

Summary
Membrane bound mucins constitute a large portion of the periciliary layer of lung epithelial surfaces, and thus play an important role in many aspects of innate defense. The biophysical and biochemical properties of the membrane bound mucins have important implications for mucociliary clearance, viral penetration, and potential therapeutics delivered to the airway surface. Hence, isolating them and determining these properties is important in understanding airways disease and ultimately in developing treatments. Here, we describe a method using isopycnic centrifugation to enrich and isolate shed membrane bound mucins from the washings of human bronchial epithelial cell cultures
Keywords: Membrane bound mucins, Human Bronchial Epithelial Cells, MUC1, MUC4, MUC16
1. Introduction
The airway epithelial surface can be described as a gel-on-brush model: the overlying mucus gel consisting of the major gel-forming mucins, MUC5B and MUC5AC, and their interacting proteins; and the “brush”, the periciliary layer (PCL), consisting of the membrane bound mucins (MBMs) and other glycoproteins on the epithelial surface [1]. Though the overlying mucus layer deservedly draws a lot of attention, the role of the PCL and its constituents cannot be overlooked. The PCL functions as a barrier; specifically, the membrane bound mucins have been shown to exclude some pathogens from entering the PCL and entrapping others [2, 3]. Likewise, inhaled intracellular therapeutics must successfully penetrate the PCL barrier to reach their targets. Therefore, isolating and characterizing MBMs and determining their interactions with pathogens, as well as therapeutic vehicles and vectors, is an essential area of study. MBMs can be acquired from the washings of well-differentiated human bronchial epithelial (HBE) cell cultures. HBE cell cultures maintained under air-liquid interface conditions shed MBMs apically along with cell secretions [4]. These MBMs can then be isolated from HBE cell culture washings [5–7]. Here, we describe a protocol for isolating membrane bound mucins MUC1, MUC4, and MUC16 from human bronchial epithelial cell washings. We collect and pool the HBE washings and then use a ‘dissociative’ isopycnic gradient to separate the MBMs from the gel forming mucins and other non-interacting proteins. We then take advantage of the keratan sulfate glycosylation found on MBMs to identify the MBM rich fractions and pool them together [8], so that they are ready for further purification and analysis.
2. Materials
2.1. Collecting and Storing Washings
Well-differentiated human bronchial epithelial (HBE) cultures.
Sterile Phosphate Buffer Saline (PBS).
Sterile 50-mL centrifuge tubes (with a screw cap).
70% Ethanol.
Water bath.
Biosafety cabinet.
2.2. Cesium Chloride Isopycnic Density Gradient Centrifugation
Cesium chloride.
Guanidine hydrochloride (8M) buffer: Prepare an 8 M guanidine HCl buffer (pH 8.0). 8M guanidine hydrochloride, .1 M Tris-HCl, 5 mM EDTA. Prepare using ultrapure water and adjust pH to 8 using hydrochloric acid. Filter the final buffer twice through a .2 um cellulose nitrate membrane filter.
Guanidine hydrochloride (4M) buffer. The same as the buffer prepared above but diluted 1:1 with de-ionized water.
Ultracentrifuge tubes (40-ml) (Polypropylene Quick-Seal centrifuge Tubes #342414, Beckman Coulter, Ca, USA) or an equivalent.
Ultracentrifuge and a fixed angle rotor capable of spinning 40 mL tubes at 145,500 × gavg.
Large plastic syringe (30-mL) and 18-gauge needles.
2.3. Unloading the Gradient and Running a Slot/dot Blot
Five-mL syringes and 18-gauge needles.
Slot/dot blot apparatus.
0.45 um Nitrocellulose blotting membrane cut to the dimensions of the slot/dot blot apparatus.
Grade 3MM chromatography filter paper cut to the dimensions of the slot/dot blot apparatus.
Plastic trays that accommodate the dimensions of the cut blotting membrane.
Tris-buffered saline and TWEEN (TBST) solution: Prepare a TBST buffer (pH 8.0). 10 mM Tris base, 150 mM NaCl, and 0.05% TWEEN. Prepare using ultrapure water and adjust the pH to 8 using hydrochloric acid.
