Phospho-eIF2α (Ser51) (cell signaling #53085), ATF-4 (cell signaling #11815), β-actin (Sigma-Aldrich A5316), p53 (Leica, NCL-L-p53-CM5p), alpha-tubulin (Cell Signaling #2125), and histone H3 (Cell Signaling #3638). IRDye 680LT Donkey anti-Mouse IgG and 800CW Donkey anti-Rabbit IgG (LI-COR Biosciences) were used as secondary antibodies.
Evaluation of cytotoxicity and synergism in AML cell lines
AML cell lines (MOLM13, OCIAML2 and OCIAML3) were seeded at a density of 15,000 cells per well in 96 well plate containing complete RPMI-1640 medium supplemented with 10% fetal calf serum. The cells were treated for 48 h with Atorvastatin at doses ranging from 0 to 10 μM and Nutlin (N3a) at doses ranging from 0 to 5 μM, resulting in a 6 × 6 matrix of treatment. Each experiment was conducted independently three times. Viable cell numbers were assessed by quantifying ATP using the CellTiter-Glo Luminescent Cell Viability Assay (Promega). The results were normalized to the values obtained for DMSO-treated cells and expressed as a percentage of control. Dose-response curves were analyzed using a curve-fitting routine based on nonlinear regression to calculate the EC50 value. Additionally, synergistic effects of Atorvastatin with Nutlin were determined based on the mean values obtained from three independent experiments. Synergy evaluation was performed by collecting, processing, and visualizing the data using the BLISS Index method with COMBENEFIT software.
Immunohistochemical analysis
Immunohistochemical analysis was performed to investigate the expression of p53 protein in formalin-fixed paraffin-embedded tissue sections. The VECTASTAIN Elite ABC HRP Kit (Vector Laboratories, Pk-6101) was used for the immunohistochemical staining. Formalin-fixed paraffin-embedded tissue sections were deparaffinized by immersion in two changes of xylene, followed by two changes of 100% ethanol. Subsequently, the sections were passed through 95%, 75%, and 50% ethanol for 5 min per change. To block endogenous peroxidase activity, the tissue sections were immersed in a 3% H2O2 solution in methanol for 10 min. The antigen retrieval process was performed by immersing the slides in Coplin jars containing antigen retrieval solution. The slides were then placed in an IHC-Tek Epitope Retrieval Steamer (IHC World, IW-1102) and steamed for 10 min. Afterward, the slides were allowed to cool down for 20 min at room temperature. To prevent non-specific binding, the slides were blocked for 60 min with 10% serum in 0.1% TBST (Tris-buffered saline with Tween 20). The primary antibody against p53 (CM5, Leica P53-CM5P-L) was diluted in 2% FBS (fetal bovine serum) in 0.1% TBST and incubated overnight at 4°C in a light-protected hydration chamber. Following the primary antibody incubation, the slides were washed twice for 5 min each in Tris-buffered saline (TBS). The slides were incubated with the biotinylated secondary antibody solution for 30 min at room temperature. Slides were washed twice for 5 min each in TBS. Slides were incubated with ABC solution for 30 min and washed twice for 5 min each in phosphate-buffered saline (PBS). The slides were incubated in the 3,3′-Diaminobenzidine (DAB) solution until the desired signal intensity was achieved. Excess DAB solution was washed off with deionized water, and the slides were counterstained with hematoxylin for 80 s. Slides were immersed in lithium carbonate solution for 60 s, followed by immersion in deionized water for 10 min. The slides were sequentially immersed in 95% ethanol and 100% ethanol for 90 s per change (two changes for each). Slides were immersed three times for 5 min each in xylene. Finally, the slides were mounted with Richard-Allan Scientific Mounting Medium (Thermo Scientific, 4112) for preservation and analysis. The immunohistochemically stained slides were then examined under a microscope to assess the expression pattern and intensity of p53 protein in the tissue samples.
Cell blood counts
Mouse peripheral blood (50 μL) was collected into BD Microtainer vials (Fisher, 02-669-33) via retro-orbital bleeding. Blood samples were analyzed on the Horiba ABX SAS PENTRA 60 C+ cell blood counter to identify hematopoietic irregularities.
