

Summary
Photoperiod insensitivity has been selected by breeders to help adapt crops to diverse environments and farming practices. In wheat, insensitive alleles of Photoperiod-1 (Ppd-1) relieve the requirement of long daylengths to flower by promoting expression of floral promoting genes early in the season; however, these alleles also limit yield by reducing the number and fertility of grain-producing florets through processes that are poorly understood. Here, we performed transcriptome analysis of the developing inflorescence using near-isogenic lines that contain either photoperiod-insensitive or null alleles of Ppd-1, during stages when spikelet number is determined and floret development initiates. We report that Ppd-1 influences the stage-specific expression of genes with roles in auxin signaling, meristem identity, and protein turnover, and analysis of differentially expressed transcripts identified bZIP and ALOG transcription factors, namely PDB1 and ALOG1, which regulate flowering time and spikelet architecture. These findings enhance our understanding of genes that regulate inflorescence development and introduce new targets for improving yield potential.
Keywords: wheat, inflorescence, spike, flowering time, Photoperiod-1, ALOG1, transcriptome
Graphical abstract
Highlights
-
•
Ppd-1 significantly influences the transcriptome of a developing wheat inflorescence
-
•
PDB1 regulates photoperiod-insensitive flowering and spikelet termination
-
•
ALOG1 suppresses spikelet branching and delays flowering
-
•
ALOG1 shares a conserved role in the Triticeae crops wheat and barley
Gauley et al. investigate the influence of Photoperiod-1 (Ppd-1) during early wheat inflorescence development and identify two transcription factors that are regulated by Ppd-1 to control flowering time and inflorescence architecture.
Introduction
Flowering time is a key adaptative trait that contributes substantially to the reproductive fitness of plants by aligning fertilization and seed production with favorable environmental conditions. Ancient and modern breeders have used genetic variation for photoperiod responsiveness to expand the geographic distribution of crop cultivation by accelerating flowering, which can limit yield potential by reducing the number of flowers available for grain or fruit production.1,2,3,4,5,6,7,8,9 With global demands for food rising, an improved understanding of processes that act downstream of photoperiod-responsive genes in inflorescences is required to identify strategies for optimizing yield potential in fast-flowering genotypes.
In long-day plants, such as bread wheat (Triticum aestivum L.), flowering is promoted by the extending days of spring.1,10 The responsiveness of wheat to long daylengths is determined largely by allelic variation for Photoperiod-1 (Ppd-1), which encodes a pseudo-response regulator.1,9 Photoperiod-insensitive alleles of Ppd-1 (e.g., Ppd-D1a) are used widely in breeding to reduce the requirement for long daylengths, promoting flowering earlier in the season, relative to sensitive alleles; early flowering alleles help ensure grain is set in marginal environments where seasonal conditions limit the duration of plant growth.11,12,13,14 Photoperiod-insensitive alleles accelerate flowering by hastening and increasing the expression of FLOWERING LOCUS T1 (FT1), a conserved floral activator, which is expressed in leaves and transported to the shoot apical meristem to promote inflorescence development.1,10,15,16 While the effect of photoperiod-insensitive alleles on FT1 activity in leaves and the consequences for flowering time are well characterized, very little is known about their influence on gene expression in the developing inflorescence, particularly when spikelets and florets begin to form.1,10,15,17,18 It is crucial that we learn more about the effect of Ppd-1 on the developing inflorescence transcriptome because photoperiod-insensitive alleles significantly reduce spikelet number, floret number, and fertility, which are key yield determinants.7,8,13 Knowledge of genes that act downstream of Ppd-1 in the inflorescence could provide new breeding strategies to improve yield potential by balancing the effects of photoperiod insensitivity on flowering time and grain production.7,8,15
To investigate the effect of Ppd-1 on gene expression in the inflorescence, we performed RNA sequencing (RNA-seq) transcriptome analysis during early developmental stages using near isogenic lines (NILs) that contain either photoperiod-insensitive or -sensitive alleles of Ppd-1, or are ppd-1 null mutants across all three genomes.10,11,17 Our analysis shows that Ppd-1 modifies the activation and suppression of gene activity during early stages of inflorescence development and influences the expression of genes involved in core biological processes including translation and protein degradation. Through analysis of differentially expressed transcripts (DETs), we identified a bZIP and an ALOG transcription factor that repress flowering and modify spikelet number and architecture, providing new insights about the genes that act downstream of Ppd-1 to control key agronomic traits.
Results
Transcriptome analysis of early wheat inflorescence development
To investigate the influence of Ppd-1 during early inflorescence development, we first analyzed the transcriptome of cv. Paragon (herein referred to as wild type [WT]), which is the genetic background of the NILs that contain photoperiod-insensitive and null alleles of Ppd-1.11,17,18 We performed RNA-seq transcriptome analysis on developing inflorescences from field-grown plants at four developmental stages: vegetative (VG), double ridge (DR), lemma primordium (LP), and terminal spikelet (TS). These stages were selected because they mark key events of the vegetative-to-reproductive transition, including the initiation of spikelet and floret development, and they are modified by allelic variation for Ppd-1.7,10,15,17,19 Our analysis detected a similar number of transcripts at each of the four stages, based on a threshold value of >0.5 transcripts per million (TPM): 76,439 transcripts (64,550 unique genes, 54,227 high-confidence [HC] and 22,212 low-confidence [LC] transcripts) were expressed at VG, 77,215 at DR (65,070 unique genes, 53,787 HC and 23,428 LC), 79,240 at LP (66,567 unique genes, 55,435 HC and 23,805 LC), and 78,119 (66,097 unique genes, 54,717 HC and 23,402 LC) at TS (Data S1). The proportion of genes expressed from each genome was consistent across the four stages, with the A, B, and D genomes contributing approximately 33.5%, 30.5%, and 36% of the detected transcripts, respectively (Figure 1A; Data S1). The stronger contribution of transcripts from the A and D genomes shown here is consistent with analyses of spike transcriptomes from Chinese Spring and Azhurnaya20; however, the bias toward the D genome of Paragon is stronger than that detected in the other genotypes.21,22
Figure 1.

The transcriptional landscape of early inflorescence development in wheat
(A) Ternary plots showing relative transcript abundance of 21,627 gene triads during vegetative (VG), double ridge (DR), lemma primordium (LP), and terminal spikelet (TS) stages. Each corner represents the A (purple arrow), B (purple arrow), or D genome (yellow arrow), with the scale representing percentage contribution by that genome: lighter shades of blue represent higher transcript density.
(B) A summary of transcripts that are significantly up- (red) or downregulated (blue) between successive stages of inflorescence development in wild type (cv. Paragon).
(C) Transcript clusters that show stage-specific profiles at VG (C2), DR (C5 and C14), LP (C8 and C0), and TS (C13).
(D–G) Heatmaps displaying transcripts from the C2 (D), C5 (E), C8 (F), and C13 (G) that encode proteins enriched in the GO terms identified in the respective clusters. In C2, transferases (indigo text) and kinases (red); C5, auxin signaling/transport (black, also for C13), meristem maintenance (blue, also for C13), and flowering (green); C8, ubiquitination/SUMOylation (orange) and transcriptional process (purple); C13, cell-cycle/chromatin regulation (pink). Data are normalized TPM values of three biological replicates shown as a log2 fold changes, relative to either TS (D) or VG (E–G) stages.
See also Figures S1 and S2 and Data S1 and S2.
Having detected transcripts present at each stage, we asked how gene expression changed during early inflorescence development by identifying DETs between consecutive stages (Figure 1B; Data S2A). We detected 3,562 and 5,423 transcripts that were significantly up- and downregulated, respectively, between the VG and DR stages, 1,993 and 917 transcripts between DR and LP, and 1,219 and 1,035 transcripts between LP and TS (q < 0.05). These results indicate there is a more pronounced change in the transcriptome during the vegetative-to-reproductive transition relative to later stages, and that inflorescence development involves coordinated repression and activation of gene expression.
To further investigate stage-specific changes in gene expression, we clustered the 30,000 most abundant transcripts based on their expression profiles across the four stages. The analysis identified 15 clusters with unique expression profiles that included 14,535 transcripts, with the remaining 15,465 transcripts falling into the non-clustered category (Figure S1; Data S2B). Among the clusters, we detected profiles that displayed specific up- or downregulation at each developmental stage (Figure 1C). For example, cluster 2 (C2) included 322 transcripts that peaked at VG, followed by significant downregulation at DR and remaining stages (Figure 1C; Data S2B). Gene ontology (GO) term analysis showed these transcripts were enriched for transcripts encoding proteins with transferase, catalytic, and protein serine/threonine kinase activity, including glucosyltransferases, sulfotransferases, and methyltransferases, and calcium-dependent and mitogen-activated protein kinases (Figures 1D and S2; Data S2C and S2D). Several C2 genes perform roles in cell wall biosynthesis, suggesting the vegetative-to-reproductive transition involves reconfiguration of cell walls in the developing inflorescence.23,24
Cluster 5 (C5) contained 434 transcripts that peaked at DR, relative to VG, LP, and TS. C5 transcripts were enriched for GO terms involved in auxin-activated signaling, floral development, and DNA/RNA binding (Figures 1C, 1E, and S2; Data S2B, S2C, and S5). Auxin signaling genes included those involved in auxin transport (e.g., PIN-FORMED; PIN), regulation of auxin homeostasis (e.g., Gretchen Hagen 3.5; GH3.5), auxin-response factors (ARFs; e.g., ARF1, ARF2, ARF3, and ARF5), and homologs of Arabidopsis genes that respond to auxin (e.g., IAA27)25,26,27,28,29 (Figures 1E and S2; Data S2D). These results indicate auxin plays an important role during the vegetative-to-reproductive transition and establishment of an inflorescence meristem or axillary meristems that will form spikelets, which is consistent with reports in maize and that auxin treatment modified wheat inflorescence architecture when applied during early reproductive stages.25,30 Floral development transcripts included those with roles in meristem growth (e.g., BARELY ANY MERISTEMS1), differentiation of floral organs, and flowering-time regulation31,32,33 (Figures 1E and S2; Data S2D). The profile of C5 transcripts was mirrored by those of C14, which displayed substantially lower expression at DR relative to the other stages (Figure 1C). C14 transcripts were enriched for genes encoding proteins with roles in chromatin assembly and organization (Data S2B and S2C), suggesting there may be a pause in chromatin remodeling at DR.
LP was defined by C8 and C0, which contained transcripts that were substantially up- or downregulated, respectively, relative to the other three stages; C0 and C8 contained the most transcripts of the 15 identified clusters (Figures 1C, 1F, S1, and S2; Data S2B and S2D). The 3,278 transcripts of C8 were enriched for ubiquitination/SUMOylation and transcription-related GO terms (Data S2B and S2C). For example, C8 transcripts encode ubiquitin- and SUMO-conjugating enzymes, ubiquitin and SUMO E3 ligases, and components of the COP9 signalosome (CONSTITUTIVE PHOTOMORPHOGENESIS 9) and SCF (Skp, Cullin, F-box) complexes that facilitate protein degradation (Figures 1F and S2, Data S2C and S2D).34 C8 also included transcripts encoding RNA polymerase subunits, components of the Mediator complex, and histone acetyltransferases (Figures 1F and S2; Data S2C and S2D). C9 transcripts shared a similar profile to C8 and included ubiquitin-related genes, supporting the idea that protein degradation is a core process of LP (Figure S1; Data S2B). The 5,176 transcripts of C0 were enriched for GO terms related to translation and RNA processing (Data S2C). Transcripts included those encoding small and large ribosome subunits, translation initiation and elongation factors, transfer RNA (tRNA) synthases and ligases, poly(A)-binding proteins, and pre-mRNA splicing factors (Figure S2; Data S2B and S2D). Along with the enrichment of ubiquitin-related genes in C8, these results indicate LP marks a key transition stage for transcript and protein turnover in the developing inflorescence.
