

Abstract
Mucopolysaccharidosis type I (MPS I) is a rare genetic disorder characterized by the deficiency of the alpha-l-iduronidase enzyme necessary for the degradation of glycosaminoglycans (GAG) in the lysosome. Hurler syndrome is the most severe form of MPS I, manifesting as multiorgan dysfunction, cognitive delay, and death, usually within ten years if left untreated. Hematopoietic stem cell transplantation (HSCT) is the optimal treatment option, providing a permanent solution to enzyme deficiency and halting cognitive decline; however, the HSCT complications transplantation-associated thrombotic microangiopathy (TA-TMA) and graft-versus-host disease (GVHD) are known risk factors for bloodstream infection (BSI). BSI is a serious complication of HSCT, contributing to poor outcomes and transplantation-related morbidity. There are little data evaluating BSI after HSCT in the Hurler syndrome population. We performed a retrospective analysis of patients with Hurler syndrome who underwent HSCT at our center between 2013 and 2020 to determine the incidence of BSI within the first year post-transplantation. Patient BSI data were collected through the first year post-HSCT. Variables including patient demographics and transplantation-related characteristics were collected, including information on BSI and mortality. Twenty-five patients with a total of 28 HSCTs were included in the analysis; the majority (n = 17; 68%) were male, with a median age of 1.1 years (interquartile range, .35 to 1.44 years) at the time of transplantation. The most common graft source was cord blood (n = 15; 54%), followed by bone marrow (n = 13; 46%), with the majority from matched unrelated donors (n = 14; 52%) and mismatched unrelated donors (n = 13; 44%). Sixteen BSIs were diagnosed in 12 patients (48%). Most infections (n = 7; 43.8%) were diagnosed in the first 20 days post-transplantation, with fewer infections observed at later time points. Seven of the 9 Hurler patients diagnosed with TA-TMA (78%) also had a BSI. The incidence rate of BSIs in Hurler patients (n = 12; 48%) was higher than the rates reported in the general pediatric HSCT population at 1-year post-transplantation (15% to 35%). Given the high rate of both TA-TMA and a BSI in Hurler patients, we suspect a possible correlation between the 2. Additionally, due to the time it takes for GAG levels to normalize post-HSCT in Hurler patients, it is reasonable to suspect that the high BSI rates in these patients are linked to their Hurler diagnosis. These findings bring awareness to possible disease-related factors contributing to high BSI rates in the Hurler population post-HSCT.
Keywords: Hurler syndrome, Hematopoietic stem cell, transplantation, Bloodstream infections, CLABSI
INTRODUCTION
Mucopolysaccharidosis type I (MPS I) is a rare genetic disorder characterized by the deficiency of the alpha-l-iduronidase enzyme (IDUA) necessary for the degradation of glycosaminoglycans (GAG) heparan and dermatan sulfate, leading to their accumulation in the lysosome and extracellular matrix. The buildup of GAG in various tissues and organs perturbs molecular processes and results in a wide range of symptoms of varying severity [1,2]. Hurler syndrome is the most severe form of MPS I due to neurologic and non-neurologic symptoms [3,4]. Hurler syndrome manifests during the first year of life with significant abnormalities in stature, coarse facial features, cognitive impairment, and multiorgan dysfunction, resulting in mortality usually within the first 10 years of life in patients without treatment [5,6].
Hematopoietic stem cell transplantation (HSCT) and enzyme-replacement therapy (ERT) are approved treatments for MPS I. However, HSCT is the only treatment that successfully halts multisystem dysfunction by replacing hematopoietic cell lineages with IDUA enzyme-containing cells from a donor, penetrating the blood-brain barrier to stop cognitive decline. In contrast, ERT addresses only non-neurologic symptoms and requires repeated, regular enzyme infusions [7–9]. Previous studies have shown that although GAG levels are rapidly reduced in Hurler patients post-HSCT, achieving normal reference ranges for GAG levels can take time, signifying residual disease activity despite successful transplantation [10,11].
