

Abstract
Osteogenesis imperfecta (OI) is a heterogeneous genetic disorder of bone and connective tissue, also known as brittle bone disease. Null mutations in SERPINF1, which encodes pigment epithelium-derived factor (PEDF), cause severe type VI OI, characterized by accumulation of unmineralized osteoid and a fish-scale pattern of bone lamellae. Although the potent anti-angiogenic activity of PEDF has been extensively studied, the disease mechanism of type VI OI is not well understood. Using Serpinf1(−/−) mice and primary osteoblasts, we demonstrate that loss of PEDF delays osteoblast maturation as well as extracellular matrix (ECM) mineralization. Barium sulfate perfusion reveals significantly increased vessel density in the tibial periosteum of Serpinf1(−/−) mouse compared with wild-type littermates. The increased bone vascularization in Serpinf1(−/−) mice correlated with increased number of CD31(+)/Endomucin(+) endothelial cells, which are involved in the coupling angiogenesis and osteogenesis. Global transcriptome analysis by RNA-Seq of Serpinf1(−/−) mouse osteoblasts reveals osteogenesis and angiogenesis as the biological processes most impacted by loss of PEDF. Intriguingly, TGF-β signaling is activated in type VI OI cells, and Serpinf1(−/−) osteoblasts are more sensitive to TGF-β stimulation than wild-type osteoblasts. TGF-β stimulation and PEDF deficiency showed additive effects on transcription suppression of osteogenic markers and stimulation of pro-angiogenic factors. Furthermore, PEDF attenuated TGF-β-induced expression of pro-angiogenic factors. These data suggest that functional antagonism between PEDF and TGF-β pathways controls osteogenesis and bone vascularization and is implicated in type VI OI pathogenesis. This antagonism may be exploited in developing therapeutics for type VI OI utilizing PEDF and TGF-β antibody.
Keywords: OSTEOGENESIS IMPERFECTA, PEDF, TGF-β, MATRIX MINERALIZATION, BONE VASCULARIZATION
Introduction
Osteogenesis imperfecta (OI), also known as brittle bone disease, is a group of phenotypically and genetically heterogeneous connective tissue disorders with an incidence of approximately 1 in 15,000 to 20,000 births. OI has its primary impact on bone, with low bone mass, uncoupled bone remodeling, and compromised bone quality, which together lead to frequent low-impact bone fractures and skeletal deformities. OI displays a wide clinical spectrum of disease severities, ranging from nearly asymptomatic or mild to perinatal lethality. Different types of OI have been identified, based on clinical phenotypes and causative genes. The majority (80% to 85%) of OI cases are caused by autosomal dominant mutations in the genes encoding type I collagen (COL1A1 and COL1A2), the major component of bone matrix. The remaining 15% to 20% of OI cases have autosomal or X-linked recessive inheritance of mutations in the genes encoding non-collagenous proteins responsible for type I procollagen posttranslational modification (CRTAP, LEPRE, PPIB), trafficking, processing or secretion (SERPINH1, PLOD2, FKBP10, BMP1), or are involved in bone tissue homeostasis (IFITM5, SERPINF1, SP7, TMEM38B, WNT1, CREB3L1, SPARC, MBTPS2).(1)
PEDF is a secreted glycoprotein of the serine proteinase inhibitor (serpin) superfamily and widely expressed in various tissues. PEDF has pleiotropic functions in multiple tissues, including eyes, vascular system, and the nervous system, and its expression is reduced in various pathological conditions, such as inflammation-associated diseases and cancers.(2) PEDF binds to extracellular matrix proteins, including type I collagen and glycosaminoglycans.(3) Identification of homozygous inactivating mutations in the SERPINF1 gene as causative of type VI OI added OI to the list of human diseases in which PEDF plays a role.(4) Most individuals with type VI OI have loss-of-function SERPINF1 mutations and undetectable plasma level of PEDF protein resulting from a premature stop codon or in-frame deletions or insertions.(5–8)
Recessive OI type VI is a severe progressive deforming bone dysplasia with distinctive diagnostic features including accumulation of unmineralized osteoid seams due to prolonged mineralization lag time, a fish-scale pattern of lamellae presumably from loss of the normal orientation of bone matrix, and elevated childhood plasma alkaline phosphatase.(9) However, type VI OI displays no abnormality in type I collagen folding, posttranslational modification or secretion.(8) Individuals with type VI OI present with relatively late onset of fractures and deformities compared with other severe forms of OI and show poor response to bisphosphonate treatment.(7) Cortical bone structure was found to be abnormally “porous” in type VI OI.(10) However, the molecular basis by which loss of PEDF results in type VI OI phenotypes remains to be elucidated.
TGF-β is deposited into the bone matrix and released during bone resorption by osteoclasts, thereby coupling bone resorption and bone formation during bone remodeling. Excessive TGF-β signaling has been proposed as a contributory factor to the OI phenotype. Increased TGF-β activation and SMAD2 phosphorylation were reported in calvaria of Crtap(−/−) mice, a model for recessive type VII OI, along with higher expression of TGF-β signaling targets, Cdkn1a and Serpine1.(11) An intercross of Crtap(−/−) mice with a TGF-β signaling reporter mouse displayed increased bioluminescence.(11)
For murine models of dominant OI, calvaria of Col1a2+/p.G610C mice also had higher expression of TGF-β signaling targets and SMAD2 phosphorylation.(11) In osteoblasts from Brtl mouse model, which has discrete moderate and perinatal lethal phenotypes from a dominantly inherited Col1a1 G349C substitution, TGF-β is increased in cells from lethal, but not non-lethal, mice on Western blot, and SMAD2 phosphorylation was increased in osteoblasts from non-lethal Brtl mice.(12) Thus, the contribution of TGF-β signaling to the pathogenic mechanism of OI is still under investigation. Importantly, functional interaction of PEDF and TGF-β pathways has not been examined. To elucidate the underlying mechanism of type VI OI pathogenesis, we investigated how loss of PEDF is responsible for the bone phenotype. Examining cells from a type VI OI patient and Serpinf1(−/−) mouse, which recapitulated some phenotypic features of type VI OI, we found that PEDF stimulates osteogenesis and impairs bone vascularization, whereas TGF-β showed the opposite trends in the two biological processes. The reciprocal inhibitory actions of PEDF and TGF-β pathways may be linked to the phenotype of type VI OI.
Materials and Methods
Animals
Serpinf1(−/−) mice were the gift of Dr Susan Crawford.(13) Progeny were genotyped by PCR for verification of Serpinf1 gene deficiency. Heterozygous males and heterozygous females were crossed to maintain the mouse colony and generate offspring mice for analysis. Serpinf1(−/−) mice were viable and fertile with litters of normal size. Animal care and experiments were performed in accordance with a protocol approved by the NICHD Animal Care and Use Committee (ACUC).
Fibroblasts from type VI OI proband
Fibroblasts were cultured from a dermal punch biopsy of a proband with type VI OI caused by compound heterozygous mutations in SERPINF1 (IVS3+1G>A; c.271_279dup). The biopsy was obtained under an NICHD, NIH-approved protocol with parental informed consent. Cells were harvested for RNA extraction and transcript analysis (see below) just before confluence.
Mouse calvarial osteoblast isolation and mineralization assay
Mouse calvarial osteoblasts were isolated from newborn mice (3 to 5 days old) by serial digestion of calvarial bones with collagenase II (Worthington Biochemical Corp., Lakewood, NJ, USA). For osteoblast differentiation, cells were plated (50,000 cells/well) in 12-well tissue culture plates and cultured to confluency in α-MEM supplemented with 10% FBS (Gemini Bio-Products, West Sacramento, CA, USA) and antibiotics (penicillin (100 U/mL) and streptomycin (100 μg/mL, Thermo Fisher Scientific, Waltham, MA, USA) at 37°C and 8% CO2. Post-confluence, cells were further cultured in osteogenic media (50 μg/mL L-ascorbic acid and 2.5 mM β-glycerophosphate, all from Sigma, St. Louis, MO, USA), refreshing osteogenic media every 3 days. To visualize extracellular matrix (ECM) mineralization, cells were washed once with 1x PBS and fixed with 4% paraformaldehyde for 10 minutes at room temperature. Fixed cells were washed 3 times with 1x PBS and stained with 2% Alizarin Red S solution (pH 4.2) for 10 minutes at room temperature. Excess Alizarin Red S stain was removed by washing 5 times with distilled water. Cells were air-dried in the dark before imaging.
CRISPR/Cas9-mediated editing of the IFITM5 gene in human dermal normal fibroblasts
To generate CRISPR/Cas9-edited clones containing the IFITM5 (c.119C>T, p.S40L) mutation, human dermal normal fibroblasts were electroporated to introduce CRISPR/Cas9 vectors and HDR template using Amaxa Nucleofector (Lonza, Basel, Switzerland). Cells positive for GFP (marker for transfectants) were sorted by FACS and subjected to clonal expansion in DMEM-10 media. Genomic DNAs from 72 CRISPR-edited clones were isolated and genotyped by PCR to identify IFITM5 (S40L/+) CRISPR clones. Confirmed clones were expanded for further analysis.
RNA extraction and real-time qPCR
Total RNA was extracted from primary osteoblasts using the RNeasy mini kit (Qiagen, Valencia, CA, USA) following the manufacturer’s instructions. Genomic DNA was removed from isolated total RNA by on-column DNase digestion. Then, cDNA was synthesized using 500 ng of total RNA and iScript reverse transcription Supermix (Bio-Rad, Hercules, CA, USA), following the manufacturer’s instructions. Comparative real-time qPCR was performed in TaqMan universal PCR master mix in triplicate using Applied Biosystems Prism 7500 Fast Sequence Detection System (Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer’s protocol. TaqMan probes and primers were manufactured by Applied Biosystems (Thermo Fisher Scientific): Mouse Gapdh (Mm99999915_g1) and mouse Tbp (Mm00446971_m1) were used as endogenous controls for normalization of real-time qPCR. Relative expression was calculated using the comparative ΔΔCt method.
