

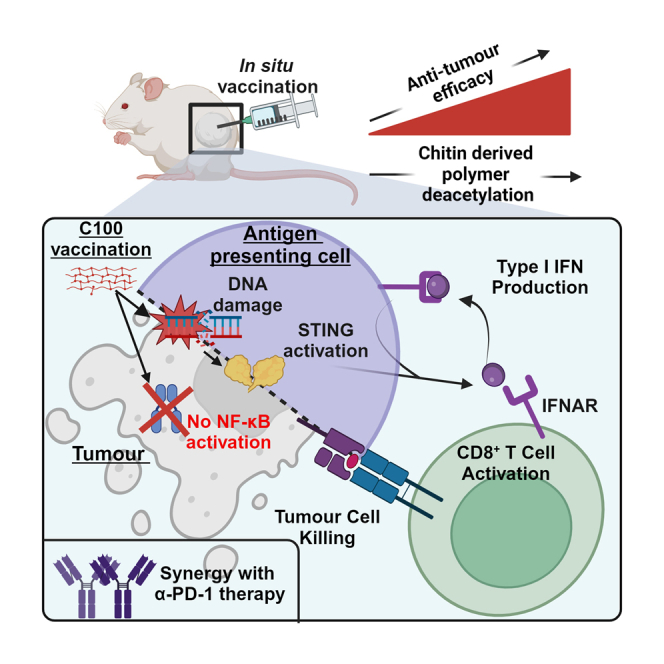
Summary
Stimulator of IFN genes (STING) is a promising target for adjuvants utilized in in situ cancer vaccination approaches. However, key barriers remain for clinical translation, including low cellular uptake and accessibility, STING variability necessitating personalized STING agonists, and interferon (IFN)-independent signals that can promote tumor growth. Here, we identify C100, a highly deacetylated chitin-derived polymer (HDCP), as an attractive alternative to conventional STING agonists. C100 promotes potent anti-tumor immune responses, outperforming less deacetylated HDCPs, with therapeutic efficacy dependent on STING and IFN alpha/beta receptor (IFNAR) signaling and CD8+ T cell mediators. Additionally, C100 injection synergizes with systemic checkpoint blockade targeting PD-1. Mechanistically, C100 triggers mitochondrial stress and DNA damage to exclusively activate the IFN arm of the cGAS-STING signaling pathway and elicit sustained IFNAR signaling. Altogether, these results reveal an effective STING- and IFNAR-dependent adjuvant for in situ cancer vaccines with a defined mechanism and distinct properties that overcome common limitations of existing STING therapeutics.
Keywords: cancer vaccine, adjuvant, cancer immunotherapy, STING, DNA sensing, CD8+ T cell, interferon, chitosan, chitin derived polymer
Graphical abstract
Highlights
-
•
Intratumoral injection of the adjuvant C100 promotes potent anti-tumor immunity
-
•
Adjuvant functionality requires STING and IFNAR signaling and CD8+ T cells
-
•
C100 injection synergizes with systemic checkpoint blockade with anti-PD1
-
•
C100 triggers DNA damage to selectively activate the IFN arm of the cGAS-STING pathway
STING is a promising adjuvant target for in situ cancer vaccination approaches. Turley, Ward, et al. demonstrate that the chitin-derived polymer adjuvant C100 selectively activates the IFN arm of the cGAS-STING pathway. Injection of C100 elicits CD8+ T cell-dependent anti-tumor immunity via STING and IFNAR signaling.
Introduction
The cGAS-STING signaling axis is a crucial regulator of type I interferon (IFN) responses and anti-tumor immunity. Upon recognition of tumor-derived, damaged double-stranded DNA (dsDNA), cGAS catalyzes the synthesis of the cyclic di-nucleotide (CDN) 2′3′-cGAMP, which serves as a second messenger that binds to and activates stimulator of IFN genes (STING). STING is an endoplasmic reticulum (ER)-associated protein that activates a TANK-binding kinase 1 (TBK1)-interferon regulatory factor 3 (IRF3)-dependent type I IFN response and nuclear factor κB (NF-κB)-mediated pro-inflammatory cytokine response. Type I IFNs stimulate the presentation of tumor antigens and the mobilization of anti-tumor immunity, in particular CD8+ cytotoxic T lymphocytes.1 The role of STING-induced NF-κB activation in cancer therapy remains poorly defined, with both pro- and anti-tumorigenic roles suggested.2,3,4
Despite the plethora of STING agonists that have been and are being developed, clinical translation has resulted in disappointingly modest efficacy, and to date, no cGAS-STING-targeted therapy has achieved clinical success. The small-molecule STING ligand DMXAA achieved robust anti-tumor immune responses in murine B16 melanoma models but failed in phase 3 trials (ClinicalTrials.gov: NCT00738387 and NCT00662597), likely due to its inability to bind human STING.5 Intratumoral (i.t.) administration of natural CDNs such as mammalian 2′3′-cGAMP and c-di-GMP, c-di-AMP, and 3′3′-cGAMP from prokaryotes was effective in mouse models of colon, brain, skin, pancreatic, breast, and B cell malignancies. However, intrinsic physicochemical characteristics such as their large size and electronegative charge render these molecules poorly membrane permeable and prone to rapid extracellular enzymatic degradation, resulting in low drug bioavailability in tumor tissues and rapid absorption into the plasma.6,7 Various approaches are under evaluation to address these limitations, such as the development of CDN drug delivery systems (e.g., exoSTING)8 and non-CDN small-molecule STING agonists with greater affinity for human STING (e.g., GSK3745417, ClinicalTrials.gov: NCT03843359).
To the best of our knowledge, there are no STING agonists under evaluation that do not result in the concomitant activation of the NF-κB pathway. In a tumor setting, the induction of pro-inflammatory signaling downstream of NF-κB could be detrimental for the establishment of anti-tumor immunity and possibly favor tumor progression. Intrinsic NF-κB signaling in tumor cells can favor proliferation, inhibit the induction of cell death pathways, upregulate coinhibitory receptor expression,9 and promote epithelial-mesenchymal transition (EMT)10 and angiogenesis in the tumor. The pro-inflammatory cytokines downstream of NF-κB activation can also have detrimental effects on anti-tumor immunity, promoting anergic responses. Indeed, inhibition of NF-κB signaling in tumor cells and i.t. myeloid cells favors tumor regression in pre-clinical models.11 Therefore, therapeutic adjuvants that exclusively activate the type 1 IFN arm of the cGAS-STING pathway and not NF-κB may be of clinical benefit.
Chitin-derived polymers are incorporated in biomedical and pharmaceutical formulations, including prolonged- or controlled-release drug delivery systems,12 wound dressings, and bone tissue engineering scaffolds, and have been investigated as vaccine adjuvants.13 We have previously demonstrated that the adjuvanticity of highly deacetylated chitin-derived polymers (HDCPs) relies on the activation of the cGAS-STING and NLRP3 inflammasome pathways via the induction of mitochondrial reactive oxygen species (mtROS) and DNA release.13 Subsequently we demonstrated that this ability to activate cGAS-STING and the NLRP3 inflammasome depended critically on the degree and pattern of polymer deacetylation, which controlled the extent of mtROS production.14 In addition, these HDCPs exclusively drove type I IFN release in the absence of pro-inflammatory cytokine production.14 Altogether, the results of this work have characterized a biocompatible polymer with a distinct means of cGAS-STING activation; therefore, this current study highlights the potential of HDCPs in the context of therapeutic cancer vaccines.
Immune checkpoint inhibitors, such as anti-PD-1 and anti-PD-L1, are frequently employed as a standard treatment for various cancers. Yet, their effectiveness is limited in numerous cancers, prompting the exploration of combination approaches to improve the efficacy and expand the range of patients and tumor types suitable for such treatments. In situ vaccination with immunogenic compounds presents a promising avenue for addressing the limitations of checkpoint inhibitors, as it has the potential to transform “cold” tumors into “hot” ones by promoting the infiltration of tumor-specific T cells.15,16
Here, we demonstrate the effectiveness of C100 (a chitin polymer deacetylated to <99.5%) as a cGAS-STING targeting i.t. therapeutic that can be used as a monotherapy or synergistically with checkpoint blockade. We have characterized the mechanism of action of this polymer, demonstrating exclusive activation of the IFN arm of STING signaling through the release of both nuclear and mitochondrial DNA (mtDNA), downstream of mitochondrial stress induction, circumventing the need for direct adjuvant-STING binding. Furthermore, we demonstrate that the therapeutic efficacy of C100 is dependent on STING and type I IFN signaling and identify CD8+ T cells as crucial mediators of C100-induced anti-tumor immunity. In summary, we characterize a STING-targeting i.t. therapeutic with advantages over alternative molecules and provide key insights into its mode of action.
