

Abstract
The emergence and rapid spread of SARS-CoV-2 prompted the global community to identify innovative approaches to diagnose infection and sequence the viral genome because at several points in the pandemic positive case numbers exceeded the laboratory capacity to characterize sufficient samples to adequately respond to the spread of emerging variants. From week 10, 2020, to week 13, 2023, Slovenian routine complete genome sequencing (CGS) surveillance network yielded 41 537 complete genomes and revealed a typical molecular epidemiology with early lineages gradually being replaced by Alpha, Delta, and finally Omicron. We developed a targeted next-generation sequencing based variant surveillance strategy dubbed Spike Screen through sample pooling and selective SARS-CoV-2 spike gene amplification in conjunction with CGS of individual cases to increase throughput and cost-effectiveness. Spike Screen identifies variant of concern (VOC) and variant of interest (VOI) signature mutations, analyses their frequencies in sample pools, and calculates the number of VOCs/VOIs at the population level. The strategy was successfully applied for detection of specific VOC/VOI mutations prior to their confirmation by CGS. Spike Screen complemented CGS efforts with an additional 22 897 samples sequenced in two time periods: between week 42, 2020, and week 24, 2021, and between week 37, 2021, and week 2, 2022. The results showed that Spike Screen can be applied to monitor VOC/VOI mutations among large volumes of samples in settings with limited sequencing capacity through reliable and rapid detection of novel variants at the population level and can serve as a basis for public health policy planning.
Keywords: SARS-CoV-2, COVID-19, next-generation sequencing, complete-genome sequencing, molecular surveillance, spike gene
Introduction
After the first reported case of SARS-CoV-2 in Slovenia—a central European country with a population of 2.1 million—on 4 March 2020, by November 2020 the number of cases had skyrocketed, overwhelming the diagnostic capabilities of laboratories. According to the World Health Organization (WHO), between 4 March 2020 and 19 April 2023, there were 1342 787 laboratory-confirmed cases of COVID-19 in Slovenia and 9267 deaths. At the peak of incidence in early 2022, Slovenia faced up to 28 000 new cases weekly (https://covid19.who.int/region/euro/country/si, accessed 6 November 2023). Although we were able to increase diagnostic capacities, the emergence of SARS-CoV-2 variants revealed another deficit: the need for efficient and rapid genome sequencing.
Although the SARS-CoV-2 virus with proofreading mechanisms enhances genome fidelity, mutations are a natural part of the replication cycle of any virus [1]. Mutations in the SARS-CoV-2 genome have led to different variants with different properties, including increased transmissibility, virulence, and immune or vaccine escape [2–6]. The WHO’s guidance for surveillance of SARS-CoV-2 variants categorizes the public health risks of known and emerging variants of interest (VOIs) and variants of concern (VOCs) as increased transmissibility, more severe clinical course, failure of detection by diagnostic tests, escape from natural or vaccine-derived immunity, and reduced susceptibility to therapeutic agents [7]. This guidance also highlights the importance of genomic surveillance and encourages countries with limited sequencing capacity to facilitate access to regional or international sequencing collaborators or to increase the capacity of existing infrastructure.
Real-time surveillance of SARS-CoV-2 variants is important for understanding the potential public health impact and evolution of the virus. This surveillance involves next-generation sequencing (NGS) of clinical samples, which can provide information on the prevalence and spread of different variants [8]. Data generated from variant surveillance can inform public health policy and interventions such as vaccine development and distribution to control the spread of the virus and mitigate its impact on global health, as was the case with previous epidemic infectious diseases caused by viruses such as human immunodeficiency virus (HIV), Ebola virus (EBOV), Zika virus (ZIKV), and monkeypox virus (MPXV) [9–15]. The main objectives of routine surveillance are to detect low-level variants circulating in the population and to monitor the relative prevalence of variants in different times and geographic areas [7]. However, the outcome of surveillance efforts largely depends on the sampling strategy [16]. The European Centre for Prevention and Disease Control has provided guidance on genomic SARS-CoV-2 surveillance, recommending sufficient sample size to ensure detection limits at 1, 2.5, and 5% prevalence of a given variant within a time unit [17].
The target detection limit, combined with the number of positive cases, drove the need for innovative cost-, logistics-, and labour-effective strategies for screening emerging variants. Routine genomic surveillance of the COVID-19 epidemic by complete-genome sequencing (CGS) of SARS-CoV-2 was unable to keep pace with the number of positive cases that would need to be characterized each week to be able to confidently reflect the presence/absence of monitored VOC/VOI variants.
As a complementary strategy to CGS for VOC/VOI detection, we developed a capacity increasing, two-step sequencing strategy dubbed Spike Screen. Here we demonstrate Spike Screen’s reliability, cost-effectiveness, and feasibility for real-time monitoring of the emergence and abundance of VOCs/VOIs in the population.
Materials and Methods
Sample collection and SARS-CoV-2 detection
Nasopharyngeal swab samples were received from general population COVID-19 testing points, and from patients treated at the Ljubljana University Medical Centre and regional hospitals as part of routine testing for SARS-CoV-2 at the Institute of Microbiology and Immunology, Faculty of Medicine, University of Ljubljana, Slovenia. We included all samples collected between 4 March 2020 and 31 March 2023. After April 2023, surveillance activities at the national level were significantly reduced due to the improvement of the epidemiological situation in the country.
RNA was isolated and tested with specific assays, based on real-time reverse transcription polymerase chain reaction (rtRT-PCR) for SARS-CoV-2 RNA, as previously published [18–20]. A flowchart outlining the steps in Spike Screen sequencing protocol is shown in Fig. 1.
Figure 1.
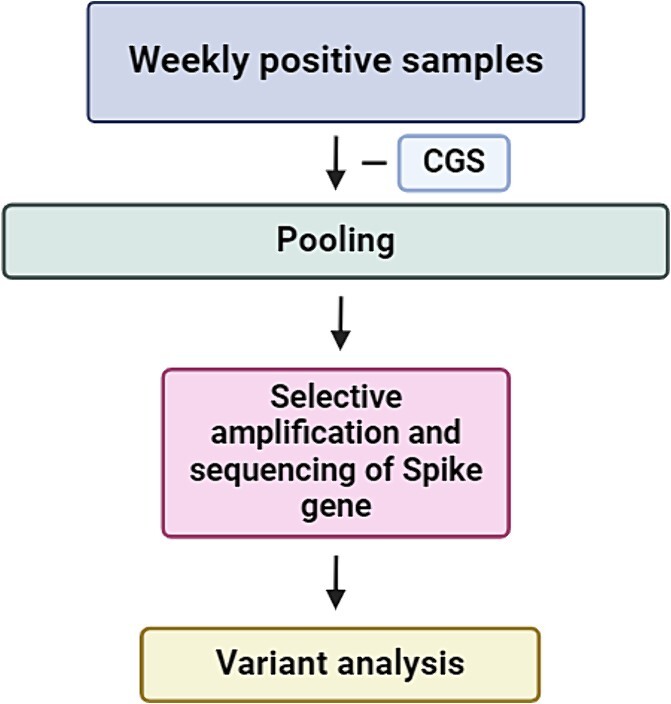
Spike Screen sequencing protocol.
Routine CGS genomic surveillance
After removal of duplicate and follow-up samples, the maximum number of positive samples was selected for routine CGS genomic surveillance according to the capacity of the MiSeq or NextSeq 550 sequencers (both Illumina, San Diego, CA). At time points when the number of total positive SARS-CoV-2 samples exceeded available sequencing platforms capacities, individual samples were randomly selected for CGS surveillance to minimize sampling bias [21]. The cycle threshold (Ct) value cut-off was set at 29 cycles for inclusion.