Bloodstream infection (BSI) is a serious complication of HSCT, contributing to poor outcomes and transplantation-related morbidity and mortality, occurring in approximately 15% to 35% of the pediatric HSCT recipients [12–15]. Previous studies in pediatric HSCT recipients have shown strong associations between the development of BSI and several HSCT complications, including transplantation-associated thrombotic microangiopathy (TA-TMA) and graft-versus-host disease (GVHD) [12,16,17]. The intestinal epithelium can be damaged in TA-TMA and GVHD, leading to bacterial translocation and subsequent BSI. The use of immunosuppressants to treat GVHD and eculizumab to treat TA-TMA also may contribute to increased susceptibility to BSI in HSCT recipients [13,18]; however, there is a lack of data on BSI in patients with Hurler syndrome. We conducted a case review of Hurler syndrome patients undergoing HSCT to better understand the incidence of BSI in these patients.
METHODS
We conducted a retrospective case-cohort study of 25 patients with Hurler syndrome who underwent allogeneic HSCT at Cincinnati Children’s Hospital Medical Center between 2013 and 2020. Patient BSI data were collected through the first year post-HSCT. Variables, including patient demographics and transplantation-related characteristics, were collected. Data on BSI and mortality were also collected. Antimicrobial prophylaxis reflected our center’s usual transplantation practice, including levofloxacin for antibacterial prophylaxis through neutrophil engraftment, antifungal prophylaxis with voriconazole or posaconazole, and acyclovir prophylaxis for HSV prophylaxis and pentamidine for Pneumocystis pneumonia prophylaxis. All patients received calcineurin-based GVHD prophylaxis.
The currently accepted clinical criteria were used to diagnose acute GVHD, TA-TMA, and engraftment syndrome in HSCT recipients [16,17,19]. The National Healthcare Safety Network definitions were used for BSI classification [20].
RESULTS
Twenty-five patients with a total of 28 HSCTs were included in the analysis; the majority (n = 17; 68%) were male, with a median age of 1.1 years (IQR, .35 to 1.44 years). All patients underwent allogeneic HSCT. Patient demographics and transplantation-related characteristics are summarized in Table 1. The majority of patients received a myeloablative conditioning regimen (n = 18; 64%), with the rest receiving a reduced-intensity regimen (n = 10; 36%). The most common graft source was cord blood (n = 15; 54%), followed by bone marrow (n = 13; 46%), with the majority from matched unrelated donors (n = 16; 57%) and mismatched unrelated donors (n = 11; 39%). The overall survival (OS) rate at 1 year post-transplantation was 100%. BSI outcomes are summarized in Table 2. Three patients (12%) had 2 HSCTs; none had a BSI after their second transplantation.
Table 1.
Demographic Data of the Study Patients (N = 25)
| Demographic | Value |
|---|---|
| Male sex, n/N (%) | 17/25 (68) |
| Age, yr, median (IQR) | 1.07 (.35–1.44) |
| Total number of transplantations | 28 |
| Patients with 2 transplantations | 3 |
| HLA match (including both bone marrow and cord blood), n/N (%) | |
| Matched unrelated | 16/28 (57) |
| Mismatched unrelated | 11/28 (39) |
| Matched related | 1/28 (4) |
| Stem cell source, n/N (%) | |
| Cord blood | 15/28 (54) |
| Bone marrow | 13/28 (46) |
| Conditioning regimen, n/N (%) | |
| Myeloablative | 18/28 (64) |
| Reduced intensity | 10/28 (36) |
| Conditioning regimen, n/N (%) | |
| Busulfan, cyclophosphamide | 13/28 (46) |
| Campath, fludarabine, melphalan | 8/28 (28) |
| Busulfan, cyclophosphamide, ATG | 3/28 (11) |
| Busulfan, fludarabine, ATG | 3/28 (11) |
| Busulfan, Campath, fludarabine | 1/28 (4) |
| GVHD prophylaxis, n/N(%) | |
| Calcineurin-based | 28/28 (100) |
| GVHD post-transplantation, n/N (%) | |
| GVHD at day 100 | 6/25 (24) |
| GVHD grade 1–2 | 4/6 (57) |
| GVHD grade 3–4 | 2/6 (33) |
| GVHD skin | 5/6 (83) |
| Stage 1–2 | 3/5 (60) |
| Stage 3–4 | 2/5 (40) |
| GVHD gut | 1/6 (17) |
| Stage 1–2 | 1/1 (100) |
| Stage 3–4 | 0/1 (0) |
| Hurler patients with both GVHD and a BSI | 2/6 (33) |
| Grade 1 | 1/2 (50) |
| Grade 4 | 1/2 (50) |
| Other transplantation complications, n/N (%) | |
| Engraftment syndrome | 8/25 (32) |
| Patients with engraftment syndrome and a BSI | 2/8 (25) |
| Patients with engraftment syndrome and TA-TMA | 0/8 (0) |
| TA-TMA | 9/25 (36) |
| Patients with TMA and a BSI | 7/9 (78) |
| Patients with TMA and a BSI treated with eculizumab | 3/7 (43) |
| Patients with GVHD and TA-TMA | 1/7 (14) |
ATG, antithymocyte globulin; MBI, mucosal barrier injury; LCBI, laboratory-confirmed bloodstream infection.