Results
C100 is the most effective HDCP as an i.t. therapeutic
Pitfalls of current STING-based therapies contributing to poor clinical translation include rapid, transient, and excessive immune activation at the site of injection coupled to a reliance on direct binding to STING itself.6,7,17,18 Given the distinctive profile of HDCPs as vaccine adjuvants that promote the STING pathway via induction of ROS and self-DNA-mediated cGAS-STING activation,13,14 a therapeutic B16F10 tumor challenge model was used to investigate the anti-tumor potential of HDCPs when administered i.t. (Figure 1A). Tumor cell lines can render the cGAS-STING pathway defective by hypomethylating promoter regions of pathway components and decreasing protein expression or by blocking STING trafficking to sites required to promote the induction of IFN responses.19 As such, we firstly assessed the expression and functionality of cGAS and STING in B16F10 cells by immunoblot using DMXAA, a small-molecule STING agonist that directly binds and activates murine STING.17 Both proteins were present under basal conditions (Figures S1A and S1B), and functional STING activity was demonstrated by its reduced expression after DMXAA treatment compared to the untreated control (Figure S1A). Following i.t. injection, C100 was the most potent adjuvant, with treatment significantly reducing tumor growth (Figures 1B and S1C) and enhancing survival compared to control mice (Figure 1C). To validate this efficacy in a second tumor model, i.t. injections were carried out following implantation of MC38 cells. As before, the expression and functionality of cGAS and STING proteins in the MC38 line were confirmed before tumor implantation (Figures S1D and S1E). Next, mice bearing MC38 tumors were treated in an identical manner to the B16 model. A more pronounced inhibition in growth and prolonged survival after C100 treatment was observed in MC38 compared to B16F10 tumors (Figures 1D, 1E, and S1F). This is in line with existing data showing increased responsiveness to immunotherapeutic interventions in MC38 tumors, owing to a higher degree of tumor immunogenicity.20,21 In addition, C100 dose optimization studies were carried out in the B16 and MC38 tumor models, with a polymer dose of 100 μg exerting the most potent anti-tumor effects, outperforming the higher (Figure S1G) and lower doses (Figure S1H) tested i.t. Overall, these results indicate that C100 is an effective polymeric adjuvant for in situ vaccination against cancer and outperforms less deacetylated alternative polymers, in line with our previous observations that the degree of deacetylation is instructive in a chitin-derived polymer’s adjuvanticity.14
Figure 1.

C100 significantly improves the survival of mice bearing MC38 colon carcinoma or B16 melanoma
(A) Schematic of B16 melanoma and MC38 carcinoma experimental design. In brief, C57BL/6 mice were injected subcutaneously (s.c.) on the flank with B16 melanoma or MC38 carcinoma cells. Tumor-bearing mice were randomized and injected i.t. with PBS or HDCPs on days 0, 4, and 8.
(B and C) B16 melanoma growth rate (B) and Kaplan-Meier survival analysis (C) in mice treated with PBS or HCDPs (100 μg) on days 0, 4, and 8.
(D and E) MC38 carcinoma growth rate (D) and Kaplan-Meier survival analysis (E) in mice treated with PBS or C100 (100 μg) on days 0, 4, and 8.
(B and D) Data are expressed as mean tumor volume ± SEM. (B and D) Unpaired two-tailed t tests or (C and E) Mantel-Cox Tests were used to determine statistical significance between treatments, ∗∗p < 0.001 and ∗∗∗p < 0.001. (B and C) n = 7 mice per group. (D and E) PBS n = 6, C100 n = 5 mice per group.
The anti-tumor efficacy of C100 is dependent on STING expression and host immune cell IFNAR signaling
We suspected that STING and IFN alpha/beta receptor (IFNAR) signaling would be critical for the anti-tumor properties of C100 given their central role in adjuvant-induced dendritic cell (DC) maturation and promoting antigen-specific Th1 responses.14 To investigate, wild-type (WT) and Tmem173−/− (STING knockout [KO]) mice were injected with WT B16F10 cells or STING KO B16F10 cells and treated with PBS or C100 as before. Injection of C100 had no therapeutic benefit in STING-KO mice bearing STING-KO tumors (Figures 2A, 2B, and S2A), indicating an essential role for STING in C100-induced protection. However, WT mice bearing STING-KO tumors and STING-KO mice bearing WT tumors responded to C100 treatment, highlighting a flexibility in the cellular source of STING activation (Figures 2A, 2B, and S2A). Indeed, the cytosolic accumulation of damaged DNA within tumor cells has been shown to promote STING signaling and trigger anti-tumor immune responses.22,23 Supporting a potential role for tumor-derived DNA in the efficacy of injected C100, treatment of B16 and MC38 cells with C100 drove high levels of mtROS (Figures S2D and S2F) that were previously demonstrated to be indicative of STING activation by HDCPs in bone marrow-derived DCs (BMDCs).14 Moreover, exposure to C100 reduced STING protein levels compared to medium controls in both B16 and MC38 cells, albeit to a lesser extent than DMXAA and 2′3′-cGAMP controls (Figures S2E and S2G). Confirming the role of STING signaling in C100-induced tumor protection, C100 treatment had no therapeutic benefit in IFNAR1−/− mice (Figures 2C, 2D, and S2B). In fact, tumor growth in IFNAR1−/− mice after C100 treatment was equivalent to untreated WT and IFNAR controls. C100 can activate the NLRP3 inflammasome in addition to cGAS-STING signaling. While NLRP3 was required for C100-induced Th1 responses against purified proteins, it remained to be determined whether NLRP3 activation contributed to C100-induced anti-tumor effects. This is an important consideration, as NLRP3 inflammasome activation has been demonstrated to play a dichotomous role in cancer, capable of mediating both pro- and anti-tumorigenic effects.24 To determine if the NLRP3 inflammasome was required for or potentially limited C100-induced anti-tumor protection, WT and NLRP3−/− mice were inoculated with MC38 tumors and treated with PBS or C100 as before. Importantly, there was no difference in growth or survival between NLRP3-deficient and WT mice treated with C100 (Figures 2E, 2F, and S2C), showing a non-essential contribution of NLRP3 activation to the anti-tumor induced response.
Figure 2.

STING and IFNAR are required for the therapeutic efficacy of C100
(A and B) C57BL/6 WT or Tmem173−/− mice were injected s.c. on the flank with WT or Tmem173−/− B16F10 cells. Tumor-bearing mice were randomized and injected i.t. with PBS or C100 (100 μg) on days 0, 4, and 8. (A) B16 melanoma growth rates. (B) B16 melanoma Kaplan-Meier survival analysis.
(C‒F) C57BL/6 WT, Ifnar1−/−, or Nlrp3−/− mice were injected s.c. on the flank with MC38 carcinoma cells. Tumor-bearing mice were randomized and injected i.t. with PBS or C100 (100 μg) days 0, 4, and 8. (C) MC38 carcinoma growth rate in WT and Ifnar−/− mice. (D) MC38 carcinoma Kaplan-Meier survival analysis in WT and Ifnar−/− mice. (E) MC38 carcinoma growth rate in WT and Nlrp3−/− mice. (F) MC38 carcinoma Kaplan-Meier survival analysis in WT and Nlrp3−/− mice.
(A, C, and E) Data are expressed as mean tumor volume ±SEM. (A and C) Unpaired two-tailed t test or (D) Mantel-Cox tests were used to determine statistical significance between treatments. (B) PBS vs. treatment. (C) WT vs. Ifnar1−/−. (E) WT vs. Nlrp3−/−. ∗p < 0.05, ∗∗p < 0.001, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001. (A and B) n = 5 mice per group. (C and D) WT PBS n = 8, C100 n = 12. Ifnar1−/− PBS n = 6, C100 n = 6. (E and F) WT PBS + C100 n = 9. Nlrp3−/− PBS n = 7, C100 n = 8.
STING activation by C100 differs from HDCPs with a lower degree of deacetylation (DDA)
Next, we sought to determine the source of ROS and DNA underlying the engagement of cGAS-STING responses. Consistent with mtROS serving as the trigger for STING activation, ROS levels were comparable in both WT and STING-KO BMDCs (Figure 3A). C100 treatment had no impact on CellROX fluorescence compared to the medium control despite high levels of mitochondrial stress (Figure S3A). Next, we monitored BMDCs from WT mice cultured with or without low-dose ethidium bromide (EtBr) to deplete mtDNA (Figure S3B). mtDNA depletion was confirmed by qPCR analysis of mitochondrial gene expression compared to nuclear gene expression (Figure S3D). As expected, EtBr had no impact on DMXAA-induced Ifnb mRNA (Figure 3B). However, EtBr abolished and dramatically reduced protasan CL213 and C90-induced Ifnb mRNA, respectively. In contrast, C100-induced ifnb expression was significantly increased in EtBr DCs, with mRNA levels increasing more than 3-fold compared to control DCs (Figure 3B), suggesting that C100 differs from chitin-derived polymers with a lower DDA in mediating STING activation. Of note, the amplified Ifnb mRNA expression in EtBr DCs stimulated with C100 was higher than DMXAA-induced Ifnb expression in control DCs. While the use of EtBr to deplete mtDNA is an established means to implicate mtDNA in an immune response, it is not without caveats. EtBr intercalates all DNA and RNA species within the cell, which, in theory, could lead to nuclear mutations that alter immune responses. To ensure that the amplified IFN responses observed in EtBr DCs treated with C100 were STING dependent and not an off-target effect, WT and Tmem173−/− BMDCs were cultured with or without EtBr and stimulated with C100 or the direct STING agonist DMXAA. EtBr-induced depletion of mtDNA was identical in WT and Tmem173−/− KO BMDCs (Figure S3E). DMXAA-induced Ifnb mRNA expression was not impacted by EtBr (Figure S3F). In contrast, C100-induced Ifnb mRNA expression in WT DCs and amplification in EtBr-treated BMDCs were entirely STING dependent.
Figure 3.

C100-induced STING activation requires nuclear DNA damage
(A) BMDCs from WT or Tmem173−/− mice were treated with 8 μg/mL HDCPs for 2 h. Histogram overlay of MitoSOX fluorescence in single live cells. Data are representative of two independent experiments.
(B) WT bone marrow precursors were cultured to BMDCs as usual (control [Ctrl]) or in low-dose ethidium bromide (EtBr). Ctrl and EtBr BMDCs were treated with 8 μg/mL HDCPs for 24 h or DMXAA for 3 h. Ifnb mRNA levels were calculated by qPCR with respect to Actb and Rps18. Data are expressed as mean ± SD for technical triplicates with respect to Actb and are representative of 3 independent experiments. Statistical analysis was performed by two-tailed unpaired Student’s t tests with the Holm-Sidak method for multiple comparisons. ∗∗p < 0.01 and ∗∗∗p < 0.001.