RNA sequencing
RNA was extracted utilizing the Direct-Zol RNA Microprep kit (Zymo Research, R2060). Barcoded Illumina-compatible stranded total RNA libraries were prepared using the TruSeq Stranded Total RNA kit (Illumina) as previously described.19 Library pools were quantified by qPCR and sequenced on the HiSeq 4000 sequencer using the 75-bp paired-end format. The raw RNA-seq readouts were subsequently mapped to the mouse mm10 assembly reference genome using TopHat2 and analyzed with DESeq2 (Bioconductor package).
Whole exome sequencing
Dual-indexed libraries compatible with Illumina were created using 15ng–50ng of genomic DNA, which had been sheared by a Biorupter Ultrasonicator (Diagenode) and treated with RNase. This DNA was processed using the Twist Library Preparation Kit and the Twist Universal Adapter System, following the manufacturer’s instructions. The libraries, indexed and prepared for capture, underwent 6 cycles of PCR amplification with Twist UDI primers. Their fragment size distribution was then evaluated using the 4200 TapeStation High Sensitivity D1000 ScreenTape (Agilent Technologies), and their concentration was determined with the Qubit dsDNA HS Assay Kit (ThermoFisher). After normalizing the concentrations, equal amounts of the uniquely dual-indexed libraries were pooled together, with 7 libraries in each pool. For exon targeting, the Twist Mouse Core Exome kit was employed. Post-capture, the enriched exon library pools were further amplified with 6 PCR cycles. Size distribution was again assessed with the 4200 TapeStation High Sensitivity D1000 ScreenTape (Agilent Technologies), and concentration measurements were taken using the Qubit dsDNA HS Assay Kit. Quantification of the exon-enriched library pools was performed by qPCR utilizing the KAPA Library Quantification Kit (KAPABiosystems). Finally, these libraries were sequenced on a NovaSeq6000 SP-200 flow cell in a 100 nt PE format.
Copy number analysis
Copy number detection was conducted using the latest version of Control-FREEC.54 This process involves using aligned reads (bam files) as input, from which Control-FREEC creates copy number and B-allele frequency profiles, incorporating tumor purity estimates and corrections. These profiles undergo normalization, segmentation, and analysis to assign copy number events to specific genomic regions. With the inclusion of a matched normal sample, Control-FREEC is able to distinguish between somatic and germline events. A threshold representing 50% of a chromosome arm’s length serves as the criterion for differentiating between broad and focal events. To mitigate false positives from hyper-segmentation, segments underwent filtering with an amplitude threshold set at a copy-difference of 0.35 on a log scale. The frequency of broad copy number changes was quantified, followed by an analysis of focal copy number changes using the GISTIC methodology, which identifies significant peaks of amplifications and deletions. The identified copy number events were classified into two categories based on their observed frequency: focal events, which are significantly smaller than a chromosome arm, and broad events, encompassing a chromosome arm or an entire chromosome.
MDM2SNP309 exploration
We used alleleCount (https://github.com/cancerit/alleleCount) on PCAWG tumor/germline BAM files at the MDM2SNP309 position (chr12:69,202,580 in hg19 coordinates) to compute variant allele frequency (VAF) (minimum count ≥5). In germlines, genotypes were directly assessed using VAF (T/T: VAF = 0; T/G: 0.35 ≤ VAF ≤0.65; G/G: VAF = 1). Copy number profiles and whole-genome doubling (WGD) information were retrieved from consensus copy number data; https://dcc.icgc.org/releases/PCAWG/consensus_cnv). In tumors, the retained allele (either T or G) was assessed in cases with MDM2 allelic imbalance where VAF was compared to copy number states of major and minor alleles, corrected for tumor purity. MDM2 copy-number statuses (normal, gain, loss, and copy-neutral) were defined by considering baseline ploidy (adjusted for WGD). TP53 mutation status was obtained from the PCAWG Drivers and Functional Interpretation Group. TP53 single nucleotide variations and insertion/deletions were functionally annotated (e.g., missense, nonsense, frameshift) using ANNOVAR.55
DNA isolation and SNP genotyping
Genomic DNA was extracted from the BM samples isolated from AML patients at diagnosis using QIAamp DNA Blood Midi kit (Qiagen). Genotyping of SNP rs2279744 (Mdm2SNP309) in intron 1 of the MDM2 gene was performed with 50 ng of genomic DNA using TaqMan SNP Genotyping Assay, (human-SNP ID:rs2279744) and TaqMan Genotyping Mastermix (ThermoFisher). PCR reactions were carried out on a Quantstudio 6 Flex instrument and analyzed using TaqMan Genotyper Software (Applied Biosystems Life Technologies).