Table 2.
BSI Outcomes
| BSI Information | Value |
|---|---|
| Hurler patients with a BSI, n/N (%) | 12/25 (48) |
| Total BSIs | 16 |
| BSIs by stem cell source, n/N (%) | |
| Cord blood | 10/16 (63) |
| Bone marrow | 6/16 (37) |
| BSIs by conditioning regimen, n/N (%) | |
| MAC | 11/16 (69) |
| RIC | 5/16 (31) |
| BSIs by HLA matching, n/N (%) | |
| Matched related | 1/16 (6) |
| Matched unrelated | 11/16 (69) |
| Mismatched unrelated | 4/16 (25) |
| Infection type, n/N (%) | |
| Monomicrobial infections | 3/16 (81) |
| Polymicrobial infections | 3/16 (19) |
| Organisms identified, n (%) | 20 |
| Staphylococcus aureus | 4 (20) |
| Klebsiella pneumoniae | 3 (15) |
| Escherichia coli | 2 (10) |
| Staphylococcus epidermidis | 2 (10) |
| Actinomyces odontolyticus | 1 (5) |
| Enterococcus faecalis | 1 (5) |
| Moraxella osloensis | 1 (5) |
| Pseudomonas aeruginosa | 1 (5) |
| Coagulase-negative Staphylococcus | 1 (5) |
| Staphylococcus haemolyticus | 1 (5) |
| Streptococcus agalactiae | 1 (5) |
| Streptococcus mitis/oralis | 1 (5) |
| Streptococcus viridans | 1 (5) |
| Time to infection, d, median (IQR) | 33 (6.5–128.5) |
MAC indicates myeloablative conditioning; RIC, reduced-intensity conditioning.
Sixteen BSIs were diagnosed in 12 out of the 25 patients (48%). Nine patients (36%) had 1 BSI, and 3 patients (12%) had more than 1 BSI. Of the 16 infections, 3 (18.8%) were polymicrobial. Thirteen different organisms were identified, with Staphylococcus aureus (n = 4; 25%) and Klebsiella pneumoniae (n = 3; 12%) reported most often. The median time to infection was 52.5 days (IQR, 6.5 to 128.5 days). Seven infections (43.8%) were diagnosed in the first 20 days post-transplantation, with fewer infections being observed at later time points. Further information on the time to diagnosis for BSI is summarized in Figure 1.
Figure 1.
