

Abstract
The rapid progression of genomics and proteomics has been driven by the advent of advanced sequencing technologies, large, diverse, and readily available omics datasets, and the evolution of computational data processing capabilities. The vast amount of data generated by these advancements necessitates efficient algorithms to extract meaningful information. K-mers serve as a valuable tool when working with large sequencing datasets, offering several advantages in computational speed and memory efficiency and carrying the potential for intrinsic biological functionality. This review provides an overview of the methods, applications, and significance of k-mers in genomic and proteomic data analyses, as well as the utility of absent sequences, including nullomers and nullpeptides, in disease detection, vaccine development, therapeutics, and forensic science. Therefore, the review highlights the pivotal role of k-mers in addressing current genomic and proteomic problems and underscores their potential for future breakthroughs in research.
Keywords: K-mers, Nullomers, Nullpeptides, Primes, Neomers, Sequence Analysis
Graphical Abstract
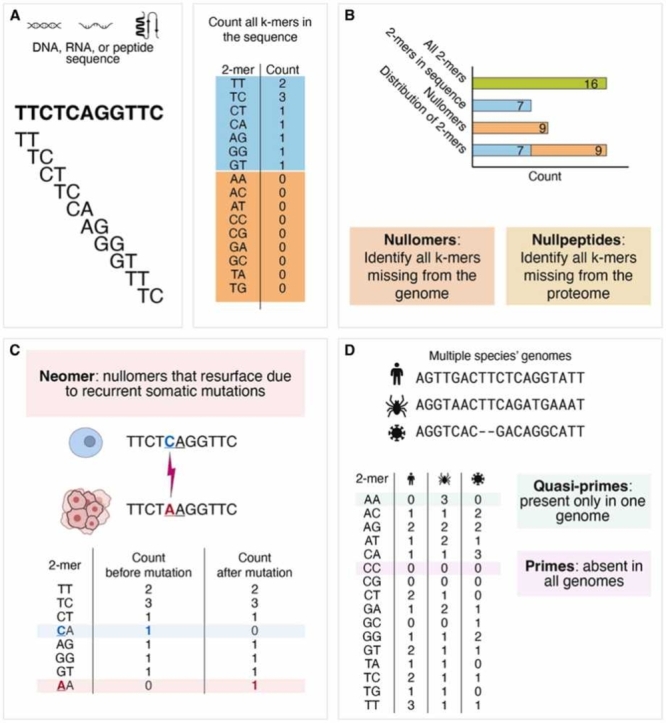
Fig. 1: Introduction to k-mers. A. All possible 2-mers, or k-mers with two nucleotides, are listed. In a specific DNA sequence, all 2-mers are recorded for frequency analysis. B. Nullomers, or possible 2-mers not in the genome, are counted by subtracting the observed 2-mers from all possible 2-mers. Nullpeptides are k-mers missing from proteomes. C. In a mutated sequence, neomers, or nullomers that resurface due to somatic mutations, can occur. AA is a neomer in this mutated sequence. D. When analyzing multiple genomes or sequences, primes, k-mers not present in any of the sequences, can be identified. There is one prime (CC) in these three sequences. Quasi-primes, or k-mers that only occur in one sequence (AA), can be identified.
1. Introduction
In recent years, the field of genomics has undergone a significant transformation due to advances in sequencing technologies and the generation of both large and diverse datasets [1], [2]. The advancements have enabled the efficient acquisition of vast amounts of genomic information within a short timeframe [3]. However, this also poses the challenge of deriving meaningful insights from complex, high-dimensional datasets to address sequence analysis problems. Conventional analytical approaches have struggled with the unprecedented scale and complexity of genomic datasets [4], [5], necessitating the development of efficient algorithms for extracting relevant information.
K-mers, defined as contiguous nucleotide or amino acid sequences of fixed length k (Table 1; Fig. 1A), have become integral in addressing these challenges. K-mer-based algorithms are widely utilized across genomic and proteomic applications and offer several advantages as fundamental units for analyses [6], [7]. First, they enable the fast retrieval of targeted sequences from next-generation sequencing data, facilitating efficient exploration and downstream analysis-based tasks [8], [9]. Moreover, k-mers can have intrinsic biological significance, and their distribution and frequency can provide insights into genomic characteristics such as repetitive elements, functional regions, genomic variation, and DNA damage and repair mechanisms [8], [9], [10], [11], [12], [13], [14], [15], [16].
Table 1.
Relevant definitions.
| Term | Definition |
|---|---|
| Absent Word | Word that does not occur in a given genome or proteome; also see definitions for Nullomer and Nullpeptide[165] |
| First Order Nullomer | Nullomer where any single base substitution at any position along the k-mer still yields a nullomeric sequence[164] |
| Frequentmer | Short sequence that is specific and recurrently observed in either patient or healthy control samples, but not in both[146] |
| High Order Nullomer | Nullomer whose mutated sequence is still a nullomer[164] |
| K-mer | Contiguous subsequence of length k derived from a longer sequence |
| Minimal Absent Word | Absent word where removing the leftmost or rightmost nucleotide results in a sequence that is no longer an absent word[165] |
| Minimizer | Minimum value k-mer selected to represent a longer k-mer or group of k-mers to reduce memory consumption and run-time[199] |
| Neomer | Nullomer that resurfaces due to somatic mutations in cancer[29] |
| Nullomer | K-mer sequence absent from a specific genome[159], [164] |
| Nullpeptide | K-mer that does not exist within a proteome[163] |
| Prime | K-mer that does not exist in any genome or proteome[159] |
| Quasi-Prime | K-mer that is only found in one species or sequence[172] |
| Strobemers | Two or more linked shorter k-mers, where the combination of linked k-mers is determined by a hash function[200] |
| Syncmers | Set of k-mers defined by the position of the smallest-valued substring of length s < k within the k-mer[199] |
| Super K-mer | Substring of maximal length wherein all k-mers within it share the same minimizer[9], [45] |
Fig. 1.

Introduction to k-mers. A. All possible 2-mers, or k-mers with two nucleotides, are listed. In a specific DNA sequence, all 2-mers are recorded for frequency analysis. B. Nullomers, or possible 2-mers not in the genome, are counted by subtracting the observed 2-mers from all possible 2-mers. Nullpeptides are k-mers missing from proteomes. C. In a mutated sequence, neomers, or nullomers that resurface due to somatic mutations, can occur. AA is a neomer in this mutated sequence. D. When analyzing multiple genomes or sequences, primes, k-mers not present in any of the sequences, can be identified. There is one prime (CC) in these three sequences. Quasi-primes, or k-mers that only occur in one sequence (AA), can be identified.
K-mers can serve as valuable clinical biomarkers for detecting pathogens [17], [18], [19], antimicrobial resistance [17], [20], [21], and human diseases [16], [22], [23], [24]. Additionally, k-mers identified from genomic or transcriptomic data from tumor samples or liquid biopsies prove valuable in cancer diagnosis, prognosis, and treatment [16], [22], [25], [26], [27]. K-mers can be categorized into various subtypes, each with distinct applications. Notably, nullomers and nullpeptides are k-mers missing from a genome or proteome, respectively (Fig. 1B). The emergence of nullomers and nullpeptides during cancer development can be used as a biomarker for cancer detection (Fig. 1C) [22], [28], [29], with certain nullpeptides even exhibiting cancer cell-killing properties [30], [31].
This review provides a comprehensive overview of the applications of k-mers in addressing analytic challenges across genomics and proteomics. It explores the development of k-mer-based tools and reviews research on the utilization of subsets of k-mers as informative indicators that reveal insights into genome variation, population genetics, and disease associations. As new k-mer-based algorithms are developed that broaden the applications for sequence analysis, the biological implications of k-mers will continue to grow.
2. The selection of k
Any genomic sequence can be fragmented into consecutive k-mers of length k. The selection of k can vary significantly depending on both the dataset and application. For example, in de novo genome assembly, shorter k-mer lengths may reduce the quality of the resulting contigs, whereas longer k-mers have a higher chance of including errors [32]. The complexity of the k-mer set, or all possible k-mers, increases exponentially with k and is equal to 4k with the four base pair alphabet for DNA and RNA. For smaller values of k, the complexity may be insufficient to represent or distinguish long sequences because the possible k-mers may appear in a genome many times and be observed in a multitude of species.
Alternatively, longer k-mers can lead to a sparser representation of the dataset in the k-mer space, allowing better differentiation between samples due to k-mers appearing in a very small number of species [33]. However, this also increases computational complexity and can result in too few common k-mers, rendering some applications, such as phylogenetic analysis or biomarker detection, impossible. For instance, in a three Gbp genome, the probability of observing a given 16-mer is 0.5. However, due to the exponential growth of the k-mer space, this probability drops to 0.01 at k = 19 [34].
This conundrum emphasizes one of the inherent limitations of some k-mer-based approaches. Although longer k-mers may more accurately reflect the biological function of sequences such as transcription factor binding sites, they can result in severe overfitting problems due to sparse k-mer counts. To address this issue, some methods using gapped k-mers as features have been introduced [13], [35].
3. Applications of k-mers
The introduction of k-mers has made it possible to process large and complex genetic data with reasonable time complexity. This improvement has allowed for the extraction of essential patterns and characteristics within both genomes and proteomes, tasks that were previously inefficient or unfeasible. As a result, the past decade has seen the development of numerous applications that utilize the capabilities of k-mers. These include methods for k-mer counting and frequency analysis, sequence alignment, genome assembly, comparative genomics, metagenomics, metaproteomics, and protein structure prediction (Table 2; Supplementary Table 1).
Table 2.
Current k-mer-based software methods and databases.