Western blot analysis
Cell lysates were analyzed by Western blotting using standard methods and indicated antibodies. Briefly, mouse osteoblasts were grown in complete culture media: α-MEM supplemented with 10% FBS (Gemini Bio-Products, West Sacramento, CA, USA) and antibiotics (penicillin [100 U/mL] and streptomycin [100 μg/mL], Thermo Fisher Scientific). Murine calvarial osteoblasts and human dermal fibroblasts were seeded at 80,000 cells/well in 12-well and at 200,000 cells/well in 6-well tissue culture plates containing complete media, respectively. Cells were then stimulated with TGF-β1 (5 ng/mL for mouse cells and 10 ng/mL for human cells) for 30 and 60 minutes, before lysis in cell lysis buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1% IGEPAL, 0.5% sodium deoxycholate, 0.1% sodium disodium sulfate, 1× protease inhibitor cocktail, and 1× phosphatase inhibitor cocktail). Cell lysates were cleared by centrifugation at 20,000g for 20 minutes at 4°C after brief sonication. Soluble cell lysates were mixed with SDS-PAGE sample buffer (50 mM Tris, pH 6.8, 2% SDS, 10% glycerol, 0.1% bromophenol blue, and 100 mM dithiothreitol) and denatured at 95°C for 5 minutes. For secreted PEDF, conditioned media (CM) was collected after 24-hour incubation in serum-free α-MEM and supplemented with protease inhibitors. CM was concentrated using Amicon Ultra-15 centrifugal filter unit (EMD Millipore, Billerica, MA, USA) and normalized to cell counts for equal loading. Proteins were resolved in 4% to 15% mini-PROTEAN precast gels (Bio-Rad) at 150 V for 50 to 60 minutes. The resolved proteins were transferred to nitrocellulose (NC) membrane using iBLOT2 dry blotting system (Thermo Fisher Scientific) following the manufacturer’s instructions. NC membranes were then immersed in 5% bovine serum albumin (BSA) in Tris-buffered saline (TBS) for 1 hour at room temperature to block non-specific binding of primary antibodies. Subsequently, NC membranes were probed with the following primary antibodies: PEDF goat polyclonal antibody (#AB-PEDF4, BioproductsMD, Middletown, MD, USA), phospho-Smad3 (Ser423/425) rabbit polyclonal antibody (#600–401–919, Rockland Inc., Limerick, PA, USA), Smad2/3 (D7G7) XP rabbit monoclonal antibody (#8685, Cell Signaling Technology, Cambridge, MA, USA), β-Actin (AC-15) mouse monoclonal antibody (#A5441, Sigma-Aldrich, St. Louis, MO, USA), and histone H3 (D1H2) XP rabbit monoclonal antibody (#4499, Cell Signaling Technology). Secondary antibodies (LI-COR Biosciences, Lincoln, NE, USA): IRDye 680RD anti-rabbit IgG (#926–68071) or IRDye 800CW anti-mouse IgG (#926–32210). After incubating NC membranes with primary antibodies for 16 hours at 4°C, membranes were washed in TBS-T (1x TBS containing 0.5% Tween-20) three times for 5 minutes each at room temperature. Protein bands were visualized by incubating membranes for 1 hour at room temperature with the following secondary antibodies (LI-COR Biosciences): IRDye 680RD anti-rabbit IgG (#926–68071) or IRDye 800CW anti-mouse IgG (#926–32210) or IRDye 800CW anti-goat IgG (#926–32214). The membranes were washed in TBS-T three times for 5 minutes each at room temperature before scanning with Odyssey Infrared Imaging Systems (LI-COR Biosciences).
Bone vascularization
Eight-week-old mice were perfused with barium sulfate (BaSO4) and euthanized before long bones were dissected and scanned as previously described.(14,15) Briefly, bones were fixed, ethanol-dehydrated, and scanned with a high-resolution μCT (μCT40; Scanco Medical, Southeastern, PA, USA) as previously described.(15) Data were acquired at 55 KeV with 18 μm cubic resolution for the entire bone assessment and at 6 μm for specific regions. Representative 3D images of the vascular bed in the tibia and surrounding tissue were provided for visualization purposes. To exclude any artifacts in the quantification of vasculature, ie, including bone tissue in the analyzed, only blood vessels in the periosteal region were quantified in a 300 μm region surrounding the bone. Briefly, the outer region of the tibia was traced and the contours expanded by 300 μm. Images were segmented using a stable threshold set at 200 to 1000 per mille.
FACS analysis
To isolate the endothelial cells, juvenile (2- to 4-week-old) mice were euthanized, long bones were collected, epiphysis at the end and the muscles and periosteum around the bone were removed, and then the bones were crushed in ice-cold PBS. The whole bone marrow was digested with collagenase A (Roche, Indianapolis, IN, USA; #10103578001) at 37°C for 20 minutes to obtain single-cell suspensions. Cells were labeled with the following antibodies and washed three times: Anti-mouse CD31-PE (Rat IgG2a,κ, Biolegend, San Diego, CA, USA), anti-mouse endomucin-FITC (V.7C7 rat monoclonal IgG2a, Santa Cruz, Dallas, TX, USA), and EasySep Mouse FITC Positive Selection Kit and EasySep Mouse PE Positive Selection Kit (STEMCELL Technologies, Cambridge, MA, USA). Labeled cells were analyzed in FACSAria system (BD Biosciences, San Jose, CA, USA).
RNA-Seq
Total RNA samples were purified by Poly-A extraction method and purified mRNAs were used to construct RNA-Seq libraries with specific barcodes using Illumina TruSeq Stranded mRNA Library Prep Kit (Illumina, San Diego, CA, USA). All the RNA-Seq libraries were pooled together and sequenced using Illumina HiSeq 2500 to generate ~40 million 2×100 paired-end reads for each sample. Demultiplexed 100 bp paired-end fastq reads were aligned to mouse reference genome GRCm38/mm10 using GENCODE release 27 gene definitions using RNA STAR v2.5.4a.(16) After alignment, reads were quantitated using the subread package featureCounts v1.5.2(17) against GENCODE release 27 gene definitions, counting multi-mapping and multi-overlapping reads at the level of gene names. All downstream analyses were performed in R v3.5.1, Ingenuity Pathway Analysis (Qiagen Bioinformatics, Hilden, Germany) and Metascape (metascape.org). The RNA-Seq data of this study are available in BioProject database at https://www.ncbi.nlm.nih.gov/bioproject/756256, reference number PRJNA756256.
Statistical analysis
Statistical analysis was performed using Microsoft Excel. Differences between groups were evaluated with Student’s t test using a two-tailed distribution. Results were considered significant when the p value is <0.05. Quantitative data are presented as mean ± standard deviation.
Results
PEDF deficiency impairs osteogenic differentiation and ECM mineralization
Accumulation of unmineralized osteoid and fish-scale organization of bone lamellae are among the most remarkable phenotypic features of type VI OI. Although Serpinf1(−/−) mice do not display a severe bone phenotype and have normal movement and size,(13) they recapitulate most of the clinical features of type VI OI.(18) To define the molecular mechanisms that lead to defective mineralization in type VI OI, primary osteoblasts were isolated from calvarial bones of newborn Serpinf1(−/−) mice. Gene expression analysis by real-time qPCR demonstrated that Serpinf1 transcript expression increased in osteoblasts from wild-type mice during early differentiation and then decreased in terminally differentiated osteoblasts (Fig. 1A). Levels of PEDF protein secreted into culture media showed a similar trend (Fig. 1B). Gene expression analysis by real-time qPCR demonstrated that expression of type I collagen (Col1a1) and alkaline phosphatase (Alpl), co-expression of which is necessary and sufficient for bone mineralization,(19) was significantly reduced in PEDF-deficient cells (Fig. 1C). In addition, expression of Sp7, an essential transcription factor for osteoblast specification,(20) was also decreased as a result of loss of PEDF. Several other markers of osteogenic differentiation and ECM mineralization (Ifitm5, Bglap, Sost, Phospho1, Phex, and Dmp1) were also less abundant in Serpinf1(−/−) cells compared with wild-type cells (Fig. 1C). Phospho1 is a phosphatase that initiates deposition of hydroxyapatite within the lumen of matrix vesicles and contributes to bone mineralization.(21) DMP1 (dentin matrix protein 1) is an acidic phosphorylated ECM protein known to promote osteoblast maturation and mineralization.(22) Reduced expression of the mineralization markers clearly reflects the cellular phenotype in vitro. A similar reduction in expression of Alpl, Ifitm5, Bglap, and Sost was observed in long bones of Serpinf1(−/−) mice (Fig. 1D). These data indicate that osteoblast maturation, matrix deposition, and ECM mineralization are diminished in the absence of PEDF.
Fig. 1.