C13 contained transcripts that were upregulated specifically at TS and were enriched for genes encoding proteins with roles in nuclear division, cell-cycle control, and inositol metabolism (Figures 1C, 1G, and S2; Data S2B–S2D). C13 transcripts also encode proteins that regulate cell proliferation and meristem maintenance and perform roles in auxin signaling and floral development (e.g., PISTILLATA; Figure 1G).35,36,37,38,39,40 Together, the cluster analysis indicates that coordination of the reproductive transition and initiation of spikelet and floret development involves stage-specific up- and downregulation of genes that perform roles in diverse biological processes, including auxin signaling, meristem maintenance, floral development, transcription, and protein turnover.
Photoperiod-1 regulates the transcriptome landscape of early inflorescence development
Having established a reference inflorescence transcriptome, we asked how allelic variation for Ppd-1 influences gene expression. This analysis was performed using NILs that contain either the photoperiod-insensitive Ppd-D1a allele or loss-of-function alleles for Ppd-1 on all three genomes (ppd-1 null).11,17,18 We detected a similar number of transcripts for each NIL across the four developmental stages as those identified in WT, with approximately 64,000–68,000 unique genes (53,000–56,000 HC and 23,000–24,000 LC) expressed in both genotypes during the four stages (Data S3 and S4). Similarly, the proportion of genes contributed by each genome remained stable (33.5%, 30.5%, and 36% from the A, B, and D genome), indicating Ppd-D1a alleles do not uniquely modify the D genome relative to the other two genomes (Figures 2A and 2B). Most transcripts that were differentially expressed (q < 0.05) in the Ppd-D1a NIL, relative to WT, were detected during VG, LP, and TS (3,274/3,451; 94.9%; Figure 2C; Data S5), which is consistent with rapid onset of the reproductive transition and spikelet termination of photoperiod-insensitive lines. Interestingly, a higher proportion of DETs were downregulated (2,173/3,451; 63%), relative to those that were upregulated (1,278; 37%), indicating the accelerated inflorescence development and flowering of photoperiod-insensitive lines may be as much due to suppression of genes that maintain a vegetative state as the activation of transcripts that promote spikelet and floret development (Figure 2C; Data S5). For the late-flowering ppd-1 NIL, we found an equal proportion of DETs were up- (13,123/29,835; 44%) and downregulated (16,172; 56%), relative to WT, with a greater number of DETs (29,835) compared to the Ppd-D1a NIL (3,451; Figure 2C; Data S5). A substantial proportion of the DETs (62%) were detected at TS, consistent with spikelet development terminating later in ppd-1, compared to WT. The higher number of DETs in ppd-1 was supported by principal component analysis, which showed ppd-1 libraries at each stage clustered away from those of WT and the photoperiod-insensitive NIL (Figure 2D). Similarly, comparisons to WT clusters showed fewer transcripts exhibited the same profile in ppd-1 (640 transcripts), relative to those expressed in Ppd-D1a (838 transcripts), and gene-network analysis showed that no transcripts shared the same profile as Ppd-B1 and Ppd-D1 in the ppd-1 NIL, unlike WT and the photoperiod-insensitive line, where multiple genes are co-expressed with Ppd-B1 (827 and 1,846 genes, respectively) and Ppd-D1 (827 and 2,383 genes, respectively; Figure S3; Data S2B and S2E–S2G). Together, these results indicate genetic variation for Ppd-1 substantially modifies the transcriptome of early inflorescence development in wheat, and that absence of Ppd-1 function has a more pronounced effect than photoperiod-insensitive alleles.
Figure 2.

Ppd-1 allelism influences the transcriptome of early wheat inflorescence development
(A and B) Ternary plots showing relative expression abundance of 21,627 gene triads during vegetative (VG), double ridge (DR), lemma primordium (LP), and terminal spikelet (TS) stages in photoperiod-insensitive (A, Ppd-D1a) and null (B, ppd-1) lines. Lighter shades of blue represent higher transcript density.
(C) A summary of transcripts that are significantly up- and downregulated between WT and the Ppd-D1a (green) and ppd-1 (magenta) NILs at each of the four stages.
(D) Principal component analysis of transcript libraries from the three genotypes (WT, Ppd-D1a, and ppd-1) across all four stages, with each condition containing three replicates.
Next, we asked how Ppd-1 expression in the inflorescence changed in the two NILs, relative to WT. In the photoperiod-insensitive line, Ppd-B1 and D1 expression increased significantly at TS, relative to WT, while Ppd-A1 transcript levels were similar (Figure S3). In ppd-1, Ppd-D1 was upregulated like it is in the leaf, indicating Ppd-D1 expression responds to absence of Ppd-B1 (Figure S3).10 Expression of Ppd-A1 remained low in ppd-1, and there were no Ppd-B1 transcripts. Thus, Ppd-1 is expressed in the developing inflorescence, and variant Ppd-1 alleles influence expression of their homeologs during inflorescence development.10,15
To further investigate the influence of Ppd-1 on stage-specific gene expression, we asked how insensitive and null alleles modify the transcript clusters detected in WT. Of the 15 clusters, 10 and 13 comparable clusters were identified in the Ppd-D1a and ppd-1 NILs, respectively (Figure 3A; Data S2E and S2F). A substantial proportion of genes that did not cluster in WT formed distinct profiles in the Ppd-D1a and ppd-1 lines (57.7% and 49.5%, respectively), such that 15 and 17 unique clusters were detected in the photoperiod-insensitive and null NIL, respectively (Figure 3). Regarding stage-specific clusters, transcripts of C2 were expressed lower at VG in the photoperiod-insensitive line, relative to WT, such that genes were not downregulated at DR, indicating a shift of Ppd-D1a VG inflorescences toward being more like DR of WT. In the null line, most C2 transcripts showed a similar profile to WT but were expressed less at VG, with multiple transcripts shifted to the non-clustered category for ppd-1 (Figures 3B and S2; Data S2D). The advanced progression of inflorescence development in the photoperiod-insensitive line was supported by analysis of transcripts from C5 of WT, where transcripts peaked specifically at DR; many transcripts displayed strong VG expression in the Ppd-D1a NIL (Figures 3B and S2; Data S2D). In ppd-1, many C5 transcripts displayed a similar profile to WT but were either up- or downregulated, with BAM, ARF2, GH3.5/6, and PIN expressed higher in ppd-1 than WT, while fewer ARF3 and ARF5 transcripts were detected (Figures 3B and S2; Data S2D).
Figure 3.

Photoperiod-insensitive and null alleles of Ppd-1 modify stage-specific expression of transcripts during wheat inflorescence development
(A) A comparison of the transcript clusters detected in WT, photoperiod-insensitive (Ppd-D1a), and null (ppd-1) lines across early stages of inflorescence development (VG, DR, LP, and TS). Clusters include the 30,000 most abundant transcripts, and each cluster contains at least 20 genes.
(B) An alluvial plot demonstrating the shift in transcript expression profiles of Ppd-D1a and ppd-1 lines, relative to WT—the cluster numbers and colors correspond to those shown in (A), and line thickness indicates transcript numbers. NC, no cluster; transcripts do not alter during the analyzed stages.
The genotype-dependent shifts in expression profiles relative to development continued during later stages (Figure 3B). For example, C8 transcripts encoding proteins with roles in ubiquitination/SUMOylation peaked earlier at DR in Ppd-D1a, with some sustaining expression through to TS (Figure S2; Data S2D). In ppd-1, transcripts encoding SUMOylation and ubiquitination proteins peaked earlier at DR and dipped at LP, except for ubiquitin E3 ligases, which climaxed at LP but were more abundant in ppd-1 than WT. Similarly, transcripts encoding proteins that perform roles in transcription peaked earlier at DR and to lower levels in the photoperiod-insensitive line, relative to WT, while these transcripts climaxed at DR or TS in ppd-1. For C0 genes that were downregulated at LP, transcripts encoding ribosomal proteins did not fall at LP in the photoperiod-insensitive line as much as they did in WT, nor did they show strong upregulation at TS (Figure S2; Data S2D). In ppd-1, transcripts encoding ribosome subunits dropped between VG and DR and continued to fall through LP and TS. Similar trends were observed for genes encoding eukaryotic translation initiation factors, poly(A)-binding proteins, and cap-binding proteins. C0 genes encoding splicing factors displayed stable expression between DR and TS in Ppd-D1a, with no drop at LP, while these transcripts were stable between DR and LP in ppd-1 but trended downward at TS, which is opposite to WT (Figure S2; Data S2D). At TS, C13 transcripts that encode proteins involved in nuclear division and cell-cycle control, auxin signaling, and meristem maintenance dipped earlier at DR in Ppd-D1a, rather than at LP as they did in WT, but they maintained a peak at TS (Figure S2; Data S2D). In ppd-1, these transcripts peaked at LP rather than TS. Taken together, these data indicate Ppd-1 substantially influences stage-specific expression of transcripts during inflorescence development, with gene expression profiles shifting earlier in the photoperiod-insensitive line, relative to WT, while they are often delayed or reduced in ppd-1.
Ppd-1 is required for correct expression of spikelet development genes
To investigate the effect of Ppd-1 on the activity of genes that influence spikelet and floret formation, we examined transcripts encoding MADS-box transcription factors, including genes that regulate spikelet architecture and floral development41,42,43,44,45,46 (Figure 4A; Data S2H). Transcripts of these genes grouped into eight clusters in WT, with each resolving largely into functional classes of floral development (Figure 4A). Two clusters identified transcripts encoding APETALA1-like (VRN1, AP1-2, and AP1-3) and SEPALLATA-like (SEP1 and SEP3) transcription factors of the A and E classes, which are expressed at a relatively low level during VG before rising during DR and LP.44,45,47 Transcripts of E class SEP3-like genes were expressed at a low level until LP, before increasing significantly at TS. Two independent clusters included the AG-like and PI-like genes of the B and C classes, for which expression decreased between VG and LP, before increasing again at TS. Transcripts encoding the D class transcription factors were represented by two clusters: expression of the STK-like and AGL12 genes peaked at DR before declining during LP and TS, while ALG6 was expressed at a low level from VG to LP before peaking strongly at TS. Transcripts of Bsister-like genes peaked at VG and DR, before reducing significantly during the LP and TS stages,42 and those encoding SHORT VEGETATIVE PHASE (SVP) and FLOWERING LOCUS C (FLC) transcription factors peaked at VG before falling during successive stages (Figure 4A).41,43,46,48
Figure 4.