| Application | Description | Tools(reference) |
|---|---|---|
| K-mer Counting | K-mer counting is preliminary to many applications such as genome assembly, error correction, sequence alignment, and classification. Common approaches include hash tables with lock-free-based tools, hash tables with lock-based tools, disk-based tools, bloom-filter tools, quotient filtering, and burst-tree tools. | BFCounter[201], CHTKC[202], Discount[203], DSK2[204], GECKO[205], Gerbil[206], Jellyfish2[8], KAnalyze[207], KCMBT[208], KCOSS[209], KHMer[210], KMC3[45], KmerAnalysis.jl[211], Kmerator[212], Kounta[213], Krust[214], Meryl[46], MSPKmerCounter[215], REINDEER[47], RNA-Skim[60], Salmon[59], SEEKR[134], SeqKit[216], Seqtrie[217], Squeakr[48], SWAPCounter[36], Tallymer[218], Turtle[219], VLmer[220] |
| Frequency Analysis | K-mer spectra can be used to estimate genome size and complexity prior to assembly. This information can then be used to optimize the assembly process, detect and correct errors in the sequencing reads, and evaluate the quality of a genome assembly. | GenomeScope[221], GenomeTester4[222], KAnalyze[207], KAT[223], KHMer[210] KmerGenie[224], KmerStream[225], ntCard[226], Squeakr[48] |
| Sequence Indexing | K-mer searches serve as effective proxies for both exact and approximate sequence searches in unassembled datasets. Current methods are able to determine the presence or absence of any k-mer within collections of up to ∼2500 datasets. | COBS[49], DiscoverY[227], HowDeSBT[228], KmerGO[229], Kmerind[230], Mantis[231], MetaGraph[232], PAC[233], RecoverY[234], SBT[235], SeqOthello[236], SSHash[237], VarGeno[238] |
| Genome Assembly | In de novo genome assembly, sequencing reads undergo fragmentation into k-mers, and their overlaps are employed to assemble longer contiguous sequences. This process commonly utilizes k-mers to build De Bruijn graphs or implements overlap-layout-consensus methods. | Allpaths-LG[239], Bifrost[240], Canu[241], Cortex[242], ELBA[243], KAT[223], MEGAHIT[244], Merqury[46], QUAST-LG[245], SKESA[246], SPAdes[75], TandemQUAST[247], TandemMapper[248] |
| Sequence Comparison | Alignment-free methods are increasingly used for DNA and protein sequence comparison since they are much faster than traditional alignment-based approaches. Most alignment-free algorithms are based on the word or k-mer composition of the sequences under study.[249] | BBMap[250], Bowtie2[251], [252], BWA[253], [254], [255], iMOKA[256], MiniMap2[71] |
| Taxonomic Classification | In sequence composition-based methods, the frequency and distribution of k-mers in metagenomic data are analyzed to assess genome similarity across various taxonomic ranks.[257], [258] | ARK[259], BinDash[122], Bracken[260], CDKAM[261], CLARK[262], Dashing[124], fmh-funprofiler[128], Genometa[263], Kaiju[138], KMCP[264], KmerFinder[265], Kraken2[136], KrakenUniq[19], LMAT[266], Mash[72], Mash Screen[34], Matchtigs[267], MetaCache[268], MetaPalette[269], MetaProFi[50], NIQKI[126], SEK[270], StrainSeeker[271], SuperSampler[127], TACOA[272], Taxonomer[273], TETRA[274], VirFinder[18], WGSQuikr[275] |
| Phylogeny Reconstruction | Pairwise evolutionary distances between protein or nucleic acid sequences and phylogenetic distances can be estimated from the number of k-mer matches between two sequences. Alignment-free sequence comparison quantifies distance using the decay of the number of k-mer matches between two sequences and compares the results to known phylogenetic trees.[249] | AAF[276], FSWM[277], PhyloPythia[278], Skmer[279], Slope-SpaM[280], SlopeTree[281] |
| Protein Sequence Searching and Alignment | Sequence match is determined by aligning translated DNA sequences to a reference protein database. | BLAT[65], BLAST[68], BLASTX[64], [282], DIAMOND[283], MMSeqs2[284], PAUDA[285], RAPSearch2[286], USEARCH & UBLAST[287] |
| Databases | K-mer databases provide k-mer sequences that are present or absent in each species. | kmerDB.com[171], nullomers.org[162] |
3.1. K-mer counting
Counting the occurrence of all distinct k-mers in biological sequences is a crucial step in many bioinformatic applications such as genome assembly, sequence alignment, sequence clustering, error correction of sequencing reads, and genome size estimation (Fig. 1A; Fig. 2A; Table 2) [36]. Pattern recognition tasks in sequence analysis can be efficiently reduced to counting k-mers of length k. For example, k-mer counting tools are often used in the identification of repetitive elements, such as transposable elements and tandem repeats. Repetitive elements are characterized by high k-mer counts [37], [38]. In contrast, variable genomic regions, including single nucleotide polymorphisms [39] and structural variations associated with phenotypic differences or diseases, may exhibit differences in k-mer counts during alignment across various genomes (Fig. 2B) [25], [40]. Unique genomic regions are defined by lower k-mer counts [25], [41], [42], [43], [44].
Fig. 2.

Applications of k-mers. A. K-mer counting and frequency analysis are crucial steps in various bioinformatic applications, including detecting sample contamination. B. K-mers are used in graph-based genome assembly and identification of genetic variants. C. In sequence assembly, k-mers are utilized for sequence alignment. Sequencing reads are fragmented into k-mers, and overlaps between k-mers are identified to reconstruct the original sequence. D. In adaptive sequencing, k-mer counting allows for the identification of unique sequences. K-mer based variant-filtering methods are used for improving accuracy in genome assembly; algorithms will filter false positives from alignments. E. K-mers are utilized in genome editing to identify suitable target sites and design guide RNAs, and efficient k-mer indexing enables primer candidates to be identified with low off-target site potential. F. K-mers are used for taxonomic profiling and classification in comparative genomics and metagenomics.
In 2018, Manekar and Sathe performed a benchmark study comparing several common k-mer counting methods [10]. Bioinformatic tools such as Jellyfish [8], KMC3 [45], Meryl [46], REINDEER [47], and Squeakr [48] leverage optimized data structures and algorithms and are commonly written in low-level programming languages to process large-scale genomic datasets and accurately determine k-mer abundance (Supplementary Table 1) [9]. Jellyfish is a simple k-mer counter toolkit for lengths up to 31 bases that utilizes a combination of a Bloom filter and lock-free hash table, but is slower than more recent applications [8]. Squeakr uses a counting quotient filter, providing memory efficiency, faster processing for larger k-mers, and also the option to obtain approximate counts at a much lower computational cost for applications where exact counts are not required [48]. REINDEER was constructed to efficiently store and query exact k-mer abundances across multiple datasets, utilizing De Bruijn graphs for optimal indexing. Other tools include COBS, an inverted index based on Bloom filters [49], and MetaProFi, which utilizes k-mers to profile and query protein and nucleotide sequence data based on k-mer signatures [50]. MetaProFi uses Bloom filters for rapid searches in large sequence datasets and builds indexes of nucleic acid or amino acid sequences, enabling protein-level sequence comparison.
3.2. K-mer distribution and frequency analysis
A sequencing dataset can be characterized using the distribution of k-mers, and this is referred to as the k-mer spectrum or histogram. Different taxonomic domains are known to have differing distributions of genomic k-mer spectra [51]. Chor et al. discovered that archaeal and bacterial species display a unimodal spectra, while tetrapods are represented by multimodal spectra [51]. Tetrapods’ spectra are characterized by specific GC content and CpG suppression. Notably, the protozoa Entamoeba histolytica exhibits CpG suppression but lacks multimodal k-mer spectra, suggesting CpG suppression alone does not determine modality [51]. Typically, a moderately heterozygous diploid organism has a k-mer spectrum with four apparent coverage peaks: sequencing errors (low coverage), unique genomic sequences from heterozygous loci, all homozygous loci in the genome, and genomic duplications, which is usually a smaller peak. In terms of the first peak, k-mer spectrum-based algorithms have been developed to identify and remove sequencing read errors based on the infrequency of these k-mers [52], [53], [54].
The frequency of specific k-mers can reveal various aspects of the genomic structure and complexity of a biological sample (Fig. 1A; Fig. 2A) [9], [10], [51], [55]. By examining the empirical frequencies of k-mers, the GC-content, CpG suppression, repeat content, heterozygosity, and sequencing coverage of the sample can be inferred [51], [56], [57]. Bussi et al. analyzed k-mer frequency patterns in over 5000 archaea, bacteria, and eukaryotic complete genomes and found that sequence space coverage (SSC) depends heavily on both genome length and GC-content [56], a result that was consistent with findings by Liu et al. [56], [57]. Bussi et al. also found that maintaining a non-0.5 GC-content was shown to influence the prevalence of specific k-mers, leading to decreased sequence entropy and SSC [56].
Analyzing transcriptomic data, especially in non-model organisms or meta-transcriptomes, can be challenging due to the levels of complexity, variability, and noise. In such cases, k-mer frequency analysis can be utilized to quantify and detect subtle differences in gene expression or organism composition that may be missed by other methods [58]. K-mer-based software such as Salmon [59] and RNA-Skim [60] can accurately quantify transcript abundance levels from RNA-Seq data. Importantly, Salmon, the successor of Sailfish [61], corrects for fragment GC-content bias to improve the accuracy of abundance estimates [59].
In a proteomic analysis, Poznanski et al. utilized k-mer frequency analysis to analyze five-mer peptide frequencies in proteins and highlight deviations from expected patterns [62]. Enrichment was noted in the majority of permutation groups with numerous outlier sequences; this aligned across protein families and evolutionary lineages, suggesting non-random variations. Interestingly, the identified outlier sequences often contained known motifs, and over-represented five-mer peptides were significantly related to known functional motifs. Therefore, k-mer frequency analysis has numerous advantages for interpreting biologically relevant information from sequence analysis across the genome, transcriptome, and proteome.
Despite varied methodologies for analyzing k-mer frequency, approaches were limited in that they did not provide a measure of the rarity of nucleic or peptide sequences. In response, Chantzi et al. estimated the rarity of nucleic and peptide k-mers across organismal genomes and proteomes and introduced an index of k-mer rarity across organisms [63]. Ultimately, the study found that the rarity of k-mers can be inferred by their amino acid and nucleotide composition.
3.3. Sequence alignment and genome assembly
3.3.1. Sequence alignment
One of the challenges of analyzing biological sequences is finding the optimal alignment between two or more sequences that may have variations such as insertions, deletions, or substitutions. Programs such as Basic Local Alignment Search Tool (BLAST) are sequence similarity search programs that compare combinations of nucleotide or peptide sequence queries with genomic, transcriptomic, or protein databases [64]. Subsequently, they find short matches between two sequences and attempt to start alignments based on a measure of local similarity, the maximal segment pair (MSP) score.
Similar in many ways to BLAST, BLAT is a mRNA/DNA and translated protein alignment algorithm, and its speed originates from an index of all nonoverlapping k-mers in the genome [65]. BLAT utilizes the index to identify potentially homologous regions, performs an alignment between them, assembles these aligned segments into larger alignments, and then revisits small internal exons while adjusting large gap boundaries with canonical splice sites when possible. Recently, k-mer-based alignment algorithms have been utilized in detecting and filtering out contamination by comparing observed k-mers against a reference database of potential contaminants (Fig. 2D) [66], [67].
In sequence alignment algorithms (Fig. 2B) [68], k-mers can serve as effective minimizers to reduce memory and computational requirements for genomic data analysis (Table 1) [69]. The basic idea behind minimizers, or k-mers that represent groups of k-mers, is that it is not necessary to consider every possible k-mer in a sequence but that it is often sufficient to focus on a smaller, representative subset for an analysis. The selection of the minimizer k-mer is done through a deterministic procedure; traditionally, the smallest k-mer in lexicographical ordering is chosen, but more recently, other alternatives such as ordering based on universal hitting sets have also been proposed [70]. This approach allows indexing a single minimizer as a representative of multiple k-mer sequences, significantly reducing memory requirements as well as the time taken to compare subsets. Currently, minimizers are used to accelerate aligners such as Minimap2 [71] and assist algorithms such as Mash [72] in estimating the similarity between genomes.
3.3.2. Genome assembly
Genome assembly is the process of reconstructing the original DNA sequence of an organism from sequencing reads. It is a crucial step in bioinformatic analyses, but accurate assembly is often challenging due to sequencing errors, repetitive regions, and structural variations in DNA. There are two forms of genomic assembly: reference-based and de novo, which does not utilize a reference genome for alignment.
Reference-based genome assembly can be utilized when a high quality reference genome is available [73]. Tools include RaGOO [74], which utilizes Minimap2 [71] alignments to a closely related reference genome with a k-mer size of 19 base pairs to cluster, order, and orient genome assembly contigs into pseudomolecules. While reference-based methods might introduce a bias towards the reference genome, it typically offers a significantly quicker and more cost-effective alternative to de novo methods.