(C) BMDCs were treated with 5 μg/mL HDCPs for 9 h or 100 μM etoposide for 24 h and then monitored for DNA fragmentation with the APO-BrdU (bromodeoxyuridine) TUNEL kit. Data are representative of two independent experiments.
(D) Ctrl and EtBr BMDCs were left untreated or treated with 5 μg/mL C100 for 8 h and then monitored for DNA fragmentation. Data are representative of two independent experiments.
(E and F) C57BL/6 mice were injected s.c. on the flank with B16F10 cells. Tumor-bearing mice were randomized and injected i.t. with C100 or intraperitoneally with cisplatin on days 0, 4, and 8. (E) Unpaired two-tailed t test or (F) Mantel-Cox tests were used to determine statistical significance between treatments.
∗p < 0.05 and ∗∗p < 0.001. (E and F) n = 7 mice per group.
HDCP-induced DNA damage is dependent on the degree of polymer deacetylation
Intracellular ROS can damage organelles, disrupt the nuclear compartment, and lead to the release and damage of DNA.25,26,27,28 A TUNEL assay was performed to investigate whether C100 exerts damage to nuclear DNA. This assay relies on the detection of dsDNA breaks, which facilitates the incorporation of the synthetic thymidine analog bromodeoxyuridine via an enzymatic reaction catalyzed by exogenous terminal deoxynucleotidyl transferase. In line with the ifnb mRNA data, HDCP-induced DNA damage was dependent on polymer DDA, with C100 the only HDCP capable of inducing high amounts of DNA fragmentation (Figure 3C). Furthermore, DNA damage was amplified in EtBr DCs treated with C100 compared to untreated EtBr cells or WT cells treated with C100 (Figure 3D). These results identify C100 as a DNA-damaging agent, akin to EtBr and chemotherapeutics known to activate cGAS-STING, a prime example being the platinum-based agent cisplatin.29 A study was designed to compare the therapeutic efficacy of C100 and cisplatin. i.t. C100 therapy was shown to exert comparable therapeutic efficacy to cisplatin treatment, demonstrated through inhibition of tumor growth (Figure 3E) and prolongation of survival (Figure 3F). These results successfully benchmark C100’s therapeutic efficacy with a clinically relevant chemotherapeutic.
C100 exclusively activates type I IFN signaling downstream of STING
In contrast to certain chemotherapeutics and, crucially, other STING agonists, C100 does not promote pro-inflammatory cytokine release downstream of STING activation.13,14 In fact, unlike other STING agonists that drive IRF3 and NF-κB activation, HDCP-induced DC activation is entirely IFNAR dependent.14 Furthermore, building on previously published data, a time course experiment was set up to monitor NF-κB activation (Figure 4A). IκBα is one of three components in the IκB complex that interacts with NF-κB transcription factor dimers, being responsible for their retention in the cytoplasm. Upon phosphorylation by IκB kinases (IKKs), IκB undergoes proteasome-dependent degradation, and NF-κB is translocated to the nucleus, where it drives inflammatory gene expression. As expected, DMXAA treatment reduced the relative expression of IκBα compared to the medium control. In contrast, C100 stimulation did not reduce IκBα levels at any time point tested (Figure 4A), demonstrating that it promotes type 1 IFNs in the absence of NF-κB activation. In addition, in contrast to 2′3′-cGAMP and DMXAA, C100 stimulation of BMDCs did not result in the induction of il6 or tnfa expression (Figure 4B). While these data demonstrate that C100 does not promote NF-κB activation and associated pro-inflammatory cytokine induction in vitro, it was crucial to validate this in vivo. As such, mice were treated with i.t. delivery of PBS, C100, or DMXAA, and 24 h later, the tumor was isolated for gene expression analysis via NanoString (Figure 4C). The induction of gene expression in treatment groups was compared to PBS-treated control mice, and differentially expressed genes were identified and grouped into genes predominantly activated via NF-κB or IRF activation. While the induction of IRF target gene expression was comparable between C100- and DMXAA-treated mice, there was a higher activation of NF-κB target genes in DXMAA-treated mice, with minimal activation observed in C100-treated mice (Figures 4D and 4E). There was a significantly higher relative fold induction of NF-κB-dependent genes in the DMXAA treatment group compared to genes associated with IRF induction (Figure 4F). These results highlight the properties of C100 that differentiate it in terms of its mechanism of action from alternative STING agonists.
Figure 4.

C100 preferentially activates IRF and not NF-κB signaling
(A) BMDCs were stimulated with C100 (5 μg/mL) or DMXAA (10 μg/mL) for indicated times. Cells were lysed and IκBα levels determined by immunoblot.
(B) BMDCs were stimulated with C100 (5 μg/mL), DMXAA (10 μg/mL), or 2′3′-cGAMP (1 μM) for indicated times. mRNA levels were calculated by qPCR for il6 and tnfa with respect to actb. Data show technical triplicate mRNA levels with respect to actb and are representative of three independent experiments.
(C‒F) C57BL/6 mice were injected s.c. on the flank with 5 × 105 B16F10 cells. Tumor-bearing mice (volume: 40–60 mm3) were randomized and injected i.t. with PBS, DMXAA, or C100. 24 h later, tumors were isolated with RNA subsequently extracted for gene expression analysis via NanoString. (C) Schematic of study. (D) Heatmaps displaying the relative induction of expression of IRF or NF-κB target genes in treatment vs. PBS Ctrl groups (log2 fold change relative to PBS) measured via NanoString. (E) Representative induction of IRF and NF-κB genes from NanoString dataset. (F) Overall IRF and NF-κB gene fold induction (log2 fold change relative to PBS − C100 values/DMXAA values) from NanoString dataset. (C‒F) n = 5 mice per group.
i.t. injection of C100 synergizes with checkpoint blockade therapy for enhanced therapeutic efficacy
As highlighted previously, response rates to checkpoint blockade therapies remain suboptimal, particularly in tumors where there is a low immune infiltrate, and as a result, there has been a movement toward combination strategies utilizing i.t injection of adjuvants to promote immune infiltrate and transition tumors from immunologically cold to hot.16 STING-activating adjuvants have been identified as candidates for i.t. therapeutics and have been trialed as monotherapies or in conjunction with checkpoint blockade.5 To explore whether i.t. injection of C100 could synergize with systemic anti-PD-1, a therapeutic MC38 tumor challenge model was utilized (Figure 5A). As before, mice received three i.t. injections of C100 and, in addition, received intraperitoneal injections with monoclonal α-PD-1 therapy on the same days. As previously, injection of C100 alone prolonged survival (Figure 5C) and inhibited tumor growth (Figures 5B and S4A). Synergy was observed between i.t. C100 injection and systemic anti-PD-1 delivery, significantly reducing the tumor growth rate when compared to administration of PBS, C100, or PD-1 alone (Figures 5B and S4A). Crucially, 50% of mice in the combination group had cleared their tumors/were alive at study endpoint with higher median survival time when compared to mice receiving α-PD-1 or C100 alone (Figure 5C). This synergistic effect is more evident when a single dose of α-PD-1 is utilized in combination with C100 treatment (Figure S4D). This synergistic effect with checkpoint blockade was further confirmed in a therapeutic B16 challenge model, which typically responds poorly to anti-PD-1 injection.30,31 Synergistic effects on the inhibition of tumor growth (Figures 5D and S4B) and survival (Figure 5E) were observed in mice treated with C100 and α-PD-1 combination therapy. Altogether, these results strongly support the use of C100 as part of a combinatorial treatment approach with checkpoint blockade.
Figure 5.

i.t. C100 therapy synergizes with systemic α-PD-1 therapy
(A‒C) C57BL6/J mice were injected s.c. with 3.5 × 105 MC38 tumor cells. Tumor-bearing mice (volume: 40–60 mm3) were randomized and injected i.t. with PBS or C100 (100 μg) and received either PBS or 250 μg of a monoclonal anti-PD-1 antibody on days 0, 4, and 8. (A and B) MC38 tumor growth rates. (C) MC38 Kaplan-Meier survival analysis.
(D‒E) C57BL6/J mice were injected s.c. with 5 × 105 B16F10 melanoma cells. Tumor-bearing mice (volume: 40–60 mm3) were randomized and injected i.t. with PBS or C100 (100 μg) and received either PBS or 250 μg of a monoclonal anti-PD-1 antibody on days 0, 4, and 8. (D) B16 tumor growth rates. (E) B16 Kaplan-Meier survival analysis. An unpaired two-tailed t test was used to determine statistical significance between treatments.
(B) Unpaired two-tailed t tests were used to determine statistical significance between treatments. ∗p < 0.05, ∗∗p < 0.001, ∗∗∗p < 0.001, and ∗∗∗∗p <0 .0.0001. (E) A Mantel-Cox test was used to determine statistical significance between the survival curves of α-PD-1 alone vs. C100- and α-PD-1-treated mice, where ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. (B and C) PBS, PBS/PD-1 n = 9. C100, C100/PD-1 n = 7. (D and E) PBS, C100, C100 + PD-1 n = 7. PBS + PD-1 n = 6.