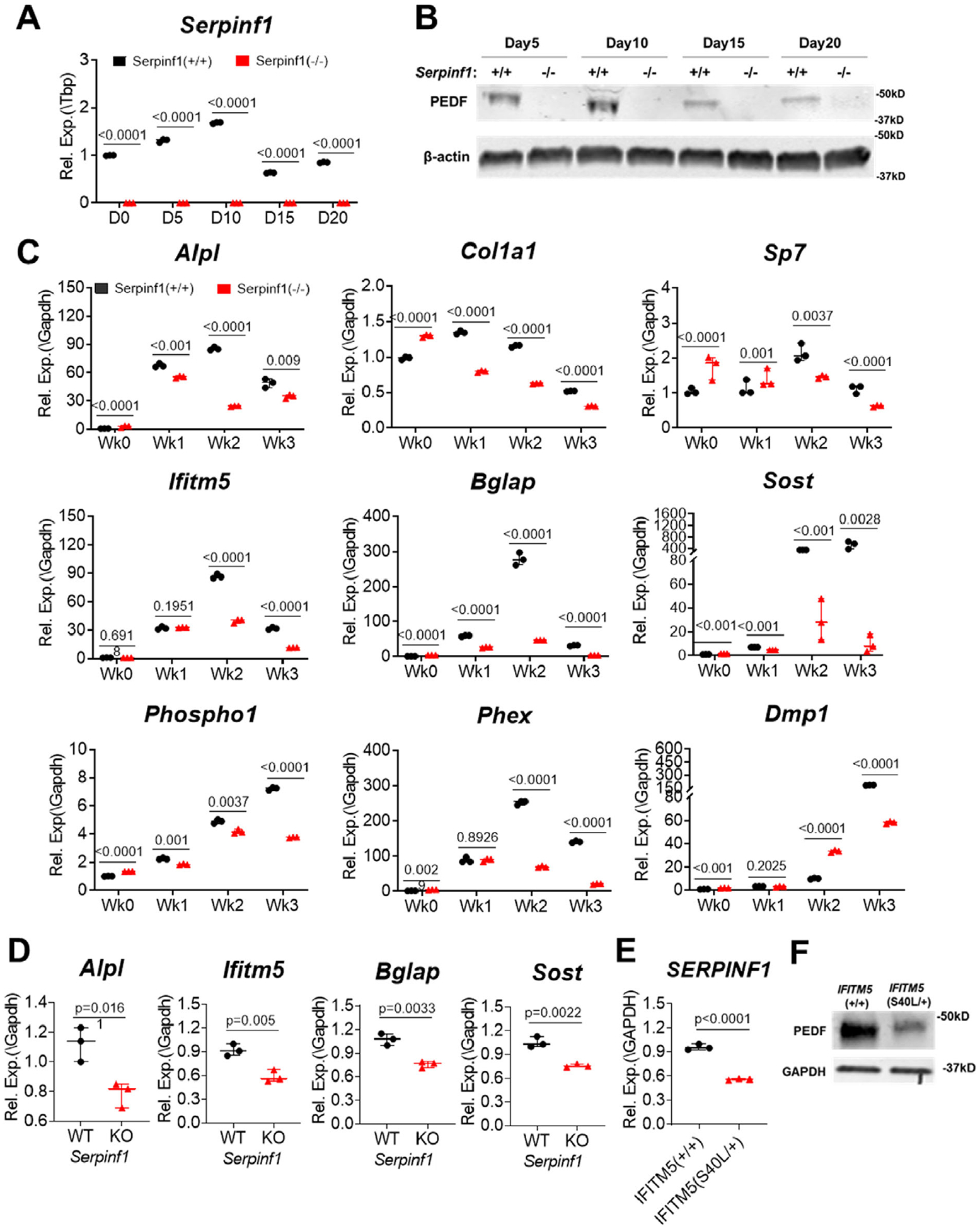
PEDF deficiency impairs osteogenic differentiation. (A) Real-time qPCR analysis of Serpinf1 transcript level during osteogenic differentiation of murine calvarial cells in culture. N = 3, Student’s t test. (B) Western blot analysis of PEDF protein secreted from osteoblasts. (C) Real-time qPCR analysis of osteogenic marker expression using RNA extracted from cultured primary osteoblasts. Serpinf1(−/−) osteoblasts expressed lower level of osteogenic marker genes than Serpinf1(+/+) osteoblasts. N = 3, Student’s t test. (D) Real-time qPCR analysis of osteogenic marker expression using RNA extracted from mouse bones. Serpinf1(−/−) mouse bone showed lower level of osteogenic marker expression than Serpinf1(+/+) mouse bone. N = 3, Student’s t test. (E) Real-time qPCR analysis of SERPINF1 transcript level. A CRISPR clone of IFITM5 (S40L/+) displayed reduced level of SERPINF1 expression. N = 3, Student’s t test. (F) Western blot analysis of PEDF protein secreted from CRISPR clone (IFITM5 (S40L/+)) of human dermal normal fibroblasts. Normal fibroblasts (IFITM5(+/+)) were a control for comparison. GAPDH is used as an internal control for equal sample loading in each lane. IFITM5 (S40L/+) clone shows reduced PEDF protein secretion compared with normal fibroblasts.
A recurring heterozygous mutation in the 5′ UTR of IFITM5 (c.−14C>T) creates an in-frame start codon adding 5 amino acids to the IFITM5 (BRIL, bone-restricted IFITM-like) protein and causes autosomal dominant type V OI, which presents with a discrete phenotype, including hyperplastic callus formation without evidence of mutations in type I collagen.(23,24) However, a heterozygous mutation within the coding region of IFITM5 (c.119C>T, p.Ser40Leu) leads to a phenotype resembling type VI OI with no mutation in SERPINF1.(25) Intriguingly, our previous studies found that IFITM5 (c.119C>T; p.S40L) results in decreased SERPINF1 transcript expression, whereas the type V OI IFITM5 mutation (c.−14C>T) increased SERPINF1 transcript expression and PEDF protein secretion.(26) These results suggest a potential positive feedback relationship between SERPINF1 (PEDF) and IFITM5 (BRIL) in bone formation. We generated CRISPR clones with IFITM5 (c.119C>T, p.Ser40Leu) mutation, a cellular model of atypical type VI OI. SERPINF1 transcripts and PEDF protein secretion were reduced in mutant IFITM5 CRISPR clones compared with wild-type control cells (Fig. 1E, F). In agreement with the finding with OI patient cells, we also found that Serpinf1(−/−) osteoblasts exhibit lower expression of ifitm5 transcripts (Fig. 1A), corroborating the positive feedback relationship between BRIL (IFITM5) and PEDF (SERPINF1) in bone and phenotypes of OI types V and VI. Lack of circulating PEDF can be used as a specific marker to diagnose type VI OI.(7) However, atypical type VI OI patients with an IFITM5 (c.119C>T, p.Ser40Leu) mutation have normal serum level of PEDF despite reduced production of SERPINF1 transcripts and PEDF secretion from osteoblasts,(25) suggesting that local PEDF deficiency is responsible for the defective bone matrix mineralization.
PEDF deficiency increases bone vascularization
PEDF is a ubiquitously expressed protein originally identified as a neurotrophic factor in retinal pigment epithelium and well described for its anti-angiogenic activity.(27) The cortical bone structure of individuals with type VI OI is abnormally porous to the extent that the outer and inner cortices cannot be distinguished,(10) suggesting increased vascularity in type VI OI bones. Elevated vascular porosity was also observed in other types of OI specimens, and this parameter was associated with reduced bone material strength.(28) Bone vascularization is crucial for bone modeling and remodeling. In addition to bone development, close interaction between angiogenesis and osteogenesis was also evidenced during aging and in various skeletal pathologies that are associated with altered vasculature. Especially, CD31(+)/Endomucin(+) endothelial cells secrete factors that affect osteoprogenitor differentiation and play a critical role in coupling osteogenesis and vascularization in bone.(29) Previous studies showed that Serpinf1(−/−) mice have an increased stromal vasculature and epithelial tissue growth in mouse prostate and pancreas.(13)
To assess whether high cortical porosity in type VI OI is associated with bone vascularity and to explore the role of PEDF in bone vascularization, we utilized whole animal vascular perfusion with barium sulfate (BaSO4) and FACS analysis of CD31+/Endomucin+ endothelial cells isolated from long bone. In vivo, PEDF deletion resulted in a significant increase in vessel density by 33% (p < 0.05) in the tibial periosteum of BaSO4 perfused Serpinf1(−/−) mice (Fig. 2A, B). Increased vascular permeability was also detected in Serpinf1(−/−) bone, with absence of the anti-permeability action of PEDF.(30) In agreement, the number of CD31+/Endomucin+ endothelial cells is increased in Serpinf1(−/−) mice by 18% (p < 0.05) in FACS analysis (Fig. 2C). RT-qPCR analysis further demonstrated that endothelial cells from Serpinf1(−/−) mice have increased expression level of vascular endothelial growth factor (VEGF)-R (Kdr) and VEGFα (Fig. 2D). These data suggest an overall stimulatory effect of PEDF deficiency in bone vascularization.
Fig. 2.
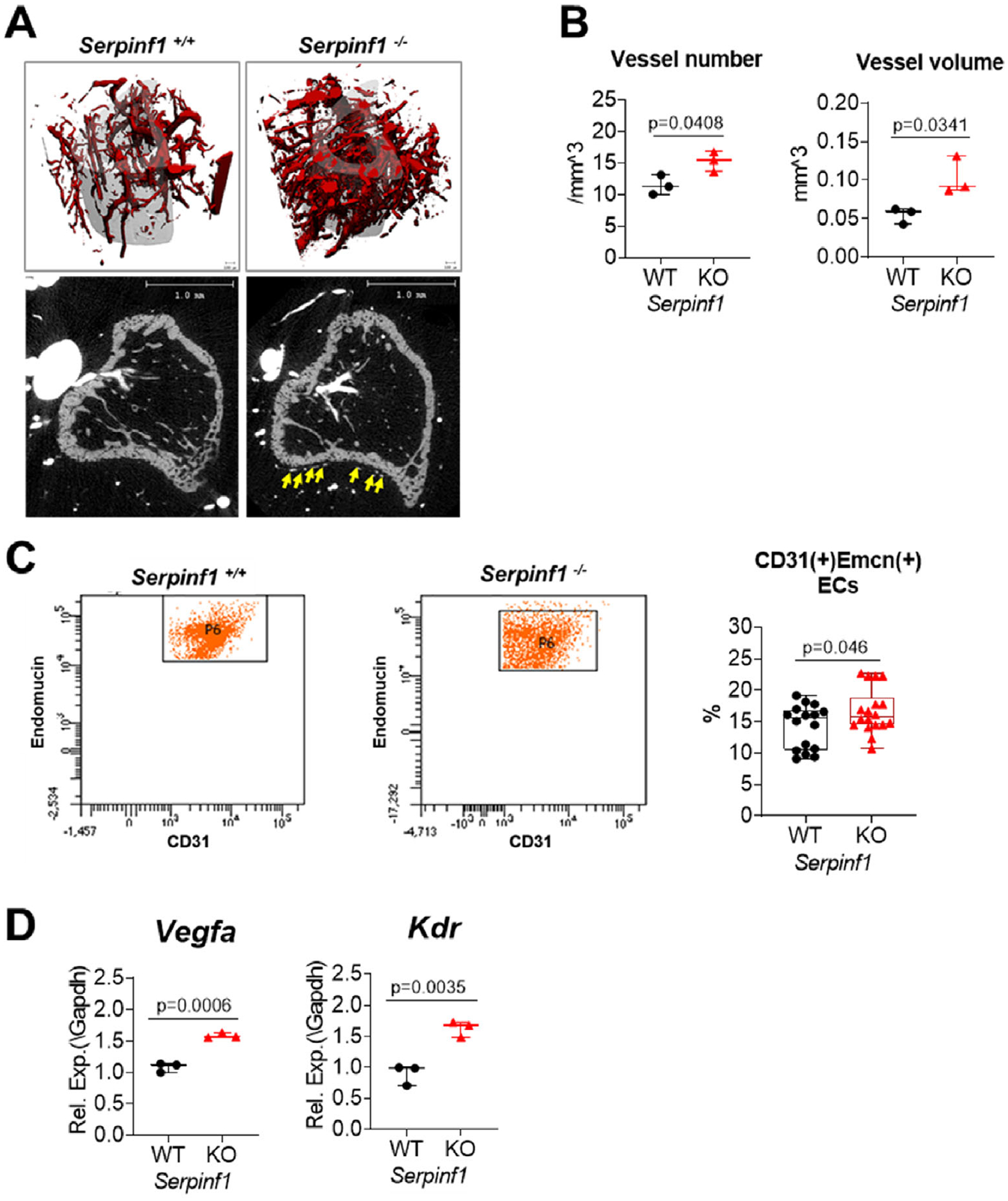
PEDF deficiency increases bone vascularization. (A) Bone vessels were visualized by microCT scan after barium sulfate perfusion. (B) Serpinf1(−/−) mouse bone showed increased vessel numbers and volume compared with Serpinf1(+/+) mouse bone. (C) FACS analysis of CD31(+)Endomucin(+) endothelial cells isolated from long bones of juvenile (2- to 4-week-old) mice. Serpinf1(−/−) mouse bones have higher level of CD31(+)Endomucin(+) endothelial cells than Serpinf1(+/+). N = 3, Student’s t test. (D) Real-time qPCR analysis showed that CD31(+)Endomucin(+) endothelial cells from Serpinf1(−/−) mouse express higher level of VEGFa and VEGF-receptor. N = 3, Student’s t test.