In de novo genome assemblers such as SPAdes [75] and multiple others [76], a common approach is to first divide sequencing reads into k-mers and use each unique k-mer to represent a node in a De Brujin Graph [77]. K-mers with an overlap of k-1 bases are represented as adjacent nodes. Valid paths through the graph are then used to identify longer contiguous sequences or contigs, which are then arranged and oriented to create larger units called scaffolds. Ultimately, the scaffolds are utilized to reconstruct the original genomic sequence (Fig. 2C) (Fig. 2C) [78], [79], [80], [81].
In genome assembly, pangenome graphs, which model direct relationships between all genomes in the analysis, can be utilized to capture genetic diversity and understand genetic variations across different populations and species [82]. K-mers are utilized for genotyping known variants directly from raw sequencing reads without alignment. Algorithms such as Pangenome-based Genome Inference or PanGenie use haplotype-resolved pangenome references and k-mer counts from short-read sequencing data to analyze genetic variation [83].
3.3.3. Error correction
Short-read sequencing technologies such as Illumina allow for the high-throughput production of short reads at a low cost, but produced reads can contain various errors [84]. However, k-mers can play a significant role in error correction for de novo genome assembly and short-read mapping. Short-read error correction algorithms include RECKONER [85], [86] and RACER [87], which correct substitution errors, are dedicated to Illumina reads, and rely on k-mer counting. The workflow of RECKONER, whose new version also corrects indel errors, specifically includes k-mer counting, determining the threshold of number of k-mer appearances, removing untrusted k-mers from the database, and correcting the reads [85], [86]. Other algorithms for short-read error correction include the k-mer spectrum-based error correctors Musket [52] and BLESS [53], which utilize both k-mer counting and a Bloom filter. Interestingly, the tool Lighter fully avoids k-mer counting and relies instead on a pair of Bloom filters [88], while Karect [89], SAMDUDE [90], and CARE [91] utilize alignment-based approaches. Lastly, Rcorrector is a k-mer-based method that relies on De Brujin graphs for correcting random sequencing errors in Illumina RNA-seq reads specifically [92].
Long-read sequencing technologies such as those developed by Pacific Biosciences (PacBio) and Oxford Nanopore provide long reads, which, unlike short-reads, are able to span repeated elements or repetitive regions in de novo genome assembly and structural variant calling [93]. One caveat of these methods is that they suffer from significantly higher error rates, necessitating novel methods of error correction [94], [95], [96]. In response to these issues, multiple hybrid approaches based on k-mers were proposed, utilizing long reads for scaffolding and short Illumina reads for correcting errors [97], [98], [99], [100]. For a detailed comparison, readers can also refer to a number of publications evaluating the strengths of individual approaches [101], [102], [103].
3.4. Genome editing
CRISPR-Cas9 (Clustered Regularly Interspaced Short Palindromic Repeats) enables precise and efficient genome editing, but encounters various analytical challenges such as understanding off-target effects and improving delivery efficiency [104], [105]. K-mers have been employed as a strategy to improve the performance and accuracy of CRISPR-Cas9 technology [106], [107], [108], [109]. This is achieved by refining the specificity of guide RNA design, reducing false positives of target detection, and identifying genomic variation (Fig. 2E). For example, k-mer-based technologies like Redk-mer and BoostMEC analyze k-mer distributions to identify unique target sites with a low likelihood of off-target effects [106], [107]. JACKIE, a k-mer-based tool, was developed to improve the creation of guide RNAs (gRNAs) complementary to specific target sequences [108]. In addition, a web application named KmerKeys, which facilitates rapid querying of k-mers in genome assemblies, holds potential for CRISPR/Cas9 target design [110]. Its efficient k-mer indexing enables the identification of primer candidates with off-target site potential and allows for the representation of variants at the population level.
3.5. Comparative genomics
The study of genomic differences and similarities across and within taxonomic levels has benefited from k-mer-based approaches (Fig. 2D) [56]. These methods enable comparative analysis of k-mer occurrences and distributions across genomes, facilitating the identification of conserved regions, gene families, regulatory motifs, and genomic rearrangements to better understand evolutionary relationships [51], [111], [112], [113]. One primary application of k-mers in comparative genomics is constructing phylogenetic trees by quantifying the genetic distance between species based on k-mer frequencies [114], [115], [116], [117], [118]. Recent phylogenetic analyses have led to discoveries like the novel picorna-like viral sequences found in gut metagenomes [119], [120].
Kmer-db is an efficient tool for estimating evolutionary relationships between pathogens based on derived k-mers [121]. This makes it well-suited for processing large and diverse bacterial genome datasets, enabling more accurate comparisons for phylogeny reconstruction even among distantly related genomes with limited shared k-mers. Mash, which extends the MinHash dimensionality-reduction technique, has been used for phylogeny reconstruction and allows for efficient clustering, search, and mutation distance estimation in large sequence collections [72]. However, Kmer-db is roughly 26 times faster than Mash and is subsequently better equipped to process larger datasets [121]. Additional sketch-based methods that utilize k-mers in comparative genomics include Bindash 1.0 [122] and 2.0 [123], Dashing 1.0 [124] and 2.0 [125], NIQKI [126], SuperSampler [127], and fmh-funprofiler [128] (Table 2).
3.6. Metagenomics
Determining the abundance of taxonomic communities within diverse environments has important implications for agriculture, wildlife conservation, and healthcare improvements [129]. However, metagenomic profiling, such as the analysis of entire microbial communities, encounters challenges associated with the diversity, complexity, and quality of the data (Fig. 2D) [119]. By efficiently characterizing genetic material with k-mer-based approaches, diverse taxonomic groups can be identified and profiled [130]. Low abundance species in metagenomics data containing tens of millions of reads can be detected (Fig. 2F) [19].
K-mer-based methods for profiling taxa consist of three steps: first, k-mer profiles are compared to reference datasets, the individual sequences are then labeled, and lastly, the abundance of microbial taxa in large-scale metagenomic datasets is estimated [118], [131], [132]. K-mers can be utilized to label these individual sequences, facilitating metabarcoding across various organisms or individual cells [133]. Additionally, to understand the functional potential of metagenomic data, the distribution of k-mers associated with functional genes or metabolic pathways can be analyzed [134], [135].
Several k-mer-based approaches have been developed for quantifying and classifying taxa in metagenomic samples. Notable examples include Kraken2 [136], KrakenUniq [19], YACHT [137], and Kaiju [138], which assign reads to taxa without alignment based on approximate matches to a reference database of genomic or proteomic sequences. Despite their efficiency and sensitivity, these methods are limited by the quality and completeness of the reference database, an inability to detect novel taxa, and susceptibility to false positives due to horizontal gene transfer or contamination. In large-scale prokaryote and virus projects, k-mer frequency-based tools like VirFinder are used to identify viral sequences in mixed metagenomic datasets containing both viral and host contigs [18]. Additionally, k-mer-based genome binning techniques, such as Phages from Metagenomics Binning (PHAMB), facilitate the extraction and categorization of thousands of viral genomes from bulk metagenomics datasets [139]. These tools cluster viral genomes into taxonomic viral populations and have been instrumental in understanding viral-microbial host interactions from the Human Microbiome Project 2 dataset [139]. Furthermore, k-mer-based approaches, such as metaSPades, prove valuable for reconstructing individual genomes from metagenomic datasets [140].
K-mer-based approaches in clinical metagenomics hold the potential for rapid diagnosis and monitoring of infectious diseases [141]. With the reduced cost of high-throughput or next generation sequencing, clinical applications of sequencing are on the rise [142]. Researchers can employ k-mers to directly characterize microbial pathogens from patient samples, infer antibiotic resistance profiles for treatment guidance, and identify novel or emerging pathogens not yet present in reference databases. The development of machine learning models for predicting antimicrobial resistance faces challenges stemming from the high-dimensional and strong correlations of k-mer-based representations [20], [143], [144].
To enhance the interpretability of genomic signatures in k-mer-based predictive models, Jaillard et al. introduced an adaptive cluster lasso strategy, which identified sparse, meaningful, and interpretable genomic signatures and improved clinical utility [145]. Additionally, Wang et al. developed MetaGO, which uses group-specific metagenomic sequences to highlight differences between patients affected by liver cirrhosis and healthy cohorts [23]. This method aimed to predict clinical disease outcomes using long k-mer (k ≥ 30 bps) sequence signatures.
Similarly, Mouratidis et al. introduced “frequentmers,” short sequences that are specific and recurrently observed in either patient or healthy control samples, but not in both [146]. Using metagenomic NGS data from liver cirrhosis patients and healthy controls, machine learning models trained with frequentmers (k = 16) outperformed previous models and achieved an AUC of 0.91 in liver cirrhosis detection. Interestingly, the authors also identified microbial organisms in liver cirrhosis samples associated with the most predictive frequentmer biomarkers, potentially offering insight into the role of the gut microbiome in disease.
3.7. Metaproteomics
Metaproteomic datasets contain a vast array of protein sequences from diverse and unlabeled organisms, posing significant challenges for accurate identification and functional interpretation [147], [148]. Similar to metagenomics, k-mers aid in identifying proteins and understanding the functional potential of microbial communities [149]. Protein sequences are broken down into overlapping k-mers, and the generated peptide fragments are utilized for protein database searching [150]. Subsequently, researchers can detect proteins from less-known organisms and identify closely related species with the closest matching and similar sequences. Quantifying shared and unique k-mers across metaproteomic samples helps identify similarities and differences in protein expression profiles, enabling comparisons of microbial communities under different environmental conditions or between healthy and diseased states [151]. The utilization of k-mers in metaproteomics has even led to the identification of virion-associated protein annotations in “viral dark matter” genomic sequences [152].
3.8. Modeling protein structure
Protein structures can be modeled and predicted using k-mer-based algorithms that analyze the distribution of specific k-mers within protein sequences. K-mers prove effective in capturing and predicting information about protein folding patterns [153], secondary structure elements [153], and tertiary structures [154]. Additionally, k-mers are utilized to identify antigenic regions and potential epitopes within protein sequences [155], [156]. Analyzing k-mer distribution in known epitopes aids in developing models to predict the likelihood of specific regions acting as antigenic determinants or antimicrobial peptides [157], [158]. This approach enhances our understanding of antibody-epitope interactions and proves valuable in the design of diagnostics, therapeutic antibodies, and vaccines [157].
4. Absent sequences from genomes and proteomes
4.1. Overview of nullomers, nullpeptides, MAWs, primes, and quasi-primes
Nullomers, defined initially as the shortest k-mers absent from a specific genome (Table 1; Fig. 1B) [159], have gained attention across various fields due to their usefulness in different applications. They serve as distinctive markers for pathogens [160] and cancer [22], are used in barcoding [161], and have potential applications in drug discovery [31]. Their absence has been attributed to detrimental effects on an organism, higher mutation rates, and stochastic effects [162], [163], [164]. Research into the potentially deleterious effects of absent sequences has also included investigation into nullpeptides, which are peptide sequences that do not exist within a given proteome (Table 1; Fig. 1B) [159], [162], [163].