Ppd-1 influences the expression of transcripts encoding genes that regulate spikelet and floret development
(A) Eight clusters of transcripts encoding MADS-box transcription factors that are expressed during the vegetative (VG), double ridge (DR), lemma primordium (LP), and terminal spikelet (TS) stages of wheat inflorescence development.
(B) Heatmaps display transcripts of MADS-box transcription factors that are expressed differentially during each of the four analyzed stages of inflorescence development. Data are normalized TPM values of three biological replicates shown as log2 fold changes, relative to WT.
(C) Transcript profiles of genes that regulate spikelet architecture and number, which are expressed differentially in either Ppd-D1a (cyan) or ppd-1 (magenta) NILs, relative to WT (orange). Data are presented as ribbon plots that show transcript levels (TPM, solid line with data points) ± SEM (shaded region) of three biological replicates. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Given earlier analyses showed that AP1- and SEP1-like genes are downregulated in inflorescences of ppd-1 mutants, we predicted that multiple genes encoding MADS-box transcription factors would be mis-regulated in the null and photoperiod-insensitive lines.10,15 Consistent with previous studies, AP1-, SEP1-, and SUPPRESSOR OF CONSTANS1-like (SOC1) genes were significantly downregulated at TS in ppd-1, relative to WT (Figures 4B and S4; Data S2H and S2I).15 Significantly fewer transcripts were detected for homeologs of SVP (SVP1 and VRT2) and FLC1 at VG in ppd-1, relative to WT, while substantially more transcripts were detected for FLC3, FLC4, and FLC6 at LP or TS (Figures 4B and S4; Data S2H and S2I). For other MADS-box genes, the level and profile of transcripts were maintained in ppd-1, relative to WT, despite FT1 being expressed significantly lower in the null line (Data S2H and S2I).10 In the photoperiod-insensitive line, expression of AP1-2, AP1-3, and SEP1-6 was significantly higher at VG or TS, relative to WT, as were homeologs of VRT2 and SOC1-3 (Figures 4B and S4; Data S2H and S2I). However, no other MADS-box genes were significantly upregulated at DR or LP (Data S2I). PI, AP3-1, and FLC4 were downregulated significantly at TS in the photoperiod-insensitive line, relative to WT, consistent with the cluster analysis. These results indicate photoperiod-insensitive Ppd-1 alleles do not increase transcript levels of multiple MADS-box genes, relative to WT, even though FT1 is expressed significantly higher in these genotypes. Gene-network and comparative transcript analyses indicate that the effect of the Ppd-D1a allele, instead, is to shift the pattern of expression of transcription factors that perform roles in spikelet and floret development, with 14 MADS-box genes being co-expressed with Ppd-D1 in the photoperiod-insensitive NIL that were not detected to be so in WT inflorescences (Figure S3; Data S2G). In ppd-1, transcripts of multiple MADS-box genes were dampened or delayed relative to WT (Figure S4; Data S2H). Together, these results indicate Ppd-1 is required for robust expression of meristem identity genes, and photoperiod-insensitive alleles affect activity of MADS-box genes by modifying the seasonal timing of expression, rather than altering transcript levels.10
Next, we asked how Ppd-1 influences genes that regulate spikelet architecture and number. Transcripts of spikelet architecture genes, including TEOSINTE BRANCHED-D1 (TB-D1), DUO-B1, DUO-D1, and WHEAT FRIZZY PANICLE-B1 (WFZP-B1), were significantly higher in ppd-1 at TS relative to WT, as were those of DUO-B1 at VG; however, no significant differences were detected for HB-2 (all homeologs), TB-A1, TB-B1, DUO-A1, WFZP-A1, and WFZP-D1 (Figure 4C; Data S2H).49,50,51,52,53 In the photoperiod-insensitive line, only HB-B2 was significantly different, with more transcripts detected at VG relative to WT. Regarding spikelet number genes, transcripts of FLOWERING LOCUS T-A2 (FT-A2) and FT-B2 were significantly downregulated in ppd-1 at TS, relative to WT, while FT-A2 transcripts were significantly upregulated in the Ppd-D1a NIL at LP (Figure 4C; Data S2H).10,54,55,56 COL-D5 was significantly downregulated in both the photoperiod-insensitive and null lines at VG, while COL-B5 and WAPO1 homeologs were expressed similarly in all three genotypes. Together, these results indicate Ppd-1 is required for correct expression of genes that control spikelet development, and that identification of transcripts mis-regulated in ppd-1 could help discover genes that regulate spikelet number and architecture.
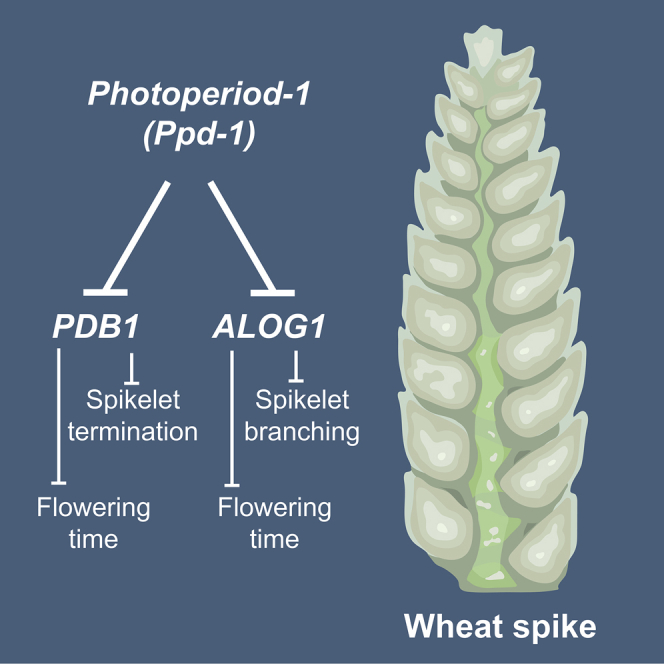
PBD1 and ALOG1 regulate inflorescence architecture and flowering time
To investigate genes influenced by Ppd-1 allelism that regulate inflorescence architecture, we analyzed the top 100 most significant DETs in the Ppd-D1a and ppd-1 NILs relative to WT (Data S5). We identified two genes for which expression of all three homeologs was modified in the NILs (Figures 5A and 5B). These genes included TraesCS6A02G096300, TraesCS6B02G124700, and TraesCS6D02G087400, which encode a basic-leucine zipper (bZIP) transcription factor, and TraesCS6A02G139700, TraesCS6B02G168300, and TraesCS6D02G129400, which encode an ALOG domain (Arabidopsis LSH1, Oryza G1) transcription factor.
Figure 5.

Identification of PDB1 and ALOG1 as transcription factors influenced by Ppd-1 allelism
(A and B) PDB1 (A) and ALOG1 (B) homeolog expression is influenced by photoperiod-insensitive (Ppd-D1a, cyan) and null (ppd-1, magenta) alleles of Ppd-1, relative to WT (orange), during early inflorescence development. Data are presented as ribbon plots that show transcript levels (TPM, solid line) ± SEM (shaded region) of three biological replicates.
(C and D) Unrooted maximum-likelihood phylogenetic trees of (C) group S bZIP and (D) ALOG transcription factors from wheat (Ta), rice (Os), maize (Zm), and Arabidopsis (At). Branches containing PDB1 and ALOG1 are highlighted in green.
(E–I) Expression analysis of PDB1 and ALOG1, including (E) in situ hybridization of PDB1 (AS, anti-sense; DR, double ridge stage) and tissue-specific expression analysis of (F) PDB1 and (G) ALOG1 in inflorescence (IM), flag leaf (FL), emerging leaves (LF), and roots (RT) of WT. Data are the average ± SEM of four biological replicates. In the boxplot, each box is bound by the lower and upper quartiles, the central bar represents the median, and the whiskers indicate the minimum and maximum values. Individual data points are shown; n.d., not detected. (H) A heatmap showing transcript levels of homeologs for ALOG transcription factors with TPM > 5 in at least one stage and (I) in situ hybridization of ALOG1. Red arrows indicate spikelet primordia and black arrows indicate direction of inflorescence apex. ∗∗p < 0.01; ∗∗∗p < 0.001. Scale bars, 100 μm (E and I).
Phylogenetic analysis showed the bZIP gene encodes a group S bZIP transcription factor homologous to bZIP60 of maize (Zea mays) and bZIP11/14 of rice (Oryza sativa); other members include LIP19 and OBF-1 (ocs-element binding factor-1; Figure 5C; Data S2J). Given the inconsistent naming of rice (either bZIP11 or bZIP14) and maize (bZIP60) homologs, we named the wheat gene based on the molecular phenotype of it being a photoperiod-1-dependent bZIP transcription factor, or PDB1 (homeologs: PDB-A1, PDB-B1, and PDB-D1). The gene network analysis supported PDB1 as being regulated by Ppd-1 in WT, as PDB-B1 and -D1 were detected in the same sub-network as Ppd-D1 (Figure S3; Data S2G). ALOG-domain transcription factors include the LIGHT-DEPENDENT SHORT HYPOCOTYL (LSH) proteins of Arabidopsis and OsG1-like proteins of rice.57,58,59 The wheat sequence identified here is orthologous to OsG1L1 (Figure 5D); we named the wheat gene TaALOG1 (homeologs named ALOG-A1, ALOG-B1, and ALOG-D1) to be consistent with the wheat gene nomenclature guidelines, with the other members named TaALOG2 to TaALOG9 and TaGL1 (G1-Like-1; Data S2K).60 ALOG1 was the only member for which all three homeologs were mis-regulated in the Ppd-1 NILs; G1-A1 transcripts were lower in Ppd-D1a inflorescences at TS, while ALOG-A3 (DR), ALOG-A4 (VG), and ALOG-A5 (VG) transcripts were higher in ppd-1 (Data S2K).
In WT, PDB1 expression increased significantly between VG and DR, before declining at LP and remaining low at TS; PDB1 grouped with cluster C7 (Figure 5A; Data S2B and S2J). As well as being expressed in developing inflorescences, where transcripts were detected throughout the inflorescence, PDB1 transcripts were detected by qRT-PCR in root tips, but not in leaves (Figures 5E and 5F). PDB-A1 and PDB-B1 were expressed comparably and higher than PDB-D1, consistent with public transcriptome data (Figure 5A).20,61 Stage-wise PDB1 expression profiles were maintained in both NILs; however, in ppd-1, all three PDB1 homeologs were expressed significantly higher at DR and TS, relative to WT, and there were significantly fewer transcripts in the photoperiod-insensitive line at DR (Figure 5A; Data S2J). Together, these results indicate Ppd-1 suppresses PDB1 expression, particularly at DR.