Quantification and statistical analysis
Means and standard deviations were calculated using GraphPad Prism 6 software. The Student’s t test was used for comparative analysis between two groups. Analysis of variance was used to compare multiple groups. p < 0.05 was set as statistically significant. A p value of less than 0.05 was considered statistically significant. In figure legends, symbols ∗, ∗∗, ∗∗∗, and ∗∗∗∗ denote p values of less than 0.05, 0.01, 0.001, and 0.0001, respectively, unless stated otherwise. All experimental procedures were replicated three times, and error bars in the figures denote mean ± standard deviation (SD) values, unless mentioned otherwise.
Acknowledgments
We express our gratitude to the staff of the Research Animal Support Facility and to Nalini Patel, Nguyen Nguyen, and Nicole R. Vaughn in the Flow Cytometry and Cellular Imaging Core (supported by NIH grant P30 CA016672). Special thanks go to Dr. Gigi Lozano for providing Trp53wmR172H mice and to Sean Post for critically reading the manuscript. We extend our appreciation to Mahesh Basyal, Edward Ayoub, Erika Thompson, Sunita Patterson, Sara McCracken, Sherry Pierce, and Jairo Matthews for their invaluable assistance. Additionally, we would like to thank Dr. Arnold J. Levine for insightful discussions. This research was made possible through funding from the Paul and Mary Haas Chair in Genetics (to M.A.), a CPRIT MIRA (to M.A.), The Ladies Leukemia League research grant (to R.P.), NIH SPORE Career Enhancement Award (to R.P.), and the University of Texas MD Anderson Cancer Center MDS/AML Moon Shot (to M.A.). T.L. and P.V.L. are supported by the Francis Crick Institute, which receives core funding from Cancer Research UK (CC2008), the UK Medical Research Council (CC2008), and the Wellcome Trust (CC2008). P.V.L. is a CPRIT Scholar in Cancer Research and acknowledges CPRIT grant support (RR210006).
Author contributions
Conceptualization: R.P. and M.A. Methodology: R.P. and M.G. Software: B.L., M.M., T.L., and P.V.L. Investigation: R.P., R.H.M., H.A., L.O., S.K., R.Z., J.D.K., N.B., and M.G. Resources: M.A. and R.P. Writing – original draft: R.P. Writing – review & editing: M.A. Supervision: P.V.L. and M.A. Project administration: R.P. Funding acquisition: R.P. and M.A.
Declaration of interests
The authors declare no potential conflicts of interest.
Published: May 10, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101558.
Supplemental information
References
- 1.Daver N.G., Maiti A., Kadia T.M., Vyas P., Majeti R., Wei A.H., Garcia-Manero G., Craddock C., Sallman D.A., Kantarjian H.M. TP53-Mutated Myelodysplastic Syndrome and Acute Myeloid Leukemia: Biology, Current Therapy, and Future Directions. Cancer Discov. 2022;12:2516–2529. doi: 10.1158/2159-8290.CD-22-0332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jones S.N., Roe A.E., Donehower L.A., Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378:206–208. doi: 10.1038/378206a0. [DOI] [PubMed] [Google Scholar]
- 3.Montes de Oca Luna R., Wagner D.S., Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378:203–206. doi: 10.1038/378203a0. [DOI] [PubMed] [Google Scholar]
- 4.Adams C.M., Mitra R., Xiao Y., Michener P., Palazzo J., Chao A., Gour J., Cassel J., Salvino J.M., Eischen C.M. Targeted MDM2 Degradation Reveals a New Vulnerability for p53-Inactivated Triple-Negative Breast Cancer. Cancer Discov. 2023;13:1210–1229. doi: 10.1158/2159-8290.CD-22-1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levine A.J. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 6.Oliner J.D., Kinzler K.W., Meltzer P.S., George D.L., Vogelstein B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature. 1992;358:80–83. doi: 10.1038/358080a0. [DOI] [PubMed] [Google Scholar]