The therapeutic efficacy of i.t. C100 injection is dependent on CD8+ T cells
To investigate the cellular mediators underlying the effectiveness of C100, either alone or in therapeutic combination strategies, cell depletion assays were performed. Natural killer (NK) cells have potent anti-tumor functionality, and their activity has been shown to be mediated by type I IFN signaling.32,33 Indeed, therapy with STING-activating CDNs has been shown to promote NK cell-mediated tumor clearance in some pre-clinical models, downstream of DC activation.34,35 As a result, a therapeutic study involving the depletion of NK cells was carried out (Figure S5A). NK cells were successfully depleted from experimental mice, as shown through peripheral blood sampling and fluorescence-activated cell sorting (FACS) analysis (Figures S5B and S5C). No impact on therapeutic benefit was observed in mice depleted of NK cells when comparing those given i.t. PBS vs. C100 (Figures S5D and S5E). In addition to NK cells, STING agonists have also been shown to elicit anti-tumor CD8+ T cell responses.34,36 Following i.t. therapy with STING agonists including ADU-S100 and cGAMP, CD8+ T cell responses have been shown to be elicited via promotion of DC-mediated cross-priming downstream of type I IFN signaling.34,36 Therefore, a therapeutic study involving i.t. C100 injection, systemic anti-PD-1 therapy, and CD8+ T cell depletion was utilized (Figure 6A). Successful depletion of CD8+ T cells was confirmed in experimental mice using peripheral blood sampling and FACS staining (Figure 6B). CD8+ T cell depletion was shown to completely abrogate the therapeutic benefit of i.t. C100 injection alone or in conjunction with systemic anti-PD-1 therapy, as reflected in tumor growth kinetics (Figures 6C and S4C) or survival analysis (Figures 6D and 6E). These results identify CD8+ T cells as the crucial mediators of C100-induced anti-tumor immunity when used alone or in conjunction with checkpoint blockade therapy. Finally, C100 was demonstrated to be able to mediate the induction of mitochondrial stress and the activation of STING in the human THP-1 cell line (Figures 6F–6H). These observations are crucial, as poor human-mouse translatability has led to the suboptimal clinical results of previous STING-targeted i.t. therapeutics,5,34 and the induction of mitochondrial stress and STING activation have been shown to be essential for C100’s adjuvanticity.14
Figure 6.

The therapeutic efficacy of i.t. C100 monotherapy or in combination with systemic α-PD-1 therapy is dependent on CD8+ T cells
(A) C57BL6/J mice were injected s.c. with 3.5 × 105 MC38 tumor cells. Tumor-bearing mice (volume: 40–60 mm3) were randomized and injected i.t. with PBS or C100 (100 μg) and received either PBS or 250 μg of a monoclonal anti-PD-1 antibody on days 0, 4, and 8. In addition, mice were injected intraperitoneally with either an anti-CD8α-targeted monoclonal antibody to deplete CD8α+ T cells or a relevant isotype Ctrl.
(B) CD8+ T cells were successfully depleted from experimental mice.
(C) MC38 tumor growth rates.
(D) MC38 Kaplan-Meier survival analysis.
(E) Median survival time of experimental mice post-initial treatment.
(F) THP-1 cells were stimulated with media, rotenone (5 μM), C100 (5 μg/mL), cGAMP (CDN) (1 μM), or lipopolysaccharide (LPS; 10 ng/mL), for 4 h. Cells were assayed for induction of mitochondrial stress via MitoSOX, with a histogram overlay of MitoSOX fluorescence in single live cells displayed.
(G and H) Cells were lysed and measured for STING and β-actin levels by immunoblot. (G) Mean fold STING densitometry compared to β-actin densitometry, n = 3.
(C and D) Iso PBS, Iso C100, α-CD8α C100 + PD-1 n = 7. Iso C100 + PD-1, CD8α PBS n = 8. CD8α C100 n = 9.
Discussion
Despite significant advances, there are no cGAS-STING-targeting cancer immunotherapeutics currently approved for use, and no candidate agonist has made it successfully past phase 3 clinical trials. To address these shortcomings, this work aims to characterize the therapeutic efficacy and mechanism of action of a cGAS-STING-activating chitin-derived polymer adjuvant that possesses distinct advantages over conventional agonists. Building on previous work,14 we have confirmed the value of measuring mitochondrial stress induction and the expression of DC costimulatory molecules as an in vitro readout for HDCP adjuvanticity in a therapeutic context. C100 was previously identified as the most potent HDCP in driving mitochondrial stress and BMDC maturation,14 and this work demonstrates that C100 possesses the greatest anti-tumor functionality, clearly demonstrating that the degree of polymer deacetylation is the key parameter in dictating efficacy as an i.t. cancer immunotherapeutic.
Mechanistically dissecting this C100-induced anti-tumor response, it was found that STING expression and IFNAR signaling were essential for the adjuvanticity of C100 both in vitro and in vivo. Crucially, C100-mediated activation of STING does not result in concomitant activation of NF-κB signaling, identifying the polymer as a distinctive STING activator in comparison to widely used molecules such as DMXAA and cGAMP. Activation of these pathways is regarded as being intricately tied. Although the underlying mechanisms are not fully defined, roles have been suggested for TBK1 and IKKε.37 STING-mediated activation of NF-κB is thought to predominantly occur at the ER, as blockade of STING translocation to the endoplasmic reticulum-Golgi intermediate compartment (ERGIC)/perinuclear compartment successfully blocks optimal type I IFN production but not NF-κB activation.38,39,40 This suggests that C100 may mediate rapid/early translocation of STING to the ERGIC, circumventing NF-κB activation while retaining intact type I IFN induction. This may be of significant value in an i.t. therapeutic setting, where activation of NF-κB signaling in tumor cells may exert pro-tumorigenic effects through inhibition of cell death pathways, upregulation of checkpoint molecules,9 and promotion of tumor angiogenesis and EMT.10 Crucially, C100 does not rely on direct binding to cGAS or STING and instead relies on the release and accumulation of endogenous DNA, both mitochondrial and nuclear in origin, which activate cGAS, leading to the subsequent production of 2′3′-cGAMP and STING activation. This avoids issues that have plagued STING agonists clinically, whereby mouse-human translation in terms of STING binding is challenging,17 and differences in STING haplotypes in human populations can reduce the affinity of STING binding.18,41 In addition, CDN STING agonists are large, electronegative molecules that are not conducive to cellular uptake42 and consequently can be further compromised by susceptibility to extracellular degradation by phosphodiesterases e.g., ENPP1, raising concerns regarding stability and bioavailability. In comparison, C100 is taken up into cells, releasing self-DNA, and in addition, chitin-derived polymers with high degrees of deacetylation are less susceptible to enzymatic degradation,43 potentially giving C100 a favorable stability profile.
Intact type I IFN signaling in the host was essential for the therapeutic efficacy of C100 following i.t. injection. IFNAR-KO mice have been documented to fail to adequately reject immunogenic tumors, correlating with impaired tumor infiltration of DCs and T cells,1,44 and type I IFN signaling in DCs has been shown to promote maturation,45,46 migration,47,48 antigen cross presentation, and successful priming of anti-tumor T cell responses.49 In addition, type I IFNs have been shown to potentiate the activation and cytotoxic potential of NK and T cells in the context of anti-tumor immunity. The success of C100 therapy in WT mice challenged with STING-KO tumors is not surprising, as DCs can successfully take up DNA or cGAMP from damaged or dying tumor cells or even through the transfer of cGAMP through tight junctions.50 This indicates that C100 could activate cGAS in either tumor cells or responding immune cells to elicit anti-tumor immune responses. Furthermore, STING expression in host endothelial cells positively correlates with tumor-infiltrating CD8+ T cells and enhanced survival,51 and STING activation in these cells can promote effective anti-tumor immune responses,36 identifying another cell type that C100 could be targeting in the tumor microenvironment (TME). Similar therapeutic observations have been observed using other STING agonists in comparable experimental designs.52 The success of C100 therapy in STING-KO mice challenged with WT tumors was a more surprising observation, as previous studies have demonstrated that STING-KO mice lack the capacity to mount spontaneous immune responses,53 with diminished antigen-specific CD8+ T cell responses observed.54 However, it has been documented that STING expression in tumor cells not only correlates with increased CD8+ T cells in the TME but can promote susceptibility to NK and CD8+ T cell-mediated killing.55 This likely relies on the promotion of type I IFN signaling by the tumor cells56 and associated immune cell recruitment to the TME. However, other groups have observed that despite production of type I IFN by tumor cells, the activation of STING by host immune cells was required to mount anti-tumor T cell responses.57 In addition, STING-deficient tumors have been shown to express less CCL5 and CXCL10, which are essential for the infiltration of both CD4+ and CD8+ T and NK cells.58,59
CD8+ T cells were identified as crucial mediators of C100-induced anti-tumor immunity, and this is in line with the literature surrounding the use of STING agonists therapeutically in a cancer setting including cGAMP,36 other synthetic CDNs,52 and non-CDN agonists.60 Altogether, our data suggest that C100-mediated activation of the cGAS-STING pathway in tumor cells or host immune cells is sufficient to promote anti-tumor immune responses via type I IFN production. Fundamentally, this requires IFNAR expression on host cells to ultimately prime anti-tumor CD8+ T cells.
The capacity of C100 to enhance the therapeutic efficacy of PD-1 blockade is particularly promising. Despite increases in the cohorts of patients eligible for checkpoint blockade therapy, there remains significant room for improvement in terms of response rates, as currently only roughly 20%–30% of patients respond to immune checkpoint blockade (ICB), with many patients developing resistance to therapy and subsequently relapsing.16,61 Response to ICB correlates with a number of factors including the immune contexture of tumors, which provides prognostic value.16,62 This largely hinges on the presence of an existing, intact tumor-intrinsic or circulating anti-tumor adaptive immune response involving CD8+ T cells or Th1 cells.16 Accordingly, patient/tumor stratification has been proposed utilizing immune parameters, and this has had success clinically in predicting response rates to ICB. According to this scoring method, immunologically cold tumors, with low levels of tumor-infiltrating lymphocytes (TILs), typically have lower response rates to ICB, while immunologically hot tumors, with a significant population of TILs, have optimal response rates to ICB. Therefore, therapeutic regimens that culminate in the immunological heating up of tumors may synergize with ICB.16 We propose that i.t. C100 therapy could be used both as a monotherapy but also in combination strategies aiming to sensitize tumors to respond to ICB through priming anti-tumor CD8+ T cell responses. This could be a broadly applicable treatment option, as expression of STING in the tumor cells themselves, or in host cells, is sufficient for therapeutic efficacy once IFN signaling pathways in the host remain intact. In addition, the local delivery of C100 to the tumor may prove advantageous over systemic immunotherapies where treatment-related adverse effects can be severe.63
In summary, this work characterizes the anti-tumor efficacy of C100, a cGAS-STING-activating adjuvant with critical advantages over conventional agonists, while providing key insights into its mechanism of action.