ALOG1 expression peaked at VG and DR (cluster C1) and was lower at LP and TS; of the highly expressed ALOG transcription factors, ALOG1 is the only member that is expressed equally or higher at DR as VG, with transcripts encoding the other ALOG genes being substantially lower at DR, relative to VG (Figures 5B, 5G, and 5H; Data S2B and S2K). qPCR indicated ALOG1 is expressed exclusively in the developing inflorescences, and in situ hybridization localized transcripts to the lower region of the lateral meristem that subtends the spikelet primordia; no transcripts were detected in root tips or leaves, consistent with public transcriptome data (Figures 5G and 5I).20,61 ALOG-A1 and -D1 were expressed comparably and higher than ALOG-B1 (Figure 5B). The profile of each ALOG1 transcript was maintained in the two NILs, but ALOG-A1 was expressed significantly higher in the null line at DR and TS, and ppd-1 contained more ALOG-B1 and -D1 transcripts at TS, relative to WT (Figure 5B; Data S2K). Upregulation of ALOG1 in the ppd-1 NIL was not mirrored by downregulation in the photoperiod-insensitive line, indicating Ppd-1 negatively regulates ALOG1 expression but does not cause further suppression when expressed constitutively.
Based on the Ppd-1-dependent expression profiles of PBD1 and ALOG1, we hypothesized that lines carrying mutations in these genes would display spikelet and/or flowering-time phenotypes. To test this hypothesis, we generated CRISPR/Cas9 gene-edited lines for PDB1 and ALOG1 in the cultivar Fielder (Figure 6); no deleterious mutants were detected for these genes in the Cadenza TILLING population. The PDB1 gene-edited lines (pdb1_m1 and pdb1_m2) contain deletions in PDB-B1 (31 bp) and PDB-D1 (30 bp) or PDB-A1 (30 bp) and PDB-D1 (30 bp), while those of ALOG1 (alog1_m1 and alog1_m2) carry deletions in each of ALOG-A1 (2 bp + 2 bp, or 2 bp), ALOG-B1 (3 bp), and ALOG-D1 (58 bp + 1 bp, or 1 bp); edits in each gene render transcripts that encode proteins with premature stop codons or lack amino acids that are highly conserved in ALOG transcription factors (Figure S5). To test the effect of these mutations on flowering time and inflorescence architecture, we grew the lines under long days (16 h light/8 h dark). Flowering time was accelerated by approximately 25 days in the PDB1 mutants (44.4 ± 0.9 and 45.6 ± 0.8 days after germination [DAG]) relative to WT (71.5 ± 4.1 DAG), while flowering occurred approximately 13 days earlier in the alog1 mutants (56.8 ± 1.2 and 57.8 ± 1.7 DAG; Figures 6A–6C). Both pdb1 and alog1 mutants produced inflorescences with significantly fewer rachis nodes relative to WT, and the alog1 lines formed multiple paired spikelets (Figures 6D–6F); paired spikelets are composed of two spikelets at one rachis node, where a secondary spikelet forms immediately below the regular primary spikelet.15 The secondary spikelets of alog1_m1 formed predominantly in the central region of the inflorescence (Figure S6). Many secondary spikelets formed fertile florets, such that alog1_m1 produced significantly more grain per inflorescence than WT, while pdb1_m1 formed significantly fewer grains per inflorescence (Figure S6). The grains of alog1_m1 and pdb1_m1 were of similar size and weight to WT, while the grains from secondary spikelets of alog1_m1 plants were smaller and lighter than those of primary spikelets (Figure S6). Given the pdb1 lines reproduced the flowering time and spikelet phenotypes of photoperiod-insensitive wheat, we asked if pdb1_m1 flowers under non-inductive short-day conditions (SD).1,10,15 Remarkably, pdb1_m1 displayed photoperiod insensitivity by flowering under extreme short daylengths (8 h light/16 h dark); neither WT nor alog1_m1 flowered under these conditions (Figure 6C). The pdb1_m1 line produced 13.4 ± 0.27 spikelets under SD, indicating the termination of spikelet development also occurred rapidly under these conditions. Together, these results demonstrate that flowering time, spikelet number, and inflorescence architecture can be altered by modifying the function of Ppd-1-dependent genes expressed in the inflorescence.
Figure 6.

PDB1 and ALOG1 influence flowering time and inflorescence architecture
(A–C) The pdb1 (A and C) and alog1 (B and C) gene-edited lines flower earlier than WT (cv. Fielder) under long days (LD) and pdb1_m1 flowers rapidly under short days (SD).
(D–F) Analysis of spikelet number and architecture phenotypes in the pdb1 (pink) and alog1 (green) lines, relative to WT (white). The secondary spikelets of alog1_m1 are highlighted in pink.
In the boxplots (C–E), each box is bound by the lower and upper quartiles, the central bar represents the median, and the whiskers indicate the minimum and maximum values of 4–5 biological replicates. Boxes that do not share a lowercase letter in the plots indicate a significant difference, p < 0.001.
See also Figures S5 and S6.
Discussion
Our transcriptome analysis shows that inflorescence development involves the coordinated up- and downregulation of gene expression as spikelet and floret development initiates. Recent evidence indicates that correct regulation of gene expression during inflorescence development is required to produce a wheat spike of the correct form. For example, failure to suppress VRT2 and SVP1 after DR causes abnormal floral organ growth and reduced spikelet fertility, while upregulation of VRN1, FUL2, and FUL3 during early stages is required to complete spikelet meristem development and prevent paired spikelet formation.15,41,43,44,46 Our cluster analysis indicates that stage-specific regulation of gene expression may also influence the timing of key biological processes that coordinate inflorescence development. For example, DR was enriched for genes involved in auxin transport and signaling, indicating this stage is important for the establishment of auxin maxima that initiate axillary meristem formation; these results are consistent with auxin transport helping to define boundary regions between lateral meristems of maize inflorescences and with auxin treatment disrupting spikelet formation when applied to immature wheat inflorescences.25,30 At LP, we detected increased expression of ubiquitin/SUMOylation- and transcription-related genes and suppression of genes involved in translation and RNA processing. These results suggest LP is a key transition stage that involves protein turnover; our previous analyses support LP marking an important transition stage of wheat inflorescence development, as it aligns with a substantial increase of FT1 and the detection of differences in spikelet number.10 We propose that stage-specific regulation of genes at DR and LP is required to define the number and arrangement of spikelets that form on a wheat inflorescence—this conclusion is consistent with spikelet number genes such as WAPO1, DUO1, and FT2 shifting their expression at LP.10,53,54,55
Our analysis showed that Ppd-1 influences gene expression substantially during early inflorescence development and contributes to both the activation and suppression of transcripts during inflorescence development. Analysis of transcripts detected within clusters showed that, in general, gene expression profiles shift to occur earlier in the photoperiod-insensitive line, relative to WT, while they are often delayed or dampened in the absence of Ppd-1 function. The principal component, network, and differential transcript analyses showed that loss of Ppd-1 function has a more pronounced effect on the developing inflorescence transcriptome than the Ppd-D1a allele, which is consistent with ppd-1 plants displaying more severe inflorescence architecture phenotypes than photoperiod-insensitive lines.11,15,17,18 In support of these trends, transcripts encoding proteins that influence spikelet number (e.g., FT2, COL5), architecture (e.g., TB1, WFZP), and fertility (e.g., SVP1, VRT2) are expressed differentially more often in the ppd-1 null than the photoperiod-insensitive line, relative to WT.10,43,49,51,52,55,56 Similarly, more MADS-box genes are mis-expressed in ppd-1 than in the Ppd-D1a NIL, contrary to our expectations that higher FT1 expression in the Ppd-D1a NIL would boost transcripts of genes that promote spikelet and floret development.15 Taken together with seasonal expression analysis of FT1 in these genotypes, our transcriptome and gene network data indicate that photoperiod-insensitive Ppd-1 alleles promote early flowering and termination of spikelet development by accelerating the onset of expression for MADS-box and spikelet identity genes in the developing inflorescence (e.g., SEP4- and AP1-like genes), rather than increasing transcript levels.10 This information suggests the productivity of photoperiod-insensitive genotypes could be enhanced by delaying or extending the duration of MADS-box gene expression—such an approach is supported by prolonged and higher expression of AP1-like genes increasing the yield potential of maize.62
Our analysis of transcripts expressed differentially in developing inflorescences of photoperiod-insensitive and ppd-1 lines identified two flowering-time repressors, PDB1 and ALOG1, which regulate spikelet number and architecture. Mutations in PDB1 facilitated early and photoperiod-insensitive flowering and the formation of inflorescences with fewer spikelets. The pdb1 mutants, therefore, reproduce flowering time and spikelet number traits of Ppd-D1a genotypes, consistent with PDB1 being downregulated in the photoperiod-insensitive NIL.10,15,18,63 The photoperiod insensitivity indicates flowering can be induced under short days by altering the activity of a gene expressed in the developing inflorescence, which is unique from early flowering being promoted by upregulating FT1 expression in leaves.1,10,18 The formation of fewer spikelets is consistent with other early flowering genotypes and indicates that spikelet development terminates earlier in pdb1 mutants, relative to WT.1,10,17,64 Interestingly, a group C bZIP transcription factor expressed similarly to PDB1 reduces spikelet number in tetraploid wheat but does not accelerate flowering.65 While the production of fewer spikelets is not favorable for breeding high-yielding cultivars, the accelerated flowering provides an opportunity to generate rapid maturing varieties that suit multiple cropping farming systems.66 The alog1 mutants also flowered earlier than WT under long daylengths and produced inflorescences with fewer rachis nodes. These phenotypes are consistent with ALOG1 being expressed lower in the Ppd-D1a NIL, relative to WT, and higher in the late-flowering ppd-1 line. In addition, the alog1 mutants produced paired spikelets, with multiple secondary spikelets forming fertile florets. Development of paired spikelets in an early flowering genotype is surprising because other secondary spikelet-producing genotypes flower simultaneously or later than WT siblings, and strong floral-promoting signals can reduce paired spikelet formation.15,49,50 Together with it being expressed below the spikelet primordia, we propose that ALOG1 helps define the region of a lateral meristem that will form a spikelet, such that a single spikelet forms rather than a pair. Indeed, such a role is consistent with the function of ALOG1 and ALOG2 in barley, in which alog1/2 mutants form extra spikelets comparable to the secondary spikelets of alog1 wheat mutants.67 The striking similarity of paired spikelet development in the wheat and barley alog1 mutants provides a unique example where a modified spikelet phenotype is shared in these two members of the Triticeae tribe, indicating ALOG1 performs a conserved role in maintaining the unbranched form of a spike inflorescence. ALOG transcription factors, including G1L1, G1L2, and TAWAWA1, have been investigated in rice, and although loss-of-function mutants produce fewer secondary branches than WT, which contrasts the formation of supernumerary spikelets in wheat and barley, the proposed role of these proteins as maintainers of inflorescence meristem activity and suppressors of spikelet meristem identity may explain phenotypes of the wheat and barley mutants.57,59 For example, perturbed maintenance of inflorescence meristems may help accelerate the onset of spikelet termination and reduce rachis nodes, while impaired suppression of spikelet meristems would facilitate secondary spikelet outgrowth. Further work is required to define the roles of PDB1 and ALOG1 during inflorescence development; nonetheless, our research highlights the potential to identify genes that regulate flowering and spikelet architecture by comparing the inflorescence transcriptomes of lines with modified Ppd-1 activity.