Library preparation for CGS
After single-stranded cDNA synthesis with Super Script IV reverse transcriptase (Thermo Fisher Scientific, Waltham, MA), whole genome spanning PCR amplicons were prepared and cleaned using the ARTIC V2 primer scheme, according to the nCoV-2019 sequencing protocol (https://www.protocols.io/view/ncov-2019-sequencing-protocol-v2-bp2l6n26rgqe/v2?version_warning=no, accessed 6 November 2023). DNA concentration was measured using the Qubit dsDNA High Sensitivity Assay Kit on a Qubit 3.0 (Thermo Fisher Scientific). We used cleaned PCR amplicons for NGS library preparation using the Nextera XT library preparation kit or the Illumina COVIDSeq Test protocol (both Illumina, San Diego, CA). After measuring library concentration (Qubit dsDNA HS Assay Kit) and fragment size (Agilent High Sensitivity DNA Kit, Agilent Technologies, Santa Clara, CA), sequencing was performed using MiSeq Reagent Kit v3 (600 cycles) on a MiSeq sequencer or NextSeq 500/550 High Output Kit v2 (300 cycles) on a NextSeq 550 sequencer (both Illumina).
Selective amplification and sequencing of the spike gene in sample pools (Spike Screen)
In addition to the number of positive samples already selected for routine CGS surveillance at any given time, a larger subsample of rtRT-PCR-confirmed clinical samples was selected that would not have been included because of sequencer capacity limitations. In total, 29 897 samples distributed between 825 pools were additionally included. Samples were selected based on available metadata to minimize inclusion bias. The selected samples were parcelled into individual pools according to their geographical origin, based on the distribution of Slovenian municipalities, and according to their respective Ct values. Pooling of samples with respect to their Ct values was performed in such a manner that the standard deviation of Ct values in each pool did not exceed 1. This ensured a comparable read depth for each pooled sample.
A pool covered 1 to 2 days of the epidemic and usually contained 40 samples per pool (range 7–53). Pool sizes were designed to allow detection of circulating variants at <5% prevalence based on the total number of positive SARS-CoV-2 cases for each week in the timeframe. The median number of samples sequenced per week was 576 (range 17–3076). The strategy was implemented in two time periods, from week 42, 2020, to week 24, 2021, and from week 37, 2021, to week 2, 2022, when the numbers of positive samples were greatest or when extended monitoring was required due to the emergence of a new VOC (Alpha and Omicron, respectively).
Library preparation for Spike Screen
Library preparation for selective amplification of the spike gene followed the same procedure as library preparation for amplicon-based CGS, with minor modifications. Selective amplification of the spike gene was performed using 14 oligonucleotide pairs (from pair 71 to pair 84) from the Artic nCoV-2019 primer scheme version V3. The selected oligonucleotides cover the region from nucleotides 21 357 to 25 673 of the SARS-CoV-2 genome (NCBI accession number NC_045512.2).
Data analysis
The in-house developed bioinformatics pipeline consisted of reads trimming using BBDuk (v38.96), quality control of reads using FastQC (v0.11.5) and reads mapping to the Wuhan Hu-1 reference genome (NCBI accession number NC_045512.2) using BWA-MEM (v0.7.17-r1188) [22]. Subsequent data processing was performed using Samtools (v1.9) [23] and variant calls were generated with iVAR (v1.3.1) [24]. We used SnpEff (v5.0e) for variant annotation and effect prediction [25]. The threshold for allele frequency cut-off was set at 1%.
We assigned lineages using Pangolin (v4.2) [26]. Mutations were classified as characteristic if their prevalence in a defined lineage was >75% [27]. To assess the effectiveness of proposed approach on Omicron lineage, we extracted the mutational profile of each detected Omicron sub lineage present in Slovenia up to June 2023 and performed overlap analysis to determine the uniqueness of each mutation profile. Formal data analysis was performed using R statistical software (version 4.2.3, R Foundation for Statistical Computing, Vienna, Austria).
Data and code availability
The genomic data used in this study is available at GISAID: EPI_SET_230505ny, https://doi.org/10.55876/gis8.230505ny. The code and ready-to-deploy pipeline is available on GitHub at https://github.com/IMIMF-UNILJSI/scov2-spikeScreen,
Results
Overview of CGS-based monitoring of SARS-CoV-2 variants
From early March 2020 to late March 2023, we sequenced 41 537 complete SARS-CoV-2 genomes. The temporal distribution of specific variants determined by CGS served as a basis for evaluating Spike Screen strategy, presented in Fig. 1. The relative distribution of chronological variants and the absolute number of sequenced SARS-CoV-2 genomes, aggregated at the VOC/VOI level, is shown in Fig. 2.
Figure 2.
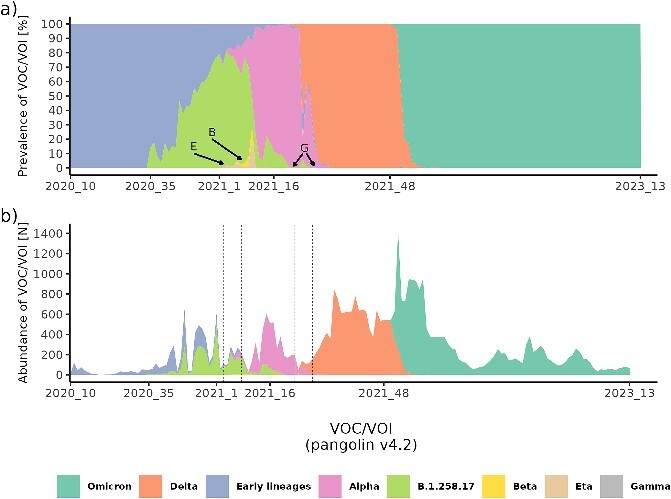
Temporal lineage distribution from the beginning of March 2020 to the end of March 2023 (161 weeks) as determined by CGS. The annotation of week on the x-axis corresponds to the points of transition from the predominance of the previous to the next VOC/VOI and the delineation of the entire timeframe of CGS surveillance of SARS-CoV-2 variants. Panel (A) shows the temporal relative prevalence of major VOCs/VOIs, and panel (B) shows the absolute abundance of the major VOCs/VOIs. In panel (A) rare variants Eta (E), Beta (B), and Gamma (G) are highlighted due to low prevalence. The corresponding temporal positions of these variants in panel (B) are denoted by dashed lines.
At the beginning of the pandemic, the most abundant early lineages were B.1.160 (21.8% of all early lineages), B.1.258 (16.0% of all early lineages), and B.1.1.70 (14.9% of all early lineages). The remaining 47.3% of early lineages were distributed among 74 distinct lineages, with prevalence ranging from 0.03 to 9.0%.
The emergence of the B.1.258.17 variant marked the first major wave of infections in Slovenia. The transition to the next wave was dominated by the emergence of Alpha. We also detected a few cases of Beta, Eta, and Gamma, which appeared in Slovenia only as individual cases, or small clusters after being imported by travellers. In the Delta wave, we detected 88 sub-lineages of Delta, with AY.43 (34.4% of all Deltas), AY.122 (11.2% of all Deltas), and AY.98.1 (9.4% of all Deltas) being the most abundant. The remaining 45.0% of Delta was distributed among 85 distinct Delta sub-lineages. In the last and largest wave, characterized by a rapid emergence of Omicron, the most abundant Omicron sub-lineages were BA.1.1 (34.4%), BA.2 (13.2%), and BA.1 (5.8%). Because the wide distribution of Omicron resulted in more fragmentation of lineages, the remaining 46.6% were distributed among 302 sub-lineages. Table 1 provides an overview of major lineages, as determined by routine CGS surveillance.