RNA-Seg-based transcriptome analysis
To gain further insight into the biological pathways affected by the loss of PEDF, RNA transcripts from Serpinf1(−/−) mouse osteoblasts were subjected to RNA-Seq. Differential expression analysis revealed that 100 genes were downregulated (log2FC < 1, false discovery rate [FDR] < 0.05) and 77 genes were upregulated at week 1 (log2FC > 1, FDR < 0.05) in Serpinf1(−/−) cells compared with wild-type cells. Gene ontology (GO) term analyses (Metascape and Reactome) of these two cohorts of genes revealed an enrichment for a series of distinct annotation clusters. As summarized in Fig. 3A, genes downregulated in Serpinf1(−/−) cells cultured for 1 and 2 weeks in osteogenic condition were highly enriched in transcripts encoded by genes involved in osteoblast differentiation, bone mineralization, bone morphogenesis, ECM organization, positive regulation of protein secretion, regulation of cell communication, and regulation of response to cell stimulus. Specifically, genes associated with RUNX2-regulated bone development include Bglap, Satb2, and Sp7. The genes critical for ECM organization (Col22a1, Col11a2, Col24a1, Col13a1, Ntm, Mmp17, Dmp1, Col9a1, Mmp9, Adamts18, Acan, Ibsp, Vash1, Aldh1l2, Col10a1, and A2m) were also downregulated in Serpinf1(−/−).
Fig. 3.
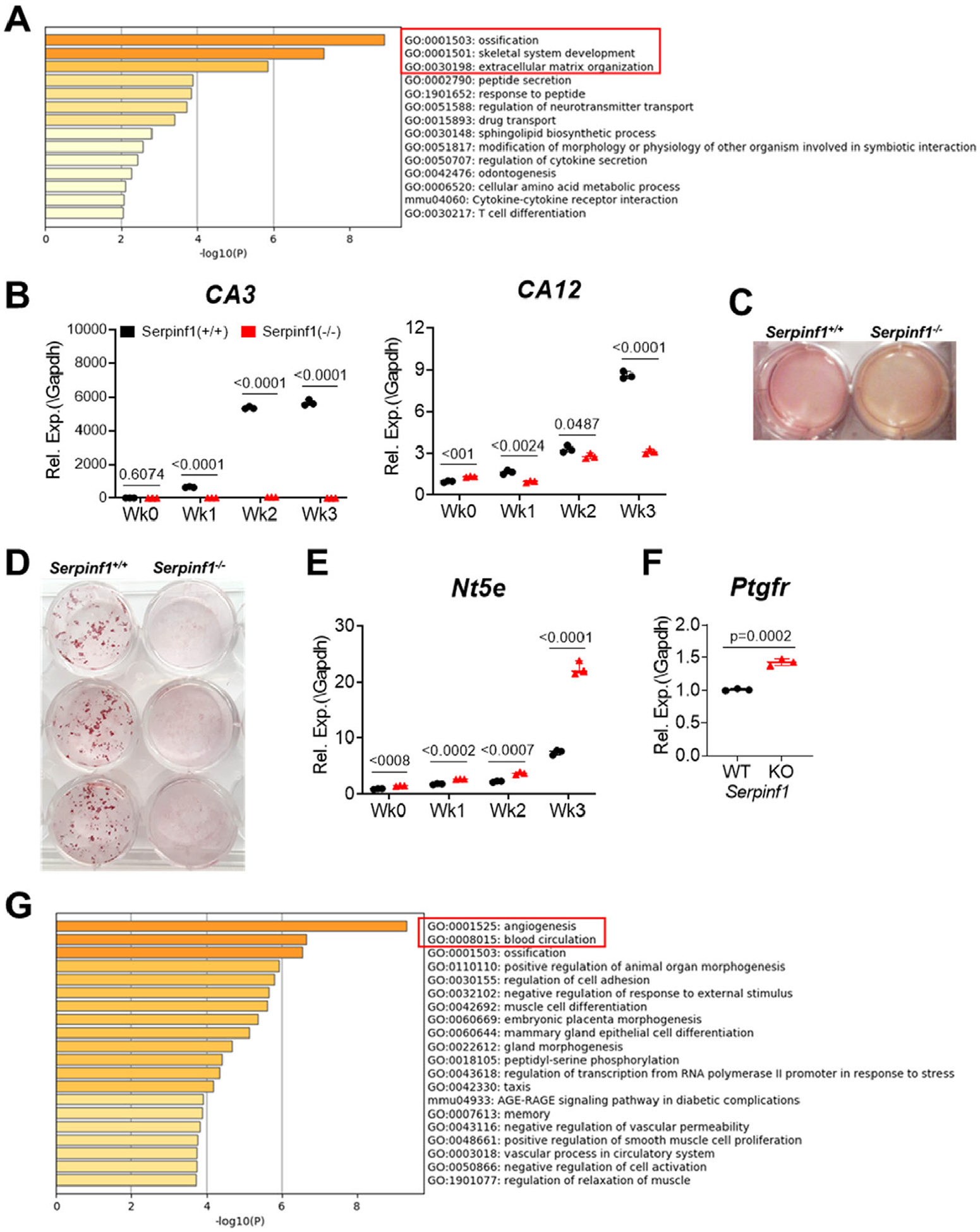
Global transcriptome analysis by RNA-seq reveals decreased osteogenesis and increased angiogenesis in Serpinf1(−/−) cells. (A) Metascape analysis of downregulated DEGs in Serpinf1(−/−) osteoblasts at week 1 in osteogenic culture. (B) Real-time qPCR analysis of carbonic anhydrases expression. Serpinf1(−/−) osteoblasts expressed significantly lower level of CA3 and CA12 than Serpinf1(+/+) osteoblasts. N = 3, Student’s t test. (C) Primary cultures of Serpinf1(+/+) and Serpinf1 (−/−) cells in osteogenic condition. Culture media of Serpinf1 (−/−) is more acidic as indicated by more yellow-orange color than in Serpinf1(+/+) cell culture. (D) Osteoblasts stained with Alizarin red S to detect calcium nodule formation (mineralization). (E) Real-time qPCR analysis of Nt5e (CD73) expression. Serpinf1(−/−) osteoblasts expressed significantly higher level of Nt5e than Serpinf1(+/+) osteoblasts during differentiation. N = 3, Student’s t test. (F) Real-time qPCR analysis of Ptgfr (prostaglandin F receptor) expression. N = 3, Student’s t test. (G) Metascape analysis of upregulated DEGs in Serpinf1(−/−) osteoblasts at week 1 in osteogenic culture. N = 3, Student’s t test.
Genes for reversible hydration of carbon dioxide, such as cytosolic carbonic anhydrase 3 (CA3) and membrane-associated carbonic anhydrases (CA12), were downregulated in Serpinf1(−/−) cells. These carbonic anhydrases are induced by osteogenic stimulation and reduced in Serpinf1(−/−) cells (Fig. 3B). Carbonic anhydrases catalyze direct conversion of carbon dioxide and water to bicarbonate ions (HCO3−), which have a stimulatory effect on mineralization by neutralizing acidic condition.(31) Reduced bicarbonate level results in decreased pH, inhibiting mineralization, and increases dissolution of mineralization from bone matrix.(32,33) Consistently, conditioned media of Serpinf1(−/−) cell culture was more acidic than wild-type cell culture (Fig. 3C). Serpinf1(−/−) mouse osteoblasts also exhibited delayed nodule formation and significant decrease in calcified mineral production than wild-type osteoblasts, as visualized by Alizarin red staining (Fig. 3D).
Alkaline phosphatase (Alpl), ectonucleotide pyrophosphatase/phosphodiesterase 1 (Enpp1), and ecto-5′-nucleotidase (Nt5e, CD73) are enzymes that play critical roles for regulating mineralization by modulating levels of orthophosphate (Pi) and pyrophosphate (PPi).(34) The CD73 converts AMP to adenosine, inhibiting ectopic tissue calcification. Homozygous loss-of-function mutations in the NT5E gene lead to the calcification of joints and arteries.(35) Serpinf1(−/−) mouse osteoblasts expressed increased level of Nt5e, which converts AMP to adenosine, at week 3 in osteogenic culture condition (Fig. 3E). These data suggest that upregulation of a negative regulator for mineralization (Nt5e) and downregulation of alkaline phosphatase (Alpl) synergistically contribute to defective mineralization in Serpinf1(−/−) cells.