In summary, our data provide important insights into genes and biological processes that act downstream of Ppd-1 in the developing inflorescence, which can be modified to change spikelet number, architecture, and flowering time. Given the frequent use of photoperiod-insensitive Ppd-1 alleles in global wheat breeding, this information introduces genetic targets that could help optimize flowering time and inflorescence development to generate higher yielding cultivars.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Critical commercial assays | ||
| RNeasy Plant Mini Kit | Qiagen | 74904 |
| TURBO DNA-free Kit | ThermoFisher Scientific | AM1907 |
| NEBNext Ultra RNA Library Prep Kit for Illumina | New England Biolabs | E7770 |
| Deposited data | ||
| Raw data from RNA-seq analysis | This paper | SRA: PRJNA1081669 |
| Experimental models: Organisms/strains | ||
| Hexaploid wheat (Triticum aestivum), cv. Paragon | Bentley et al.11; Shaw et al.17 | N/A |
| Hexaploid wheat, near-iosgenic lines: Ppd-D1a and ppd-1 null | Bentley et al.11; Shaw et al.17 | 3A-7 and 3C-17 |
| ALOG1 CRISPR lines: alog1_m1 and alog1_m2 | This paper | N/A |
| PDB1 CRISPR lines: pdb1_m1 and pdb1_m2 | This paper | N/A |
| Oligonucleotides | ||
| sgRNAs used to generate ALOG1 CRISPR lines: AGCGCGGTGGACAGCCCTGG (sgRNA_1), GCAGGTACGAGTCGCAGAAGCGG (sgRNA_2), GCACCGCGCCAGCTCCAGCGGG (sgRNA_3), GCCCCCCGCTGGAGCTGGCGCGG (sgRNA_4) | This paper | N/A |
| sgRNAs used to generate PDB1 CRISPR lines: ATGGCGTCCTCCAGCGGGAGCGG (sgRNA_1), CACGGGCTCGCTGTCGACGGCGG (sgRNA_2), TGGAGCAGCGCCGGGCCAAGCGG (sgRNA_3), GCGCGGCGAGGTCGTCGAGGTGG (sgRNA_4) | This paper | N/A |
| Oligonucleotides to amplify across gene-edited sites: CTTGATCTGCCATAGCTAGAATC and TGGTCTTGCCGAACTGGTC (ALOG-A1), GCTGAATCCTGATATGCCATG and TGGTCTTGCCGAACTGGTC (ALOG-B1), CTGAATCCTCATCTGCCGTAG and TGGTCTTGCCGAACTGGTC (ALOG-D1), AAGGAAAGCAGGGAGTGCC and GCTGGGAACATGAACATCTC (PDB-A1), CTCGCTTACTCTCTCTCTCTCGTC and GCTGGGAACATGAACATCTC (PDB-B1), TGCCTGCTCGCTTGGTG and GCTGGGAACATGAACATCTC (PDB-D1) | This paper | N/A |
| Oligonucleotides used to generate probes for in situ hybridisation: CGCACTACCTGTTCCCCAT and TAATACGACTCACTATAGGGCTAGGGGTGTTC AAAT-GGCG (ALOG1, AS), CTAGGGGTGTTCAA ATGGCG and TAATACGACTCACTATAGGGCGC ACTACCTGTTCCC-CAT (ALOG1, S); GGCACGGA GGAGGAGATG and TAATACGACTCACTATAGGGC TCATGCAGGCGATGA-TGTC (PDB1, AS); CTCATGCAGGCGATGATGTC and TAATACGACTCACTATAGGGGGCACGG AGGAGGAG-ATG (PDB1, S). |
This paper | N/A |
| Oligonucleotides used for qRT-PCR: AGCTCGACGCTGAGAATTAAG and GCGAGATGACCAAGCCAAG (ALOG1), GACTTCCTCTTCAGATCCTCC and TGAAACACCAGAAGCATCAG (PDB1) | This paper | N/A |
| Software and algorithms | ||
| Kallisto | Bray et al.68 | v0.42.3 |
| Sleuth | Pimentel et al.69 | 0.28.0 |
| GOseq | Young et al.70 | v.3.0.4 |
| ggplot2 R package | Hamilton et al.71 | N/A |
| Clust | Abu-Jamous and Kelly72 | N/A |
| WGCNA R package | Langfelder et al.73 | N/A |
| DESeq2 | Love et al.63 | N/A |
| Cytoscape | Shannon et al.74 | 3.10.1 |
| Other | ||
| IWGSC Reference genome | IWGSC75 and Ramirez-Gonzalez et al.20 | IWGSC_v1.1_ALL_20170706_transcripts |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to the lead contact, Scott Boden (scott.boden@adelaide.edu.au).
Materials availability
The genetic resources generated in this study are available from the corresponding author or the Germplasm Resource Unit (John Innes Centre, Norwich, UK) upon request.
Data and code availability
-
•
The RNA-seq sequencing data used for the transcriptome analyses performed here have been deposited at NCBI Sequence Read Archive (SRA) and are publicly available. The project number is listed in the key resources table.
-
•
The paper does not report original code.
-
•
Any additional information required to reanalyze the data reported here is available from the lead author upon request.
Experimental model and study participant details
Plant materials
Hexaploid wheat genotypes (Triticum aestivum) used here included: wild-type photoperiod-sensitive cv. Paragon; Paragon near-isogenic lines (NILs) containing the Ppd-D1a photoperiod-insensitive allele or null ppd-1 alleles on the A, B and D genomes11,17,18; and transgenic lines containing edits in all three homeologs of PDB1 and ALOG1 generated in cv. Fielder (see details below).
Growth conditions
The three Ppd-1 NILs were grown at field sites based at Church Farm, John Innes Centre, Bawburgh, Norfolk, UK (52°62′25.7″N, 1°21′83.2″E) in 1 m2 plots. Seeds were sown in week 2 of October 2017. Wild-type Fielder and the pdb1_m1, pdb1_m2, alog1_m1, alog1_m2 lines used for phenotype analysis were grown in controlled growth cabinets under short-day (8 h/16 h light/dark) or long-day (16 h / 8 h light/dark) photoperiods at 300 μmol/m2/s (using Plantastar 400-W HQI bulbs [Osram] and Maxim 60-W tungsten bulbs) and 20°C/15°C (day/night) temperatures. The gene edited lines used for phenotype analysis were from the T3 generation and traits were verified using T4 generation plants.
Method details
RNA extraction, sequencing, and expression analysis
For RNA-seq transcriptome analysis, inflorescences were collected from wild-type, Ppd-D1a and ppd-1 NILs (cv. Paragon) at the vegetative (VG), double ridge (DR), lemma primordium (LP) and terminal spikelet (TS) stages. Three biological replicates were collected per stage, with each replicate being a pool of 5 – 15 inflorescences. RNA was extracted using the RNeasy Plant Mini Kit (Qiagen, The Netherlands) and treated with TURBO DNA-free Kit (Thermo Fischer Scientific, USA). RNA was examined by gel electrophoresis; RNA purity was checked using the NanoPhotometer spectrophotometer (IMPLEN, USA), and RNA integrity examined using the RNA Nano 6000 Assay Kit and Bioanalyzer 2100 system (Agilent Technologies, USA). Library construction and RNA-seq were performed by Novogene (Novogene HK Company Ltd., Hong Kong). Sequencing libraries were generated using the NEBNext Ultra RNA Library Prep Kit for Illumina (NEB), and index codes were added to attribute sequences to each sample. For sequencing, clustering of the index-coded samples was performed on the cBot Cluster Generation System using the TruSeq PE Cluster Kit v3-cBot-HS (Illumina, USA). After cluster generation, the libraries were sequenced on an Illumina NovaSeq platform to generate 150-bp paired-end reads.
For the quantitative real-time PCR analysis (qRT-PCR), RNA was extracted from the following tissues of wild-type cv. Paragon: young emerging leaves (lamina; harvested at LP stage); flag leaf (lamina; harvested after ear emergence); inflorescences at LP (pool of 10 inflorescences), and root tips (pool of 10 root tips per replicate) from wheat seedlings. RNA was extracted using the RNeasy Plant Mini Kit and treated with TURBO DNA-free Kit before cDNA synthesis. Synthesis of cDNA and qRT-PCR were performed as described previously.49 Oligonucleotides for qRT-PCR analysis are provided in the key resources table. Expression of candidate genes was normalized using TraesCS6D02G145100 and TraesCS5A02G015600,60 and data are average of four biological replicates and two technical replicates.
Read alignment and expression analyses
Read alignment and differential expression analysis was carried out as described previously.76 Reads were aligned to the IWGSC Chinese Spring reference genome model index v1.1.75 Read alignment and expression quantification of transcripts were completed using kallisto-0.42.3 with default parameters,68 30 bootstraps (-b 30) and the –pseudobam option as used previously.20,50 Differential expression analysis was performed for the stage specific samples of wild-type and between samples of each genotype using sleuth-0.28.0,69 with default parameters, and transcripts with a mean abundance of < 0.5 TPM in all three genotypes in all stages were excluded from further analysis. Transcripts that had a false-discovery rate adjusted p-value (q value) < 0.05 and a difference of > 0.5 TPM were considered to be differentially expressed.76 For each condition, the mean TPM of all three biological replicates was calculated ± standard error of the mean (s.e.m.).
Predicted functional annotation of transcripts was performed using Ensembl Plants Biomarts, with transcripts selected based on 2 or 3 homeologs displaying similar profiles and being homologs of genes with known functions in Arabidopsis thaliana, maize (Zea mays) and rice (Oryza sativa). Gene ontology (GO) term enrichment analysis was performed using the R package GOseq v.1.40.0.70 Significantly enriched GO terms were those that had adjusted p values of <0.05.
For data visualization, normalized mean TPM of each transcript is shown as log2 fold-changes, relative to wild-type (Figure 4) or vegetative and terminal spikelet stages (Figures 1 and 5), were visualized as a heatmap using the heatmap.2 function in the R package gplots v.3.0.4, as described previously.50 For the MADS-box genes, TPM data for each transcript was normalized using a combination of quantile normalization, log2 and Z score normalization, so transcripts with different abundances could be plotted on the same graph. Ribbon plots of transcript expressions were plotted using the R package ggplot2 as described previously,10 showing average TPM (solid line) and s.e.m. (shaded region) of three biological replicates.