The potential applications of nullomers and nullpeptides have stimulated the development of various related k-mer concepts and algorithms to prioritize absent sequences. For example, Vergni & Santoni introduced a subset of nullomers termed high order nullomers, nullomers whose mutated sequences are still nullomers [164]. The nth order nullomer is one where any n base substitutions across each base in the sequence still yield a nullomer; first-order nullomers are the subset of nullomers where any single base substitution at any position along the k-mer still yields a nullomeric sequence (Table 1). In addition, Pinho et al. defined minimal absent words (MAWs) as absent sequences that lose the property of being absent from a genome (or sequence space by extension) if a character is removed from either end (Table 1) [162], [165]. The growth rate of the set of MAWs is linear with respect to the length of the string, unlike the exponential growth rate of nullomers. Therefore, the identification and classification of MAWs is more manageable when accounting for high computational costs in analysis [165]. MAWs have been proposed as potential tools for drug discovery, design, and delivery as they may have specific binding affinities and interactions with other molecules [162], [166], [167], [168], [169].
Recent work characterized significant MAWs, which were statistically expected to exist yet absent in the genomes and proteomes of different species [162]. The researchers found that substitution mutations in human protein coding genes can cause the appearance of MAWs and that over 25% of human proteins have the potential to generate a significant MAW through a single substitution mutation [162]. MAWs can also expand phylogenetic inference by providing novel insights into the evolutionary relationships and divergence times of different taxa [30], [167]. For example, the intra-species variation in the number and content of MAWs is generally less pronounced than inter-species variation, suggesting that MAWs are relatively conserved and stable within species [162], [169].
Extending on the derivation of nullomers and nullpeptides, which are absent from one genome or proteome, primes are designated as sequences that are universally absent from every sequenced genome or proteome [159]. Nucleic primes are k-mers absent from every genome, while peptide primes are the nullpeptides absent from every proteome (Table 1; Fig. 1D) [163]. In 2009, 417 five-amino acid primes not found in the universal proteome were identified [170]. An analysis across the genome of over twelve species (human, chimp, and ten other non-primates) identified 60,370 nullomers absent across them for 15 bp length. Work in 2021 expanded the identification and functional characterization of peptide primes; 140,308,851 nullpeptide primes absent from all known species were identified using the UniParc database (which had 1,030,456,800 proteins) [163]. Recently, kmerDB was made public containing 5,186,757 nucleic and 214,904,089 peptide primes isolated from the examination of 45,785 complete organismal genomes and 22,386 reference organismal proteomes [171].
The identification of organisms at the species level is often challenging due to the limitations of molecular markers that can discriminate between closely related taxa. A novel approach based on k-mers is to use quasi-primes, which are the shortest k-mers that are unique to a single species and absent from any other sequenced genome or proteome. Mouratidis et al. cataloged quasi-prime peptides (for k-mer lengths up to seven amino acids) from 21,875 species and identified quasi-primes (for k-mer lengths of six and seven amino acids) for 21 human pathogens, 8 model organisms, humans, Bonobo, and Gorilla [172]. The identification of quasi-primes may offer insights into species-specific traits, trait acquisition, evolutionary relationships, and mechanistic processes and may be applied for highly sensitive and specific real-time organismal detection (Table 1; Fig. 1D). For example, in the human genome, nucleic quasi-prime loci are predominantly linked to genes involved in brain development and cognitive function [173].
4.2. Nullomers, nullpeptides, and selection constraints
Nullomers have been viewed as potential signatures of natural selection against deleterious sequences [162], [163], [164]. Acquisti et al. challenged this idea and suggested that the mutational dynamics of the genome, namely the hypermutability of CpG dinucleotides in vertebrates, likely account for the emergence of certain short sequence motifs as nullomers (k = 11 bp) [174]. Their analysis revealed that many reported human nullomers differ by only one nucleotide, implying the significant role of mutations in the evolution of nullomers. Further emphasizing the insufficiency of the CpG hypermutability model, Vergini & Santoni found that nullomers from the human genome exhibit different statistical properties compared to those expected from random sequences [164]. The study demonstrated that CpG dinucleotide frequencies in genomes cluster into homogenous groups based on their CpG frequency profiles, with close species sharing similar patterns as depicted in phylogenetic trees. Lastly, the authors found that nullomers display higher mean helical rise values, suggesting a potential interaction with histone complexes that could explain their removal from DNA pending experimental validation [175], [176].
In contrast, many studies have suggested that the hypermutability of CpG dinucleotides is an insufficient explanation for nullomer origin [164], [167]. Garcia et al. examined 22 organisms from various domains and found that mutational biases are not uniform across different sets of MAWs of increasing length [169]. They suggested that MAWs may have been inherited from a common ancestor, in addition to lineage-specific inheritance. Subsequently, they advocated for the utility of MAWs in inferring genomic homology, understanding genome evolution, and addressing limitations in existing methods that often overlook non-protein-coding regions. Koulouras and Frith introduced an open-source software tool, Nullomers Assessor, to identify statistically significant MAWs in biological sequences [162]. Their analysis of over 147,000 viral sequences utilizing Nullomers Assessor revealed that the most frequent significant absent motifs in viral genomes corresponded to restriction recognition sites. This evidence supports the hypothesis that MAWs are absent due to negative selection and implies that they may have been replaced by specialized sequences with similar or optimized functions.
In 2021, Georgakopoulos-Soares et al. investigated the occurrence of both putative and germline nullomer-emerging mutations, which are genetic changes that introduce nullomers into a genome [163]. A significantly higher number of mutations than expected by chance for specific nullomer sequences within transposable elements were found, potentially due to their suppression and their role in the jumping activity of these elements. Genes with high-density nullomer-emerging mutations were involved in epigenetic regulation and DNA organization, while genes with low-density mutations were linked to processes like cell-to-cell contact and chemical stimuli detection. An enrichment of these mutations in promoters and enhancers suggested putative functions of nullomers in gene regulation. When examining nullomers across different species, nullomers were used for phylogenetic classification in vertebrate evolution. The human genome, in particular, was found to contain a higher number of nullomers than expected based on simulated genomes. This evidence supports that nullomers are typically under negative selection, with a subset of nullomers either not resurfacing or resurfacing with low probability through common variants, indicating their deleterious nature.
4.3. Applications of absent sequences
4.3.1. Pathogen and cancer detection
Nullomers have been used in examining sequence composition differences between organisms, profiling the evolutionary pressures of prokaryotic and eukaryotic genomes, and constructing phylogenetic frameworks (Fig. 3A) [167], [177]. In the context of the 2014 Ebola virus outbreak, Silva et al. introduced a novel subset of MAWs termed minimal relative absent words (RAWs) that are derived from a pathogen genome, but absent in its host, to analyze distinctions among Ebola virus sequences associated with the outbreak, sequences from other Ebola virus species, historical outbreak sequences, and the human genome [160]. Three minimal RAWs were identified as conserved pathogen-specific signatures with implications for rapid diagnostics and targeted therapeutic interventions. The authors suggested using these specific RAWs to design primers for identifying Ebola virus infections or distinguishing between Ebola virus species or outbreaks, highlighting the potential use of k-mer-based solutions for enhancing the precision of pathogen detection. Similarly, Pratas & Silva analyzed SARS-CoV-2 genomes from the outbreak in late 2019 and revealed the presence of RAWs of 12 or 13 base pairs persistent across all SARS-CoV-2 genomes and absent from the human genome and transcriptome [178]. These are minimal signatures of the SARS-CoV-2 genome that distinguish it from other human coronavirus species.
Fig. 3.

Applications of absent sequences. A. The nullomer profile of individuals may be used to characterize populations. B. Nullomers have been detected in cell-free DNA and utilized for cancer detection. C. Immunogenicity is associated with nullomers and nullpeptides, and both have been used in vaccines to increase efficacy and decrease autoimmune reactions. D. Certain nullpeptides exhibit cytotoxic effects and have cancer-killing properties. E. Nullomers and primes can be used for barcoding purposes to label or differentiate samples, as they are absent from one or more organisms. F. Quasi-primes can serve as species-specific biomarkers. Their use as universal fingerprints has potential for real-time organismal detection, understanding evolution as well as for biosecurity.
Beyond pathogen detection, nullomers and nullpeptides have emerged as diagnostic tools for cancer detection and show promise as sensitive and highly specific biomarkers (Fig. 3B). A study by Georgakopoulos-Soares et al. introduced neomers, a subset of nullomers largely absent from healthy human genomes but emerging recurrently in the tumor genome due to somatic mutations, as a potential tool for early cancer detection using cell-free DNA (Table 1; Fig. 1C) [29]. The authors analyzed over 2500 whole-genome sequencing tumor samples and demonstrated that a neomer-based classifier can accurately distinguish the tissue of origin between twenty-one tumor types. Neomers were utilized to sensitively and specifically identify cancer from a limited amount of plasma in four cancer types (prostate, lung, ovarian, colorectal) and therefore offer a potential tool for liquid biopsies and early-stage cancer detection.
In an analysis of over 10,000 Whole Exome Sequencing tumor samples, Montgomery et al. identified nullomer emerging mutational hotspots within cancer genes and used cell-free RNA from peripheral blood samples to demonstrate the utility of nullomers in classifying diverse tumor types (AUC > 0.90) [22]. Tsiatsianis et al. examined the emergence of nullpeptides during cancer development, finding multiple recurrently emerging nullpeptides in cancer genes in patients [28]. The study identified specific hotspots for nullpeptides within the loci of oncogenes and tumor suppressors and observed that recurrent nullpeptides are more frequently associated with neoantigens, implicating them as potentially effective targets for immunotherapy. This suggests that nullpeptides could serve as valuable indicators for prioritizing candidates for immunotherapeutic interventions in cancer treatment.
4.3.2. Vaccine development
In light of the observed correlation between the strength of immune response and the prevalence of specific amino acid sequences in nature [179], [180], it is hypothesized that nullomers and nullpeptides display heightened immunogenicity and therefore reinforce host defenses against pathogens (Fig. 3C) [181], [182], [183]. The absence of specific five and six amino acid peptide combinations in publicly available proteome sequences has previously been identified [184], [185], and there is potential enhancement of immunomodulatory effects and immunogenicity when incorporating these rare sequences into the development of vaccines.
Patel et al. found that rare five-mer peptides induced stronger cellular responses than common sequences [186]. Integration of these peptides into an H5N1 hemagglutinin antigen in a DNA vaccine improved immune responses, with several five-mer peptides enhancing clinical outcomes against lethal influenza virus challenges in mice and ferrets. Also, combining a five-mer peptide with a commercial Hepatitis B vaccine significantly increased anti-HBsAg antibody production in mice. The study suggested that rare or non-existent oligopeptides could be developed as immunomodulators and encouraged further evaluation of specific five-mer peptides as potential vaccine adjuvants. In pursuit of potential peptides for a SARS-CoV-2 vaccine, Santoni et al. opted for third-order nullpeptides [187]. This decision aimed to address concerns related to cross-reactivity and mitigate the risks of autoimmunity, given their lower similarity to human self.