Table 1.
Abundance, percentage, and characteristic spike gene mutations of each early lineage/VOI/VOC detected
| VOC/VOI | Week of detection | Count [n] | Percentage [%] | Characteristic spike gene mutations |
|---|---|---|---|---|
| Early lineages | 2020_10 | 3263 | 7.8 | D614G, S477N, A222V |
| B.1.258.17 | 2020_35 | 3651 | 8.8 | L189F, N439K, D614G, H69_V70del |
| Alpha | 2021_1 | 3912 | 9.4 | N501Y, A570D, D614G, P681H, T716I, S982A, D1118, H69_V70del, Y145del |
| Beta | 2021_6 | 22 | 0.05 | D80A, D215G, K417N, E484K, N501Y, D614G, A701V |
| Eta | 2021_3 | 29 | 0.07 | Q52R, A67V, E484K, D614G, Q677H, F888L |
| Gamma | 2021_22 | 3 | 0.007 | L18F, T20N, P26S, D138Y, R190S, K417T, E484K, N501Y, D614G, H655Y, T1027I, V1176F |
| Delta | 2021_16 | 13 184 | 31.7 | T19R, G142D, R158G, L452R, T478K, D614G, P681R, D950N |
| Omicron | 2021_48 | 17 473 | 42.1 | T19I, L24S, G124D, V213G, G339, S371F, S373P, S375F, T376A, D405N, R408S, K417N, N440K, S477N, T478K, E484A, Q493R, Q498R, N501Y, Y505H, D614G, H655Y, N679K, P681H, N764K, D796Y, Q954H, N969K, H69_V70del |
Overview of variant monitoring by Spike Screen
We sequenced 825 pools of amplified spike gene, encompassing a total of 29 216 samples. This approach allowed us to expand the surveillance of circulating variants by an additional 22 897 samples that were not included in routine CGS surveillance (Fig. 3). This samples expansion along with the incorporation of a higher throughput sequencer in the surveillance workflow, secured reliable detection of any circulating variant in the population during a given timeframe, at theoretical prevalence levels of 2.5% or even 1.0%. The difference of 6319 samples represents the proportion of samples that were sequenced with both approaches. The Spike Screen strategy allowed us to increase the numbers of monitored samples by an additional 55.0%, while utilizing only ≈3% of additional resources.
Figure 3.
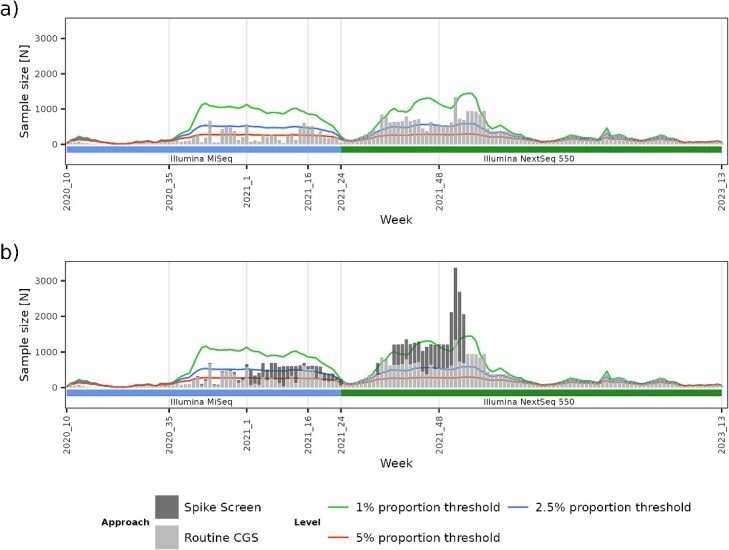
Overview of the increase in SARS-CoV-2 sequencing volume by adding a higher throughput instrument and the contribution of the Spike Screen strategy to the overall CGS samples monitored for VOCs/VOIs. Panel (A) shows the sequencing volume attained with routine CGS per week. Coloured lines represent sample size threshold requirements to secure circulating variant detection at 1, 2.5, and 5% prevalence. Panel (B) shows the contribution of the Spike Screen capacity expansion compared with the baseline CGS.
Sequencing of the pooled spike gene resulted in excellent coverage depth along the entire stretch of the gene (Fig. 4). The sharp decrease in coverage depth at nucleotide position 21 764 was due to deletion H69_V70del. Global median coverage was 6135 reads per position (range 0–36 866), and the median coverage for each individual position was 4996 reads (range 273–8059).
Figure 4.
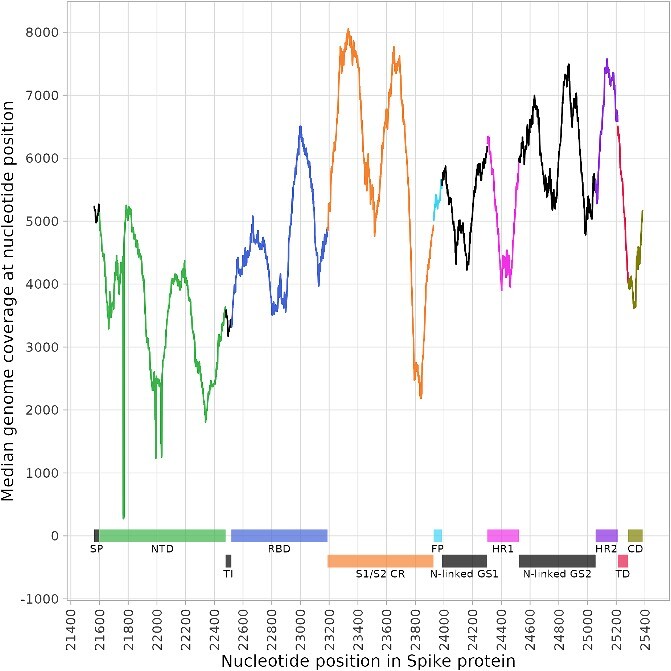
Median sequencing coverage per individual nucleotide position in the spike protein gene. The x-axis represents global nucleotide coordinates, based on the SARS-CoV-2 reference genome: NC_045512.2. SP = signal peptide, NTD = N-terminal domain, TI = trimer interface, RBD = receptor-binding domain, S1/S2 CR = S1/S2 cleavage region, FP = fusion peptide, N-linked GS1 = N-linked glycosylation Site 1, HR1 = heptapeptide repeat sequence 1, N-linked GS2 = N-linked glycosylation site 2, HR2 = heptapeptide repeat sequence 2, TD = terminal domain, CD = cytoplasm domain.
Monitored frequencies show excellent agreement with the population prevalence of VOCs/VOIs
We can observe in Fig. 5 that the frequencies of the characteristic mutations in the spike gene of lineage B.1.258.17 (L189F, N439K, and V772I) correspond almost perfectly with the variant prevalence in the population as determined by CGS. The frequency of mutation D614G remains largely constant since this mutation was already found in earlier lineages. A similar effect is observed in the emergence of Alpha, where characteristic spike mutations follow almost identical dynamics to the prevalence of Alpha in the population. We can also observe the consistent presence of D614G mutation, which has been carried over into the emergence of Delta. On the other hand, during the emergence of Delta, we can also observe a decrease in the frequency of deletion H69_V70del, which is not a characteristic mutation of Delta. We detected the emergence of Delta by a steep increase in frequencies of mutations T19R, T478K, and L452R, which corresponded to the population dynamics of Delta. We can observe the fixation of the D950N mutation with a peak prevalence of ≈70% in Delta and a lower but consistent fixation of G142D with a peak prevalence of ≈20% in Delta. Other characteristic mutations in the spike gene of Delta were fixed at 100%, along with mutation D614G. Tracking the appearance of Omicron, we observed the lagging G142D mutation and fixed D614G and T478K mutations. The remaining characteristic mutations in the spike gene of Omicron responded uniformly, with the exception of deletion H69_V70del, which along with the G142D mutation exhibited the earliest signal for the presence of Omicron in the population before it was detected by routine CGS. The fluctuating frequency of deletion H69_V70del could be explained by several factors, including the sampling strategy, the sporadic presence of deletion H69_V70del in each Omicron sub lineage, the lower coverage of N-terminal domain (NTD) region harbouring this deletion and the co-occurrence of different Omicron variants that do not carry the deletion in question.