Importantly, toll-like receptor cascade, including Ptgfr, prostaglandin F receptor (Fig. 3F), was upregulated at week 0. Previously, IFITM5 was demonstrated to form a complex with FKBP11-CD81-FPRP for expression of interferon-induced genes in osteoblasts. FPRP (prostaglandin F2 receptor inhibitor) inhibits the binding of prostaglandin F2-alpha (PGF2-alpha) to its specific receptor (PTGFR) by decreasing the receptor number.(36) Thus, upregulated Ptgfr expression can be linked to reduced Ifitm5 expression in Serpinf1(−/−) cells, and the regulation of PTGFR by PEDF may be the missing link in the positive feedback loop of IFITM5 and PEDF.
At week 1, Serpinf1(−/−) cultures also showed increased expression of multiple genes involved in angiogenesis, including Grem1, Amot, Klf2, Id1, Adamts1, CD34, Adm, and Ptgs2 (Fig. 3G). In addition, genes in the pathways for response to wounding, cell adhesion, and regulation of ossification were upregulated (Fig. 3G). In aggregate, these data highlight that PEDF is a common mediator for osteogenesis and bone vascularization.
TGF-β signaling pathway is stimulated in type VI OI
Overly active TGF-β signaling contributes to phenotypes of some types of OI, as demonstrated with Crtap(−/−) mice, a mouse model of recessive OI type VII, and Col1α2+/G610C knock-in mouse, a mouse model of dominant OI.(11) To explore the possibility that TGF-β signaling is also implicated in the pathogenesis of type VI OI, we assessed the TGF-β signaling pathway in dermal fibroblasts from an individual with type VI OI and found significantly higher expression of TGF-β1 and its signaling target genes (Fig. 4A). Consistently, when cells were stimulated with TGF-β1, the major TGF-β isoform in bone, type VI OI dermal fibroblasts displayed higher level of phosphorylated SMAD3 than normal fibroblasts (Fig. 4B). In agreement with human cells, Serpinf1(−/−) mouse osteoblasts showed increased level of phosphorylated SMAD3 compared with wild-type mouse osteoblasts (Fig. 4C). The increased TGF-β pathway activation in Serpinf1(−/−) mouse osteoblasts was also reflected in increased expression of TGF-β1 responsive genes (Fig. 4D). Although basal level SMAD3 phosphorylation did not differ significantly between Serpinf1(−/−) and wild-type cells, RT-qPCR assay of gene expression revealed increased TGF-β activation in Serpinf1(−/−) even in the absence of TGF-β stimulation. TGF-β expression was also found to be higher in Serpinf1(−/−) than in wild-type during weeks 2 and 3 of osteogenic differentiation in vitro (Fig. 4E). Thrombospondin-1 (TSP-1, encoded by Thbs1) is a physiologic regulator of TGF-β bioactivation.(37) Increased TGF-β signaling in Serpinf1(−/−) was corroborated by upregulated TSP-1 expression in Serpinf1(−/−) (Fig. 4F). These data suggest that loss of PEDF augments transduction of TGF-β signaling pathway in osteoblasts and that increased TGF-β signaling contributes to the type VI OI phenotype.
Fig. 4.
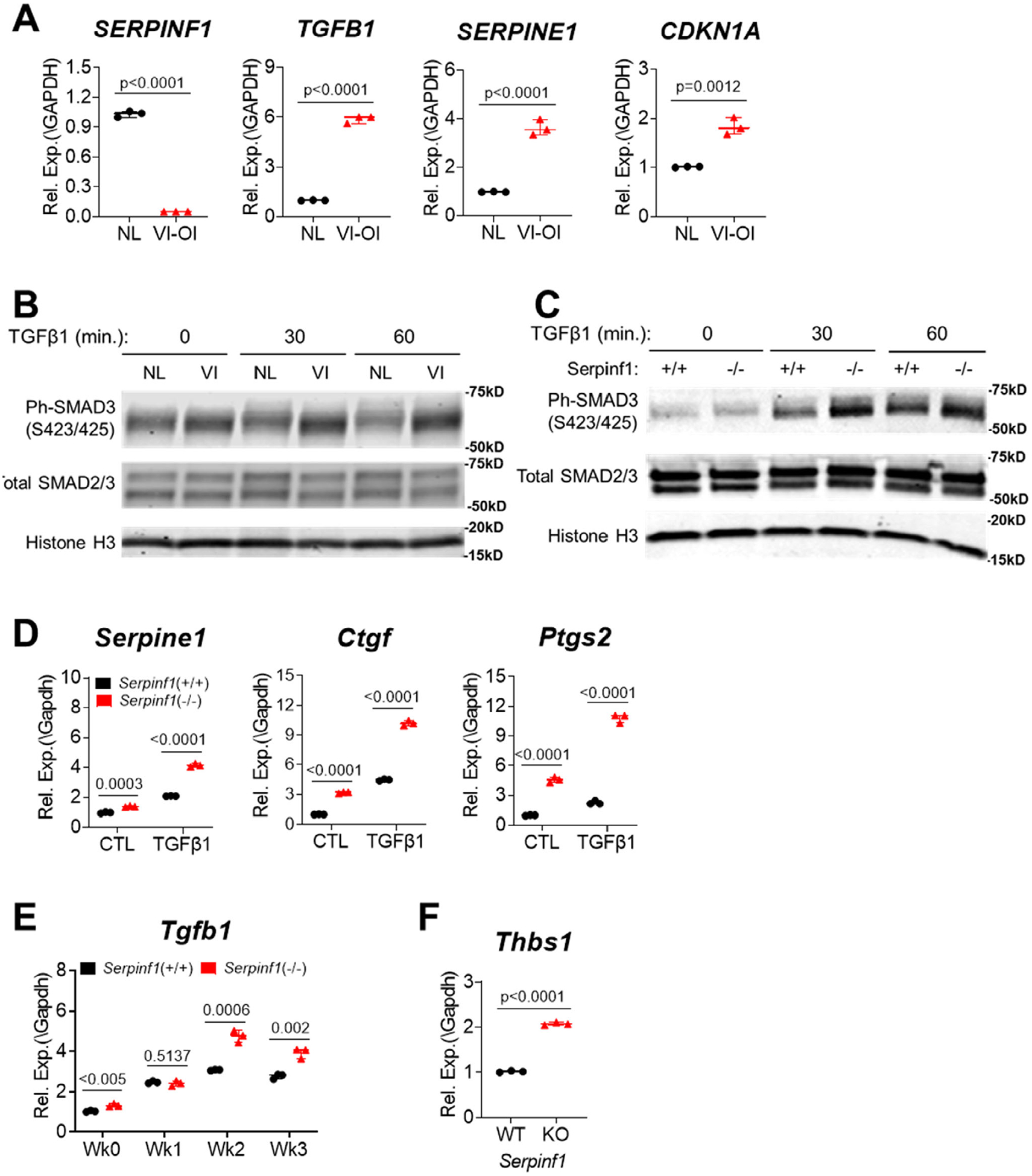
TGF-β signaling pathway is stimulated in type VI OI. (A) Real-time qPCR analysis of TGF-β signaling target gene expression in dermal fibroblasts from individual with type VI OI. N = 3, Student’s t test. (B) Human dermal fibroblasts of normal (NL) and type VI OI (VI) were stimulated with recombinant TGF-β1 for indicated time and TGF-β signaling activation was assessed by increased ph-SMAD3 in Western blot analysis. (C) Primary osteoblast cultures of Serpinf1(+/+) and Serpinf1(−/−) were stimulated with recombinant TGF-β1 for indicated time and TGF-β signaling activation was assessed by increased ph-SMAD3 in Western blot analysis. (D) Primary osteoblast cultures of Serpinf1(+/+) and Serpinf1(−/−) were stimulated with recombinant TGF-β1, and TGF-β signaling target gene expression was examined by real-time qPCR analysis. N = 3, Student’s t test. (E) Real-time qPCR analysis of Tgfb1 transcript. Serpinf1(−/−) osteoblasts expressed higher level of Tgfb1 than Serpinf1(+/+) osteoblasts during differentiation. N = 3, Student’s t test. (F) Real-time qPCR analysis of Thbs1 (TSP-1) transcript. N = 3, Student’s t test.
Antagonistic effects of PEDF and TGF-β in osteogenesis and bone vascularization
As shown in Fig. 5A, Serpinf1(−/−) decreased osteogenic marker gene expression and TGF-β1 stimulation exacerbated the reduced osteogenic marker gene expression, indicating the antagonistic relationship between PEDF pathway and TGF-β pathway in osteogenesis.
Fig. 5.