Blocking solution: 1 g of Non-Fat Dry Milk mixed into 50 mL of TBST.
MUC5B antibody solution (HPA08246, MilliporeSigma): Prepare in TBST diluted 1:500.
Keratan sulfate antibody (MAB2022, MilliporeSigma): Prepare in TBST diluted 1:500.
Secondary antibodies for detection (matched to scanner).
Scanner/Biomolecular imager.
3. Methods
Carry out all procedures at room temperature unless specified otherwise.
3.1. Collecting HBE Cell Culture Washings
All of the techniques in this subsection will use sterile technique. Before placing items into the biosafety cabinet, they should be sprayed with 70% ethanol. All of the buffers, pipettes, and pipette tips should be sterile.
Determine the total amount of PBS needed to collect the culture washings. If using 12 mm diameter inserts, each insert will require 250 μL of PBS. If using 30 mm diameter inserts, each insert will require 1 mL of PBS. Using sterile technique, transfer the PBS into an appropriately sized centrifuge tube with a screw top lid.
Using a water bath, warm the sterile PBS to 37°C.
Remove the PBS from the water bath and place the tube in a holder inside of a biosafety cabinet.
Remove the cells from the incubator and place the culture plates into the biosafety cabinet.
For each plate, open the plate and pipette PBS onto the apical side of the insert. For 12 mm inserts dispense 250 μL, for 30 mm inserts, dispense 1 mL. Close the plate.
Return the culture plates to the incubator and let them sit for 30 min.
Place the cultures plates back into the biosafety cabinet.
Place a sterile 50-mL centrifuge tube into the bio safety cabinet into a test tube stand and remove the lid.
For each plate, open the plate and tilt it slightly towards you so that the apical liquid collects between the insert surface and insert wall (as shown in Fig. 1.). Collect the apical liquid by gently pipetting the liquid up, and then pipetting it into the centrifuge tube.
Repeat this for all of the plates. Once finished, return the plates to the incubator, cap the collection centrifuge tube, and store the tube at 4°C. (see Note 1)
Continue the collection once weekly, until there is enough material to run an isopycnic gradient (ideally at least 20 ml of material). (see Note 2)
Fig. 1.
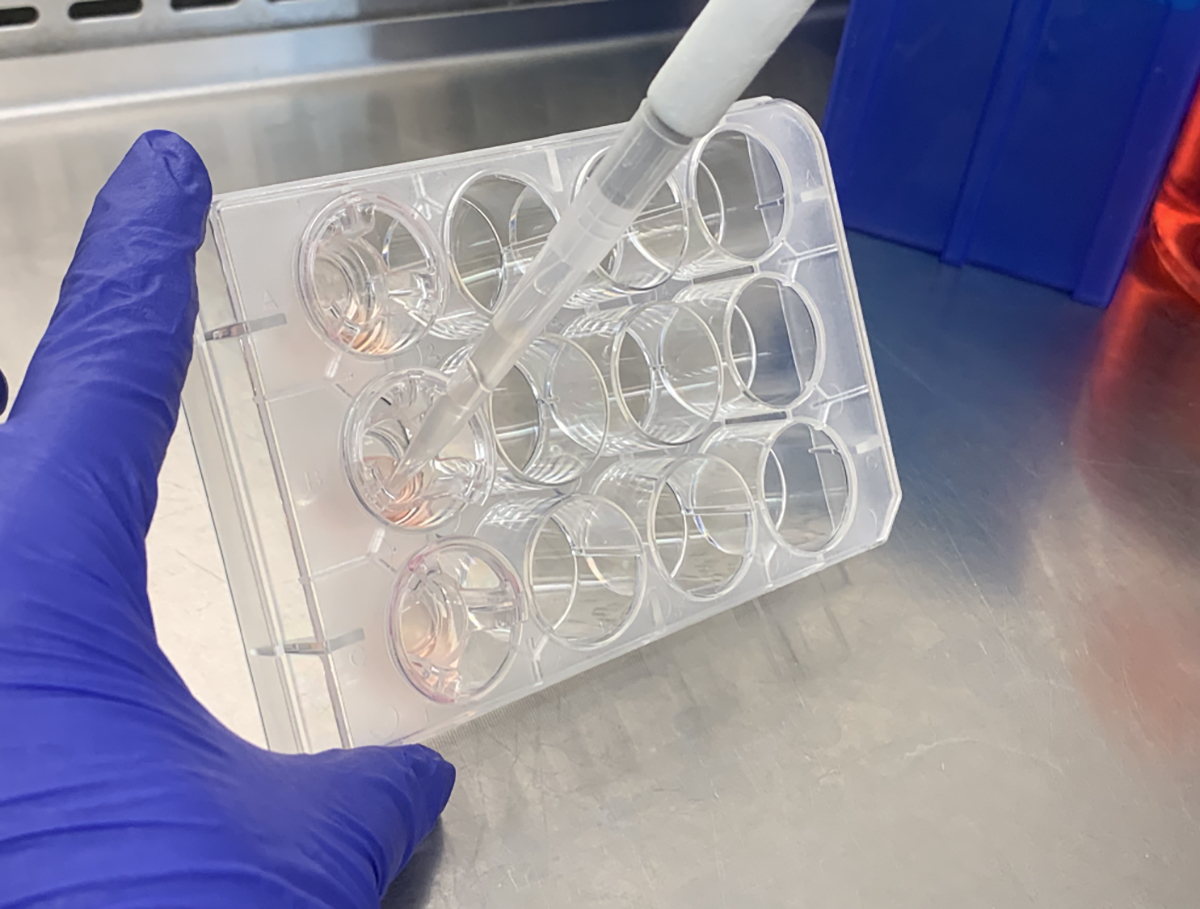
Removing the apical washings from the culture. Tilt the culture plate towards you so that you can retrieve all of the apical fluid without damaging the cultures.