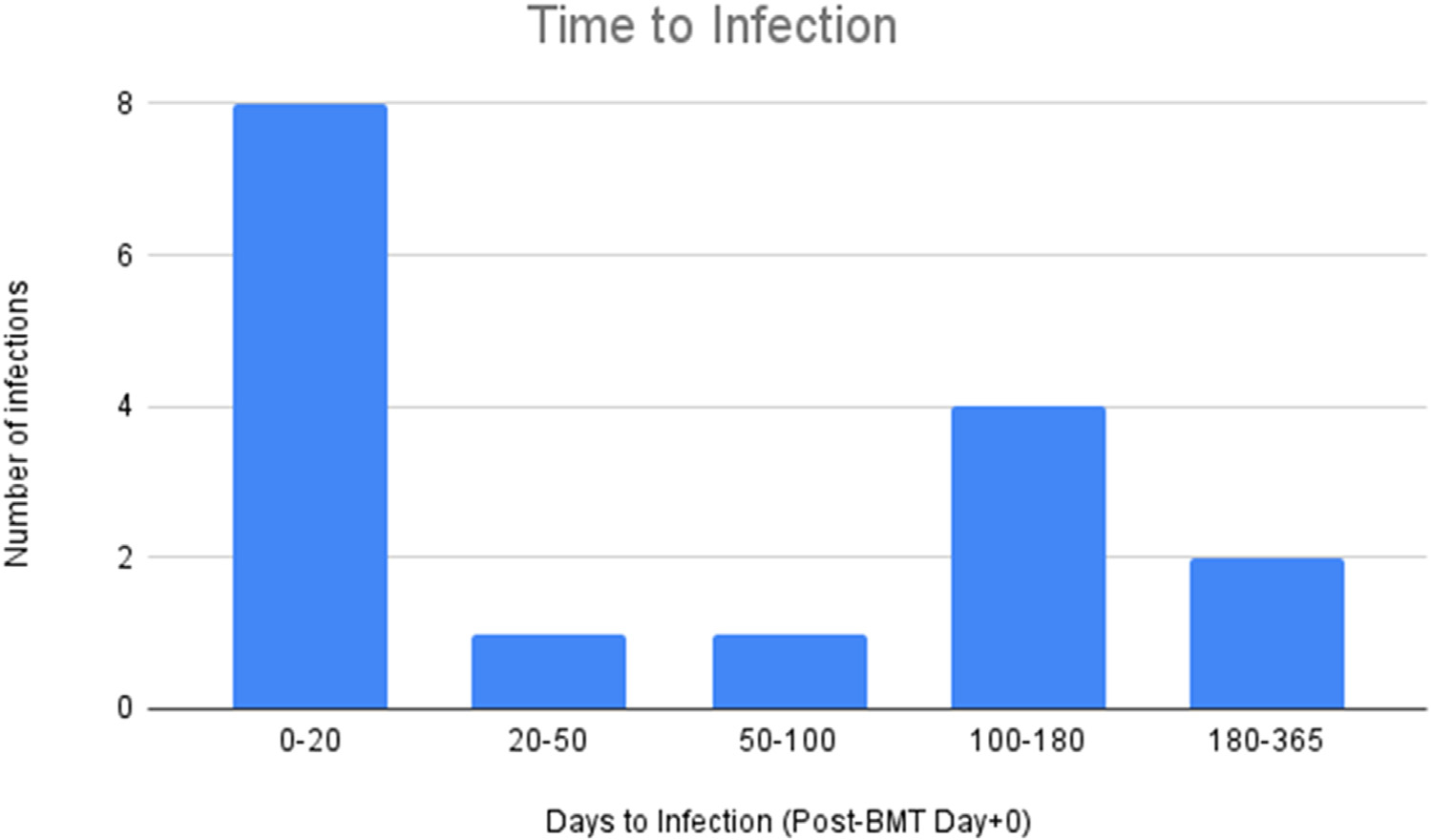
Time to infection post-transplantation.
The majority of BSIs occurred in patients who underwent HSCT with a matched unrelated donor (n = 11; 69%), followed by a mismatched unrelated donor (n = 4; 25%) and a matched related donor (n = 1; 6%). Furthermore, more BSIs were diagnosed in patients who received cord blood grafts (n = 10; 63%) than in patients who received bone marrow grafts (n = 6; 37%). Eleven patients (69%) received a myeloablative conditioning regimen, and 5 (31%) received a reduced-intensity conditioning regimen.
Six of 25 patients (24%) were diagnosed with GVHD at day +100. Of the 6 patients with GVHD, only 2 also had a BSI. Of those 2, 1 patient had grade I GVHD (50%), and the other had grade IV (50%). Additionally, 9 of the 25 (36%) were diagnosed with TA-TMA. Seven of these patients (78%) had both TA-TMA and a BSI, 3 of who were treated with eculizumab. These 3 patients had a BSI diagnosis before their TA-TMA diagnosis and eculizumab therapy. The other 4 patients with TA-TMA and a BSI did not receive eculizumab therapy. Only 1 patient was diagnosed with both GVHD and TA-TMA (14%).