4.3.3. Cancer therapeutics
Recent clinical investigations highlight the potential of peptide-based drugs in cancer therapy, emphasizing advantages such as their small size, specificity, efficacy across a broad spectrum of cancers, potentially selective cytotoxicity, and cost-effectiveness [188], [189]. The rationale behind k-mer-based drug design approaches proposes that injecting the smallest absent sequences into hosts can induce toxic or immune-stimulatory reactions (Fig. 3D) [31]. Alileche et al. first demonstrated the anti-neoplastic benefits of nullpeptides, including 9R and 9S1R [31]. These nullpeptides proved lethal to cancer cells. 9R and 9S1R induced mitochondrial impairment as evidenced by increased reactive oxygen species production, ATP depletion, cell growth inhibition, and cell death. Remarkably, while normal cells exhibited diminishing sensitivity to nullpeptides over time, the impact on cancer cells intensified over the same duration. In a subsequent study, Alileche et al. investigated the effects of nullpeptides 9R, 9S1R, and 124R (five-amino acid) on the NCI-60 panel of cancer cells from nine organs and four normal cell lines. The research revealed that these nullpeptides demonstrated a broad lethal effect on cancer cells, including drug- and hormone-resistant cell lines and cancer stem cells [30]. 9R and 9S1R, unlike 124R, displayed a broader activity spectrum than several existing cancer drugs. Within a three-hour timeframe, they induced substantial cellular ATP depletion, while exhibiting lower toxicity towards normal cells. These findings highlight the potential of nullpeptides as cancer treatments capable of targeting diverse cancer types and addressing cellular heterogeneity.
Ali et al. investigated the therapeutic potential of a ten amino acid nullpeptide, 9S1R-NulloPT [190]. The investigation revealed that mice treated with the peptide, in contrast to the untreated controls, demonstrated reduced tumor size during the initial treatment phase of triple-negative breast cancer. Subsequently, in later stages and in excised tumors, there was a reduction in in-vivo bioluminescence. This observation implied the presence of metabolically inactive tumors, while secondary metastasis in the lungs remained unaffected. Furthermore, the peptide treatment induced alterations in the tumor immune microenvironment, characterized by heightened immune cell infiltration and inflammation. These changes were accompanied by shifts in gene expression, marked by the downregulation of genes associated with cellular metabolism and translation machinery and the upregulation of genes linked to developmental pathways and extracellular matrix organization.
4.3.4. Engineering barcodes, PCR primers, and fingerprinting organisms
Synthetic DNA primers and molecular barcodes derived from nullomers have been employed to prevent forensic contamination (Fig. 3E) [161]. In order to mitigate the risks of cross-contamination and misidentification of reference samples in PCR analysis, Goswami et al. proposed a method that leverages nullomer barcodes as internal amplification controls to distinguish between reference and evidentiary samples in forensic DNA analysis [161]. The results demonstrated that nullomers could be integrated into the multiplex PCR reactions of forensic profiling kits and used jointly with PCR for sequencing. KeeSeek was developed to design distant non-existing k-mers for barcodes or PCR primers [191]. Unlike previously existing tools, the software produced longer sequences (up to 31 nt) and provided the distance from the reference genome in terms of the number of mismatches. With primer-like feature filters, it was optimized for targeted genomic manipulation experiments using techniques like zinc finger nucleases, TALEN, and CRISPR.
K-mers that appear in a single protein of a particular proteome have been identified and can be utilized for protein identification [192], [193]. Research on nucleic primes and quasi-primes has emphasized their potential for molecular barcodes (Fig. 3E-F) [172], [173]. Because quasi-primes are the shortest unique sequences in a species or sequence, their distinctiveness makes them ideal for applications requiring precise and unambiguous identification of a sample (Fig. 3F). However, quasi-prime identification may currently be limited by the availability of species-specific reference sequence information, variation, and sequencing errors. Future work will aim to address these limitations and explore their relationship to uniqueness and differentiation among organisms.
5. Conclusions
K-mers have influenced the fields of genomics, transcriptomics, proteomics, and biological data analysis. In this review, we explore various k-mer-based methods while highlighting their expansive applications in basic science research, translational medicine, healthcare, agriculture, biosecurity, and conservation. The discussed applications of k-mers include k-mer counting and frequency analysis, sequence alignment, genome assembly, comparative genomics, metagenomics, metaproteomics, and protein structure prediction. This broad spectrum of applications demonstrates the versatility of k-mer-based approaches in addressing biological problems, ranging from elucidating evolutionary relationships to serving as biomarkers. We emphasize the utilization of k-mer-based tools, delineating the integration of k-mers into bioinformatics methodologies as a means to enhance the analysis of vast genomic and proteomic datasets on a large scale.
Previous reviews have focused on several of the research areas covered in this review, including sequence alignment [194], [195], genome assembly [196], indexing and querying large datasets [197], and metagenomics-based methods and pipelines [6], [198]. However, we emphasize the utility of k-mer-based approaches across biological problems and review different k-mer concepts including nullomers, nullpeptides, minimal absent words, quasi-primes, and primes, as well as their applications. The review of nullomers, nullpeptides, and primes offers valuable insights into the implications of absent words for understanding genomic variation, evolution, and human diseases. Furthermore, the use of absent sequences in pathogen and cancer detection, vaccine development, cancer therapeutics, barcoding, and DNA fingerprinting accentuates their versatility in various applications. In light of recent global health crises like the SARS-CoV-2 pandemic, the significance of efficient diagnostic methods cannot be overstated. The ability to identify absent words in the genomes of pathogens such as SARS-CoV-2 not only facilitates accurate diagnosis but also paves the way for potential therapeutic interventions [178].
In conclusion, the versatility, scalability, and efficiency of k-mer-based analysis are evident across a spectrum of applications. Considering the increasing size and complexity of genomic and proteomic datasets, k-mer-based algorithms offer substantial potential to enhance biological information extraction from genetic material. Therefore, expanding k-mer-based methodologies with robust techniques may improve our understanding of biology and accelerate genetic translation into meaningful application.
Author Contributions
I.G.S. conceived and supervised the study. C.M., M.M., M.A.K., C.C., I.M., A.M., N.C., G.A.P., and I.G.S. wrote the manuscript. C.M., M.M., M.A.K., C.C. and I.G.S. generated the figures and tables.
Author Statement
All authors contributed to the work and have approved this version of the manuscript to be submitted. The authors have no conflicts or financial interests to disclose. This work has not been previously submitted and is not under consideration elsewhere. All ethical guidelines were upheld in the preparation of this manuscript. We are not using any copyrighted information, identifiers, or other protected health information in this paper. No text, text boxes, or tables in this article have been previously published or owned by another party.
CRediT authorship contribution statement
Manvita Mareboina: Writing – original draft. Camille Moeckel: Conceptualization, Supervision, Visualization, Writing – original draft, Writing – review & editing. Candace S.Y. Chan: Visualization, Writing – review & editing. Maxwell A. Konnaris: Writing – original draft, Writing – review & editing. Georgios A. Pavlopoulos: Writing – review & editing, Investigation, Supervision. Austin Montgomery: Writing – review & editing. Ioannis Mouratidis: Conceptualization, Supervision, Writing – original draft, Writing – review & editing. Ilias Georgakopoulos-Soares: Conceptualization, Supervision, Visualization, Writing – original draft, Writing – review & editing, Resources. Nikol Chantzi: Writing – review & editing.
Declaration of Competing Interest
All authors declare that they have no conflicts of interest.
Acknowledgements
This study was funded by the startup funds of I.G.S. from the Penn State College of Medicine and by the Huck Innovative and Transformational Seed Fund (HITS) award from the Huck Institutes of the Life Sciences at Penn State University.; G.A.P was supported by: Fondation Sante; Onasis Foundation; Hellenic Foundation for Research and Innovation (H.F.R.I) under the call 'Greece 2.0 - Basic Research Financing Action, sub-action II, Grant ID: 16718-PRPFOR; Program 'Greece 2.0, National Recovery and Resilience Plan', Grant ID: TAEDR-0539180.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.csbj.2024.05.025.
Appendix A. Supplementary material
Supplementary material
.
References
- 1.Slatko B.E., Gardner A.F., Ausubel F.M. Overview of next-generation sequencing technologies. Curr. Protoc. Mol. Biol. 2018;122 doi: 10.1002/cpmb.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hu T., Chitnis N., Monos D., Dinh A. Next-generation sequencing technologies: an overview. Hum. Immunol. 2021;82:801–811. doi: 10.1016/j.humimm.2021.02.012. [DOI] [PubMed] [Google Scholar]
- 3.Dai X., Shen L. Advances and trends in omics technology development. Front. Med. 2022;9 doi: 10.3389/fmed.2022.911861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koumakis L. Deep learning models in genomics; are we there yet? Comput. Struct. Biotechnol. J. 2020;18:1466–1473. doi: 10.1016/j.csbj.2020.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.D’Argenio V. The high-throughput analyses era: are we ready for the data struggle? High-Throughput. 2018;7:8. doi: 10.3390/ht7010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Breitwieser F.P., Lu J., Salzberg S.L. A review of methods and databases for metagenomic classification and assembly. Brief. Bioinform. 2019;20:1125–1136. doi: 10.1093/bib/bbx120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leggett R.M., Ramirez-Gonzalez R.H., Clavijo B.J., Waite D., Davey R.P. Sequencing quality assessment tools to enable data-driven informatics for high throughput genomics. Front. Genet. 2013;4:288. doi: 10.3389/fgene.2013.00288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marçais G., Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27:764–770. doi: 10.1093/bioinformatics/btr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pérez N., Gutierrez M., Vera N. Computational performance assessment of k-mer counting algorithms. J. Comput. Biol. 2016;23:248–255. doi: 10.1089/cmb.2015.0199. [DOI] [PubMed] [Google Scholar]