Figure 5.
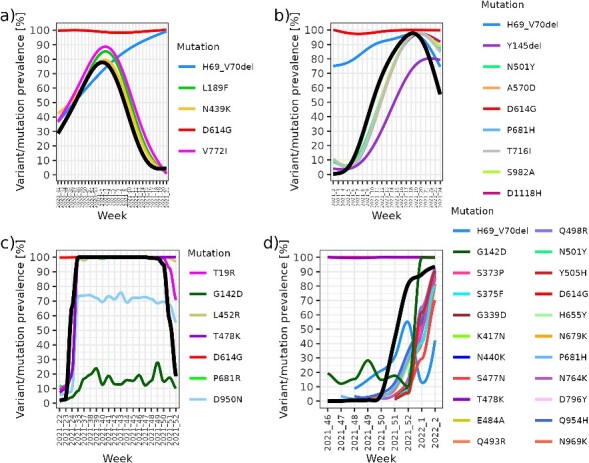
Relationships between monitored mutations and population prevalence of (A) B.1.258.17, (B) Alpha, (C) Delta, and (D) Omicron. The coloured lines represent the monitored prevalence of the characteristic mutations (obtained with the Spike Screen strategy), and the bolded black line represents the prevalence in the population (obtained with routine CGS monitoring). Early signal from Spike Screen characteristic mutation monitoring can be observed, prior to CGS confirmed occurrence of lineage as above-threshold (5%) frequency of observed mutations (panels B, C, and D).
Detection of low-level variants at high-predominance and high-variability settings with Spike Screen
Further investigation of the reliability of the Spike Screen strategy led us to examine the relationship between pools of known lineage composition and the monitored frequencies of characteristic mutations in the spike gene in these pools. Mutations are defined as nonspecific if they were harboured by multiple lineages in the same pool, and they cannot act as a ‘marker’ mutation that allows discrimination between lineages (e.g. mutation D614G, deletions H69_V70del and Y145del).
The pool presented in Fig. 6A consists of 22/25 (88.0%) samples of the B.1.258.17 lineage, and 3/25 (12%) of the B.1.146, B.1.258.24, and Eta lineages. We can observe that the frequencies of V772I and N439K (≈85%) are consistent with the percentage of the B.1.258.17 variant in the pool. The tracking of characteristic mutations enabled us to detect even low-percentage lineages (single sample) as seen for Eta (Fig. 6B). The detection of the B.1.146 and B.1.258.24 lineages, which were also present in the pool, is less straightforward because they harbour only the D614G mutation in the spike gene. The presence of such lineages can be inferred as the difference between the percentage of the majority lineage B.1.258.17 (≈85%) and the observed 100% frequency of the D614G mutation in the pool if they all harbour such a mutation.
Figure 6.
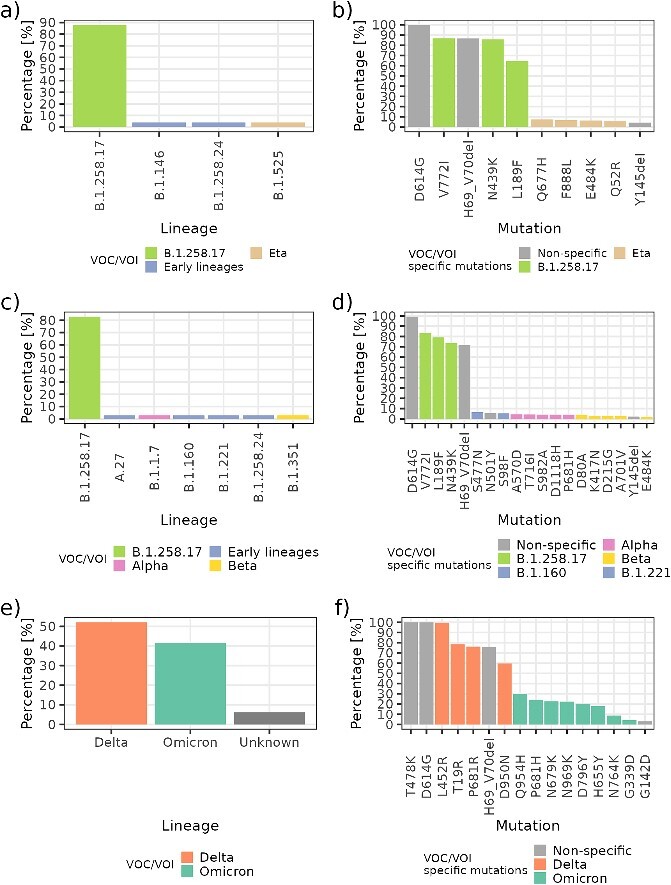
Relationship between the actual percentage of lineages in the pool and the detected frequency of characteristic mutations. The panels on the left (A, C, E) show the known lineage composition of the pool, and the panels on the right (B, D, F) show the detected frequency of characteristic mutations in the sequence. Low-level characteristic mutations of the Eta variant are detected by the Spike Screen strategy, as seen in (A) and (B). A co-detection of low-level characteristic mutations of Alpha and Beta, along with characteristic mutations (S477N, S98F) of the B.1.160 lineage and B.1.221 lineage, can be observed in (C) and (D). A slow transition to the predominance of Omicron versus Delta can be observed in panels (E) and (F).
We can also observe that co-detection of multiple low-percentage lineages can be discerned with this strategy. The pool presented in Fig. 6C consists of 29/35 (82.6%) of the B.1.258.17 lineage, and 6/35 (17.4%) of the B.1.258.24, B.1.160, B.1.211, A.27, Alpha, and Beta lineages. We detected all characteristic mutations for both Alpha and Beta. In this example, we were able to distinguish the characteristic mutations for the B.1.160 lineage (S477N) and the B.1.211 lineage (S98F). However, we were unable to detect characteristic mutations of the A.27 lineage (L18F, L452R, A653V, H655Y, D796Y, and G1219V).
The proposed strategy for variant monitoring also worked well in monitoring Omicron. Figure 6E shows a pool composed of 25/48 (52.0%) Delta, 20/48 (41.7%) Omicron, and 3/48 (6.3%) samples for which no CGS lineage information was included a priori. One can observe the emergence of Omicron as well as the actual ratio of Delta to Omicron in the pool (70:30). Spike Screen allowed not only the identification of a new variant in population, but also tracking of the progression and its relative abundance.
Spike Screen reliably predicts lineage prevalence from frequencies of characteristic mutations
To demonstrate that the Spike Screen strategy can reliably assess the percentage of a given VOC/VOI based on the characteristic mutation frequency, we performed linear regression modelling for the P681H mutation and the known percentage of Alpha in the pools presented (Fig. 7). A perfectly linear relationship can be observed with coefficient β = 0.83 (P < 0.001), meaning that a 1% increase in Alpha in the pool is associated with a 0.83% increase in P681H mutation frequency. Based on the adjusted R2 metric, the percentage of specific VOCs/VOIs explains 83.0% of the variability in P681H mutation frequency. This result indicates that there are most likely very few additional factors other than the VOC/VOI percentage in the pool that contribute to the dynamics of a mutation frequency. For two outliers that were not consistent with the calculated model, the discrepancy was attributed to a higher variance in the Ct values of samples included in these two pools.