3.2. Cesium Chloride Isopycnic Density Gradient Centrifugation
Using low speed centrifugation, spin the cell culture washings at 500 × g for 10 min.
Take the supernatant of the cell culture washings and mix 1:1 with the 8 M guanidine hydrochloride buffer. Discard the pellet. Mix using a large beaker with a stir bar. (see Note 3,4)
Determine how many centrifuge tubes will be needed: consider the current volume of material and that the added cesium chloride will account for ~ 13% of the final volume. The final volume should be the amount needed to fill this number of tubes + 5% extra material for any mistakes.
- Determine the amount of cesium chloride needed to bring the density of the solution to 1.45 g/mL (at the final volume) using the following formula.
Measure out the cesium chloride and slowly add it to supernatant/guanidine mixture stirring in the large beaker. (see Note 5)
Add 4 M guanidine hydrochloride buffer to reach the final intended volume.
Let the solution continue to mix until the cesium chloride is completely dissolved.
Let the solution mix for an additional 15 min.
Check the density of the solution by stopping the stir bar, pipetting 1 mL of material, and weighing the material in a weigh boat on a balance. The target mass for 1 mL is 1.45 g. If lower than this, adjust by adding cesium chloride to the solution, letting it stir for 5 min and rechecking the density. If higher, adjust by adding 4 M guanidine hydrochloride buffer, letting the solution stir for 5 min, then checking the density. (see Note 6)
Once satisfied with the density, fill the ultracentrifuge tubes with the solution. Using a 30-mL or larger plastic syringe and a blunt tip syringe needle, pull the solution into the syringe. Slowly inject the solution into the top of the centrifuge tube. Fill the tubes stopping just short of the neck.
Check the weight of the tubes and ensure that they can be balanced. If the number of tubes can’t be balanced, a blank tube can be quickly made by adding cesium chloride to 4 M guanidine hydrochloride buffer. As there is no protein, this can be rapidly mixed by placing into a 50-mL tube, capping the tube and then mixing using a vortex mixer. Once mixed and the density has been checked, a blank ultracentrifuge tube can be prepared as in step 10.