- 10.Manekar S.C., Sathe S.R. A benchmark study of k-mer counting methods for high-throughput sequencing. Gigascience. 2018;7 doi: 10.1093/gigascience/giy125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Georgakopoulos-Soares I., Jain N., Gray J.M., Hemberg M. MPRAnator: a web-based tool for the design of massively parallel reporter assay experiments. Bioinformatics. 2017;33:137–138. doi: 10.1093/bioinformatics/btw584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mejía-Guerra M.K., Buckler E.S. A k-mer grammar analysis to uncover maize regulatory architecture. BMC Plant Biol. 2019;19:103. doi: 10.1186/s12870-019-1693-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghandi M., Lee D., Mohammad-Noori M., Beer M.A. Enhanced regulatory sequence prediction using gapped k-mer features. PLoS Comput. Biol. 2014;10 doi: 10.1371/journal.pcbi.1003711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.di Iulio J., Bartha I., Wong E.H.M., Yu H.-C., Lavrenko V., Yang D., Jung I., Hicks M.A., Shah N., Kirkness E.F., et al. The human noncoding genome defined by genetic diversity. Nat. Genet. 2018;50:333–337. doi: 10.1038/s41588-018-0062-7. [DOI] [PubMed] [Google Scholar]
- 15.Smith R.P., Riesenfeld S.J., Holloway A.K., Li Q., Murphy K.K., Feliciano N.M., Orecchia L., Oksenberg N., Pollard K.S., Ahituv N. A compact, in vivo screen of all 6-mers reveals drivers of tissue-specific expression and guides synthetic regulatory element design. Genome Biol. 2013;14:1–15. doi: 10.1186/gb-2013-14-7-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Annapragada A.V., Niknafs N., White J.R., Bruhm D.C., Cherry C., Medina J.E., Adleff V., Hruban C., Mathios D., Foda Z.H., et al. Genome-wide repeat landscapes in cancer and cell-free DNA. Sci. Transl. Med. 2024;16 doi: 10.1126/scitranslmed.adj9283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aun E., Brauer A., Kisand V., Tenson T., Remm M. A k-mer-based method for the identification of phenotype-associated genomic biomarkers and predicting phenotypes of sequenced bacteria. PLoS Comput. Biol. 2018;14 doi: 10.1371/journal.pcbi.1006434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ren J., Ahlgren N.A., Lu Y.Y., Fuhrman J.A., Sun F. VirFinder: a novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome. 2017;5:69. doi: 10.1186/s40168-017-0283-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Breitwieser F.P., Baker D.N., Salzberg S.L. KrakenUniq: confident and fast metagenomics classification using unique k-mer counts. Genome Biol. 2018;19:198. doi: 10.1186/s13059-018-1568-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davis J.J., Boisvert S., Brettin T., Kenyon R.W., Mao C., Olson R., Overbeek R., Santerre J., Shukla M., Wattam A.R., et al. Antimicrobial Resistance Prediction in PATRIC and RAST. Sci. Rep. 2016;6:27930. doi: 10.1038/srep27930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clausen P.T.L.C., Zankari E., Aarestrup F.M., Lund O. Benchmarking of methods for identification of antimicrobial resistance genes in bacterial whole genome data. J. Antimicrob. Chemother. 2016;71:2484–2488. doi: 10.1093/jac/dkw184. [DOI] [PubMed] [Google Scholar]
- 22.Montgomery A., Tsiatsianis G.C., Mouratidis I., Chan C.S.Y., Athanasiou M., Papanastasiou A.D., Kantere V., Vathiotis I., Syrigos K., Yee N.S., et al. Utilizing nullomers in cell-free RNA for early cancer detection. medRxiv. 2023 doi: 10.1101/2023.06.10.23291228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y., Fu L., Ren J., Yu Z., Chen T., Sun F. Identifying sequences for microbial communities using long -mer sequence signatures. Front. Microbiol. 2018;9:872. doi: 10.3389/fmicb.2018.00872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.LaPierre N., Ju C.J.-T., Zhou G., Wang W. MetaPheno: a critical evaluation of deep learning and machine learning in metagenome-based disease prediction. Methods. 2019;166:74–82. doi: 10.1016/j.ymeth.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee H., Shuaibi A., Bell J.M., Pavlichin D.S., Ji H.P. Unique -mer sequences for validating cancer-related substitution, insertion and deletion mutations. NAR Cancer. 2020;2 doi: 10.1093/narcan/zcaa034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pinskaya M., Saci Z., Gallopin M., Gabriel M., Nguyen H.T., Firlej V., Descrimes M., Rapinat A., Gentien D., Taille A. de la, et al. Reference-free transcriptome exploration reveals novel RNAs for prostate cancer diagnosis. Life Sci Alliance. 2019:2. doi: 10.26508/lsa.201900449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nguyen H.T.N., Xue H., Firlej V., Ponty Y., Gallopin M., Gautheret D. Reference-free transcriptome signatures for prostate cancer prognosis. BMC Cancer. 2021;21:394. doi: 10.1186/s12885-021-08021-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsiatsianis G.C., Chan C.S.Y., Mouratidis I., Chantzi N., Tsiatsiani A.M., Yee N.S., Kantere V., Georgakopoulos-Soares I. Peptide absent sequences emerging in human cancers. Eur. J. Cancer. 2024;196 doi: 10.1016/j.ejca.2023.113421. [DOI] [PubMed] [Google Scholar]
- 29.Georgakopoulos-Soares I., Barnea O.Y., Mouratidis I., Chan C.S.Y., Bradley R., Mahajan M., Sims J., Cintron D.L., Easterlin R., Kim J.S., et al. Leveraging sequences missing from the human genome to diagnose cancer. medRxiv. 2023 doi: 10.1101/2021.08.15.21261805. [DOI] [Google Scholar]
- 30.Alileche A., Hampikian G. The effect of Nullomer-derived peptides 9R, 9S1R and 124R on the NCI-60 panel and normal cell lines. BMC Cancer. 2017;17:533. doi: 10.1186/s12885-017-3514-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alileche A., Goswami J., Bourland W., Davis M., Hampikian G. Nullomer derived anticancer peptides (NulloPs): differential lethal effects on normal and cancer cells in vitro. Peptides. 2012;38:302–311. doi: 10.1016/j.peptides.2012.09.015. [DOI] [PubMed] [Google Scholar]
- 32.Chikhi R., Medvedev P. Informed and automated k-mer size selection for genome assembly. Bioinformatics. 2014;30:31–37. doi: 10.1093/bioinformatics/btt310. [DOI] [PubMed] [Google Scholar]
- 33.Sarkar B.K., Sharma A.R., Bhattacharya M., Sharma G., Lee S.-S., Chakraborty C. Determination of k-mer density in a DNA sequence and subsequent cluster formation algorithm based on the application of electronic filter. Sci. Rep. 2021;11:1–12. doi: 10.1038/s41598-021-93154-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ondov B.D., Starrett G.J., Sappington A., Kostic A., Koren S., Buck C.B., Phillippy A.M. Mash Screen: high-throughput sequence containment estimation for genome discovery. Genome Biol. 2019;20:1–13. doi: 10.1186/s13059-019-1841-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghandi M., Mohammad-Noori M., Beer M.A. Robust k-mer frequency estimation using gapped k-mers. J. Math. Biol. 2014;69:469–500. doi: 10.1007/s00285-013-0705-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ge J., Meng J., Guo N., Wei Y., Balaji P., Feng S. Counting Kmers for biological sequences at large scale. Interdiscip. Sci. 2020;12:99–108. doi: 10.1007/s12539-019-00348-5. [DOI] [PubMed] [Google Scholar]
- 37.Titievsky A., Putintseva Y.A., Taranenko E.A., Baskin S., Oreshkova N.V., Brodsky E., Sharova A.V., Sharov V.V., Panov J., Kuzmin D.A., et al. Comparative genomics analysis of repetitive elements in ten gymnosperm species: ‘dark repeatome’ and its abundance in conifer and species. Life. 2021;11 doi: 10.3390/life11111234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu S., Zheng J., Migeon P., Ren J., Hu Y., He C., Liu H., Fu J., White F.F., Toomajian C., et al. Unbiased K-mer analysis reveals changes in copy number of highly repetitive sequences during maize domestication and improvement. Sci. Rep. 2017;7:42444. doi: 10.1038/srep42444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen M.-M., Shi G.-H., Dai Y., Fang W.-X., Wu Q. Identifying genetic variants associated with amphotericin B (AMB) resistance in via merbased GWAS. Front. Genet. 2023;14:1133593. doi: 10.3389/fgene.2023.1133593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sohn J.-I., Choi M.-H., Yi D., Menon V.A., Kim Y.J., Lee J., Park J.W., Kyung S., Shin S.-H., Na B., et al. Ultrafast prediction of somatic structural variations by filtering out reads matched to pan-genome k-mer sets. Nat Biomed Eng. 2023;7:853–866. doi: 10.1038/s41551-022-00980-5. [DOI] [PubMed] [Google Scholar]
- 41.Annalora A.J., O’Neil S., Bushman J.D., Summerton J.E., Marcus C.B., Iversen P.L. A k-mer based transcriptomics approach for antisense drug discovery targeting the Ewing’s family of tumors. Oncotarget. 2018;9:30568–30586. doi: 10.18632/oncotarget.25736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nordström K.J.V., Albani M.C., James G.V., Gutjahr C., Hartwig B., Turck F., Paszkowski U., Coupland G., Schneeberger K. Mutation identification by direct comparison of whole-genome sequencing data from mutant and wild-type individuals using k-mers. Nat. Biotechnol. 2013;31:325–330. doi: 10.1038/nbt.2515. [DOI] [PubMed] [Google Scholar]
- 43.Audemard E.O., Gendron P., Feghaly A., Lavallée V.-P., Hébert J., Sauvageau G., Lemieux S. Targeted variant detection using unaligned RNA-Seq reads. Life Sci Alliance. 2019;2 doi: 10.26508/lsa.201900336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tian S., Yan H., Klee E.W., Kalmbach M., Slager S.L. Comparative analysis of de novo assemblers for variation discovery in personal genomes. Brief. Bioinform. 2018;19:893–904. doi: 10.1093/bib/bbx037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kokot M., Dlugosz M., Deorowicz S. KMC 3: counting and manipulating k-mer statistics. Bioinformatics. 2017;33:2759–2761. doi: 10.1093/bioinformatics/btx304. [DOI] [PubMed] [Google Scholar]
- 46.Rhie A., Walenz B.P., Koren S., Phillippy A.M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 2020;21:245. doi: 10.1186/s13059-020-02134-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marchet C., Iqbal Z., Gautheret D., Salson M., Chikhi R. REINDEER: efficient indexing of k-mer presence and abundance in sequencing datasets. Bioinformatics. 2020;36:i177–i185. doi: 10.1093/bioinformatics/btaa487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pandey P., Bender M.A., Johnson R., Patro R., Berger B. Squeakr: an exact and approximate k-mer counting system. Bioinformatics. 2018;34:568–575. doi: 10.1093/bioinformatics/btx636. [DOI] [PubMed] [Google Scholar]
- 49.Bingmann T., Bradley P., Gauger F., Iqbal Z. COBS: A Compact Bit-Sliced Signature Index. String Processing and Information Retrieval. 2019 [Google Scholar]
- 50.Srikakulam S.K., Keller S., Dabbaghie F., Bals R., Kalinina O.V. MetaProFi: an ultrafast chunked Bloom filter for storing and querying protein and nucleotide sequence data for accurate identification of functionally relevant genetic variants. Bioinformatics. 2023;39 doi: 10.1093/bioinformatics/btad101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chor B., Horn D., Goldman N., Levy Y., Massingham T. Genomic DNA k-mer spectra: models and modalities. Genome Biol. 2009;10:R108. doi: 10.1186/gb-2009-10-10-r108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu Y., Schröder J., Schmidt B. Musket: a multistage k-mer spectrum-based error corrector for Illumina sequence data. Bioinformatics. 2013;29:308–315. doi: 10.1093/bioinformatics/bts690. [DOI] [PubMed] [Google Scholar]
- 53.Heo Y., Wu X.-L., Chen D., Ma J., Hwu W.-M. BLESS: bloom filter-based error correction solution for high-throughput sequencing reads. Bioinformatics. 2014;30:1354–1362. doi: 10.1093/bioinformatics/btu030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lim E.-C., Müller J., Hagmann J., Henz S.R., Kim S.-T., Weigel D. Trowel: a fast and accurate error correction module for Illumina sequencing reads. Bioinformatics. 2014;30:3264–3265. doi: 10.1093/bioinformatics/btu513. [DOI] [PubMed] [Google Scholar]