Triad analysis
Triad analysis was performed as described previously.20 Traid analysis was performed exclusively on gene triads that had a 1:1:1 correspondence across the three homoeologous subgenomes, and a gene triad was deemed to be expressed when total expression was >0.5 TPM. (21,627 gene triads were detected). To standardize expression of all genes, the TPM for each gene was represented as a percentage of total triad expression. The relative triad expressions were then plotted into ternary diagrams using the R package ggtern.71
Clustering analysis
Clustering analysis was performed using the python package clust72 on the 30,000 most abundant transcripts across the four developmental stages. TPM data for each transcript was normalized to the input data using a combination of quantile normalization, log2 and Z score. The package was run using default parameters. To confirm clusters generated using the 30,000 most abundant transcripts (including 19,804 HC genes, with 2,674 represented by more than one splice variant) provided a reliable assessment, we performed clust analysis using: 1) TPM of genes where values for multiple transcript variants of a given gene within the 30,000 transcripts were collapsed to a single value, and 2) all transcripts with a TPM value > 0.5. These analyses identified similar clusters, validating the cluster analysis presented in Figures 1 and S1. Following generation of transcript clusters for all three genotypes, transcript profiles were compared between genotypes and presented using alluvial plot visuals, created using RAWGraphs.77
Co-expression gene network construction and visualization
TPM and count values of the differentially expressed transcripts were summarized at the gene level and used for the construction of co-expression gene networks. Only genes with TPM ≥ 0.5 in at least one sample were included in this analysis. A scale-free co-expression network was constructed using the WGCNA package in R for each genotype.73 The count values of the selected genes were normalized using the varianceStabilizingTransformation function from DESeq2.63 The scale-free topology criterion was used to select the soft power threshold for adjacency calculation. The adjacency matrices were transformed into a topological overlap matrix (TOM), measuring the network connectivity of a gene defined as the sum of its adjacency with all other genes for network generation. The blockwiseModules() function was used to calculate matrices and construct blockwise networks using the following parameters: NetworkType = “signed hybrid”; maxBlockSize = 25,000 genes; power = 15, 11, and 20 for WT, Ppd-D1a, and ppd-1, respectively; corType = “bicor”; maxPOutliers = 0.05; TOMType = “unsigned”; mergeCutHeight = 0.15; and minModuleSize = 30. Average linkage hierarchical clustering was used to classify genes with similar expression profiles into gene modules. Hub genes within each module were identified using the function signedKME. Modules that contain Ppd-1 homeologs were identified and used to assess and visualize the connection to other genes, including those are known to be related to spike development in wheat. The “exportNetworkToCytoscape” function was used to create edge and node files to be used to visualize the network using Cytoscape software (version 3.10.1).74 To reduce the complexity of the visualized networks, a weight (connection strength between two genes) threshold >1 was used to filter visualized genes.
Phylogenetic analysis
Sequences for homologs of ALOG1 and PDB1 were obtained by BLAST (Basic Local Alignment Search Tool) analysis using Ensembl Plants website (https://plants.ensembl.org/index.html). Amino acid sequence alignments were performed using MUSCLE v3.8.425 with default parameters and were checked manually.78 Unrooted trees were generated using MEGAX,79 with the maximum likelihood method, 100 bootstrap replicates. The Jones-Taylor-Thornton (JTT) matrix-based model was used for both trees, using partial deletions and gamma distributed rates. For all, positions with less than 95% site coverage were eliminated, i.e., fewer than 5% alignment gaps, missing data, and ambiguous bases were allowed at any position.
Wheat transformation, DNA extractions and sequencing
Gene sequences for PDB-A1, -B1, -D1 and ALOG-A1, -B1 and -D1 were obtained from the Ensembl Plants website (https://plants.ensembl.org/index.html). Four single guide RNA (sgRNA) sequences were designed to target all three homeologs of PDB1 and ALOG1 in the respective experiments (sequences are provided in KRT). The pdb1 and alog1 gene edited lines were generated using Clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 and Agrobacterium-mediated transformation of immature embryos isolated from cv. Fielder, as described previously.80,81 Leaf tissue from seedlings of gene-edited plants (T1, T2, T3 and T4 generations) were sampled and genomic DNA was extracted.60 Clones of PDB1 and ALOG1 homeologs were amplified to detect gene edits using PrimeSTAR GXL DNA polymerase (Takara Bio) and oligonucleotides provided in KRT. DNA fragments were sequenced using the Big-Dye Terminator Sequencing v3.1 Ready Reaction Kit (PerkinElmer, Applied BioSystems, Thermo Fischer Scientific) or with Mix2Seq Kit (Eurofins), and aligned to the reference sequences using SnapGene software (www.snapgene.com). Corresponding DNA regions from other members of the group S bZIP and ALOG transcription factor families were amplified and sequenced to confirm edits only occurred in the target sequences.
Phenotype analysis of inflorescence architecture, flowering, and grain morphology
All primary and secondary spikelets (fertile and rudimentary) were counted using the inflorescence of the main shoot. Images of inflorescences from alog1 gene-edited lines highlight secondary spikelets in pink, performed using Adobe Photoshop (Adobe). Flowering-time measurements and secondary spikelet distribution were determined as described previously,15,50 with values being the average ± s.e.m. for 4-5 biological replicates. Grain morphology measurements (grain area and thousand grain weight) were recorded using the MARVIN grain analyzer (GTA Sensorik GmbH, Germany). Measurements for each genotype include 4-5 biological replicates. The grain of alog1_m1 mutants separated into those derived from either the primary or secondary spikelets.
In situ mRNA hybridization experiments
In situ hybridization was performed as described previously,82 using inflorescence samples collected at the double ridge and glume primordium stages from cv. Paragon. Probe templates for PDB1 and ALOG1 were amplified by PCR using gene-specific oligonucleotides, which were fused with the T7 promoter (KRT). Images were obtained using an optical microscope (Ni-E, Nikon).
Quantification and statistical analysis
All statistical analyses were performed using R version 4.2.0 (http://www.r-project.org/). Principal component analysis of transcript libraries from each of the three genotypes was performed using the R package DESeq2.63 For analysis of the phenotypes of the gene-edited lines, the normality of the data was confirmed using the Shapiro–Wilk test, and means were compared statistically using a one-way ANOVA with Tukey post hoc test. Data comparing the grain of primary and secondary spikelets from alog1_m1 were analyzed using a Student’s two-tailed t-test. In all boxplots, each box is bound by the upper and lower quartiles, and the center line represents the median. Whiskers show the maximum and minimum values, and filled circles or dots represent individual data points.
Acknowledgments
We acknowledge the BBSRC Norwich Research Park Doctoral Training Partnership for A.G. (BB/M011216/1), the BBSRC Designing Future Wheat programme (BB/P016855/1), the Royal Society (UF150081), and the Australian Research Council (FT210100810) for funding the research. We thank Guojing Jiang and Thorsten Schnurbusch (IPK Gatersleben, Germany) for sharing unpublished data, Chao Ma and Kara Schmidt (University of Adelaide) for assistance with the in situ hybridizations, Simon Griffiths and Richard Morris (JIC) for helpful comments during the project, and the horticultural and field services team at JIC for assistance with plant husbandry.
Author contributions
A.G., A.K.A., and S.A.B. designed experiments; A.G., M.P., G.V.Y., A.K.A., S.H., M.A.S., and S.A.B. performed experiments; L.E.D. and S.A.B. supervised and supported the project; and A.G., L.E.D., and S.A.B. wrote and commented on the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: May 22, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.cub.2024.04.029.
Supplemental information
Transcript abundance data for all genes in developing inflorescences of wildtype plants (WT, cv. Paragon).
Transcript abundance and identification tables supporting the differential transcript and whole genome network analyses, and discovery of PDB1 and ALOG1. A) Stage-wise differential transcript analysis. B) Transcript clusters detected in inflorescences of wildtype (cv. Paragon). C) A summary of GO terms detected in stage-specific clusters. D) Quantification of transcripts detected in stage-specific clusters. E) Transcript clusters detected in inflorescences of the photoperiod insensitive NIL. F) Transcript clusters detected in inflorescences of the ppd-1 null NIL. G) Gene network analysis modules. H) Transcript analysis of MADS-box transcription factor genes. I) Differentially expressed MADS-box transcription factors. J) Transcript data of group S bZIP genes in wheat. K) Transcript data of ALOG genes in wheat.
Transcript abundance data for all genes in developing inflorescences of the photoperiod insensitive (Ppd-D1a) NIL.
Transcript abundance data for all genes in developing inflorescences of the ppd-1 null.
Summary of DETs in the photoperiod insensitive (Ppd-D1a) NIL, relative to wild-type, at the VG (A), DR (C), LP (E) and TS (G) stages. Summary of DETs in the ppd-1 null NIL, relative to wild-type, at the VG (B), DR (D), LP (F) and TS (H) stages
References
- 1.Beales J., Turner A., Griffiths S., Snape J.W., Laurie D.A. A pseudo-response regulator is misexpressed in the photoperiod insensitive Ppd-D1a mutant of wheat (Triticum aestivum L.) Theor. Appl. Genet. 2007;115:721–733. doi: 10.1007/s00122-007-0603-4. [DOI] [PubMed] [Google Scholar]
- 2.Faure S., Turner A.S., Gruszka D., Christodoulou V., Davis S.J., von Korff M., Laurie D.A. Mutation at the circadian clock gene EARLY MATURITY 8 adapts domesticated barley (Hordeum vulgare) to short growing seasons. Proc. Natl. Acad. Sci. USA. 2012;109:8328–8333. doi: 10.1073/pnas.1120496109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gao H., Jin M., Zheng X.M., Chen J., Yuan D., Xin Y., Wang M., Huang D., Zhang Z., Zhou K., et al. Days to heading 7, a major quantitative locus determining photoperiod sensitivity and regional adaptation in rice. Proc. Natl. Acad. Sci. USA. 2014;111:16337–16342. doi: 10.1073/pnas.1418204111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gonzalez A.M., Vander Schoor J.K., Fang C., Kong F., Wu J., Weller J.L., Santalla M. Ancient relaxation of an obligate short-day requirement in common bean through loss of CONSTANS-like gene function. Curr. Biol. 2021;31:1643–1652. doi: 10.1016/j.cub.2021.01.075. [DOI] [PubMed] [Google Scholar]