Limitations of the study
We acknowledge the limitations of suggesting that C100 outperforms cisplatin given that the treatments are given by different administration routes and only one dose of cisplatin was tested. While we have shown that C100 activates STING in human THP-1 cells, further work is required to address the efficacy of the adjuvant in human cells as a bridge toward clinical evaluation. Taken together, while there are limitations, our work provides a strong basis to pursue C100 as a cancer immunotherapeutic agent.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD16/CD32 | BD Biosciences | Cat# 553142; RRID: AB_394657 |
| Anti-mouse NK1.1 IgG2a (PK136) | Assay Genie | Cat# IVMB0100; RRID: AB_2935827 |
| Anti-mouse CD8α IgG2B (Clone 2.43) | Assay Genie | Cat# IVMB0070; RRID: AB_2935826 |
| IgG2a Isotype control (Clone C1.18.4) | Assay Genie | Cat# IVMB0188; RRID: AB_3097702 |
| IgG2b Isotype Control (Clone MPC-11) | Assay Genie | Cat# IVMB0190; RRID: AB_3097703 |
| Anti-PD1 (Clone RMP1-14) | Assay Genie | Cat# IVMB0037; RRID: AB_2929013 |
| PE Anti-mouse CD8 (Clone 53–6.7) | Biolegend | Cat# 100707; RRID: AB_312747 |
| Alexa 488 Anti-mouse BrdU | Part of APO-BrdU™ TUNEL Assay Kit | N/A |
| BV421 Anti-mouse NK1.1 (Clone PK136) | BD Biosciences | Cat# 562921; RRID: AB_2728688 |
| FITC Anti-mouse CD4 (Clone RM4-5) | Biolegend | Cat# 100509; RRID: AB_312713 |
| BV650 Anti-mouse CD3 (Clone 17A2) | BD Biosciences | Cat# 740530; RRID: AB_2740239 |
| Anti-mouse β-actin antibody (8H10D10) | Cell Signaling | Cat# 3700; RRID: AB_2242334 |
| Anti-mouse STING antibody (D2P2F) | Cell Signaling | Cat# 13647; RRID: AB_2732796 |
| anti-mouse IKBα antibody | Cell Signaling | Cat# 9242; RRID: AB_331623 |
| Anti-mouse cGAS (D3080) | Cell Signaling | Cat# 31659; RRID: AB_2799008 |
| IRDye 800CW Goat anti-rabbit IgG (H + L) | Odyssey | Cat# 926–32211; RRID: AB_621843 |
| IRDye 680CW Goat anti-mouse IgG (H + L) | Odyssey | Cat# 926–68070; RRID: AB_10956588 |
| Chemicals, peptides, and recombinant proteins | ||
| CpG ODN 1826 | Invivogen | tlrl-1826 |
| Cisplatin | Merck-Sigma | 232120 |
| C90 | Viscogel AB | N/A |
| C100 | Viscogel AB | N/A |
| Protasan | Novamatrix, Norway | N/A |
| DMXAA | Sigma | 117570-53-3 |
| Etoposide | Merck | 341205-25MG |
| LPS | Enzo Life Sciences | Serotype R515 |
| Phorbol 12-myristate 13-acetate (PMA) | Sigma | P1585 |
| Ethidium Bromide | Sigma | E1510 |
| GM-CSF | Peprotech | 300–03 |
| Pure Ethanol | Sigma | E7148 |
| Recombinant Mouse IFN-beta Protein | R&D systems | 12410–1 |
| Trypsin-EDTA | Sigma | T4049-500ML |
| Random Hexamer | MWG Biotech | BIO-38028 |
| RT 5x Buffer | Promega | M531A |
| Deoxyribonucleotide triphosphates (dNTPs) | Biolabs | BIO-39028 |
| Ribonuclease Inhibitor (RNAse Out) | Invitrogen | 10777019 |
| -M-MLV Reverse transcriptase | Promega | M170A |
| Acrylamide/Bis-acrylamide, 30% Solution | Merck | A3699 |
| Sodium Dodecyl Sulfate | Sigma | L5750 |
| Methanol | Sigma | 34860-1L-R |
| 1% Protease Inhibitor | Biosciences | 78430 |
| PVDF Membranes | Millipore | ISEQ00010 |
| Nitrocellulose Membranes | Cytiva | 10600001 |
| Glycine | Sigma | G8898 |
| Ammonium persulfate | Merck | A3678-100G |
| RNAlater Stabilization Solution | Invitrogen | R0901 |
| Bromo-3-chloropropane (BCP) | Sigma | B9673 |
| N,N,N′,N′-Tetramethylethylenediamine (TEMED) | Sigma | 411019 |
| Protein Ladder | Thermo Fisher | 26616 |
| Lamelli Buffer | Thermo Fisher | S3401 |
| CellRox Green | Biosciences | C10492 |
| MitoSOX Red | Biosciences | M36008 |
| Fixable Viability stain 510 | BD Biosciences | 564406 |
| Fixable Viability Stain 700 | BD Biosciences | 564997 |
| Critical commercial assays | ||
| APO-BrdU™ TUNEL Assay Kit | Thermo Fisher | A23210 |
| CXCL10 ELISA | R&D Systems | DY466 |
| IL-1beta ELISA | R&D Systems | DY401 |
| DNeasy Blood and tissue kits | Qiagen | 69504 |
| RNeasy Plus Mini Kit | Qiagen | 74134 |
| NanoString Murine Inflammation V2 panel | ||
| High Pure RNA isolation Kit | Roche | 11828665001 |
| KAPA SYBR® FAST qPCR Kit | Kapa Biosystems | KR0389 |
| Experimental models: Cell lines | ||
| THP-1 | ATCC | TIB-202 |
| B16F10 Melanoma | Prof Kingston Mills (Trinity College Dublin, previously purchased from ATCC) | N/A |
| MC38 Adenocarcinoma | Prof Chris Scott, Queens University Belfast (QUB) | N/A |
| Tmem173−/− B16F10 Melanoma | Prof Roger Greenberg (University of Pennsylvania). | N/A |
| Experimental models: Organisms/strains | ||
| C57BL/6 | Comparative Medicines Unit, TCD | N/A |
| Nlrp3−/− | Prof. Jurg Tscopp (Department of Biochemistry, University of Lausanne, Switzerland) | N/A |
| Ifnar1−/− | Paul Hertzog (Centre for Innate Immunity and Infectious Diseases MIMR-PHI and Monash University, Clayton, Victoria, Australia). | N/A |
| Tmem173−/− | Dr. Lei Jin (Department of Medicine, Division of Pulmonary, Critical Care and Sleep Medicine, University of Florida, USA) | N/A |
| Oligonucleotides | Invitrogen | See Table S1 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ed Lavelle, Trinity College Dublin (lavellee@tcd.ie).
Materials availability
There is no availability of biological material. We did not generate reagents in this study.
Data and code availability
Data: All data reported in this paper will be shared by the lead contact upon request.
Code: This paper does not utilise code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
Experimental models and study participant details
Animal models
All mice (male and female, aged 8–16 weeks) were bred in house and maintained according to the regulations of the EU and the Irish Health Products Regulatory Authority (HPRA). Mice were housed in individually ventilated cages under specific-pathogen-free conditions, under a 12-h light cycle at 21°C and 50% relative humidity with water and food ad libitum. Procedures were conducted under animal license number AE19136/P172 and were approved by TCD Animal Research Ethics Committee (reference number 010722).
For in vitro experiments with B16 and MC38 tumor cells, cells were cultured in cDMEM and upon reaching 60–80% confluency, harvested using trypsin EDTA. Cells were plated at 0.5 × 106 cells/well in cDMEM. For in vivo tumor models, cells were harvested at roughly 50% confluency using Trypsin-EDTA, washed, counted, and resuspended to appropriate concentration in ice-cold HBSS. 12-week-old male and female wt, Ifnar1−/−, Tmem173−/−, Nlrp3−/− or WT C57BL6/JCR mice were injected s.c with 5 × 105 B16-F10 cells (WT or Tmem173−/−) or 3.5 × 105 MC38 cells on the right flank. Tumor growth was measured daily, and tumor volume was determined as: (Length x (Width)2)/2. Upon tumor volume reaching ≈60mm3, mice were randomly allocated to experimental groups and injected with HDCP or DMXAA intratumorally (i.t), cisplatin intraperitoneally (i.p) or anti-PD-1 i.p (concentrations specified in figure legends). See individual graphs for the number of treatments received. Mice were culled at a humane endpoint determined as reaching 15 mm in diameter.
In vitro culture
Isolated bone marrow cells were seeded at 0.45 × 106 cells/mL in complete RPMI 1640 medium supplemented with 20 ng/mL of murine GM-CSF and incubated at 37°C with 5% CO2. On day 3 of culture, cells were supplemented with 30 mL of complete RPMI 1640 medium containing 20 ng/mL GM-CSF. On day 6, non-adherent cells were removed from flasks by gentle pipetting and 30 mL of complete RPMI 1640 medium containing 20 ng/mL GM-CSF was added. Two days later, cells were supplemented for the final time with 30 mL of complete RPMI 1640 medium containing 20 ng/mL GM-CSF. On day 10, loosely adherent cells were harvested by gentle pipetting and plated in complete RPMI 1640 medium supplemented with 10 ng/mL GM-CSF.