- 7.Haupt Y., Maya R., Kazaz A., Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 8.Kubbutat M.H., Jones S.N., Vousden K.H. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- 9.Honda R., Tanaka H., Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25–27. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- 10.Swaminathan M., Bannon S.A., Routbort M., Naqvi K., Kadia T.M., Takahashi K., Alvarado Y., Ravandi-Kashani F., Patel K.P., Champlin R., et al. Hematologic malignancies and Li-Fraumeni syndrome. Cold Spring Harb. Mol. Case Stud. 2019;5 doi: 10.1101/mcs.a003210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Donehower L.A., Lozano G. 20 years studying p53 functions in genetically engineered mice. Nat. Rev. Cancer. 2009;9:831–841. doi: 10.1038/nrc2731. [DOI] [PubMed] [Google Scholar]
- 12.ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium Pan-cancer analysis of whole genomes. Nature. 2020;578:82–93. doi: 10.1038/s41586-020-1969-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feeley K.P., Adams C.M., Mitra R., Eischen C.M. Mdm2 Is Required for Survival and Growth of p53-Deficient Cancer Cells. Cancer Res. 2017;77:3823–3833. doi: 10.1158/0008-5472.CAN-17-0809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghandi M., Huang F.W., Jané-Valbuena J., Kryukov G.V., Lo C.C., McDonald E.R., 3rd, Barretina J., Gelfand E.T., Bielski C.M., Li H., et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature. 2019;569:503–508. doi: 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bond G.L., Hu W., Bond E.E., Robins H., Lutzker S.G., Arva N.C., Bargonetti J., Bartel F., Taubert H., Wuerl P., et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell. 2004;119:591–602. doi: 10.1016/j.cell.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 16.Olive K.P., Tuveson D.A., Ruhe Z.C., Yin B., Willis N.A., Bronson R.T., Crowley D., Jacks T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119:847–860. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 17.Loizou E., Banito A., Livshits G., Ho Y.J., Koche R.P., Sánchez-Rivera F.J., Mayle A., Chen C.C., Kinalis S., Bagger F.O., et al. A Gain-of-Function p53-Mutant Oncogene Promotes Cell Fate Plasticity and Myeloid Leukemia through the Pluripotency Factor FOXH1. Cancer Discov. 2019;9:962–979. doi: 10.1158/2159-8290.CD-18-1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hill R., Song Y., Cardiff R.D., Van Dyke T. Selective evolution of stromal mesenchyme with p53 loss in response to epithelial tumorigenesis. Cell. 2005;123:1001–1011. doi: 10.1016/j.cell.2005.09.030. [DOI] [PubMed] [Google Scholar]
- 19.Pourebrahim R., Zhang Y., Liu B., Gao R., Xiong S., Lin P.P., McArthur M.J., Ostrowski M.C., Lozano G. Integrative genome analysis of somatic p53 mutant osteosarcomas identifies Ets2-dependent regulation of small nucleolar RNAs by mutant p53 protein. Genes Dev. 2017;31:1847–1857. doi: 10.1101/gad.304972.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y., Xiong S., Liu B., Pant V., Celii F., Chau G., Elizondo-Fraire A.C., Yang P., You M.J., El-Naggar A.K., et al. Somatic Trp53 mutations differentially drive breast cancer and evolution of metastases. Nat. Commun. 2018;9:3953. doi: 10.1038/s41467-018-06146-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marino S., Vooijs M., van Der Gulden H., Jonkers J., Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000;14:994–1004. [PMC free article] [PubMed] [Google Scholar]
- 22.Grier J.D., Yan W., Lozano G. Conditional allele of mdm2 which encodes a p53 inhibitor. Genesis. 2002;32:145–147. doi: 10.1002/gene.10066. [DOI] [PubMed] [Google Scholar]
- 23.Lang G.A., Iwakuma T., Suh Y.A., Liu G., Rao V.A., Parant J.M., Valentin-Vega Y.A., Terzian T., Caldwell L.C., Strong L.C., et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004;119:861–872. doi: 10.1016/j.cell.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 24.Jacks T., Remington L., Williams B.O., Schmitt E.M., Halachmi S., Bronson R.T., Weinberg R.A. Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 25.Harvey M., McArthur M.J., Montgomery C.A., Jr., Butel J.S., Bradley A., Donehower L.A. Spontaneous and carcinogen-induced tumorigenesis in p53-deficient mice. Nat. Genet. 1993;5:225–229. doi: 10.1038/ng1193-225. [DOI] [PubMed] [Google Scholar]