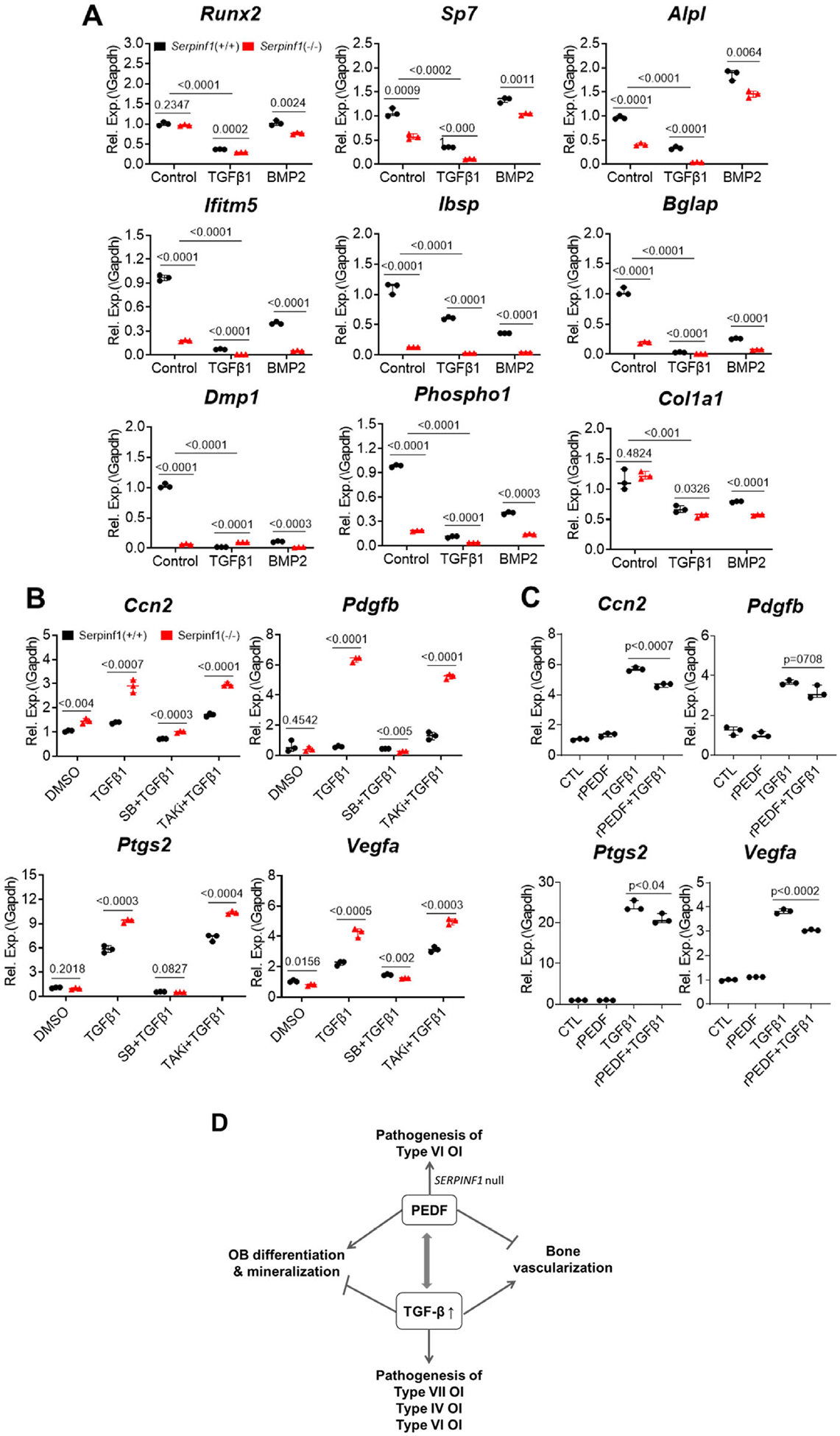
Antagonistic functions of PEDF and TGF-β in osteogenesis and bone vascularization. (A) Real-time qPCR analysis of osteogenic markers after treating cells with recombinant TGF-β1 or recombinant BMP2. TGF-β1 suppressed osteogenic gene expression, and loss of Serpinf1 furthered the suppression. N = 3, Student’s t test. (B) Real-time qPCR analysis of pro-angiogenic factor expression in Serpinf1(+/+) and Serpinf1(−/−) murine osteoblasts after treating cells with recombinant TGF-β1. TGF-β1 stimulated pro-angiogenic factor expression and loss of Serpinf1 furthered the suppression. N = 3, Student’s t test. (C) Real-time qPCR analysis of pro-angiogenic factor expression in mouse osteoblasts treated with recombinant TGF-β1 and PEDF. Stimulated expression of pro-angiogenic factor expression was suppressed by co-treatment with PEDF. N = 3, Student’s t test. (D) Proposed model of antagonistic actions of PEDF and TGF-β in osteogenesis and bone vascularization, and potential connection to human OI types.
PEDF is also well known for its potent anti-angiogenic activity in various tissues. Conversely, TGF-β promotes the expression of VEGF,(38) a master regulator of angiogenesis. However, despite these apparent opposite effects on angiogenesis, the functional interaction between PEDF and TGF-β has not yet been examined. Serpinf1(−/−) mouse osteoblasts expressed higher level of pro-angiogenic factors than wild-type osteoblasts (Fig. 5B). TGF-β1 treatment further enhanced the expression of pro-angiogenic factor genes in Serpinf1(−/−) mouse osteoblasts. Platelet-derived growth factor-B (PDGF-β) secreted by preosteoclasts stimulates MSC migration and contributes to bone formation and bone vascularization during bone remodeling.(39) The increased TGF-β signaling and its stimulatory effect on PDGF-β expression in Serpinf1(−/−) osteoblasts indicates that the increased vascularization in Serpinf1(−/−) mice may also be attributable to osteoblast-derived PDGF-β. The pro-angiogenic effect of TGF-β was abolished by pretreatment of cells with SB431542 (TGFbRI inhibitor), but not by Takinib (TAK inhibitor), suggesting that canonical TGF-β/SMAD signaling pathway is involved in the pro-angiogenic activity of TGF-β (Fig. 5B).
To further examine whether PEDF might block the pro-angiogenic effects of TGF-β, cells were co-treated with PEDF and TGF-β. RT-qPCR assay showed that recombinant PEDF partially suppressed the TGF-β-mediated stimulation of pro-angiogenic genes in Serpinf1(−/−) mouse osteoblasts (Fig. 5C). These results support the interpretation that antagonistic effects of PEDF pathway and TGF-β pathway on osteogenesis and bone vascularization contribute to type VI OI phenotype.
Discussion
OI types that are caused by mutations in the genes encoding non-collagen proteins have different pathophysiological mechanisms and clinical phenotypes. Elucidation of the cellular and molecular mechanisms underlying the pathogenesis of each OI type is critical to develop effective treatment strategies. In this study, we sought to better understand how loss of PEDF results in the pathological bone phenotype of type VI OI. Displaying reduced trabecular bone volume, the accumulation of unmineralized bone matrix, and an augmented mineral-to-matrix ratio, Serpinf1(−/−) mice partially mimicked bone structural and biochemical features found in type VI OI patients.(18) Using the Serpinf1(−/−) mice and their primary mouse osteoblasts, we found that PEDF deficiency significantly delayed osteoblast differentiation and ECM mineralization in vitro, suggesting that PEDF is involved in multiple stages of osteogenic differentiation. In addition, lack of PEDF promoted bone vascularization and increased CD31(+)/Endomucin(+) endothelial cells, suggesting PEDF involvement in coupling osteogenesis and vascularization in bone. This study also presented evidence that TGF-β signaling is augmented in Serpinf1(−/−) cells and that antagonistic action of PEDF and TGF-β is implicated in integrating osteogenesis with bone vascularization, further providing the underlying molecular mechanism of type VI OI pathogenesis (Fig. 5D).
PEDF maintains stem cell population, regulates cell cycle progression, and directs cell fate to promote neurogenesis and osteogenesis.(40) A study with recombinant PEDF and PEDF siRNA indicated that PEDF positively regulates alkaline phosphatase activity and mineral deposition in mesenchymal stem cells (MSCs) cultured in osteogenic media.(41) Another study with murine MSCs, osteoblast precursors, and human MSCs also demonstrated that PEDF directs MSCs fate toward osteoblasts and away from adipocytes through PEDF action as an agonist for the Wnt/β-catenin signaling pathway that suppresses PPARγ.(42) PEDF transduces signals via the ERK/MAPK pathway, thus activating downstream signaling cascades that lead to osteogenic differentiation. ECM deposition by osteoblasts is also modulated by the ERK/MAPK pathway.(43,44) Moreover, the ERK/MAPK pathway has a positive regulatory effect on type I collagen synthesis in human dermal fibroblasts.(45) Intriguingly, both COL1A1 transcripts and type I collagen protein secretion were decreased in osteoblasts with type V OI on days 10, 12, and 15, although not day 1, of differentiation. The same decrease in type I collagen transcripts and protein was found in osteoblasts with atypical type VI OI when examined on day 10 of differentiation.(25,26) Since type I collagen expression was also shown to increase upon stimulation with recombinant PEDF,(46) it is not unexpected that Serpinf1(−/−) murine calvarial osteoblasts have reduced Col1a1 expression at days 7, 14, and 21, although not day 0, of differentiation. Furthermore, Serpinf1(−/−) undifferentiated osteoblasts had decreased Col1a1 expression versus wild type after treatment with either BMP2 or TGF-β. Given the fact that ECM mineralization is contingent on collagen maturation and that PEDF has binding sites for type I collagen,(47) lack of PEDF may have caused incomplete maturation of bone matrix.
With this in mind, attempts to improve the bone phenotype of Serpinf1(−/−) mice by systemic reconstitution of PEDF have shown conflicting results. Intraperitoneal injection of human recombinant PEDF-containing microspheres increased trabecular bone volume and biomechanical parameters of bone plasticity in Serpinf1(−/−) mice but not in wild-type mice of young (19-day-old) and adult (6-month-old) age.(48) The same group was able to normalize bone matrix defects in iPSCs derived from an individual with type VI OI.(49) However, another group transduced a helper-dependent adenoviral vector to express human SERPINF1 in the mouse liver; although the serum PEDF level was restored, PEDF did not correct the bone phenotype of Serpinf1(−/−) mice.(50) These inconsistent outcomes point to the importance of locally produced, rather than circulating, PEDF with paracrine and/or autocrine function for proper mineralization and bone formation. This notion is corroborated by our findings in atypical type VI OI, which is caused by a missense mutation in IFITM5 (p.S40L) rather than mutations in the SERPINF1 gene. The individual with atypical type VI OI has normal serum level of PEDF but reduced PEDF production from cultured cells,(25) leading to accumulation of osteoid and severe bone dysplasia.