Seal the tubes and load them into the rotor in a balanced configuration. Load the rotor into the centrifuge and run the centrifuge with the following parameters: 145,500 × g, 14°C, and set the time to hold. (Final time can be anywhere from 60–96 h)
3.3. Unloading the Gradient and Running a Slot/dot Blot
Stop the centrifuge. Carefully remove the rotor from the centrifuge.
Open the rotor, and carefully remove the tubes from the rotor and place them onto a stable rack/stand.
For 40-mL centrifuge tubes, each tube will be fractionated from the top in 2 mL increments. Set aside 20 tubes for fractions that will be able to accommodate the volume of each fraction across all of the loaded centrifuge tubes (2 mL * number of tubes loaded) and label the tubes 1–20.
Fractionating a tube: Start by gently using a sharp tip 18-gauge syringe needle to puncture a hole in the top of the centrifuge tube. Make sure that the puncture hole is complete, but there is no need to full insert the needle into the tube.
Make a second hole at the top of the centrifuge tube.
Attach a 5-ml syringe to the needle and remove the top 2 mL of fluid from the tube. Place this into the correspondingly numbered tube. Repeat this for the next 5 fractions. (6 fractions in all)
Using a razor blade, carefully remove the top of the centrifuge tube (approximately 5mm above the liquid level after removing the top 6 fractions.
Use a pipette to remove fractions 7–19 in 2 mL increments.
Prior to collecting fraction 20, pipette vigorously to remove any material that may have pelleted to the bottom of the tube. Collect fraction 20.
Repeat steps 4–9 for any remaining tubes.
Perform slot/dot blot antibody analysis on the 20 fractions. For each tube use 20 μL of material for the blot. After blocking with milk, probe the blots with MUC5B (#HPA008246, MilliporeSigma) and keratan sulfate (#MAB2022, Millipore Sigma) antibodies to determine their, presence, distributions, and abundance across the fractions. In our lab, we image using an Odyssey scanner and the corresponding secondary antibodies (IRDYE 680LT and IRDYE 800LT) (Licor Biosciences, NE, USA) but any equivalent scanner/secondary antibody combination can be used. (See Note 7,8)
For each antibody, plot the signal vs. the fraction to generate an antibody concentration profile (as shown Fig. 2).
Identify the keratan sulfate peak.
Pool the fractions at the keratan sulfate rich peak together as enriched membrane bound mucin pool. These fractions are dominated by the MBMs as compared to the MUC5B dominant fractions, as assessed by proteomic analysis in Fig. 3. Atomic force microscopy imaging of these fractions shows the presence of membrane bound mucins (Fig. 4).
Fig. 2.

Antibody intensity plot of the isopycnic fractions. In the plot above the antibody signal for keratan sulfate (black) and MUC5B (gray) are plotted for each fraction. The values have been normalized to the peak value for each antibody, thus the peak has a value of 1. MUC5B has a peak in fraction 11, while KS peaks in fraction 17.
Fig. 3.

Total precursor intensity comparison of MUC5B and the membrane bound mucins in the MUC5B rich fraction vs. the keratan sulfate rich fraction. MUC5B is 10× more abundant in the MUC5B rich fraction as compared to the KS rich fraction. MUC1, MUC4 and MUC16 are more abundant in the KS rich fraction by factors of ~2×, 4×, and 4× respectively.
Fig. 4.

Atomic Force Microscope image produced from a dilute preparation of the keratan sulfate rich peak showing a collection of membrane bound mucins (Scale bar = 1 um).
4. Notes
It is fine to collect samples over the course of several weeks, extending out towards a few months. We haven’t seen evidence of degradation at that time length, provided that the washings are stored at 4°C and the cultures are free of contamination.
This protocol is design around using 40-mL centrifuge tubes because we get better separation at this size than with smaller volumes. This procedure can be done for smaller (or larger) sized tubes, but the spin time, rate, and fraction sizes will all need to be adjusted appropriately.
We typically avoid using a vortex mixer when trying to mix mucin solutions.