- 55.Yang Z., Li H., Jia Y., Zheng Y., Meng H., Bao T., Li X., Luo L. Intrinsic laws of k-mer spectra of genome sequences and evolution mechanism of genomes. BMC Evol. Biol. 2020;20:157. doi: 10.1186/s12862-020-01723-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bussi Y., Kapon R., Reich Z. Large-scale k-mer-based analysis of the informational properties of genomes, comparative genomics and taxonomy. PLoS One. 2021;16 doi: 10.1371/journal.pone.0258693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu Z., Venkatesh S.S., Maley C.C. Sequence space coverage, entropy of genomes and the potential to detect non-human DNA in human samples. BMC Genomics. 2008;9:509. doi: 10.1186/1471-2164-9-509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baizan-Edge A., Cock P., MacFarlane S., McGavin W., Torrance L., Jones S. Kodoja: a workflow for virus detection in plants using k-mer analysis of RNA-sequencing data. J. Gen. Virol. 2019;100:533–542. doi: 10.1099/jgv.0.001210. [DOI] [PubMed] [Google Scholar]
- 59.Patro R., Duggal G., Love M.I., Irizarry R.A., Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang Z., Wang W. RNA-Skim: a rapid method for RNA-Seq quantification at transcript level. Bioinformatics. 2014;30:i283–i292. doi: 10.1093/bioinformatics/btu288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Patro R., Mount S.M., Kingsford C. Sailfish enables alignment-free isoform quantification from RNA-seq reads using lightweight algorithms. Nat. Biotechnol. 2014;32:462–464. doi: 10.1038/nbt.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Poznański J., Topiński J., Muszewska A., Dębski K.J., Hoffman-Sommer M., Pawłowski K., Grynberg M. Global pentapeptide statistics are far away from expected distributions. Sci. Rep. 2018;8:15178. doi: 10.1038/s41598-018-33433-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chantzi N., Mouratidis I., Mareboina M., Konnaris M.A., Montgomery A., Georgakopoulos-Soares I. The determinants of the rarity of nucleic and peptide short sequences in nature. bioRxiv. 2023 doi: 10.1101/2023.09.24.559219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McGinnis S., Madden T.L. BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004;32:W20–W25. doi: 10.1093/nar/gkh435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kent W.J. BLAT--the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cornet L., Meunier L., Van Vlierberghe M., Léonard R.R., Durieu B., Lara Y., Misztak A., Sirjacobs D., Javaux E.J., Philippe H., et al. Consensus assessment of the contamination level of publicly available cyanobacterial genomes. PLoS One. 2018;13 doi: 10.1371/journal.pone.0200323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Allesøe R.L., Lemvigh C.K., Phan M.V.T., Clausen P.T.L.C., Florensa A.F., Koopmans M.P.G., Lund O., Cotten M. Automated download and clean-up of family-specific databases for kmer-based virus identification. Bioinformatics. 2021;37:705–710. doi: 10.1093/bioinformatics/btaa857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 69.Roberts M., Hayes W., Hunt B.R., Mount S.M., Yorke J.A. Reducing storage requirements for biological sequence comparison. Bioinformatics. 2004;20:3363–3369. doi: 10.1093/bioinformatics/bth408. [DOI] [PubMed] [Google Scholar]
- 70.Marçais G., Pellow D., Bork D., Orenstein Y., Shamir R., Kingsford C. Improving the performance of minimizers and winnowing schemes. Bioinformatics. 2017;33:i110–i117. doi: 10.1093/bioinformatics/btx235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sahlin K., Baudeau T., Cazaux B., Marchet C. A survey of mapping algorithms in the long-reads era. Genome Biol. 2023;24:133. doi: 10.1186/s13059-023-02972-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ondov B.D., Treangen T.J., Melsted P., Mallonee A.B., Bergman N.H., Koren S., Phillippy A.M. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016;17:132. doi: 10.1186/s13059-016-0997-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Martin J.A., Wang Z. Next-generation transcriptome assembly. Nat. Rev. Genet. 2011;12:671–682. doi: 10.1038/nrg3068. [DOI] [PubMed] [Google Scholar]
- 74.Alonge M., Soyk S., Ramakrishnan S., Wang X., Goodwin S., Sedlazeck F.J., Lippman Z.B., Schatz M.C. RaGOO: fast and accurate reference-guided scaffolding of draft genomes. Genome Biol. 2019;20:224. doi: 10.1186/s13059-019-1829-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bankevich A., Nurk S., Antipov D., Gurevich A.A., Dvorkin M., Kulikov A.S., Lesin V.M., Nikolenko S.I., Pham S., Prjibelski A.D., et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sohn J.-I., Nam J.-W. The present and future of de novo whole-genome assembly. Brief. Bioinform. 2018;19:23–40. doi: 10.1093/bib/bbw096. [DOI] [PubMed] [Google Scholar]
- 77.Compeau P.E.C., Pevzner P.A., Tesler G. How to apply de Bruijn graphs to genome assembly. Nat. Biotechnol. 2011;29:987–991. doi: 10.1038/nbt.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pootakham W., Sonthirod C., Naktang C., Kongkachana W., U-Thoomporn S., Phetchawang P., Maknual C., Jiumjamrassil D., Pravinvongvuthi T., Tangphatsornruang S. A de novo reference assembly of the yellow mangrove Ceriops zippeliana genome. G3. 2022;12 doi: 10.1093/g3journal/jkac025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shen A., Luo C., Tan Y., Shen B., Liu L., Li J., Tan Z., Zeng L. A high-quality genome assembly of Lactarius hatsudake strain JH5. G3. 2022;12 doi: 10.1093/g3journal/jkac262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang X., Chen S., Zhang Y., Xiao Y., Qin Y., Li Q., Liu L., Liu B., Chai L., Yang H., et al. Draft genome of the medicinal tea tree Melaleuca alternifolia. Mol. Biol. Rep. 2023;50:1545–1552. doi: 10.1007/s11033-022-08157-8. [DOI] [PubMed] [Google Scholar]
- 81.Tang H., Zhang X., Miao C., Zhang J., Ming R., Schnable J.C., Schnable P.S., Lyons E., Lu J. ALLMAPS: robust scaffold ordering based on multiple maps. Genome Biol. 2015;16:3. doi: 10.1186/s13059-014-0573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Eizenga J.M., Novak A.M., Sibbesen J.A., Heumos S., Ghaffaari A., Hickey G., Chang X., Seaman J.D., Rounthwaite R., Ebler J., et al. Pangenome Graphs. Annu. Rev. Genomics Hum. Genet. 2020;21:139–162. doi: 10.1146/annurev-genom-120219-080406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ebler J., Ebert P., Clarke W.E., Rausch T., Audano P.A., Houwaart T., Mao Y., Korbel J.O., Eichler E.E., Zody M.C., et al. Pangenome-based genome inference allows efficient and accurate genotyping across a wide spectrum of variant classes. Nat. Genet. 2022;54:518–525. doi: 10.1038/s41588-022-01043-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Salmela L., Schröder J. Correcting errors in short reads by multiple alignments. Bioinformatics. 2011;27:1455–1461. doi: 10.1093/bioinformatics/btr170. [DOI] [PubMed] [Google Scholar]
- 85.Dlugosz M., Deorowicz S. RECKONER: read error corrector based on KMC. Bioinformatics. 2017;33:1086–1089. doi: 10.1093/bioinformatics/btw746. [DOI] [PubMed] [Google Scholar]
- 86.Długosz M., Deorowicz S. Illumina reads correction: evaluation and improvements. Sci. Rep. 2024;14:2232. doi: 10.1038/s41598-024-52386-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ilie L., Molnar M. RACER: Rapid and accurate correction of errors in reads. Bioinformatics. 2013;29:2490–2493. doi: 10.1093/bioinformatics/btt407. [DOI] [PubMed] [Google Scholar]
- 88.Song L., Florea L., Langmead B. Lighter: fast and memory-efficient sequencing error correction without counting. Genome Biol. 2014;15:509. doi: 10.1186/s13059-014-0509-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Allam A., Kalnis P., Solovyev V. Karect: accurate correction of substitution, insertion and deletion errors for next-generation sequencing data. Bioinformatics. 2015;31:3421–3428. doi: 10.1093/bioinformatics/btv415. [DOI] [PubMed] [Google Scholar]
- 90.Fischer-Hwang I., Ochoa I., Weissman T., Hernaez M. Denoising of aligned genomic data. Sci. Rep. 2019;9:15067. doi: 10.1038/s41598-019-51418-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kallenborn F., Cascitti J., Schmidt B. CARE 2.0: reducing false-positive sequencing error corrections using machine learning. BMC Bioinformatics. 2022;23:227. doi: 10.1186/s12859-022-04754-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Song L., Florea L. Rcorrector: efficient and accurate error correction for Illumina RNA-seq reads. Gigascience. 2015;4:48. doi: 10.1186/s13742-015-0089-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Amarasinghe S.L., Su S., Dong X., Zappia L., Ritchie M.E., Gouil Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020;21:30. doi: 10.1186/s13059-020-1935-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Goodwin S., Gurtowski J., Ethe-Sayers S., Deshpande P., Schatz M.C., McCombie W.R. Oxford nanopore sequencing, hybrid error correction, and de novo assembly of a eukaryotic genome. Genome Res. 2015;25:1750–1756. doi: 10.1101/gr.191395.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Koren S., Phillippy A.M. One chromosome, one contig: complete microbial genomes from long-read sequencing and assembly. Curr. Opin. Microbiol. 2015;23:110–120. doi: 10.1016/j.mib.2014.11.014. [DOI] [PubMed] [Google Scholar]
- 96.Loman N.J., Quick J., Simpson J.T. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat. Methods. 2015;12:733–735. doi: 10.1038/nmeth.3444. [DOI] [PubMed] [Google Scholar]
- 97.Myers E.W., Jr A history of DNA sequence assembly. it - Information Technology. 2016;58:126–132. [Google Scholar]
- 98.Berlin K., Koren S., Chin C.-S., Drake J.P., Landolin J.M., Phillippy A.M. Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat. Biotechnol. 2015;33:623–630. doi: 10.1038/nbt.3238. [DOI] [PubMed] [Google Scholar]
- 99.Chin C.-S., Alexander D.H., Marks P., Klammer A.A., Drake J., Heiner C., Clum A., Copeland A., Huddleston J., Eichler E.E., et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods. 2013;10:563–569. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- 100.Carvalho A.B., Dupim E.G., Goldstein G. Improved assembly of noisy long reads by k-mer validation. Genome Res. 2016;26:1710–1720. doi: 10.1101/gr.209247.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fu S., Wang A., Au K.F. A comparative evaluation of hybrid error correction methods for error-prone long reads. Genome Biol. 2019;20:26. doi: 10.1186/s13059-018-1605-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dohm J.C., Peters P., Stralis-Pavese N., Himmelbauer H. Benchmarking of long-read correction methods. NAR Genom Bioinform. 2020;2 doi: 10.1093/nargab/lqaa037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhang H., Jain C., Aluru S. A comprehensive evaluation of long read error correction methods. BMC Genomics. 2020;21:889. doi: 10.1186/s12864-020-07227-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Allen F., Crepaldi L., Alsinet C., Strong A.J., Kleshchevnikov V., De Angeli P., Páleníková P., Khodak A., Kiselev V., Kosicki M., et al. Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol. 2018 doi: 10.1038/nbt.4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kosicki M., Tomberg K., Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018;36:765–771. doi: 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Papathanos P.A., Windbichler N. Redkmer: an assembly-free pipeline for the identification of abundant and specific x-chromosome target sequences for x-shredding by CRISPR endonucleases. CRISPR J. 2018;1:88–98. doi: 10.1089/crispr.2017.0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Alkhnbashi O.S., Meier T., Mitrofanov A., Backofen R., Voß B. CRISPR-Cas bioinformatics. Methods. 2020;172:3–11. doi: 10.1016/j.ymeth.2019.07.013. [DOI] [PubMed] [Google Scholar]