BMDCs were grown as normal until day 6, when media was removed and replaced with 30 mL fresh cRPMI containing 20 ng/mL GM-CSF and 150 ng/mL EtBr. On Day 7, 30 mL of cRPMI containing 20 ng/mL GM-CSF and 150 ng/mL EtbR was added to each T175 flask. On day 10, loosely adherent cells were removed by gentle repeat pipetting. Cells were transferred to a 50 mL falcon tube and spun down (1200 rpm, RT, 5 min). Cells were resuspended and DNA from 5 × 106 cells was isolated using DNeasy Blood and tissue kits. Relative total mtDNA amounts were then quantified by qPCR with primers specific for the mitochondrial D loop region (dloop) or a region of mtDNA that is not inserted into nuclear DNA (non-numt) and primers specific for nuclear DNA telomerase reverse transcriptase (tert) and β2 microglobulin (β2m).
THP1 cells were cultured in cRPMI. Upon reaching 70% confluency, cells were plated in fresh cRPMI containing 80 nM PMA to promote monocyte to macrophage differentiation. Three days later, the PMA was removed, and cells were given fresh cRPMI. Cells were left to rest for an additional 24 h before stimulation. All cells were cultured and maintained in cell culture incubators set to 37°C with 95% humidity and 5% CO2.
Method details
Detection of mitochondrial and cellular ROS
Cells were incubated with CellROX Green and MitoSOX Red for 30 min at 37 °C at 5% CO2 following stimulation. Cell culture plates were then centrifuged at 1200 rpm for 5 min at room temperature, and supernatants were discarded. Cells were removed from wells using ice-cold PBS, transferred to labeled FACs tubes, and washed with 2 mL PBS. Once washed, cells were stained with BV510 viability stain for 15 min at RT. Cells were centrifuged as before and were washed twice in PBS and re-suspended in 200μL FACS buffer and acquired immediately on the BD FACSCANTO II with data analyzed using FlowJo software.
Detection of DNA damage
The Apo-TUNNEL DNA damage kit was used to measure DNA fragmentation according to the manufacturer’s instructions. Briefly, cells were stained with A700 viability dye for 15 min at RT. Cells were then fixed in 1% PFA for 15 min and permeabilized overnight in pure ethanol at −20°C. The next morning, cells were stained with the DNA labeling solution containing reaction buffer (10 μL/sample), TdT enzyme (0.75 μL/sample), BrdUTP (8 μL/sample) and dH20 (31.25 μL/sample) for 1 h at 37°C. After 1 h, cells were washed with rinse buffer (provided in kit) and stained with 100 μL of the antibody staining solution (10 μL Alexa 488 anti-BrdU Ab and 90 μL rinse buffer) for 30 min at RT. Without washing, 200 μL PI/RNase A staining buffer was added to each FACS tube already containing the antibody staining solution. Cells were analyzed 30 min after the addition of the PI/RNase A buffer and the data analyzed using FlowJo software (Treestar, Oregon).
Q-PCR
RNA was isolated using Isolation Kits and reverse transcribed into complementary DNA (cDNA) with an M-MLV reverse transcriptase, RNase H minus, point mutant.Quantitative PCR was performed using KAPA SYBR FAST qPCR Kit Master Mix (2X) in accordance with the instructions provided by the manufacturer using Aligent Technologies Stratagene Mx3005P technology.
Western Blot
Cells were lysed in Laemmli Buffer containing 1% Protease inhibitor (1/100 dilution) and heated for 5 min at 95°C in a heating block. Equal volumes (20–30 μL) of the Laemmli-cell lysate were loaded into 12% gels and ran at 100 V for roughly 1.5 Hrs. Gels were transferred onto PVDF or nitrocellulose membranes using Biometra Fastblot Transfer Machine and PowerStation 200 powerpack with the number of mAmps dependent on the size of the protein of interest for 1.5 h. Following transfer, membranes were blocked in TBST containing 3% BSA for 1 h at RT and then incubated with primary antibodies at 4°C overnight PVDF/Nitrocellulose membranes were washed three times in TBST for 5 min each. Membranes were incubated with secondary antibody solutions for 1 h at RT, washed as before and then analyzed on an Odyssey LICOR machine. Images were analyzed using Image Studio Lite or Empiria Studio Software.
In vivo CD8 & NK cell depletion
For the CD8α T cell/NK cell depletion experiments, upon the appearance of tumors (>20 mm3), mice were randomly allocated into two groups and injected i.p with anti-mouse CD8α or the isotype control, or in the case of NK cell depletion, mice were injected i.p with anti-mouse NK1.1 or isotype control. Two days later, mice were treated with C100 and/or anti-PD1 as before. Cell depletion was maintained through additional antibody injections every 5 days throughout the experiment with half the initial dose of antibody used for subsequent injections. Cell depletion was confirmed by tail bleed, in brief, 100μL of blood was collected into tubes containing 100μL of 3 mg/mL EDTA. Samples were then transferred to FACs tubes and 0.88% NH4Cl was used to lyse RBC for 2 min, with reaction quenched with cRPMI. FACs tubes were centrifuged at 1200rpm for 5 min at room temperature, supernatants were removed, and cells were stained in 100μL BV510 fixable viability stain (1/500 dilution in PBS) on ice for 30 min in the dark. Cells were then washed as before and were resuspended in PBS supplemented with purified anti-mouse CD16/CD32 to block FcγRII/III for 10 min in the dark on ice. Cells were then surface stained through addition of 10 μL of fluorochrome-conjugated antibodies against: CD8α, CD3 #, CD4 to confirm CD8 T cell depletion andCD3, NK1.1 to confirm NK cell depletion. Samples were acquired using an LSR Fortessa cytometer FACSCANTO II with FACS Diva software and the data analyzed using Flowjo software (Treestar, Oregon).
RNA extraction from B16 tumors
24 h following i.t. treatment, tumors were collected in RNAlater Stabilization Solution and stored following manufacturer’s instructions. Tumors were homogenised using the Qiagen TissueLyser at max speed (30 Hertz) for 2 min. 100μL of 1—BCP were added to the homogenate, samples were shaken for 15 s and allowed to sit for 3min. After that time, samples were centrifuged at 12,000 x g for 15 min at 4°C. Then, 450μL from the upper aqueous layer was transferred to labeled tubes, followed by the addition of 450μL of 70% ethanol. After this, the RNA isolation from the samples was performed using the Qiagen RNA tissue extraction kit according to the manufacturer’s instructions. RNA quantity was measured using Nanodrop and quality assessed using the Agilent Bioanalyzer RNA 6000 system. Samples were stored at −80°C until performing NanoString gene expression analysis for the study of inflammation-associated genes.
NanoString gene expression analysis
The NanoString Murine Inflammation V2 panel (CodeSet) was selected for the study. NanoString’s nCounter MAX Analysis System (School of Veterinary Medicine, UCD) employs the use of digital barcode technology for directly measuring multiple analytes, allowing a high sensitivity of <1 copy per cell. The hybridization reaction was set up at RT. The master mix was made up by adding 70μL of hybridization buffer to the Reporter CodeSet tube and inverting it repeatedly. This mastermix was added to each RNA sample. The Capture Probeset was then mixed and spun down and 2μL was added to each tube. The tubes were mixed by repeated inversion, spun down and placed in a thermal cycler at 65°C overnight. The NanoString Prep Station is a multi-channel pipetting robot that processes samples to prepare them for data collection on the Digital Analyzer. After sample processing and hybridization at 65°C overnight, the hybridized samples were inserted into the Prep Station for purification and immobilization onto the internal surface of the sample cartridge (cartridge for nCounter MAX Analysis System). Following purification and immobilization steps, the sample cartridge was then transferred to the Digital Analyzer for imaging the immobilized fluorescent reporters in the cartridge with a CCD camera through a microscopic lens and analysing the fields of view (FOV) collected for each sample and counting hundreds to thousands of target molecules. The images were then exported to a comma separated value (CSV) file and analyzed using NanoString’s nSolver Analysis Software and Advanced Analysis add-on. QC was carried out using nSolver QC tools highlighting targets with an absolute copy number of 20 or lower. Internal reference genes were selected based on nSolver criteria including comparable expression between treatment and control groups and a wide range of Ct values to analyze variabilities in each candidate reference gene. Gene induction was compared between PBS, C100 and DMXAA treated groups.
Quantification and statistical analysis
Statistical analysis was performed using GraphPad Prism 5 software. Specific tests performed are specified in figure legends for individual experiments.
Acknowledgments
We thank Prof. Roger Greenberg for the Tmem173−/− B16F10 cells utilized in this study. We thank Dr. Barry Moran, manager of the Flow Cytometry Facility at the School of Biochemistry and Immunology, and Comparative Medicine Unit staff at the Trinity Biomedical Sciences Institute for their valuable assistance. E.C.L., R.W.W., J.L.T., and N.M.W. received support from SFI investigator award 12/1A/1421 and Frontiers of the future award 19/FFP/6484. R.W.W. received support from IRC GOIPG/2019/4236. We thank Dr. John Browne and Prof. Stephen Gordon at University College Dublin for providing access to NanoString and help with sample preparation and data analysis.