Figure 7.
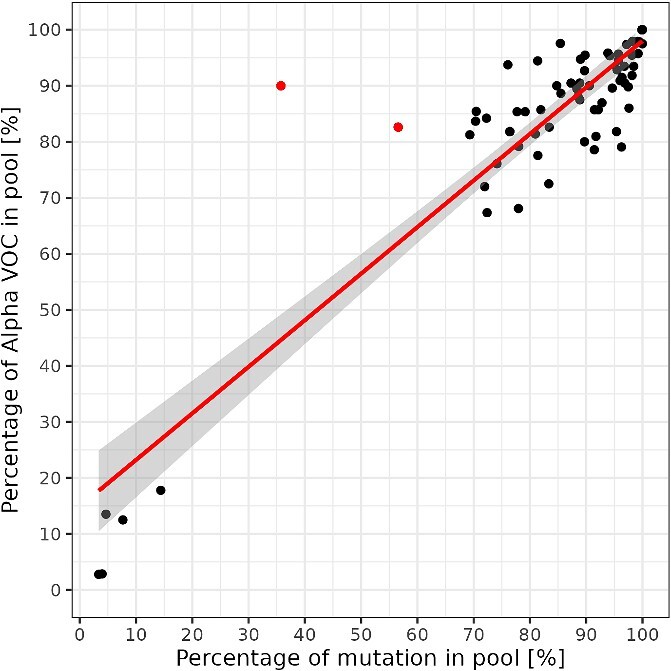
Relationship between characteristic mutation frequency and the known percentage of Alpha in a pool. The red regression line indicates the significant linear relationship between the characteristic Alpha mutation S:P681H frequency and actual Alpha prevalence. Two red dots in the upper middle part represent the outliers based on Cook’s distance. The deviation from the regression line in these two pools was due to samples with high variability of Ct values in these pools.
Spike Screen reliably infers lineage presence from the frequency of the characteristic mutations
In the case presented in Fig. 8A, for 4/40 (10.0%) samples, the CGS-determined lineage was unknown. Based on the portion of samples with known lineages, one would not expect any mutations other than the ones harboured by B.1.258.17, B.1.258, and B.1 lineages. However, we detected characteristic mutations for Alpha, including (S982A, T716I, D1118H, P681H, and A570D), whose frequencies correspond to the percentage of unknown sample lineages. Based on the mutation frequencies, it can reliably be determined that at least one unknown sample in the pool belongs to Alpha.
Figure 8.
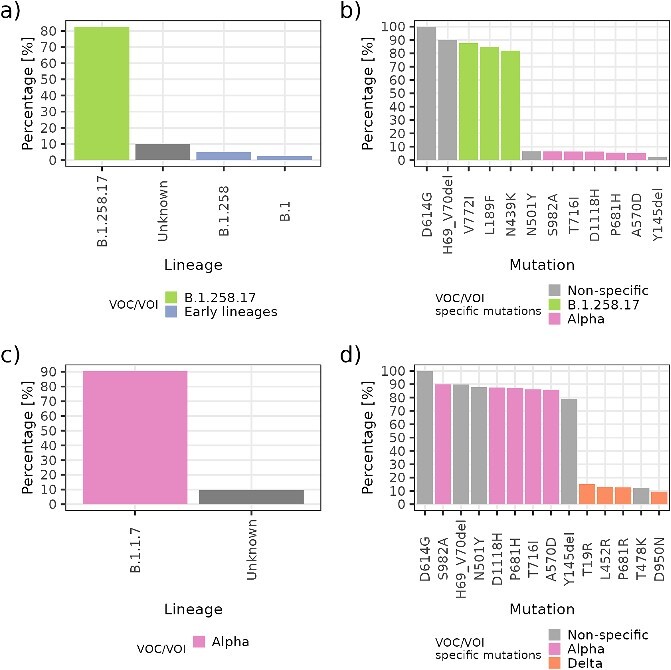
Evaluation of unknown lineage presence in the pool. The left panels, (A) and (C), show the known lineage composition of the pool, and the right panels, (B) and (D), show the detected frequency of characteristic mutations in the sequence. The characteristic mutations of Alpha and characteristic mutations of Delta can be observed in unknown samples.
Figure 8C further presents an example with similar proportions of known lineages as in the previous case, with 20/22 (91%) Alpha VOCs and 2/22 (9%) unknown lineages. One can observe the detection of ‘specific’ characteristic mutations for Delta (specific with respect to Alpha, where the emergence cannot be distinguished by the presence of D614G, N501Y, H69_V70del, and Y145del deletions because they are also harboured by Alpha). Based on the mutation frequencies, this result indicates that the two samples with unknown lineage most likely belong to Delta.
Predicted effectiveness of Spike Screen efficacy in detecting Omicron sub lineages
To assess the likely efficacy of Spike Screen in distinguishing the Omicron sub lineages, we performed a comprehensive evaluation of the mutation profile of 18 063 Omicron sequences from Slovenia. We described 370 Omicron sub lineages that harboured 273 distinct spike mutations. We found a specific mutation profile for each Omicron sub lineage (unique distribution of mutations), indicating a theoretical ability of Spike Screen to reliably discriminate between sub lineages based on specific spike mutations (Fig. 9). Screening for specific spike mutations revealed an additional advantage of monitoring the entire spike gene as opposed to monitoring a specific genomic region such as the receptor-binding domain (RBD). Although the RBD contained 63/273 (23.1%) of distinct mutations present in Omicron sub lineages, 10 of these mutations were clonal (present in at least 90% of all Omicron sub lineages) and could not serve as ‘marker’ mutations. In contrast, 124/273 (45.4%) of distinct mutations in the Omicron sub lineages and only 6 clonal mutations were identified in the NTD, allowing a higher resolution of the corresponding mutations. We also detected several lineage-defining mutations in the S1/S2 cleavage region and in the remaining spike structural proteins.
Figure 9.

Mutational profile of 370 distinct Omicron sub lineages present in the Slovenian population up to June 2023. Overlap analysis reveals sufficient differences in the mutations profiles to allow for reliable distinction between sub lineages using Spike Screen.
Discussion
In this study, we present an original variant surveillance strategy using sample pooling and selective amplification coupled with resourceful use of bioinformatic genomic variant analysis called Spike Screen. The presented framework provides reliable and timely information on circulating variants to inform policy makers on pandemic progression, using a bioinformatics pipeline specifically designed and implemented for this task, while keeping it general enough to capture variants characterized by novel mutations. We have shown that complementing CGS surveillance with Spike Screen enables detection of novel variants at lower population prevalence. This can increase performance, and cost-effectiveness of NGS variant surveillance in settings with limited sequencing resources, conferring increased epidemic preparedness. The significance of detecting of a novel variant at a prevalence of 2.5% in the population corresponds to a prediction of 7 to 8 weeks before the emerging variant becomes the dominant variant, based on the weekly growth rate of 50% as exhibited by Alpha [8]. To the best of our knowledge, so far efforts have been made to characterize only short segment of the spike gene [15]. We were the first to develop and validate a variant surveillance approach based on amplification and sequencing of the entire SARS-CoV-2 spike gene in conjunction with an integrated bioinformatics pipeline to be used with minimal effort. We also performed a comprehensive characterization of the likely efficacy of Spike Screen in discriminating between Omicron sub lineages. We have demonstrated in silico that the presented framework should be also able to discriminate between Omicron sub lineages based on the specific mutation profiles of so far detected sub lineages.