In contrast with the dramatic effects of PEDF deficiency on osteoblastogenesis, we did not detect any significant changes in osteoclast formation in Serpinf1(−/−) bone marrow-derived macrophages (BMMs), suggesting a non-cell autonomous effect of PEDF in osteoclast differentiation and maturation. In fact, PEDF expression in BMM-derived osteoclasts was approximately 50-fold less than that of primary osteoblasts.(51) Recombinant PEDF was shown to inhibit osteoclast formation via antagonizing receptor activator of NF-κB ligand (RANKL)-mediated cell survival.(52) The PEDF-mediated upregulation of osteoblast osteoprotegerin (OPG), a decoy receptor for RANKL, also contributes to the inhibitory role of PEDF on osteoclastogenesis and bone resorption.(52) In line with this notion, treatment of children with type VI OI with the RANKL antibody denosumab appeared to yield benefits of both increased bone mineral density and a reduced fracture rate.(53,54)
As exemplified in epiphyseal cartilage of developing bone, bone vascularization is a tightly regulated process in which the actions of pro-angiogenic factors (eg, VEGF) are counterbalanced by the actions of anti-angiogenic factors (eg, PEDF and TSP-1).(55,56) As presented in this study, Gattu and colleagues also reported increased TSP-1 transcripts in Serpinf1(−/−) osteoblast precursors in culture.(42) Our study with Serpinf1(−/−) murine bone revealed that loss of PEDF in bone results in increased vascularization both in metaphyseal area and periosteum and that PEDF activity counteracts the stimulatory effect of TGF-β signaling for expression of pro-angiogenic factors in bone. The excess levels of pro-angiogenic factors in Serpinf1(−/−) bone may have adverse effects on mineralization of matrix. Although TSP-1 is also known as a potent angiogenesis inhibitor,(57) augmented TSP-1 expression in Serpinf1(−/−) appears to be more reflected in the increased TGF-β signaling and inhibitory action on bone matrix mineralization.(58)
In this study, we provided evidence that TGF-β acts to counter PEDF and modulate osteogenesis and bone vascularization (Fig. 5D). An antagonistic relationship between PEDF and TGF-β was observed in other pathological conditions. In diabetic kidney disease, PEDF administration has a salutary effect against fibrotic factors, such as TGF-β and CTGF.(59) The tumor-suppressive effect of PEDF suppressed TGF-β production by pancreatic stellate cells.(60) And in nonalcoholic fatty liver disease, the antioxidative and anti-inflammatory property of PEDF also suppressed expression of TGF-β, to counter its pro-inflammatory function.(61)
TGF-β signaling has multiple functions in the ossification process.(62) Although TGF-β does not induce osteogenesis of MSCs, it increases the pool of committed osteoprogenitors by stimulating cell proliferation. In later-stage ossification, osteoblasts either undergo apoptosis or convert to osteocytes or bone lining cells. During this transdifferentiation of osteoblasts into osteocytes, TGF-β inhibits osteoblast apoptosis via MMP activation of latent TGF-β.(63) According to our transcriptome data from RNA-Seq of mouse osteoblasts, osteoblasts express relatively high levels of MMP-2, MMP-13, and MT1-MMP (MMP-14) among other MMPs. Membrane type 1-matrix metalloproteinase (MT1-MMP) was shown to activate MMP-2, −9, and −13.(64,65) Moreover, PEDF was demonstrated to effect its anti-metastatic activity by inhibiting surface localization of MT1-MMP.(66) Taken together, it is plausible to predict that upregulation of MT1-MMP may also reinforce the augmented TGF-β signaling in Serpinf1(−/−) murine osteoblasts. Whether inhibition of TGF-β signaling in type VI OI ameliorates any of its skeletal pathology remains to be examined in vivo. This study provides insight into a potentially more effective therapy for type VI OI, utilizing a combination of recombinant PEDF replacement approach and anti-TGF-β antibody treatment.
Supplementary Material
Acknowledgments
HK was supported by the Michael Geisman Fellowship from the Osteogenesis Imperfecta Foundation (OIF). This study was supported by National Institute of Diabetes and Digestive and Kidney Diseases (DK114158 to VD) and National Institute of Child Health and Human Development intramural funds (ZIA HD000408-38 to JCM). The authors thank Tianwei Li and James Iben at the Molecular Genomics Core (NICHD) for RNA sequencing and primary data processing.
Authors’ roles: HK, SAAC, AMB, AM, and VD designed and conducted experiments and analyzed data. HK, SAAC, VD, and JCM contributed to data interpretation. HK prepared the manuscript, with significant input from JCM, then edited and approved by SAAC, AMB, AM, VD, and SEC.
Footnotes
Disclosures
All authors state that they have no conflicts of interest.
Additional Supporting Information may be found in the online version of this article.
Data availability statement
The RNA-Seq data has been deposited with the NCBI BioSample repositor) (PRJNA756256) (https://www.ncbi.nlm.nih.gov/biosample) and prior to public release is available on reasonable request from the corresponding author.
References
- 1.Kang H, Aryal ACS, Marini JC. Osteogenesis imperfecta: new genes reveal novel mechanisms in bone dysplasia. Transl Res. 2017;181: 27–48. [DOI] [PubMed] [Google Scholar]
- 2.Crawford SE, Fitchev P, Veliceasa D, Volpert OV. The many facets of PEDF in drug discovery and disease: a diamond in the rough or split personality disorder? Expert Opin Drug Discov. 2013;8(7):769–792. [DOI] [PubMed] [Google Scholar]
- 3.Sekiya A, Okano-Kosugi H, Yamazaki CM, Koide T. Pigment epithelium-derived factor (PEDF) shares binding sites in collagen with heparin/heparan sulfate proteoglycans. J Biol Chem. 2011; 286(30):26364–26374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Becker J, Semler O, Gilissen C, et al. Exome sequencing identifies truncating mutations in human SERPINF1 in autosomal-recessive osteogenesis imperfecta. Am J Hum Genet. 2011;88(3):362–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Homan EP, Rauch F, Grafe I, et al. Mutations in SERPINF1 cause osteogenesis imperfecta type VI. J Bone Miner Res. 2011;26(12):2798–2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cho SY, Ki CS, Sohn YB, Kim SJ, Maeng SH, Jin DK. Osteogenesis imperfecta type VI with severe bony deformities caused by novel compound heterozygous mutations in SERPINF1. J Korean Med Sci. 2013;28(7):1107–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rauch F, Husseini A, Roughley P, Glorieux FH, Moffatt P. Lack of circulating pigment epithelium-derived factor is a marker of osteogenesis imperfecta type VI. J Clin Endocrinol Metab. 2012;97(8):E1550–E1556. [DOI] [PubMed] [Google Scholar]
- 8.Venturi G, Gandini A, Monti E, et al. Lack of expression of SERPINF1, the gene coding for pigment epithelium-derived factor, causes progressively deforming osteogenesis imperfecta with normal type I collagen. J Bone Miner Res. 2012;27(3):723–728. [DOI] [PubMed] [Google Scholar]
- 9.Glorieux FH, Ward LM, Rauch F, Lalic L, Roughley PJ, Travers R. Osteogenesis imperfecta type VI: a form of brittle bone disease with a mineralization defect. J Bone Miner Res. 2002;17(1):30–38. [DOI] [PubMed] [Google Scholar]
- 10.Fratzl-Zelman N, Schmidt I, Roschger P, et al. Unique micro- and nano-scale mineralization pattern of human osteogenesis imperfecta type VI bone. Bone. 2015;73:233–241. [DOI] [PubMed] [Google Scholar]
- 11.Grafe I, Yang T, Alexander S, et al. Excessive transforming growth factor-beta signaling is a common mechanism in osteogenesis imperfecta. Nat Med. 2014;20(6):670–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bianchi L, Gagliardi A, Maruelli S, et al. Altered cytoskeletal organization characterized lethal but not surviving Brtl+/− mice: insight on phenotypic variability in osteogenesis imperfecta. Hum Mol Genet. 2015;24(21):6118–6133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doll JA, Stellmach VM, Bouck NP, et al. Pigment epithelium-derived factor regulates the vasculature and mass of the prostate and pancreas. Nat Med. 2003;9(6):774–780. [DOI] [PubMed] [Google Scholar]
- 14.David V, Martin A, Hedge AM, Rowe PS. Matrix extracellular phosphor-glycoprotein (MEPE) is a new bone renal hormone and vascularization modulator. Endocrinology. 2009;150(9):4012–4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roche B, David V, Vanden-Bossche A, et al. Structure and quantification of microvascularisation within mouse long bones: what and how should we measure? Bone. 2012;50(1):390–399. [DOI] [PubMed] [Google Scholar]
- 16.Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–930. [DOI] [PubMed] [Google Scholar]
- 18.Bogan R, Riddle RC, Li Z, et al. A mouse model for human osteogenesis imperfecta type VI. J Bone Miner Res. 2013;28(7):1531–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murshed M, Harmey D, Millan JL, McKee MD, Karsenty G. Unique coexpression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes Dev. 2005;19(9):1093–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hojo H, Ohba S, He X, Lai LP, McMahon AP. Sp7/Osterix is restricted to bone-forming vertebrates where it acts as a dlx co-factor in osteoblast specification. Dev Cell. 2016;37(3):238–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roberts S, Narisawa S, Harmey D, Millan JL, Farquharson C. Functional involvement of PHOSPHO1 in matrix vesicle-mediated skeletal mineralization. J Bone Miner Res. 2007;22(4):617–627. [DOI] [PubMed] [Google Scholar]
- 22.George A, Sabsay B, Simonian PA, Veis A. Characterization of a novel dentin matrix acidic phosphoprotein. Implications for induction of biomineralization. J Biol Chem. 1993;268(17):12624–12630. [PubMed] [Google Scholar]