Author contributions
Conceptualization, E.C.L., J.L.T., and R.W.W.; methodology, J.L.T., R.W.W., J.H.-C., K.R., A.B., M.A., and N.M.W.; investigation, J.L.T., R.W.W., N.M.W., J.H.-C., and K.R.; writing – original draft, J.L.T., R.W.W., and E.C.L.; writing – review & editing, J.L.T., R.W.W., J.H.-C., K.R., N.M.W., A.B., and E.C.L.; funding acquisition, E.C.L., R.W.W., and N.M.W.; resources, E.C.L., M.A., L.J., and A.B.; supervision, E.C.L. and N.M.W.; project management, E.C.L.; formal analysis and validation, J.L.T., R.W.W., J.H.-C., K.R., and N.M.W.; visualization, J.L.T. and R.W.W.
Declaration of interests
E.C.L. is a co-founder and equity holder of AilseVax that aims to develop adjuvants for cancer vaccination. R.W.W., J.L.T., and N.M.W. are equity holders of AilseVax. E.C.L., R.W.W., and J.L.T. are inventors on patent WO 2022/113022 A1 – Use of C100: Immunotherapy for cancer.
Published: May 9, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101560.
Supplemental information
References
- 1.Fuertes M.B., Kacha A.K., Kline J., Woo S.R., Kranz D.M., Murphy K.M., Gajewski T.F. Host type I IFN signals are required for antitumor CD8 + T cell responses through CD8α + dendritic cells. J. Exp. Med. 2011;208:2005–2016. doi: 10.1084/jem.20101159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zheng J., Mo J., Zhu T., Zhuo W., Yi Y., Hu S., Yin J., Zhang W., Zhou H., Liu Z. Comprehensive elaboration of the cGAS-STING signaling axis in cancer development and immunotherapy. Mol. Cancer. 2020;19:133. doi: 10.1186/s12943-020-01250-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang L., Wei X., Wang Z., Liu P., Hou Y., Xu Y., Su H., Koci M.D., Yin H., Zhang C. NF-κB activation enhances STING signaling by altering microtubule-mediated STING trafficking. Cell Rep. 2023;42 doi: 10.1016/j.celrep.2023.112185. [DOI] [PubMed] [Google Scholar]
- 4.Yum S., Li M., Fang Y., Chen Z.J. TBK1 recruitment to STING activates both IRF3 and NF-κB that mediate immune defense against tumors and viral infections. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2100225118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Le Naour J., Zitvogel L., Galluzzi L., Vacchelli E., Kroemer G. Trial watch: STING agonists in cancer therapy. OncoImmunology. 2020;9 doi: 10.1080/2162402X.2020.1777624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meric-Bernstam F., Sweis R.F., Hodi F.S., Messersmith W.A., Andtbacka R.H.I., Ingham M., Lewis N., Chen X., Pelletier M., Chen X., et al. Phase I Dose-Escalation Trial of MIW815 (ADU-S100), an Intratumoral STING Agonist, in Patients with Advanced/Metastatic Solid Tumors or Lymphomas. Clin. Cancer Res. 2022;28:677–688. doi: 10.1158/1078-0432.CCR-21-1963. [DOI] [PubMed] [Google Scholar]
- 7.Motedayen Aval L., Pease J.E., Sharma R., Pinato D.J. Challenges and Opportunities in the Clinical Development of STING Agonists for Cancer Immunotherapy. J. Clin. Med. 2020;9:3323. doi: 10.3390/jcm9103323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jang S.C., Economides K.D., Moniz R.J., Sia C.L., Lewis N., McCoy C., Zi T., Zhang K., Harrison R.A., Lim J., et al. ExoSTING, an extracellular vesicle loaded with STING agonists, promotes tumor immune surveillance. Commun. Biol. 2021;4:497. doi: 10.1038/s42003-021-02004-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Antonangeli F., Natalini A., Garassino M.C., Sica A., Santoni A., Di Rosa F. Regulation of PD-L1 Expression by NF-κB in Cancer. Front. Immunol. 2020;11 doi: 10.3389/fimmu.2020.584626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huber M.A., Azoitei N., Baumann B., Grünert S., Sommer A., Pehamberger H., Kraut N., Beug H., Wirth T. NF-κB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J. Clin. Invest. 2004;114:569–581. doi: 10.1172/JCI21358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Achyut B.R., Angara K., Jain M., Borin T.F., Rashid M.H., Iskander A.S.M., Ara R., Kolhe R., Howard S., Venugopal N., et al. Canonical NFκB signaling in myeloid cells is required for the glioblastoma growth. Sci. Rep. 2017;7:13754. doi: 10.1038/s41598-017-14079-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hurler J., Škalko-Basnet N. Potentials of Chitosan-Based Delivery Systems in Wound Therapy: Bioadhesion Study. J. Funct. Biomater. 2012;3:37–48. doi: 10.3390/jfb3010037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carroll E.C., Jin L., Mori A., Muñoz-Wolf N., Oleszycka E., Moran H.B.T., Mansouri S., McEntee C.P., Lambe E., Agger E.M., et al. The Vaccine Adjuvant Chitosan Promotes Cellular Immunity via DNA Sensor cGAS-STING-Dependent Induction of Type I Interferons. Immunity. 2016;44:597–608. doi: 10.1016/j.immuni.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Turley J.L., Moran H.B.T., McEntee C.P., O'Grady K., Muñoz-Wolf N., Jin L., Follmann F., Andersen P., Andersson M., Lavelle E.C. Chitin-derived polymer deacetylation regulates mitochondrial reactive oxygen species dependent cGAS-STING and NLRP3 inflammasome activation. Biomaterials. 2021;275:120961. doi: 10.1016/j.biomaterials.2021.120961. [DOI] [PubMed] [Google Scholar]
- 15.Saxena M., Burg S.H., Melief C.J.M., Bhardwaj N. Therapeutic cancer vaccines. Nat. Rev. Cancer. 2021;21 doi: 10.1038/s41568-021-00346-0. [DOI] [PubMed] [Google Scholar]
- 16.Galon J., Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019;18:197–218. doi: 10.1038/s41573-018-0007-y. [DOI] [PubMed] [Google Scholar]
- 17.Conlon J., Burdette D.L., Sharma S., Bhat N., Thompson M., Jiang Z., Rathinam V.A.K., Monks B., Jin T., Xiao T.S., et al. Mouse, but not Human STING, Binds and Signals in Response to the Vascular Disrupting Agent 5,6-Dimethylxanthenone-4-Acetic Acid. J. Immunol. 2013;190:5216–5225. doi: 10.4049/jimmunol.1300097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yi G., Brendel V.P., Shu C., Li P., Palanathan S., Cheng Kao C. Single Nucleotide Polymorphisms of Human STING Can Affect Innate Immune Response to Cyclic Dinucleotides. PLoS One. 2013;8:e77846. doi: 10.1371/journal.pone.0077846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ng K.W., Marshall E.A., Bell J.C., Lam W.L. cGAS–STING and Cancer: Dichotomous Roles in Tumor Immunity and Development. Trends Immunol. 2018;39:44–54. doi: 10.1016/j.it.2017.07.013. [DOI] [PubMed] [Google Scholar]
- 20.Lechner M.G., Karimi S.S., Barry-holson K., Angell T.E., Murphy K.A., Church C.H., Ohlfest J.R., Hu P., Epstein A.L. Immunogenicity of murine solid tumor models as a defining feature of in vivo behavior and response to immunotherapy. J. Immunother. 2013;36:477–489. doi: 10.1097/01.cji.0000436722.46675.4a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jin Y., An X., Mao B., Sun R., Kumari R., Chen X., Shan Y., Zang M., Xu L., Muntel J., et al. Different syngeneic tumors show distinctive intrinsic tumor-immunity and mechanisms of actions (MOA) of anti-PD-1 treatment. Sci. Rep. 2022;12:3278. doi: 10.1038/s41598-022-07153-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marcus A., Mao A.J., Lensink-Vasan M., Wang L., Vance R.E., Raulet D.H. Tumor-Derived cGAMP Triggers a STING-Mediated Interferon Response in Non-tumor Cells to Activate the NK Cell Response. Immunity. 2018;49:754–763.e4. doi: 10.1016/j.immuni.2018.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schadt L., Sparano C., Schweiger N.A., Silina K., Cecconi V., Lucchiari G., Yagita H., Guggisberg E., Saba S., Nascakova Z., et al. Cancer-Cell-Intrinsic cGAS Expression Mediates Tumor Immunogenicity. Cell Rep. 2019;29:1236–1248.e7. doi: 10.1016/j.celrep.2019.09.065. [DOI] [PubMed] [Google Scholar]
- 24.Sharma B.R., Kanneganti T.D. NLRP3 inflammasome in cancer and metabolic diseases. Nat. Immunol. 2021;22:550–559. doi: 10.1038/s41590-021-00886-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Filomeni G., De Zio D., Cecconi F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015;22:377–388. doi: 10.1038/cdd.2014.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zorov D.B., Juhaszova M., Sollott S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014;94:909–950. doi: 10.1152/physrev.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paardekooper L.M., Van Vroonhoven E., Ter Beest M., Van Den Bogaart G. Radical stress is more cytotoxic in the nucleus than in other organelles. Int. J. Mol. Sci. 2019;20 doi: 10.3390/ijms20174147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Srinivas U.S., Tan B.W.Q., Vellayappan B.A., Jeyasekharan A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019;25 doi: 10.1016/j.redox.2018.101084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghosh S. Cisplatin: The first metal based anticancer drug. Bioorg. Chem. 2019;88 doi: 10.1016/j.bioorg.2019.102925. [DOI] [PubMed] [Google Scholar]