Our approach to tracking characteristic mutations demonstrates the feasibility of this surveillance strategy in all major VOCs and VOIs detected in the Slovenian population. The mutation frequencies observed with Spike Screen in comparison to overall population variant surveillance frequencies exhibit excellent temporal resolution with a reduction in cost and an increase in sample throughput. Spike Screen showed reliable detection of circulating VOCs/VOIs both at low incidence and in a high-diversity regime, indicating the relative robustness of the approach. Regression analysis showed that the characteristic mutation frequencies detected by Spike Screen corresponded well with the known prevalence of the tracked VOCs/VOIs. Spike Screen was employed in instances with either very low or very high variant predominance, usually between ‘switches’ in predominant variants (e.g. Alpha–Delta switch or Delta–Omicron switch). This approach could be further exploited and refined by clustering and analyzing the frequency of co-occurrence of mutations to reveal a specific mutational landscape of the samples analyzed. The results of such analyzes could enable the discovery and monitoring of previously unidentified mutations associated with emerging or low prevalence variants. The information gained from the Spike Screen strategy could also potentially be used for various endeavours ranging from characterizing the presence of circular RNAs and subsequently developing a circular RNA vaccine for COVID-19 and its variants [28, 29] to various machine learning approaches such as phosphorylation site prediction [30].
Similar strategies were already used for SARS-CoV-2 genomic variant surveillance in wastewater monitoring [31, 32]. These studies pointed out the difficulties in distinguishing the initial low frequency characteristic mutations from the noise in the sequencing data. The high noise is the result of technological approaches that include the quality of the initial sample, viral RNA extraction, and amplification. In addition to early detection, Spike Screen can also be used to monitor the prevalence of a variant and assess its dynamics in the population. Furthermore, these pools of samples can capture a wider range of circulating mutations that are not detected by routine CGS screening, and so this strategy can be used to detect and track ‘shadow’ variants in the population. Furthermore, the employment of Spike Screen allowed us to detect and contain the emergence and spread of several VOCs, such as Beta, Eta, and Gamma. Due to rapid detection and deployment of quarantines and complementary epidemiologic actions, epidemiologists were able to quell the initial infectious momentum of high-profile VOCs. Compared with PCR screening for characteristic spike mutations, Spike Screen provides broader detection and monitoring capacity for all occurring mutations harboured by different variants without competing for laboratory resources already committed to diagnostic purposes. In addition, Spike Screen eliminates the need to perform multiple reactions to detect specific mutations associated with a particular variant, as well as the design of primers and probes for PCR.
At the global level, Slovenian variant surveillance capabilities have achieved a sufficient percentage of sequenced COVID-19 cases to reliably monitor the dynamics of SARS-CoV-2 variant emergence and spread. According to GISAID data (https://gisaid.org/, accessed 6 November 2023), Slovenia uploaded 85 165 complete SARS-CoV-2 genomes to this public repository, representing 6.3% of the 1343 721 reported COVID-19 cases, with a 31-day median time to deposition. In comparison to global data from all 216 countries that shared SARS-CoV-2 genomes from human cases, the global median percentage of shared sequences was 1% (range 0.02–25.9%) and the global median time to deposition 60 days (range 8–644 days). This indicates that global genomic efforts for surveillance of SARS-CoV-2 are highly heterogeneous and hinge on socioeconomic factors and public health policies [33]. Expanding global genomic surveillance and sequencing capacity remains an invaluable tool for detecting and understanding variant emergence and spread [34]. There is a dire need to close the gap between lower-income countries with newer strategies, development of cost-effective kits, and protocols for various sequencing platforms.
The results indicate that Spike Screen successfully complements the individual case CGS strategy by sacrificing less informative parts of the genome [35, 36]. Similar to other coronaviruses, the spike protein of SARS-CoV-2 mediates receptor recognition, cell attachment, and cell membrane fusion during viral infection [3, 37]. This characteristic makes it a prime target for monitoring the variant dynamics, and the focus of therapy and vaccine development [38]. Two meta-analyses of mutational profiles on a large number of COVID-19 patients showed that the spike gene harbours the highest mutation density in the SARS-CoV-2 genome, followed by the nucleocapsid phosphoprotein and ORF1ab [35, 39]. Global analysis of mutations in the structural proteins of SARS-CoV-2 revealed that >95% of amino acid sites in the spike protein exhibited at least one mutation, in contrast to the envelope proteins, where only 5% do so [40].
A similar strategy is theoretically applicable to population surveillance of any viral pathogen. The selective amplification of target regions could be rapidly and reliably deployed in severe outbreaks or in settings with scarce sequencing resources. For instance, in the case of EBOV, the target for selective amplification and surveillance could be the 676-amino-acid-long transmembrane spike glycoprotein [41]. For HIV variant surveillance, the ~850-amino-acid-long envelope glycoprotein gp120 could be used as a target [42]. In the case of ZIKV, surveillance of envelope (E) glycoprotein could be employed [43]. For MPXV, the choice of the surveillance target is less straightforward because research on comprehensive proteome structure and functions is ongoing [44]. The best candidates would be the ~335-amino-acid-long hemagglutinin (H) protein and the ~247-amino-acid-long envelope proteins B5 and A33 [45].
Finally, the economic advantage of the proposed strategy compared with routine CGS mainly arises from reduction in sequenced genome length and pooling of samples. The spike gene accounts for ≈13% of the length of the entire SARS-CoV-2 genome [2], which alone allows for an ~10-fold reduction in the associated costs and hands-on time. The gain from reducing the genome length sequenced is most prominent in cases of longer genomes, as in MPXV (196 858 base pairs) [44]. Because library preparation represents the bulk of the cost in the NGS workflow, the employment of sample pooling can reduce the costs of library preparation and sequencing by ≈97%. Another advantage of sequencing a shorter section of viral genomes is the reduced computational power requirements: an important consideration in any bioinformatics workflow. This represents a tremendous advantage for lower-income countries, where access to advanced computing infrastructure is limited.
There are several limitations in the analysis and interpretation of data obtained with the strategy presented. First, if characteristic mutations reside outside the amplified region, such a strategy cannot be employed. Second, when the same mutations occur in multiple variants, this reduces the resolution of the Spike Screen strategy because mutation profiles overlap. Third, when more variants appear and coexist in the population due to convergence, homology, or pure chance, there is more overlap. Furthermore, failure to detect some characteristic mutations could also be due to the sporadic presence of mutations in a particular lineage [27]. The selection of characteristic mutations is based on the reported prevalence of mutations in each lineage. In our study, the established threshold of 75% mutation prevalence in a lineage means that 25% of the sequences do not harbour some mutations we monitored. The possibility of a degraded sample and the sporadic presence of characteristic mutations are probably the most likely reason for failure of detection of some mutations when technical factors such as coverage and initial viral copy number are adequate. Moreover, a reason for failure of detection could be the occurrence of mutations at primer target sites, which may lead to false negative results [46]. The variability of Ct values between different platforms is another factor that makes it difficult to trace a failed detection to the individual Ct value of the sample [20]. Theoretically, it would be possible to calculate the specificity and sensitivity of the Spike Screen using the 6319 samples for which CGS data are also available. However, the specific biological setting and limitations associated with wet-lab procedures would compromise the robustness of the calculations of sensitivity, specificity and ROC curve and lead to unreliable and possibly misleading results. For example, the sensitivity of D1118H, a characteristic spike mutation of Alpha, was calculated to be 96% (of the 187 pools in which CGS confirmed the presence of Alpha, there were 180 pools in which we detected Alpha with Spike Screen). However, the D1118H mutation was also carried by earlier (B.1.473, B.1.1.514, B.1.533, etc.) and later (EG.4.4, XBB.2.3.22) lineages, and not all Alpha sequences harboured this mutation (only 99%). Due to the compounding effect, the problem is magnified when different combinations of mutations are considered.