- 23.Cho TJ, Lee KE, Lee SK, et al. A single recurrent mutation in the 5′-UTR of IFITM5 causes osteogenesis imperfecta type V. Am J Hum Genet. 2012;91(2):343–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Semler O, Garbes L, Keupp K, et al. A mutation in the 5’-UTR of IFITM5 creates an in-frame start codon and causes autosomal-dominant osteogenesis imperfecta type V with hyperplastic callus. Am J Hum Genet. 2012;91(2):349–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farber CR, Reich A, Barnes AM, et al. A novel IFITM5 mutation in severe atypical osteogenesis imperfecta type VI impairs osteoblast production of pigment epithelium-derived factor. J Bone Miner Res. 2014;29(6):1402–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reich A, Bae AS, Barnes AM, et al. Type V OI primary osteoblasts display increased mineralization despite decreased COL1A1 expression. J Clin Endocrinol Metab. 2015;100(2):E325–E332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dawson DW, Volpert OV, Gillis P, et al. Pigment epithelium-derived factor: a potent inhibitor of angiogenesis. Science. 1999;285(5425):245–248. [DOI] [PubMed] [Google Scholar]
- 28.Albert C, Jameson J, Smith P, Harris G. Reduced diaphyseal strength associated with high intracortical vascular porosity within long bones of children with osteogenesis imperfecta. Bone. 2014;66:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kusumbe AP, Ramasamy SK, Adams RH. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature. 2014;507(7492):323–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang J, Duh EJ, Caldwell RB, Behzadian MA. Antipermeability function of PEDF involves blockade of the MAP kinase/GSK/beta-catenin signaling pathway and uPAR expression. Invest Ophthalmol Vis Sci. 2010;51(6):3273–3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X, Schroder HC, Schlossmacher U, et al. Modulation of the initial mineralization process of SaOS-2 cells by carbonic anhydrase activators and polyphosphate. Calcif Tissue Int. 2014;94(5):495–509. [DOI] [PubMed] [Google Scholar]
- 32.Bushinsky DA, Sessler NE. Critical role of bicarbonate in calcium release from bone. Am J Physiol. 1992;263(3 Pt 2):F510–F515. [DOI] [PubMed] [Google Scholar]
- 33.Bushinsky DA. Acidosis and bone. Miner Electrolyte Metab. 1994; 20(1–2):40–52. [PubMed] [Google Scholar]
- 34.Millan JL. The role of phosphatases in the initiation of skeletal mineralization. Calcif Tissue Int. 2013;93(4):299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.St Hilaire C, Ziegler SG, Markello TC, et al. NT5E mutations and arterial calcifications. N Engl J Med. 2011;364(5):432–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hanagata N, Li X. Osteoblast-enriched membrane protein IFITM5 regulates the association of CD9 with an FKBP11-CD81-FPRP complex and stimulates expression of interferon-induced genes. Biochem Biophys Res Commun. 2011;409(3):378–384. [DOI] [PubMed] [Google Scholar]
- 37.Crawford SE, Stellmach V, Murphy-Ullrich JE, et al. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell. 1998;93(7):1159–1170. [DOI] [PubMed] [Google Scholar]
- 38.Ferrari G, Cook BD, Terushkin V, Pintucci G, Mignatti P. Transforming growth factor-beta 1 (TGF-beta1) induces angiogenesis through vascular endothelial growth factor (VEGF)-mediated apoptosis. J Cell Physiol. 2009;219(2):449–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie H, Cui Z, Wang L, et al. PDGF-BB secreted by preosteoclasts induces angiogenesis during coupling with osteogenesis. Nat Med. 2014;20(11):1270–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brook N, Brook E, Dharmarajan A, Chan A, Dass CR. Pigment epithelium-derived factor regulation of neuronal and stem cell fate. Exp Cell Res. 2020;389(2):111891. [DOI] [PubMed] [Google Scholar]
- 41.Li F, Song N, Tombran-Tink J, Niyibizi C. Pigment epithelium-derived factor enhances differentiation and mineral deposition of human mesenchymal stem cells. Stem Cells. 2013;31(12):2714–2723. [DOI] [PubMed] [Google Scholar]
- 42.Gattu AK, Swenson ES, Iwakiri Y, et al. Determination of mesenchymal stem cell fate by pigment epithelium-derived factor (PEDF) results in increased adiposity and reduced bone mineral content. FASEB J. 2013;27(11):4384–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lai CF, Chaudhary L, Fausto A, et al. Erk is essential for growth, differentiation, integrin expression, and cell function in human osteoblastic cells. J Biol Chem. 2001;276(17):14443–14450. [DOI] [PubMed] [Google Scholar]
- 44.Wu CC, Li YS, Haga JH, et al. Roles of MAP kinases in the regulation of bone matrix gene expressions in human osteoblasts by oscillatory fluid flow. J Cell Biochem. 2006;98(3):632–641. [DOI] [PubMed] [Google Scholar]
- 45.Bhogal RK, Bona CA. Regulatory effect of extracellular signal-regulated kinases (ERK) on type I collagen synthesis in human dermal fibroblasts stimulated by IL-4 and IL-13. Int Rev Immunol. 2008;27(6): 472–496. [DOI] [PubMed] [Google Scholar]
- 46.Li F, Song N, Tombran-Tink J, Niyibizi C. Pigment epithelium derived factor suppresses expression of Sost/Sclerostin by osteocytes: implication for its role in bone matrix mineralization. J Cell Physiol. 2015; 230(6):1243–1249. [DOI] [PubMed] [Google Scholar]
- 47.Meyer C, Notari L, Becerra SP. Mapping the type I collagen-binding site on pigment epithelium-derived factor. Implications for its antiangiogenic activity. J Biol Chem. 2002;277(47):45400–45407. [DOI] [PubMed] [Google Scholar]
- 48.Belinsky GS, Sreekumar B, Andrejecsk JW, et al. Pigment epithelium-derived factor restoration increases bone mass and improves bone plasticity in a model of osteogenesis imperfecta type VI via Wnt3a blockade. FASEB J. 2016;30(8):2837–2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Belinsky GS, Ward L, Chung C. Pigment epithelium-derived factor (PEDF) normalizes matrix defects in iPSCs derived from osteogenesis imperfecta type VI. Rare Dis. 2016;4(1):e1212150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rajagopal A, Homan EP, Joeng KS, et al. Restoration of the serum level of SERPINF1 does not correct the bone phenotype in Serpinf1 null mice. Mol Genet Metab. 2016;117(3):378–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tombran-Tink J, Barnstable CJ. Osteoblasts and osteoclasts express PEDF, VEGF-A isoforms, and VEGF receptors: possible mediators of angiogenesis and matrix remodeling in the bone. Biochem Biophys Res Commun. 2004;316(2):573–579. [DOI] [PubMed] [Google Scholar]
- 52.Akiyama T, Dass CR, Shinoda Y, Kawano H, Tanaka S, Choong PF. PEDF regulates osteoclasts via osteoprotegerin and RANKL. Biochem Biophys Res Commun. 2010;391(1):789–794. [DOI] [PubMed] [Google Scholar]
- 53.Semler O, Netzer C, Hoyer-Kuhn H, Becker J, Eysel P, Schoenau E. First use of the RANKL antibody denosumab in osteogenesis imperfecta type VI. J Musculoskelet Neuronal Interact. 2012;12(3):183–188. [PubMed] [Google Scholar]
- 54.Hoyer-Kuhn H, Netzer C, Koerber F, Schoenau E, Semler O. Two years’ experience with denosumab for children with osteogenesis imperfecta type VI. Orphanet J Rare Dis. 2014;9:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Horner A, Bishop NJ, Bord S, et al. Immunolocalisation of vascular endothelial growth factor (VEGF) in human neonatal growth plate cartilage. J Anat. 1999;194(Pt 4):519–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carulli C, Innocenti M, Brandi ML. Bone vascularization in normal and disease conditions. Front Endocrinol (Lausanne). 2013;4:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Armstrong LC, Bornstein P. Thrombospondins 1 and 2 function as inhibitors of angiogenesis. Matrix Biol. 2003;22(1):63–71. [DOI] [PubMed] [Google Scholar]
- 58.Ueno A, Miwa Y, Miyoshi K, et al. Constitutive expression of thrombospondin 1 in MC3T3-E1 osteoblastic cells inhibits mineralization. J Cell Physiol. 2006;209(2):322–332. [DOI] [PubMed] [Google Scholar]
- 59.Wang JJ, Zhang SX, Mott R, et al. Salutary effect of pigment epithelium-derived factor in diabetic nephropathy: evidence for anti-fibrogenic activities. Diabetes. 2006;55(6):1678–1685. [DOI] [PubMed] [Google Scholar]
- 60.Principe DR, DeCant B, Diaz AM, et al. PEDF inhibits pancreatic tumor-igenesis by attenuating the fibro-inflammatory reaction. Oncotarget. 2016;7(19):28218–28234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yoshida T, Akiba J, Matsui T, et al. Pigment epithelium-derived factor (PEDF) prevents hepatic fat storage, inflammation, and fibrosis in dietary steatohepatitis of mice. Dig Dis Sci. 2017;62(6):1527–1536. [DOI] [PubMed] [Google Scholar]
- 62.Wu M, Chen G, Li YP. TGF-beta and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. 2016;4:16009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Karsdal MA, Larsen L, Engsig MT, et al. Matrix metalloproteinase-dependent activation of latent transforming growth factor-beta controls the conversion of osteoblasts into osteocytes by blocking osteoblast apoptosis. J Biol Chem. 2002;277(46):44061–44067. [DOI] [PubMed] [Google Scholar]
- 64.Knauper V, Will H, Lopez-Otin C, et al. Cellular mechanisms for human procollagenase-3 (MMP-13) activation. Evidence that MT1-MMP (MMP-14) and gelatinase a (MMP-2) are able to generate active enzyme. J Biol Chem. 1996;271(29):17124–17131. [DOI] [PubMed] [Google Scholar]
- 65.Esparza J, Vilardell C, Calvo J, et al. Fibronectin upregulates gelatinase B (MMP-9) and induces coordinated expression of gelatinase A (MMP-2) and its activator MT1-MMP (MMP-14) by human T lymphocyte cell lines. A process repressed through RAS/MAP kinase signaling pathways. Blood. 1999;94(8):2754–2766. [PubMed] [Google Scholar]
- 66.Ladhani O, Sanchez-Martinez C, Orgaz JL, Jimenez B, Volpert OV. Pigment epithelium-derived factor blocks tumor extravasation by suppressing amoeboid morphology and mesenchymal proteolysis. Neoplasia. 2011;13(7): 633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-Seq data has been deposited with the NCBI BioSample repositor) (PRJNA756256) (https://www.ncbi.nlm.nih.gov/biosample) and prior to public release is available on reasonable request from the corresponding author.