- 26.Velasco-Hernandez T., Säwén P., Bryder D., Cammenga J. Potential Pitfalls of the Mx1-Cre System: Implications for Experimental Modeling of Normal and Malignant Hematopoiesis. Stem Cell Rep. 2016;7:11–18. doi: 10.1016/j.stemcr.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kogan S.C., Ward J.M., Anver M.R., Berman J.J., Brayton C., Cardiff R.D., Carter J.S., de Coronado S., Downing J.R., Fredrickson T.N., et al. Bethesda proposals for classification of nonlymphoid hematopoietic neoplasms in mice. Blood. 2002;100:238–245. doi: 10.1182/blood.v100.1.238. [DOI] [PubMed] [Google Scholar]
- 28.Wu D., Prives C. Relevance of the p53-MDM2 axis to aging. Cell Death Differ. 2018;25:169–179. doi: 10.1038/cdd.2017.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheng T., Rodrigues N., Shen H., Yang Y., Dombkowski D., Sykes M., Scadden D.T. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287:1804–1808. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- 30.Cai D., Wang J., Gao B., Li J., Wu F., Zou J.X., Xu J., Jiang Y., Zou H., Huang Z., et al. RORgamma is a targetable master regulator of cholesterol biosynthesis in a cancer subtype. Nat. Commun. 2019;10:4621. doi: 10.1038/s41467-019-12529-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cafforio P., Dammacco F., Gernone A., Silvestris F. Statins activate the mitochondrial pathway of apoptosis in human lymphoblasts and myeloma cells. Carcinogenesis. 2005;26:883–891. doi: 10.1093/carcin/bgi036. [DOI] [PubMed] [Google Scholar]
- 32.Klein A.M., Biderman L., Tong D., Alaghebandan B., Plumber S.A., Mueller H.S., van Vlimmeren A., Katz C., Prives C. MDM2, MDMX, and p73 regulate cell-cycle progression in the absence of wild-type p53. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2102420118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mendrysa S.M., McElwee M.K., Michalowski J., O'Leary K.A., Young K.M., Perry M.E. mdm2 Is critical for inhibition of p53 during lymphopoiesis and the response to ionizing irradiation. Mol. Cell Biol. 2003;23:462–472. doi: 10.1128/MCB.23.2.462-473.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maetens M., Doumont G., Clercq S.D., Francoz S., Froment P., Bellefroid E., Klingmuller U., Lozano G., Marine J.C. Distinct roles of Mdm2 and Mdm4 in red cell production. Blood. 2007;109:2630–2633. doi: 10.1182/blood-2006-03-013656. [DOI] [PubMed] [Google Scholar]
- 35.Zhang Z., Wang H., Li M., Agrawal S., Chen X., Zhang R. MDM2 is a negative regulator of p21WAF1/CIP1, independent of p53. J. Biol. Chem. 2004;279:16000–16006. doi: 10.1074/jbc.M312264200. [DOI] [PubMed] [Google Scholar]
- 36.Manfredi J.J. The Mdm2-p53 relationship evolves: Mdm2 swings both ways as an oncogene and a tumor suppressor. Genes Dev. 2010;24:1580–1589. doi: 10.1101/gad.1941710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Secchiero P., Zerbinati C., Melloni E., Milani D., Campioni D., Fadda R., Tiribelli M., Zauli G. The MDM-2 antagonist nutlin-3 promotes the maturation of acute myeloid leukemic blasts. Neoplasia. 2007;9:853–861. doi: 10.1593/neo.07523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Falk I.J., Willander K., Chaireti R., Lund J., Nahi H., Hermanson M., Gréen H., Lotfi K., Söderkvist P. TP53 mutations and MDM2(SNP309) identify subgroups of AML patients with impaired outcome. Eur. J. Haematol. 2015;94:355–362. doi: 10.1111/ejh.12438. [DOI] [PubMed] [Google Scholar]