- 5.Lu S., Zhao X., Hu Y., Liu S., Nan H., Li X., Fang C., Cao D., Shi X., Kong L., et al. Natural variation at the soybean J locus improves adaptation to the tropics and enhances yield. Nat. Genet. 2017;49:773–779. doi: 10.1038/ng.3819. [DOI] [PubMed] [Google Scholar]
- 6.Murphy R.L., Klein R.R., Morishige D.T., Brady J.A., Rooney W.L., Miller F.R., Dugas D.V., Klein P.E., Mullet J.E. Coincident light and clock regulation of pseudoresponse regulator protein 37 (PRR37) controls photoperiodic flowering in sorghum. Proc. Natl. Acad. Sci. USA. 2011;108:16469–16474. doi: 10.1073/pnas.1106212108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perez-Gianmarco T.I., Slafer G.A., Gonzalez F.G. Photoperiod-sensitivity genes shape floret development in wheat. J. Exp. Bot. 2019;70:1339–1348. doi: 10.1093/jxb/ery449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prieto P., Ochagavía H., Savin R., Griffiths S., Slafer G.A. Dynamics of floret initiation/death determining spike fertility in wheat as affected by Ppd genes under field conditions. J. Exp. Bot. 2018;69:2633–2645. doi: 10.1093/jxb/ery105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turner A., Beales J., Faure S., Dunford R.P., Laurie D.A. The pseudo-response regulator Ppd-H1 provides adaptation to photoperiod in barley. Science. 2005;310:1031–1034. doi: 10.1126/science.1117619. [DOI] [PubMed] [Google Scholar]
- 10.Gauley A., Boden S.A. Stepwise increases in FT1 expression regulate seasonal progression of flowering in wheat (Triticum aestivum) New Phytol. 2021;229:1163–1176. doi: 10.1111/nph.16910. [DOI] [PubMed] [Google Scholar]
- 11.Bentley A.R., Horsnell R., Werner C.P., Turner A.S., Rose G.A., Bedard C., Howell P., Wilhelm E.P., Mackay I.J., Howells R.M., et al. Short, natural, and extended photoperiod response in BC2F4 lines of bread wheat with different Photoperiod-1 (Ppd-1) alleles. J. Exp. Bot. 2013;64:1783–1793. doi: 10.1093/jxb/ert038. [DOI] [PubMed] [Google Scholar]
- 12.Cane K., Eagles H.A., Laurie D.A., Trevaskis B., Vallance N., Eastwood R.F., Gororo N.N., Kuchel H., Martin P.J. Ppd-B1 and Ppd-D1 and their effects in southern Australian wheat. Crop Pasture Sci. 2013;64:100–114. [Google Scholar]
- 13.Fischer R.A. Wheat physiology: a review of recent developments. Crop Pasture Sci. 2011;62:95–114. [Google Scholar]
- 14.Worland A., Börner A., Korzun V., Li W., Petrovíc S., Sayers E. The influence of photoperiod genes on the adaptability of European winter wheats. Euphytica. 1998;100:385–394. [Google Scholar]
- 15.Boden S.A., Cavanagh C., Cullis B.R., Ramm K., Greenwood J., Jean Finnegan E., Trevaskis B., Swain S.M. Ppd-1 is a key regulator of inflorescence architecture and paired spikelet development in wheat. Nat. Plants. 2015;1 doi: 10.1038/nplants.2014.16. [DOI] [PubMed] [Google Scholar]
- 16.Li C., Dubcovsky J. Wheat FT protein regulates VRN1 transcription through interactions with FDL2. Plant J. 2008;55:543–554. doi: 10.1111/j.1365-313X.2008.03526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shaw L.M., Turner A.S., Herry L., Griffiths S., Laurie D.A. Mutant alleles of Photoperiod-1 in wheat (Triticum aestivum L.) that confer a late flowering phenotype in long days. PLoS One. 2013;8 doi: 10.1371/journal.pone.0079459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shaw L.M., Turner A.S., Laurie D.A. The impact of photoperiod insensitive Ppd-1a mutations on the photoperiod pathway across the three genomes of hexaploid wheat (Triticum aestivum) Plant J. 2012;71:71–84. doi: 10.1111/j.1365-313X.2012.04971.x. [DOI] [PubMed] [Google Scholar]
- 19.Kirby E., Appleyard M. 2nd Edition Edition. NAC Cereal Unit; 1987. Cereal Development Guide. [Google Scholar]
- 20.Ramirez-Gonzalez R.H., Borrill P., Lang D., Harrington S.A., Brinton J., Venturini L., Davey M., Jacobs J., van Ex F., Pasha A., et al. The transcriptional landscape of polyploid wheat. Science. 2018;361 doi: 10.1126/science.aar6089. [DOI] [PubMed] [Google Scholar]
- 21.Feng N., Song G., Guan J., Chen K., Jia M., Huang D., Wu J., Zhang L., Kong X., Geng S., et al. Transcriptome profiling of wheat inflorescence development from spikelet initiation to floral patterning identified stage-specific regulatory genes. Plant Physiol. 2017;174:1779–1794. doi: 10.1104/pp.17.00310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y., Fu X., Zhao M., Zhang W., Li B., An D., Li J., Zhang A., Liu R., Liu X. A Genome-wide view of transcriptome dynamics during early spike development in bread wheat. Sci. Rep. 2018;8 doi: 10.1038/s41598-018-33718-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Egelund J., Obel N., Ulvskov P., Geshi N., Pauly M., Bacic A., Petersen B.L. Molecular characterization of two Arabidopsis thaliana glycosyltransferase mutants, rra1 and rra2, which have a reduced residual arabinose content in a polymer tightly associated with the cellulosic wall residue. Plant Mol. Biol. 2007;64:439–451. doi: 10.1007/s11103-007-9162-y. [DOI] [PubMed] [Google Scholar]
- 24.Mortimer J.C., Miles G.P., Brown D.M., Zhang Z., Segura M.P., Weimar T., Yu X., Seffen K.A., Stephens E., Turner S.R., Dupree P. Absence of branches from xylan in Arabidopsis gux mutants reveals potential for simplification of lignocellulosic biomass. Proc. Natl. Acad. Sci. USA. 2010;107:17409–17414. doi: 10.1073/pnas.1005456107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Galli M., Liu Q., Moss B.L., Malcomber S., Li W., Gaines C., Federici S., Roshkovan J., Meeley R., Nemhauser J.L., Gallavotti A. Auxin signaling modules regulate maize inflorescence architecture. Proc. Natl. Acad. Sci. USA. 2015;112:13372–13377. doi: 10.1073/pnas.1516473112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Galweiler L., Guan C., Muller A., Wisman E., Mendgen K., Yephremov A., Palme K. Regulation of polar auxin transport by AtPIN1 in Arabidopsis vascular tissue. Science. 1998;282:226–2230. doi: 10.1126/science.282.5397.2226. [DOI] [PubMed] [Google Scholar]
- 27.Park J.E., Park J.Y., Kim Y.S., Staswick P.E., Jeon J., Yun J., Kim S.Y., Kim J., Lee Y.H., Park C.M. GH3-mediated auxin homeostasis links growth regulation with stress adaptation response in Arabidopsis. J. Biol. Chem. 2007;282:10036–10046. doi: 10.1074/jbc.M610524200. [DOI] [PubMed] [Google Scholar]
- 28.Simonini S., Bencivenga S., Trick M., Østergaard L. Auxin-induced modulation of ETTIN activity orchestrates gene expression in Arabidopsis. Plant Cell. 2017;29:1864–1882. doi: 10.1105/tpc.17.00389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ulmasov T., Hagen G., Guilfoyle T.J. ARF1, a transcription factor that binds to auxin response elements. Science. 1997;276:1865–1868. doi: 10.1126/science.276.5320.1865. [DOI] [PubMed] [Google Scholar]
- 30.Sharman B.C. Developmental anatomy of the inflorescence of bread wheat (Triticum aestivum L.) during normal initiation and when affected by 2,4-D. Ann. Bot. 1983;52:621–639. [Google Scholar]
- 31.DeYoung B.J., Bickle K.L., Schrage K.J., Muskett P., Patel K., Clark S.E. The CLAVATA1-related BAM1, BAM2 and BAM3 receptor kinase-like proteins are required for meristem function in Arabidopsis. Plant J. 2006;45:1–16. doi: 10.1111/j.1365-313X.2005.02592.x. [DOI] [PubMed] [Google Scholar]
- 32.Heisler M.G., Atkinson A., Bylstra Y.H., Walsh R., Smyth D.R. SPATULA, a gene that controls development of carpel margin tissues in Arabidopsis, encodes a bHLH protein. Development. 2001;128:1089–1098. doi: 10.1242/dev.128.7.1089. [DOI] [PubMed] [Google Scholar]
- 33.Song H.R., Song J.D., Cho J.N., Amasino R.M., Noh B., Noh Y.S. The RNA binding protein ELF9 directly reduces SUPPRESSOR OF OVEREXPRESSION OF CO1 transcript levels in Arabidopsis, possibly via nonsense-mediated mRNA decay. Plant Cell. 2009;21:1195–1211. doi: 10.1105/tpc.108.064774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wei N., Serino G., Deng X.W. The COP9 signalosome: more than a protease. Trends Biochem. Sci. 2008;33:592–600. doi: 10.1016/j.tibs.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 35.Honma T., Goto K. The Arabidopsis floral homeotic gene PISTILLATA is regulated by discrete cis-elements responsive to induction and maintenance signals. Development. 2000;127:2021–2030. doi: 10.1242/dev.127.10.2021. [DOI] [PubMed] [Google Scholar]
- 36.Krogan N.T., Hogan K., Long J.A. APETALA2 negatively regulates multiple floral organ identity genes in Arabidopsis by recruiting the co-repressor TOPLESS and the histone deacetylase HDA19. Development. 2012;139:4180–4190. doi: 10.1242/dev.085407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ranocha P., Dima O., Nagy R., Felten J., Corratgé-Faillie C., Novák O., Morreel K., Lacombe B., Martinez Y., Pfrunder S., et al. Arabidopsis WAT1 is a vacuolar auxin transport facilitator required for auxin homoeostasis. Nat. Commun. 2013;4 doi: 10.1038/ncomms3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suzuki T., Inagaki S., Nakajima S., Akashi T., Ohto M.A., Kobayashi M., Seki M., Shinozaki K., Kato T., Tabata S., et al. A novel Arabidopsis gene TONSOKU is required for proper cell arrangement in root and shoot apical meristems. Plant J. 2004;38:673–684. doi: 10.1111/j.1365-313X.2004.02074.x. [DOI] [PubMed] [Google Scholar]
- 39.Takeda S., Tadele Z., Hofmann I., Probst A.V., Angelis K.J., Kaya H., Araki T., Mengiste T., Mittelsten Scheid O., Shibahara K.i., et al. BRU1, a novel link between responses to DNA damage and epigenetic gene silencing in Arabidopsis. Genes Dev. 2004;18:782–793. doi: 10.1101/gad.295404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang W., Sijacic P., Xu P., Lian H., Liu Z. Arabidopsis TSO1 and MYB3R1 form a regulatory module to coordinate cell proliferation with differentiation in shoot and root. Proc. Natl. Acad. Sci. USA. 2018;115:E3045–E3054. doi: 10.1073/pnas.1715903115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adamski N.M., Simmonds J., Brinton J.F., Backhaus A.E., Chen Y., Smedley M., Hayta S., Florio T., Crane P., Scott P., et al. Ectopic expression of Triticum polonicum VRT-A2 underlies elongated glumes and grains in hexaploid wheat in a dosage-dependent manner. Plant Cell. 2021;33:2296–2319. doi: 10.1093/plcell/koab119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Callens C., Tucker M.R., Zhang D., Wilson Z.A. Dissecting the role of MADS-box genes in monocot floral development and diversity. J. Exp. Bot. 2018;69:2435–2459. doi: 10.1093/jxb/ery086. [DOI] [PubMed] [Google Scholar]