This protocol uses a dissociative isopycnic gradient (by using the chaotropic agent guanidine chloride) to separate out interacting proteins and improve the separation between the mucins. This protocol can also be done associatively by using PBS as a buffer instead of guanidinium, but this will result in less enrichment. If making an associative buffer, more cesium chloride is required to get to a density of 1.45 g/mL, which will affect the calculations in steps 3 and 4 in subsection 3.2.
When mixing cesium chloride, it may help to use a wider beaker so that you can minimize having a density gradient if your solution isn’t mixed well.
The density doesn’t have to be exactly 1.45 g/mL, but it is a good goal. If the density is higher, then both the gel forming mucins and MBMs will be closer to the top of the gradient, and vice versa if the density is lower. The further you get away from 1.45 g/mL in either direction, however, will limit your separation between the two mucin types.
The volume required to get a decent antibody signal will depend on the concentration of the material, but 20 μL is a good starting point for enriched cell culture washings. You may need to adjust downward if there is too much material.
For this protocol, we use the keratan sulfate antibody to identify the distribution of the membrane bound mucins within the fractions. The primary reason for doing this is that the presence of keratan sulfate can reduce the activity of some of the antibodies specifically to MBMs. This reduced activity can vary greatly depending on the specific antibody and heterogeneity between cell cultures. That said, antibodies specific for each membrane bound mucin can also be used as a probe to determine their distributions, but may not be reliable due to the presence of keratan sulfate.
Acknowledgements
We would like to thank members of the Kesimer lab as well as the Marsico Lung Institute for help and support during the development of this protocol. The protocols presented here were developed for work supported by the NIH (P01HL164320–01) and Cystic Fibrosis Foundation (CFFCARPEN19I0)
References
- 1.Button B, Cai LH, Ehre C et al. (2012) A periciliary brush promotes the lung health by separating the mucus layer from airway epithelia. Science 337(6097):937–941. doi: 10.1126/science.1223012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kesimer M, Ehre C, Burns KA et al. (2013) Molecular organization of the mucins and glycocalyx underlying mucus transport over mucosal surfaces of the airways. Mucosal Immunol 6(2):379–392. doi: 10.1038/mi.2012.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iverson E, Griswold K, Song D et al. (2022) Membrane-Tethered Mucin 1 Is Stimulated by Interferon and Virus Infection in Multiple Cell Types and Inhibits Influenza A Virus Infection in Human Airway Epithelium. mBio 13(4):e0105522. doi: 10.1128/mbio.01055-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hattrup CL, Gendler SJ (2008) Structure and function of the cell surface (tethered) mucins. Annu Rev Physiol 70:431–457. doi: 10.1146/annurev.physiol.70.113006.100659 [DOI] [PubMed] [Google Scholar]
- 5.Davies JR, Wickstrom C, Thornton DJ (2012) Gel-forming and cell-associated mucins: preparation for structural and functional studies. Methods Mol Biol 842:27–47. doi: 10.1007/978-1-61779-513-8_2 [DOI] [PubMed] [Google Scholar]
- 6.Davies JR, Kirkham S, Svitacheva N et al. (2007) MUC16 is produced in tracheal surface epithelium and submucosal glands and is present in secretions from normal human airway and cultured bronchial epithelial cells. Int J Biochem Cell Biol 39(10):1943–1954. doi: 10.1016/j.biocel.2007.05.013 [DOI] [PubMed] [Google Scholar]
- 7.Kesimer M, Kirkham S, Pickles RJ et al. (2009) Tracheobronchial air-liquid interface cell culture: a model for innate mucosal defense of the upper airways? Am J Physiol Lung Cell Mol Physiol 296(1):L92–L100. doi: 10.1152/ajplung.90388.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carpenter J, Kesimer M (2021) Membrane-bound mucins of the airway mucosal surfaces are densely decorated with keratan sulfate: revisiting their role in the Lung’s innate defense. Glycobiology 31(4):436–443. doi: 10.1093/glycob/cwaa089 [DOI] [PMC free article] [PubMed] [Google Scholar]