Conclusion
In summary, Spike Screen is a rapid, accurate, and cost-effective way to detect and track the emergence and abundance of SARS-CoV-2 VOCs/VOIs. This approach is capable of monitoring large numbers of samples in settings with limited sequencing capacity. It thus allows reliable and rapid detection of novel variants at the population level and can therefore serve as background information for informing public health policy planning, treatment choice, or application of vaccination. By embedding samples into pools, Spike Screen extends the mutation presence monitoring capacity of CGS, and indirectly variant presence monitoring capacity, with 41 537 samples characterized to the level of complete SARS-CoV-2 genomes, by an additional 55.0% of positive samples, while only requiring ≈3.0% of increase in resources, costs, and manual labour.
Key Points
The genomic surveillance of emerging SARS-CoV-2 variants is crucial for controlling public health risk due to viral evolution.
A novel cost-sparring real-time NGS strategy has been developed for rapid and reliable monitoring of emergence and spread of SARS-CoV-2 variants.
The developed strategy is capable of monitoring a large number of samples in settings with limited sequencing capacity.
Such selective amplification and sequencing of informative genomic target regions could be also rapidly developed and deployed for other viral outbreaks in settings with scarce sequencing capacity.
Acknowledgements
The authors would like to thank all colleagues who made this work possible: the laboratory staff who prepared the samples for sequencing: Sabina Beci, Gašper Grubelnik, Kaja Kotnik Koman, Manca Luštrek, Špela Pleh, Patricija Pozvek, Zala Prestor, and Jan Slunečko; and the team for NGS library preparation and sequencing: Matic Brvar, Dominika Celar Šturm, Andraž Celar Šturm, Tina Gabrovšek, Domen Lazar, Sara Štebe, and Tina Živič. We would also like to thank the whole staff of the Institute of Microbiology and Immunology, Faculty of Medicine, University of Ljubljana for their immense efforts during the COVID-19 pandemic.
Author Biographies
Alen Suljič is a post-doctoral researcher at the Institute of Microbiology and Immunology, Medical Faculty, University of Ljubljana, working in the area of bioinformatics, statistics, and data science.
Tomaž Mark Zorec is a post-doctoral researcher at the Institute of Microbiology and Immunology, Medical Faculty, University of Ljubljana, working in the area of bioinformatics and computational science.
Samo Zakotnik is a post-doctoral researcher with background in Biochemistry at the Institute of Microbiology and Immunology, Medical Faculty, University of Ljubljana, working in the area of NGS wet-lab and bioinformatics.
Doroteja Vlaj is an NGS wet-lab expert with background in Microbiology at the Institute of Microbiology and Immunology, Medical Faculty, University of Ljubljana.
Rok Kogoj is a research scientist at the Institute of Microbiology and Immunology, Medical Faculty, University of Ljubljana, working in the area of molecular diagnostics and a member of the response team for bioterrorism emergencies.
Nataša Knap is a research scientist at the Institute of Microbiology and Immunology, Medical Faculty, University of Ljubljana, working in the area of molecular diagnostics and a member of the response team for bioterrorism emergencies.
Miroslav Petrovec is a professor, head of Institute of Microbiology and Immunology, Medical Faculty, University of Ljubljana and the Vice-Dean for Specialist Fields of Healthcare at Medical Faculty, University of Ljubljana.
Mario Poljak is a professor at Institute of Microbiology and Immunology, Medical Faculty, University of Ljubljana, head of the Laboratory for molecular diagnostics of hepatitis and HIV and fellow at the American Academy of Microbiology.
Tatjana Avšič-Županc is an academic professor at the Institute of Microbiology and Immunology, Medical Faculty, University of Ljubljana and the Vice-president for Natural, Technical and Medical Sciences of the Slovenian Academy of Sciences and Arts.
Miša Korva is an assistant professor at Institute of Microbiology and Immunology, Medical Faculty, University of Ljubljana, head of the Laboratory for COVID-19 diagnostics, Laboratory for diagnostics of zoonoses, WHO laboratory with the BSL3+ facility and head of the response team for bioterrorism emergencies.
Contributor Information
Alen Suljič, Institute of Microbiology and Immunology, Faculty of Medicine, University of Ljubljana, Zaloška cesta 4, 1000 Ljubljana, Slovenia.
Tomaž Mark Zorec, Institute of Microbiology and Immunology, Faculty of Medicine, University of Ljubljana, Zaloška cesta 4, 1000 Ljubljana, Slovenia.
Samo Zakotnik, Institute of Microbiology and Immunology, Faculty of Medicine, University of Ljubljana, Zaloška cesta 4, 1000 Ljubljana, Slovenia.
Doroteja Vlaj, Institute of Microbiology and Immunology, Faculty of Medicine, University of Ljubljana, Zaloška cesta 4, 1000 Ljubljana, Slovenia.
Rok Kogoj, Institute of Microbiology and Immunology, Faculty of Medicine, University of Ljubljana, Zaloška cesta 4, 1000 Ljubljana, Slovenia.
Nataša Knap, Institute of Microbiology and Immunology, Faculty of Medicine, University of Ljubljana, Zaloška cesta 4, 1000 Ljubljana, Slovenia.
Miroslav Petrovec, Institute of Microbiology and Immunology, Faculty of Medicine, University of Ljubljana, Zaloška cesta 4, 1000 Ljubljana, Slovenia.
Mario Poljak, Institute of Microbiology and Immunology, Faculty of Medicine, University of Ljubljana, Zaloška cesta 4, 1000 Ljubljana, Slovenia.
Tatjana Avšič-Županc, Institute of Microbiology and Immunology, Faculty of Medicine, University of Ljubljana, Zaloška cesta 4, 1000 Ljubljana, Slovenia.
Miša Korva, Institute of Microbiology and Immunology, Faculty of Medicine, University of Ljubljana, Zaloška cesta 4, 1000 Ljubljana, Slovenia.
Funding
This work was funded by the Institute of Microbiology and Immunology, Faculty of Medicine, University of Ljubljana, Slovenian Research and Innovation Agency (grant number P3-0083), Network of Infrastructure Centres of the University of Ljubljana (MRIC-UL-IC-BSL3+), and the European Union’s Horizon 2020 research and innovation program – EVA GLOBAL project (Grant agreement no. 871029). The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript or in the decision to publish the results.
References
- 1. Robson F, Khan KS, Le TK, et al. Coronavirus RNA proofreading: molecular basis and therapeutic targeting. Mol Cell 2020;79:710–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Li J, Lai S, Gao GF, et al. The emergence, genomic diversity and global spread of SARS-CoV-2. Nature 2021;600:408–18. [DOI] [PubMed] [Google Scholar]
- 3. Harvey WT, Carabelli AM, Jackson B, et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat Rev Microbiol 2021;19:409–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bushman M, Kahn R, Taylor BP, et al. Population impact of SARS-CoV-2 variants with enhanced transmissibility and/or partial immune escape. Cell 2021;184:6229–6242.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Carabelli AM, Peacock TP, Thorne LG, et al. SARS-CoV-2 variant biology: immune escape, transmission and fitness. Nat Rev Microbiol 2023;21:162–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sun C, Xie C, Bu G-L, et al. Molecular characteristics, immune evasion, and impact of SARS-CoV-2 variants. Signal Transduct Target Ther 2022;7:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. World Health Organization (WHO) . Guidance for surveillance of SARS-CoV-2 variants: Interim guidance. 2021. https://iris.who.int/bitstream/handle/10665/343775/WHO-2019-nCoV-surveillance-variants-2021.1-eng.pdf?sequence=1.