- 43.Chen Y., Liu Y., Zhang J., Torrance A., Watanabe N., Adamski N.M., Uauy C. The Triticum ispahanicum elongated glume locus P2 maps to chromosome 6A and is associated with the ectopic expression of SVP-A1. Theor. Appl. Genet. 2022;135:2313–2331. doi: 10.1007/s00122-022-04114-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li C., Lin H., Chen A., Lau M., Jernstedt J., Dubcovsky J. Wheat VRN1, FUL2 and FUL3 play critical and redundant roles in spikelet development and spike determinacy. Development. 2019;146 doi: 10.1242/dev.175398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li K., Debernardi J.M., Li C., Lin H., Zhang C., Jernstedt J., Korff M.V., Zhong J., Dubcovsky J. Interactions between SQUAMOSA and SHORT VEGETATIVE PHASE MADS-box proteins regulate meristem transitions during wheat spike development. Plant Cell. 2021;33:3621–3644. doi: 10.1093/plcell/koab243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu J., Chen Z., Wang Z., Zhang Z., Xie X., Wang Z., Chai L., Song L., Cheng X., Feng M., et al. Ectopic expression of VRT-A2 underlies the origin of Triticum polonicum and Triticum petropavlovskyi with long outer glumes and grains. Mol. Plant. 2021;14:1472–1488. doi: 10.1016/j.molp.2021.05.021. [DOI] [PubMed] [Google Scholar]
- 47.Schilling S., Kennedy A., Pan S., Jermiin L.S., Melzer R. Genome-wide analysis of MIKC-type MADS-box genes in wheat: pervasive duplications, functional conservation and putative neofunctionalization. New Phytol. 2020;225:511–529. doi: 10.1111/nph.16122. [DOI] [PubMed] [Google Scholar]
- 48.Backhaus A.E., Lister A., Tomkins M., Adamski N.M., Simmonds J., Macaulay I., Morris R.J., Haerty W., Uauy C. High expression of the MADS-box gene VRT2 increases the number of rudimentary basal spikelets in wheat. Plant Physiol. 2022;189:1536–1552. doi: 10.1093/plphys/kiac156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dixon L.E., Greenwood J.R., Bencivenga S., Zhang P., Cockram J., Mellers G., Ramm K., Cavanagh C., Swain S.M., Boden S.A. TEOSINTE BRANCHED1 regulates inflorescence architecture and development in bread wheat (Triticum aestivum) Plant Cell. 2018;30:563–581. doi: 10.1105/tpc.17.00961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dixon L.E., Pasquariello M., Badgami R., Levin K.A., Poschet G., Ng P.Q., Orford S., Chayut N., Adamski N.M., Brinton J., et al. MicroRNA-resistant alleles of HOMEOBOX DOMAIN-2 modifiy inflorescence branching and increase grain protein content of wheat. Sci. Adv. 2022;8 doi: 10.1126/sciadv.abn5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dobrovolskaya O., Pont C., Sibout R., Martinek P., Badaeva E., Murat F., Chosson A., Watanabe N., Prat E., Gautier N., et al. FRIZZY PANICLE drives supernumerary spikelets in bread wheat. Plant Physiol. 2015;167:189–199. doi: 10.1104/pp.114.250043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Poursarebani N., Seidensticker T., Koppolu R., Trautewig C., Gawroński P., Bini F., Govind G., Rutten T., Sakuma S., Tagiri A., et al. The genetic basis of composite spike form in barley and 'Miracle-Wheat'. Genetics. 2015;201:155–165. doi: 10.1534/genetics.115.176628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Y., Du F., Wang J., Wang K., Tian C., Qi X., Lu F., Liu X., Ye X., Jiao Y. Improving bread wheat yield through modulating an unselected AP2/ERF gene. Nat. Plants. 2022;8:930–939. doi: 10.1038/s41477-022-01197-9. [DOI] [PubMed] [Google Scholar]
- 54.Kuzay S., Lin H., Li C., Chen S., Woods D.P., Zhang J., Lan T., von Korff M., Dubcovsky J. WAPO-A1 is the causal gene of the 7AL QTL for spikelet number per spike in wheat. PLoS Genet. 2022;18 doi: 10.1371/journal.pgen.1009747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shaw L.M., Lyu B., Turner R., Li C., Chen F., Han X., Fu D., Dubcovsky J. FLOWERING LOCUS T2 regulates spike development and fertility in temperate cereals. J. Exp. Bot. 2019;70:193–204. doi: 10.1093/jxb/ery350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang X., Jia H., Li T., Wu J., Nagarajan R., Lei L., Powers C., Kan C.-C., Hua W., Liu Z., et al. TaCol-B5 modifies spike architecture and enhances grain yield in wheat. Science. 2022;376:180–183. doi: 10.1126/science.abm0717. [DOI] [PubMed] [Google Scholar]
- 57.Beretta V.M., Franchini E., Ud Din I., Lacchini E., Van den Broeck L., Sozzani R., Orozco-Arroyo G., Caporali E., Adam H., Jouannic S., et al. The ALOG family members OsG1L1 and OsG1L2 regulate inflorescence branching in rice. Plant J. 2023;115:351–368. doi: 10.1111/tpj.16229. [DOI] [PubMed] [Google Scholar]
- 58.Takeda S., Hanano K., Kariya A., Shimizu S., Zhao L., Matsui M., Tasaka M., Aida M. CUP-SHAPED COTYLEDON1 transcription factor activates the expression of LSH4 and LSH3, two members of the ALOG gene family, in shoot organ boundary cells. Plant J. 2011;66:1066–1077. doi: 10.1111/j.1365-313X.2011.04571.x. [DOI] [PubMed] [Google Scholar]
- 59.Yoshida A., Sasao M., Yasuno N., Takagi K., Daimon Y., Chen R., Yamazaki R., Tokunaga H., Kitaguchi Y., Sato Y., et al. TAWAWA1, a regulator of rice inflorescence architecture, functions through the suppression of meristem phase transition. Proc. Natl. Acad. Sci. USA. 2013;110:767–772. doi: 10.1073/pnas.1216151110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boden S.A., McIntosh R.A., Uauy C., Krattinger S.G., Dubcovsky J., Rogers W.J., Xia X.C., Badaeva E.D., Bentley A.R., Brown-Guedira G., et al. Updated guidelines for gene nomenclature in wheat. Theor. Appl. Genet. 2023;136:72. doi: 10.1007/s00122-023-04253-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Borrill P., Ramirez-Gonzalez R., Uauy C. expVIP: a customizable RNA-seq data analysis and visualization platform. Plant Physiol. 2016;170:2172–2186. doi: 10.1104/pp.15.01667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu J., Lawit S.J., Weers B., Sun J., Mongar N., Van Hemert J., Melo R., Meng X., Rupe M., Clapp J., et al. Overexpression of zmm28 increases maize grain yield in the field. Proc. Natl. Acad. Sci. USA. 2019;116:23850–23858. doi: 10.1073/pnas.1902593116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang Y., Yu H., Tian C., Sajjad M., Gao C., Tong Y., Wang X., Jiao Y. Transcriptome association identifies regulators of wheat spike architecture. Plant Physiol. 2017;175:746–757. doi: 10.1104/pp.17.00694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Glenn P., Woods D.P., Zhang J., Gabay G., Odle N., Dubcovsky J. Wheat bZIPC1 interacts with FT2 and contributes to the regulation of spikelet number per spike. Theor. Appl. Genet. 2023;136:237. doi: 10.1007/s00122-023-04484-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Waha K., Dietrich J.P., Portmann F.T., Siebert S., Thornton P.K., Bondeau A., Herrero M. Multiple cropping systems of the world and the potential for increasing cropping intensity. Glob. Environ. Change. 2020;64 doi: 10.1016/j.gloenvcha.2020.102131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jiang G., Koppolu R., Rutten T., Hensel G., Lundqvist U., Tandron Moya Y.A., Huang Y., Rajaraman J., Poursarebani N., von Wirén N., et al. Non-cell-autonomous signaling associated with barley ALOG1 specifies spikelet meristem determinacy. Curr. Biol. 2024 doi: 10.1016/j.cub.2024.04.083. [DOI] [PubMed] [Google Scholar]
- 68.Bray N.L., Pimentel H., Melsted P., Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 69.Pimentel H., Bray N.L., Puente S., Melsted P., Pachter L. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat. Methods. 2017;14:687–690. doi: 10.1038/nmeth.4324. [DOI] [PubMed] [Google Scholar]
- 70.Young M.D., Wakefield M.J., Smyth G.K., Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11:R14. doi: 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hamilton N.E., Ferry M. Ggtern: Ternary diagrams using ggplot2. J. Stat. Softw. 2018;87:1–17. [Google Scholar]
- 72.Abu-Jamous B., Kelly S. Clust: automatic extraction of optimal co-expressed gene clusters from gene expression data. Genome Biol. 2018;19:172. doi: 10.1186/s13059-018-1536-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.International Wheat Genome Sequencing Consortium (IWGSC) Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science. 2018;361 doi: 10.1126/science.aar7191. [DOI] [PubMed] [Google Scholar]
- 76.Brinton J., Simmonds J., Uauy C. Ubiquitin-related genes are differentially expressed in isogenic lines contrasting for pericarp cell size and grain weight in hexaploid wheat. BMC Plant Biol. 2018;18:22. doi: 10.1186/s12870-018-1241-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mauri M., Elli T., Caviglia G., Uboldi G., Azzi M. Proceedings of the 12th Biannual Conference on Italian SIGCHI Chapter. Association for Computing Machinery; 2017. RAWGraphs: A Visualisation Platform to Create Open Outputs. [Google Scholar]
- 78.Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kumar S., Stecher G., Li M., Knyaz C., Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018;35:1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hayta S., Smedley M.A., Clarke M., Forner M., Harwood W.A. An efficient Agrobacterium-mediated transformation protocol for hexaploid and tetraploid wheat. Curr. Protoc. 2021;1 doi: 10.1002/cpz1.58. [DOI] [PubMed] [Google Scholar]
- 81.Smedley M.A., Hayta S., Clarke M., Harwood W.A. CRISPR-Cas9 based genome editing in wheat. Curr. Protoc. 2021;1 doi: 10.1002/cpz1.65. [DOI] [PubMed] [Google Scholar]
- 82.Li G., Kuijer H.N.J., Yang X., Liu H., Shen C., Shi J., Betts N., Tucker M.R., Liang W., Waugh R., et al. MADS1 maintains barley spike morphology at high ambient temperatures. Nat. Plants. 2021;7:1093–1107. doi: 10.1038/s41477-021-00957-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Transcript abundance data for all genes in developing inflorescences of wildtype plants (WT, cv. Paragon).
Transcript abundance and identification tables supporting the differential transcript and whole genome network analyses, and discovery of PDB1 and ALOG1. A) Stage-wise differential transcript analysis. B) Transcript clusters detected in inflorescences of wildtype (cv. Paragon). C) A summary of GO terms detected in stage-specific clusters. D) Quantification of transcripts detected in stage-specific clusters. E) Transcript clusters detected in inflorescences of the photoperiod insensitive NIL. F) Transcript clusters detected in inflorescences of the ppd-1 null NIL. G) Gene network analysis modules. H) Transcript analysis of MADS-box transcription factor genes. I) Differentially expressed MADS-box transcription factors. J) Transcript data of group S bZIP genes in wheat. K) Transcript data of ALOG genes in wheat.
Transcript abundance data for all genes in developing inflorescences of the photoperiod insensitive (Ppd-D1a) NIL.
Transcript abundance data for all genes in developing inflorescences of the ppd-1 null.
Summary of DETs in the photoperiod insensitive (Ppd-D1a) NIL, relative to wild-type, at the VG (A), DR (C), LP (E) and TS (G) stages. Summary of DETs in the ppd-1 null NIL, relative to wild-type, at the VG (B), DR (D), LP (F) and TS (H) stages
Data Availability Statement
-
•
The RNA-seq sequencing data used for the transcriptome analyses performed here have been deposited at NCBI Sequence Read Archive (SRA) and are publicly available. The project number is listed in the key resources table.
-
•
The paper does not report original code.
-
•
Any additional information required to reanalyze the data reported here is available from the lead author upon request.