Of the 7 patients diagnosed with TA-TMA and a BSI, 3 had methicillin-sensitive Staphylococcus aureus, 2 had Escherichia coli, and 2 had Klebsiella pneumoniae. The 2 BSIs that contained Escherichia coli and the 3 BSIs from methicillin-sensitive S. aureus were found only in patients who also developed TMA.
DISCUSSION
This is the first study investigating the incidence of BSI in pediatric patients with Hurler syndrome who were treated with HSCT. Compared to previous published studies, the incidence of BSI was higher in this cohort of patients compared to the general population of HSCT recipients [12,13]. Given the high incidence of BSI in patients with Hurler syndrome, it is reasonable to suspect that their higher infection rates can be attributed to some aspect of their Hurler diagnosis.
The patients with Hurler syndrome showed similar trends as seen in pediatric HSCT recipients regarding the high number of patients with both a BSI and TA-TMA [21]. Previous studies from our center found a strong association between BSI and TA-TMA [12,21]. Our study supports these findings and reports a strong association between BSI and TA-TMA in the Hurler patient population. However, the patients from our study were diagnosed with BSI before their TA-TMA diagnosis and eculizumab treatment. This finding warrants further investigation into possible Hurler-related factors contributing to BSI.
When considering the time to infection in Hurler patients, it may be essential to note how long their GAG levels take to reach normal reference levels. Previous research states that immune function is likely to be compromised in patients with MPS-I due to the lysosome’s integral role in many immune processes [4]. GAG levels may take time to normalize post-HSCT; therefore, it is reasonable to suspect that the lysosome’s role in bacterial protection is suboptimal, causing increased susceptibility to infection in patients with Hurler syndrome. Future investigation evaluating GAG levels in Hurler patients at set time points after transplantation would be useful to further evaluate their increased risk of infection.
Our findings highlight the high rate of post-HSCT BSI in the Hurler syndrome population that had not been reported previously. This study has some limitations, however. Given the rarity of Hurler syndrome, the available sample of patients to evaluate is limited. Our data come from a single center, and so our findings might not be generalizable to other institutions with varying prevalence rates HSCT in Hurler patients. Although we found higher rates of BSI in Hurler patients, our study is a retrospective observational study and cannot determine causality. Further investigation and analysis are required to determine the relationship between BSI and Hurler syndrome.
Financial disclosure:
The authors have no financial relationships or other conflicts of interest relevant to this article to disclose.
Footnotes
Conflict of interest statement: There are no conflicts of interest to report.
REFERENCES
- 1.Hampe CS, Eisengart JB, Lund TC, et al. Mucopolysaccharidosis type I: a review of the natural history and molecular pathology. Cells. 2020;9:1838. 10.3390/cells9081838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Campos D, Monaga M. Mucopolysaccharidosis type I: current knowledge on its pathophysiological mechanisms. Metab Brain Dis. 2012;27:121–129. 10.1007/s11011-012-9302-1. [DOI] [PubMed] [Google Scholar]
- 3.Nagpal R, Goyal RB, Priyadarshini K, et al. Mucopolysaccharidosis: a broad review. Indian J Ophthalmol. 2022;70:2249–2261. 10.4103/ijo.IJO_425_22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Archer LD, Langford-Smith KJ, Bigger BW, Fildes JE. Mucopolysaccharide diseases: a complex interplay between neuroinflammation, microglial activation, and adaptive immunity. J Inherit Metab Dis. 2014;37:1–12. 10.1007/s10545-013-9613-3. [DOI] [PubMed] [Google Scholar]