- 30.Pilon-Thomas S., Mackay A., Vohra N., Mulé J.J. Blockade of Programmed Death Ligand 1 Enhances the Therapeutic Efficacy of Combination Immunotherapy against Melanoma. J. Immunol. 2010;184:3442–3449. doi: 10.4049/jimmunol.0904114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Curran M.A., Montalvo W., Yagita H., Allison J.P. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. 2010;107:4275–4280. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Müller L., Aigner P., Stoiber D. Type I interferons and natural killer cell regulation in cancer. Front. Immunol. 2017;8:304. doi: 10.3389/fimmu.2017.00304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Madera S., Rapp M., Firth M.A., Beilke J.N., Lanier L.L., Sun J.C. Type I IFN promotes NK cell expansion during viral infection by protecting NK cells against fratricide. J. Exp. Med. 2016;213:225–233. doi: 10.1084/jem.20150712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Amouzegar A., Chelvanambi M., Filderman J.N., Storkus W.J., Luke J.J. Sting agonists as cancer therapeutics. Cancers. 2021;13 doi: 10.3390/cancers13112695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nicolai C.J., Wolf N., Chang I.C., Kirn G., Marcus A., Ndubaku C.O., McWhirter S.M. Raulet D.H.NK cells mediate clearance of CD8+ T cell-resistant tumors in response to STING agonist. Sci. Immunol. 2020;5:1–14. doi: 10.1126/sciimmunol.aaz2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Demaria O., De Gassart A., Coso S., Gestermann N., Di Domizio J., Flatz L., Gaide O., Michielin O., Hwu P., Petrova T.V., et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc. Natl. Acad. Sci. 2015;112:15408–15413. doi: 10.1073/pnas.1512832112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Balka K.R., Louis C., Saunders T.L., Smith A.M., Calleja D.J., D'Silva D.B., Moghaddas F., Tailler M., Lawlor K.E., Zhan Y., et al. TBK1 and IKKε Act Redundantly to Mediate STING-Induced NF-κB Responses in Myeloid Cells. Cell Rep. 2020;31 doi: 10.1016/j.celrep.2020.03.056. [DOI] [PubMed] [Google Scholar]
- 38.Ni G., Konno H., Barber G.N. Ubiquitination of STING at lysine 224 controls IRF3 activation. Sci. Immunol. 2017;2:eaah7119. doi: 10.1126/sciimmunol.aah7119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stempel M., Chan B., Juranić Lisnić V., Krmpotić A., Hartung J., Paludan S.R., Füllbrunn N., Lemmermann N.A., Brinkmann M.M. The herpesviral antagonist m152 reveals differential activation of STING -dependent IRF and NF -κB signaling and STING ’s dual role during MCMV infection. EMBO J. 2019;38:1–22. doi: 10.15252/embj.2018100983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dunphy G., Flannery S.M., Almine J.F., Connolly D.J., Paulus C., Jønsson K.L., Jakobsen M.R., Nevels M.M., Bowie A.G., Unterholzner L. Non-canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-κB Signaling after Nuclear DNA Damage. Mol. Cell. 2018;71:745–760.e5. doi: 10.1016/j.molcel.2018.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shih A.Y., Damm-Ganamet K.L., Mirzadegan T. Dynamic Structural Differences between Human and Mouse STING Lead to Differing Sensitivity to DMXAA. Biophys. J. 2018;114:32–39. doi: 10.1016/j.bpj.2017.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yan H., Chen W. The promise and challenges of cyclic dinucleotides as molecular adjuvants for vaccine development. Vaccines. 2021;9:917. doi: 10.3390/vaccines9080917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moran H.B.T., Turley J.L., Andersson M., Lavelle E.C. Immunomodulatory properties of chitosan polymers. Biomaterials. 2018;184:1–9. doi: 10.1016/j.biomaterials.2018.08.054. [DOI] [PubMed] [Google Scholar]
- 44.Diamond M.S., Kinder M., Matsushita H., Mashayekhi M., Dunn G.P., Archambault J.M., Lee H., Arthur C.D., White J.M., Kalinke U., et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J. Exp. Med. 2011;208:1989–2003. doi: 10.1084/jem.20101158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Simmons D.P., Wearsch P.A., Canaday D.H., Meyerson H.J., Liu Y.C., Wang Y., Boom W.H., Harding C.V. Type I IFN Drives a Distinctive Dendritic Cell Maturation Phenotype That Allows Continued Class II MHC Synthesis and Antigen Processing. J. Immunol. 2012;188:3116–3126. doi: 10.4049/jimmunol.1101313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Montoya M., Schiavoni G., Mattei F., Gresser I., Belardelli F., Borrow P., Tough D.F. Type I interferons produced by dendritic cells promote their phenotypic and functional activation. Blood. 2002;99:3263–3271. doi: 10.1182/blood.v99.9.3263. [DOI] [PubMed] [Google Scholar]
- 47.Nguyen-Pham T.N., Lim M.S., Nguyen T.A.T., Lee Y.K., Jin C.J., Lee H.J., Hong C.Y., Ahn J.S., Yang D.H., Kim Y.K., et al. Type i and II interferons enhance dendritic cell maturation and migration capacity by regulating CD38 and CD74 that have synergistic effects with TLR agonists. Cell. Mol. Immunol. 2011;8:341–347. doi: 10.1038/cmi.2011.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luft T., Pang K.C., Thomas E., Hertzog P., Hart D.N., Trapani J., Cebon J. Type I IFNs enhance the terminal differentiation of dendritic cells. J. Immunol. 1998;161:1947–1953. [PubMed] [Google Scholar]
- 49.Schiavoni G., Mattei F., Gabriele L. Type I interferons as stimulators of DC-mediated cross-priming: Impact on anti-tumor response. Front. Immunol. 2013;4:1–7. doi: 10.3389/fimmu.2013.00483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ablasser A., Schmid-Burgk J.L., Hemmerling I., Horvath G.L., Schmidt T., Latz E., Hornung V. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature. 2013;503:530–534. doi: 10.1038/nature12640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang H., Lee W.S., Kong S.J., Kim C.G., Kim J.H., Chang S.K., Kim S., Kim G., Chon H.J., Kim C. STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade. J. Clin. Invest. 2019;129:4350–4364. doi: 10.1172/JCI125413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sivick K.E., Desbien A.L., Glickman L.H., Reiner G.L., Corrales L., Surh N.H., Hudson T.E., Vu U.T., Francica B.J., Banda T., et al. Magnitude of Therapeutic STING Activation Determines CD8 + T Cell-Mediated Anti-tumor Immunity. Cell Rep. 2018;25:3074–3085.e5. doi: 10.1016/j.celrep.2018.11.047. [DOI] [PubMed] [Google Scholar]
- 53.Woo S.R., Fuertes M.B., Corrales L., Spranger S., Furdyna M.J., Leung M.Y.K., Duggan R., Wang Y., Barber G.N., Fitzgerald K.A., et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41:830–842. doi: 10.1016/j.immuni.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deng L., Liang H., Xu M., Yang X., Burnette B., Arina A., Li X.D., Mauceri H., Beckett M., Darga T., et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity. 2014;41:843–852. doi: 10.1016/j.immuni.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sokolowska O., Nowis D. STING Signaling in Cancer Cells: Important or Not? Arch. Immunol. Ther. Exp. 2018;66:125–132. doi: 10.1007/s00005-017-0481-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ranoa D.R.E., Widau R.C., Mallon S., Parekh A.D., Nicolae C.M., Huang X., Bolt M.J., Arina A., Parry R., Kron S.J., et al. STING promotes homeostasis via regulation of cell proliferation and chromosomal stability. Cancer Res. 2019;79:1465–1479. doi: 10.1158/0008-5472.CAN-18-1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ho S.S.W., Zhang W.Y.L., Tan N.Y.J., Khatoo M., Suter M.A., Tripathi S., Cheung F.S.G., Lim W.K., Tan P.H., Ngeow J., Gasser S. The DNA Structure-Specific Endonuclease MUS81 Mediates DNA Sensor STING-Dependent Host Rejection of Prostate Cancer Cells. Immunity. 2016;44:1177–1189. doi: 10.1016/j.immuni.2016.04.010. [DOI] [PubMed] [Google Scholar]
- 58.Takashima K., Takeda Y., Oshiumi H., Shime H., Okabe M., Ikawa M., Matsumoto M., Seya T. STING in tumor and host cells cooperatively work for NK cell-mediated tumor growth retardation. Biochem. Biophys. Res. Commun. 2016;478:1764–1771. doi: 10.1016/j.bbrc.2016.09.021. [DOI] [PubMed] [Google Scholar]
- 59.Parkes E.E., Walker S.M., Taggart L.E., McCabe N., Knight L.A., Wilkinson R., McCloskey K.D., Buckley N.E., Savage K.I., Salto-Tellez M., et al. Activation of STING-dependent innate immune signaling by s-phase-specific DNA damage in breast cancer. J. Natl. Cancer Inst. 2017;109 doi: 10.1093/jnci/djw199. djw199-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Corrales L., Glickman L.H., McWhirter S.M., Kanne D.B., Sivick K.E., Katibah G.E., Woo S.R., Lemmens E., Banda T., Leong J.J., et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015;11:1018–1030. doi: 10.1016/j.celrep.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Haslam A., Prasad V. Estimation of the Percentage of US Patients With Cancer Who Are Eligible for and Respond to Checkpoint Inhibitor Immunotherapy Drugs. JAMA Netw. Open. 2019;2 doi: 10.1001/jamanetworkopen.2019.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Galon J., Mlecnik B., Bindea G., Angell H.K., Berger A., Lagorce C., Lugli A., Zlobec I., Hartmann A., Bifulco C., et al. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J. Pathol. 2014;232:199–209. doi: 10.1002/path.4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hu W., Wang G., Wang Y., Riese M.J., You M. Uncoupling Therapeutic Efficacy from Immune-Related Adverse Events in Immune Checkpoint Blockade. iScience. 2020;23 doi: 10.1016/j.isci.2020.101580. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data: All data reported in this paper will be shared by the lead contact upon request.
Code: This paper does not utilise code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.