- 39.Adelman E.R., Huang H.T., Roisman A., Olsson A., Colaprico A., Qin T., Lindsley R.C., Bejar R., Salomonis N., Grimes H.L., Figueroa M.E. Aging Human Hematopoietic Stem Cells Manifest Profound Epigenetic Reprogramming of Enhancers That May Predispose to Leukemia. Cancer Discov. 2019;9:1080–1101. doi: 10.1158/2159-8290.CD-18-1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jaiswal S., Ebert B.L. Clonal hematopoiesis in human aging and disease. Science. 2019;366 doi: 10.1126/science.aan4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pang W.W., Price E.A., Sahoo D., Beerman I., Maloney W.J., Rossi D.J., Schrier S.L., Weissman I.L. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc. Natl. Acad. Sci. USA. 2011;108:20012–20017. doi: 10.1073/pnas.1116110108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mejia-Ramirez E., Florian M.C. Understanding intrinsic hematopoietic stem cell aging. Haematologica. 2020;105:22–37. doi: 10.3324/haematol.2018.211342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Konopleva M., Martinelli G., Daver N., Papayannidis C., Wei A., Higgins B., Ott M., Mascarenhas J., Andreeff M. MDM2 inhibition: an important step forward in cancer therapy. Leukemia. 2020;34:2858–2874. doi: 10.1038/s41375-020-0949-z. [DOI] [PubMed] [Google Scholar]
- 44.Kojima K., Konopleva M., Samudio I.J., Shikami M., Cabreira-Hansen M., McQueen T., Ruvolo V., Tsao T., Zeng Z., Vassilev L.T., Andreeff M. MDM2 antagonists induce p53-dependent apoptosis in AML: implications for leukemia therapy. Blood. 2005;106:3150–3159. doi: 10.1182/blood-2005-02-0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prives C., White E. Does control of mutant p53 by Mdm2 complicate cancer therapy? Genes Dev. 2008;22:1259–1264. doi: 10.1101/gad.1680508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Terzian T., Suh Y.A., Iwakuma T., Post S.M., Neumann M., Lang G.A., Van Pelt C.S., Lozano G. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev. 2008;22:1337–1344. doi: 10.1101/gad.1662908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yan B., Chen Q., Xu J., Li W., Xu B., Qiu Y. Low-frequency TP53 hotspot mutation contributes to chemoresistance through clonal expansion in acute myeloid leukemia. Leukemia. 2020;34:1816–1827. doi: 10.1038/s41375-020-0710-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kucab J.E., Hollstein M., Arlt V.M., Phillips D.H. Nutlin-3a selects for cells harbouring TP53 mutations. Int. J. Cancer. 2017;140:877–887. doi: 10.1002/ijc.30504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Daver N.G., Dail M., Garcia J.S., Jonas B.A., Yee K.W.L., Kelly K.R., Vey N., Assouline S., Roboz G.J., Paolini S., et al. Venetoclax and idasanutlin in relapsed/refractory AML: a nonrandomized, open-label phase 1b trial. Blood. 2023;141:1265–1276. doi: 10.1182/blood.2022016362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boettcher S., Miller P.G., Sharma R., McConkey M., Leventhal M., Krivtsov A.V., Giacomelli A.O., Wong W., Kim J., Chao S., et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science. 2019;365:599–604. doi: 10.1126/science.aax3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Z., Burigotto M., Ghetti S., Vaillant F., Tan T., Capaldo B.D., Palmieri M., Hirokawa Y., Tai L., Simpson D.S., et al. Loss-of-Function but Not Gain-of-Function Properties of Mutant TP53 Are Critical for the Proliferation, Survival, and Metastasis of a Broad Range of Cancer Cells. Cancer Discov. 2024;14:362–379. doi: 10.1158/2159-8290.CD-23-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alt J.R., Greiner T.C., Cleveland J.L., Eischen C.M. Mdm2 haplo-insufficiency profoundly inhibits Myc-induced lymphomagenesis. EMBO J. 2003;22:1442–1450. doi: 10.1093/emboj/cdg133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eischen C.M., Boyd K. Decreased Mdm2 expression inhibits tumor development and extends survival independent of Arf and dependent on p53. PLoS One. 2012;7 doi: 10.1371/journal.pone.0046148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boeva V., Popova T., Bleakley K., Chiche P., Cappo J., Schleiermacher G., Janoueix-Lerosey I., Delattre O., Barillot E. Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics. 2012;28:423–425. doi: 10.1093/bioinformatics/btr670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38 doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All primary microscopy and western blot data generated in this study are available from the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze or reproduce data reported in this paper is available from the lead contact upon request.