- 5.Beck M, Arn P, Giugliani R, et al. The natural history of MPS I: global perspectives from the MPS I Registry. Genet Med. 2014;16:759–765. 10.1038/gim.2014.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kiely BT, Kohler JL, Coletti HY, Poe MD, Escolar ML. Early disease progression of Hurler syndrome. Orphanet J Rare Dis. 2017;12:32. 10.1186/s13023-017-0583-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hampe CS, Wesley J, Lund TC, et al. Mucopolysaccharidosis type I: current treatments, limitations, and prospects for improvement. Biomolecules. 2021;11:189. 10.3390/biom11020189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wynn RF, Wraith JE, Mercer J, et al. Improved metabolic correction in patients with lysosomal storage disease treated with hematopoietic stem cell transplant compared with enzyme replacement therapy. J Pediatr. 2009;154:609–611. 10.1016/j.jpeds.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 9.Guffon N, Pettazzoni M, Pangaud N, et al. Long-term disease burden post-transplantation: three decades of observations in 25 Hurler patients successfully treated with hematopoietic stem cell transplantation (HSCT). Orphanet J Rare Dis. 2021;16:60. 10.1186/s13023-020-01644-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuiper GA, van Hasselt PM, Boelens JJ, Wijburg FA, Langereis EJ. Incomplete biomarker response in mucopolysaccharidosis type I after successful hematopoietic cell transplantation. Mol Genet Metab. 2017;122:86–91. 10.1016/j.ymgme.2017.05.009. [DOI] [PubMed] [Google Scholar]
- 11.Minami K, Morimoto H, Morioka H, et al. Pathogenic roles of heparan sulfate and its use as a biomarker in mucopolysaccharidoses. Int J Mol Sci. 2022;23:11724. 10.3390/ijms231911724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grossmann L, Alonso PB, Nelson A, et al. Multiple bloodstream infections in pediatric stem cell transplant recipients: a case series. Pediatr Blood Cancer. 2018;65:e27388. 10.1002/pbc.27388. [DOI] [PubMed] [Google Scholar]
- 13.Dandoy CE, Ardura MI, Papanicolaou GA, Auletta JJ. Bacterial bloodstream infections in the allogeneic hematopoietic cell transplant patient: new considerations for a persistent nemesis. Bone Marrow Transplant. 2017;52:1091–1106. 10.1038/bmt.2017.14. [DOI] [PubMed] [Google Scholar]
- 14.Dandoy CE, Kim S, Chen M, et al. Incidence, risk factors, and outcomes of patients who develop mucosal barrier injury-laboratory confirmed bloodstream infections in the first 100 days after allogeneic hematopoietic stem cell transplant. JAMA Netw Open. 2020;3:e1918668. 10.1001/jamanetworkopen.2019.18668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ustun C, Young JH, Papanicolaou GA, et al. Bacterial blood stream infections (BSIs), particularly post-engraftment BSIs, are associated with increased mortality after allogeneic hematopoietic cell transplantation. Bone Marrow Transplant. 2019;54:1254–1265. 10.1038/s41409-018-0401-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spitzer T Engraftment syndrome following hematopoietic stem cell transplantation. Bone Marrow Transplant. 2001;27:893–898. 10.1038/sj.bmt.1703015. [DOI] [PubMed] [Google Scholar]
- 17.Dandoy CE, Rotz S, Badia Alonso P, et al. A pragmatic multi-institutional approach to understanding transplant-associated thrombotic microangiopathy after stem cell transplant. Blood Adv. 2021;5:1–11. 10.1182/bloodadvances.2020003455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.El-Bietar J, Warren M, Dandoy C, et al. Histologic features of intestinal thrombotic microangiopathy in pediatric and young adult patients after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2015;21:1994–2001. 10.1016/j.bbmt.2015.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Filipovich AH, Weisdorf D, Pavletic S, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: I. Diagnosis and Staging Working Group report. Biol Blood Marrow Transplant. 2005;11:945–956. [DOI] [PubMed] [Google Scholar]
- 20.CDC- National Healthcare Safety Network. Bloodstream infection event. January 2023. Available at: https://www.cdc.gov/nhsn/pdfs/pscmanual/4psc_clabscurrent.pdf. Accessed May 9, 2023.
- 21.Dandoy CE, Haslam D, Lane A, et al. Healthcare burden, risk factors, and outcomes of mucosal barrier injury laboratory-confirmed bloodstream infections after stem cell transplantation. Biol Blood Marrow Transplant. 2016;22:1671–1677. 10.1016/j.bbmt.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]