- 8. European Centre for Disease Prevention and Control/World Health Organization Regional Office for Europe . Methods for the detection and characterisation of SARS-CoV-2 variants-second update. 2022. https://www.ecdc.europa.eu/sites/default/files/documents/Methods-for-the-detection-char-SARS-CoV-2-variants_2nd%20update_final.pdf.
- 9. Gardy JL, Loman NJ. Towards a genomics-informed, real-time, global pathogen surveillance system. Nat Rev Genet 2018;19:9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Grubaugh ND, Ladner JT, Lemey P, et al. Tracking virus outbreaks in the twenty-first century. Nat Microbiol 2019;4:10–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Woolhouse MEJ, Rambaut A, Kellam P. Lessons from Ebola: improving infectious disease surveillance to inform outbreak management. Sci Transl Med 2015;7:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Inzaule SC, Tessema SK, Kebede Y, et al. Genomic-informed pathogen surveillance in Africa: opportunities and challenges. Lancet Infect Dis 2021;21:e281–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rehle T, Lazzari S, Dallabetta G, et al. Second-generation HIV surveillance: better data for decision-making. Bull World Health Organ 2004;82:121–7. [PMC free article] [PubMed] [Google Scholar]
- 14. Piltch-Loeb R, Kraemer J, Lin KW, et al. Public health surveillance for zika virus: data interpretation and report validity. Am J Public Health 2018;108:1358–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tiwari A, Adhikari S, Kaya D, et al. Monkeypox outbreak: wastewater and environmental surveillance perspective. Sci Total Environ 2023;856:159166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Inward RPD, Parag KV, Faria NR. Using multiple sampling strategies to estimate SARS-CoV-2 epidemiological parameters from genomic sequencing data. Nat Commun 2022;13:5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. European Centre for Disease Prevention and Control (ECDC) . Guidance for representative and targeted genomic SARS-CoV-2 monitoring. 2021. https://www.ecdc.europa.eu/sites/default/files/documents/Guidance-for-representative-and-targeted-genomic-SARS-CoV-2-monitoring-updated-with%20erratum-20-May-2021.pdf.
- 18. Poljak M, Korva M, Knap Gašper N, et al. Clinical evaluation of the cobas SARS-CoV-2 test and a diagnostic platform switch during 48 hours in the midst of the COVID-19 pandemic. J Clin Microbiol 2020;58(6):e00599–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kogoj R, Kmetič P, Oštrbenk Valenčak A, et al. Real-life head-to-head comparison of performance of two high-throughput automated assays for the detection of SARS-CoV-2 RNA in nasopharyngeal swabs: the Alinity m and cobas 6800 SARS-CoV-2 assays. J Mol Diagn 2021;23:920–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kogoj R, Korva M, Knap N, et al. Comparative evaluation of six SARS-CoV-2 real-time RT-PCR diagnostic approaches shows substantial genomic variant–dependent intra- and inter-test variability, poor interchangeability of cycle threshold and complementary turn-around times. Pathogens 2022;11:462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wohl S, Lee EC, DiPrete BL, et al. Sample size calculations for pathogen variant surveillance in the presence of biological and systematic biases. Cell Rep Med 2023;4:101022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Danecek P, Bonfield JK, Liddle J, et al. Twelve years of SAMtools and BCFtools. Gigascience 2021;10(2):giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grubaugh ND, Gangavarapu K, Quick J, et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol 2019;20:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cingolani P, Platts A, Wang LL, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rambaut A, Holmes EC, O’Toole Á, et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol 2020;5:1403–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gangavarapu K, Latif AA, Mullen JL, et al. Outbreak.info genomic reports: scalable and dynamic surveillance of SARS-CoV-2 variants and mutations. Nat Methods 2023;20:512–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Niu M, Wang C, Chen Y, et al. Identification, characterization and expression analysis of circRNA encoded by SARS-CoV-1 and SARS-CoV-2. Brief Bioinform 2024;25(2):bbad537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Qu L, Yi Z, Shen Y, et al. Circular RNA vaccines against SARS-CoV-2 and emerging variants. Cell 2022;185:1728–1744.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jiao S, Ye X, Ao C, et al. Adaptive learning embedding features to improve the predictive performance of SARS-CoV-2 phosphorylation sites. Bioinformatics 2023;39(11):btad627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jahn K, Dreifuss D, Topolsky I, et al. Early detection and surveillance of SARS-CoV-2 genomic variants in wastewater using COJAC. Nat Microbiol 2022;7:1151–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Amman F, Markt R, Endler L, et al. Viral variant-resolved wastewater surveillance of SARS-CoV-2 at national scale. Nat Biotechnol 2022;40:1814–22. [DOI] [PubMed] [Google Scholar]
- 33. Brito AF, Semenova E, Dudas G, et al. Global disparities in SARS-CoV-2 genomic surveillance. Nat Commun 2022;13:7003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen Z, Azman AS, Chen X, et al. Global landscape of SARS-CoV-2 genomic surveillance and data sharing. Nat Genet 2022;54:499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yusof W, Irekeola AA, Wada Y, et al. A global mutational profile of SARS-CoV-2: a systematic review and meta-analysis of 368,316 COVID-19 patients. Life 2021;11:1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nasereddin A, Golan Berman H, Wolf DG, et al. Identification of SARS-CoV-2 variants of concern using amplicon next-generation sequencing. Microbiol Spectr 2022;10:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Walls AC, Park YJ, Tortorici MA, et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020;181:281–292.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Martínez-Flores D, Zepeda-Cervantes J, Cruz-Reséndiz A, et al. SARS-CoV-2 vaccines based on the spike glycoprotein and implications of new viral variants. Front Immunol 2021;12:701501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mendiola-Pastrana IR, López-Ortiz E, Río de la Loza-Zamora JG, et al. SARS-CoV-2 variants and clinical outcomes: a systematic review. Life 2022;12:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Abavisani M, Rahimian K, Mahdavi B, et al. Mutations in SARS-CoV-2 structural proteins: a global analysis. Virol J 2022;19:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jain S, Martynova E, Rizvanov A, et al. Structural and functional aspects of Ebola virus proteins. Pathogens 2021;10:1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wilen CB, Tilton JC, Doms RW. HIV: cell binding and entry. Cold Spring Harb Perspect Med 2012;2:a006866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Agrelli A, de Moura RR, Crovella S, et al. ZIKA virus entry mechanisms in human cells. Infect Genet Evol 2019;69:22–9. [DOI] [PubMed] [Google Scholar]
- 44. Rampogu S, Kim Y, Kim S-W, et al. An overview on monkeypox virus: pathogenesis, transmission, host interaction and therapeutics. Front Cell Infect Microbiol 2023;13:1076251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ghate SD, Suravajhala P, Patil P, et al. Molecular detection of monkeypox and related viruses: challenges and opportunities. Virus Genes 2023;59:343–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mentes A, Papp K, Visontai D, et al. Identification of mutations in SARS-CoV-2 PCR primer regions. Sci Rep 2022;12:18651. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The genomic data used in this study is available at GISAID: EPI_SET_230505ny, https://doi.org/10.55876/gis8.230505ny. The code and ready-to-deploy pipeline is available on GitHub at https://github.com/IMIMF-UNILJSI/scov2-spikeScreen,