- 108.Zhu J.J., Cheng A.W. JACKIE: fast enumeration of genome-wide single- and multicopy CRISPR target sites and their off-target numbers. CRISPR J. 2022;5:618–628. doi: 10.1089/crispr.2022.0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bennis N.X., Kostanjšek M., van den Broek M., Daran J.-M.G. Improving CRISPR-Cas9 mediated genome integration in interspecific hybrid yeasts. N. Biotechnol. 2023;76:49–62. doi: 10.1016/j.nbt.2023.04.001. [DOI] [PubMed] [Google Scholar]
- 110.Pavlichin D.S., Lee H., Greer S.U., Grimes S.M., Weissman T., Ji H.P. KmerKeys: a web resource for searching indexed genome assemblies and variants. Nucleic Acids Res. 2022;50:W448–W453. doi: 10.1093/nar/gkac266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ayad L.A.K., Pissis S.P., Polychronopoulos D. CNEFinder: finding conserved non-coding elements in genomes. Bioinformatics. 2018;34:i743–i747. doi: 10.1093/bioinformatics/bty601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sievers A., Sauer L., Hausmann M., Hildenbrand G. Eukaryotic Genomes Show Strong Evolutionary Conservation of -mer Composition and Correlation Contributions between Introns and Intergenic Regions. Genes. 2021;12 doi: 10.3390/genes12101571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bize A., Midoux C., Mariadassou M., Schbath S., Forterre P., Da Cunha V. Exploring short k-mer profiles in cells and mobile elements from Archaea highlights the major influence of both the ecological niche and evolutionary history. BMC Genomics. 2021;22:186. doi: 10.1186/s12864-021-07471-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Blaisdell B.E. A measure of the similarity of sets of sequences not requiring sequence alignment. Proc. Natl. Acad. Sci. U. S. A. 1986;83:5155–5159. doi: 10.1073/pnas.83.14.5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Höhl M., Ragan M.A. Is multiple-sequence alignment required for accurate inference of phylogeny? Syst. Biol. 2007;56:206–221. doi: 10.1080/10635150701294741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Forêt S., Kantorovitz M.R., Burden C.J. Asymptotic behaviour and optimal word size for exact and approximate word matches between random sequences. BMC Bioinformatics. 2006;7(Suppl 5) doi: 10.1186/1471-2105-7-S5-S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ragan M.A., Bernard G., Chan C.X. Molecular phylogenetics before sequences: oligonucleotide catalogs as k-mer spectra. RNA Biol. 2014;11:176–185. doi: 10.4161/rna.27505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bernard G., Greenfield P., Ragan M.A., Chan C.X. mer similarity, networks of microbial genomes, and taxonomic rank. mSystems. 2018;3 doi: 10.1128/mSystems.00257-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Howe A., Chain P.S.G. Challenges and opportunities in understanding microbial communities with metagenome assembly (accompanied by IPython Notebook tutorial) Front. Microbiol. 2015;6:678. doi: 10.3389/fmicb.2015.00678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kapoor A., Simmonds P., Lipkin W.I., Zaidi S., Delwart E. Use of nucleotide composition analysis to infer hosts for three novel picorna-like viruses. J. Virol. 2010;84:10322–10328. doi: 10.1128/JVI.00601-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Deorowicz S., Gudys A., Dlugosz M., Kokot M., Danek A. Kmer-db: instant evolutionary distance estimation. Bioinformatics. 2019;35:133–136. doi: 10.1093/bioinformatics/bty610. [DOI] [PubMed] [Google Scholar]
- 122.Zhao X. BinDash, software for fast genome distance estimation on a typical personal laptop. Bioinformatics. 2019;35:671–673. doi: 10.1093/bioinformatics/bty651. [DOI] [PubMed] [Google Scholar]
- 123.Zhao J., Zhao X., Pierre-Both J., Konstantinidis K.T. BinDash 2.0: new MinHash scheme allows ultra-fast and accurate genome search and comparisons. bioRxiv. 2024 doi: 10.1101/2024.03.13.584875. [DOI] [Google Scholar]
- 124.Baker D.N., Langmead B. Dashing: fast and accurate genomic distances with HyperLogLog. Genome Biol. 2019;20:265. doi: 10.1186/s13059-019-1875-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Baker D.N., Langmead B. Dashing 2: genomic sketching with multiplicities and locality-sensitive hashing. bioRxiv. 2023 doi: 10.1101/2022.10.16.512384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Agret C., Cazaux B., Limasset A. Toward optimal fingerprint indexing for large scale genomics. bioRxiv. 2022 doi: 10.1101/2021.11.04.467355. [DOI] [Google Scholar]
- 127.Rouzé T., Martayan I., Marchet C., Limasset A. Fractional hitting sets for efficient and lightweight genomic data sketching. bioRxiv. 2023 doi: 10.1101/2023.06.21.545875. [DOI] [Google Scholar]
- 128.Hera M.R., Liu S., Wei W., Rodriguez J.S., Ma C., Koslicki D. Fast, lightweight, and accurate metagenomic functional profiling using FracMinHash sketches. bioRxiv. 2024 doi: 10.1101/2023.11.06.565843. [DOI] [Google Scholar]
- 129.Dubinkina V.B., Ischenko D.S., Ulyantsev V.I., Tyakht A.V., Alexeev D.G. Assessment of k-mer spectrum applicability for metagenomic dissimilarity analysis. BMC Bioinformatics. 2016;17:38. doi: 10.1186/s12859-015-0875-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Smits S.L., Bodewes R., Ruiz-González A., Baumgärtner W., Koopmans M.P., Osterhaus A.D.M.E., Schürch A.C. Recovering full-length viral genomes from metagenomes. Front. Microbiol. 2015;6:1069. doi: 10.3389/fmicb.2015.01069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Edwards R.A., Olson R., Disz T., Pusch G.D., Vonstein V., Stevens R., Overbeek R. Real time metagenomics: using k-mers to annotate metagenomes. Bioinformatics. 2012;28:3316–3317. doi: 10.1093/bioinformatics/bts599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.LaPierre N., Alser M., Eskin E., Koslicki D., Mangul S. Metalign: efficient alignment-based metagenomic profiling via containment min hash. Genome Biol. 2020;21:242. doi: 10.1186/s13059-020-02159-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tambe A., Pachter L. Barcode identification for single cell genomics. BMC Bioinformatics. 2019;20:32. doi: 10.1186/s12859-019-2612-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kirk J.M., Kim S.O., Inoue K., Smola M.J., Lee D.M., Schertzer M.D., Wooten J.S., Baker A.R., Sprague D., Collins D.W., et al. Functional classification of long non-coding RNAs by k-mer content. Nat. Genet. 2018;50:1474–1482. doi: 10.1038/s41588-018-0207-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gilbert J.A., Blaser M.J., Caporaso J.G., Jansson J.K., Lynch S.V., Knight R. Current understanding of the human microbiome. Nat. Med. 2018;24:392–400. doi: 10.1038/nm.4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wood D.E., Lu J., Langmead B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019;20:1–13. doi: 10.1186/s13059-019-1891-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Koslicki D., White S., Ma C., Novikov A. YACHT: an ANI-based statistical test to detect microbial presence/absence in a metagenomic sample. bioRxiv: the preprint server for biology. 2023 doi: 10.1101/2023.04.18.537298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Menzel P., Ng K.L., Krogh A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016;7:1–9. doi: 10.1038/ncomms11257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Johansen J., Plichta D.R., Nissen J.N., Jespersen M.L., Shah S.A., Deng L., Stokholm J., Bisgaard H., Nielsen D.S., Sørensen S.J., et al. Genome binning of viral entities from bulk metagenomics data. Nat. Commun. 2022;13:965. doi: 10.1038/s41467-022-28581-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Nurk S., Meleshko D., Korobeynikov A., Pevzner P.A. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 2017;27:824–834. doi: 10.1101/gr.213959.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chiu C.Y., Miller S.A. Clinical metagenomics. Nat. Rev. Genet. 2019;20:341–355. doi: 10.1038/s41576-019-0113-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gu W., Miller S., Chiu C.Y. Clinical metagenomic next-generation sequencing for pathogen detection. Annu. Rev. Pathol. 2019;14:319–338. doi: 10.1146/annurev-pathmechdis-012418-012751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Drouin A., Giguère S., Déraspe M., Marchand M., Tyers M., Loo V.G., Bourgault A.-M., Laviolette F., Corbeil J. Predictive computational phenotyping and biomarker discovery using reference-free genome comparisons. BMC Genomics. 2016;17:754. doi: 10.1186/s12864-016-2889-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mahé P., Tournoud M. Predicting bacterial resistance from whole-genome sequences using k-mers and stability selection. BMC Bioinformatics. 2018;19:383. doi: 10.1186/s12859-018-2403-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Jaillard M., Palmieri M., van Belkum A., Mahé P. Interpreting k-mer–based signatures for antibiotic resistance prediction. Gigascience. 2020;9 doi: 10.1093/gigascience/giaa110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mouratidis I., Chantzi N., Khan U., Konnaris M.A., Chan C.S.Y., Mareboina M., Georgakopoulos-Soares I. Frequentmers - a novel way to look at metagenomic Next Generation Sequencing data and an application in detecting liver cirrhosis. medRxiv. 2023 doi: 10.1101/2023.09.19.23295771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Morsa D., Baiwir D., La Rocca R., Zimmerman T.A., Hanozin E., Grifnée E., Longuespée R., Meuwis M.-A., Smargiasso N., Pauw E.D., et al. Multi-enzymatic limited digestion: the next-generation sequencing for proteomics? J. Proteome Res. 2019;18:2501–2513. doi: 10.1021/acs.jproteome.9b00044. [DOI] [PubMed] [Google Scholar]
- 148.Zhang Y., Wen J., Yau S.S.-T. Phylogenetic analysis of protein sequences based on a novel k-mer natural vector method. Genomics. 2019;111:1298–1305. doi: 10.1016/j.ygeno.2018.08.010. [DOI] [PubMed] [Google Scholar]
- 149.Chang H.-H., Huber R.G., Bond P.J., Grad Y.H., Camerini D., Maurer-Stroh S., Lipsitch M. Systematic analysis of protein identity between Zika virus and other arthropod-borne viruses. Bull. World Health Organ. 2017;95:517–525I. doi: 10.2471/BLT.16.182105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Weging S., Gogol-Döring A., Grosse I. Taxonomic analysis of metagenomic data with kASA. Nucleic Acids Res. 2021;49 doi: 10.1093/nar/gkab200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Du Z., He Y., Li J., Uversky V.N. DeepAdd: Protein function prediction from k-mer embedding and additional features. Comput. Biol. Chem. 2020;89 doi: 10.1016/j.compbiolchem.2020.107379. [DOI] [PubMed] [Google Scholar]
- 152.Brum J.R., Ignacio-Espinoza J.C., Kim E.-H., Trubl G., Jones R.M., Roux S., VerBerkmoes N.C., Rich V.I., Sullivan M.B. Illuminating structural proteins in viral ‘dark matter’ with metaproteomics. Proc. Natl. Acad. Sci. U. S. A. 2016;113:2436–2441. doi: 10.1073/pnas.1525139113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Santoni D. The impact of codon choice on translation process in Saccharomyces cerevisiae: folding class, protein function and secondary structure. J. Theor. Biol. 2021;526 doi: 10.1016/j.jtbi.2021.110806. [DOI] [PubMed] [Google Scholar]
- 154.van Kempen M., Kim S.S., Tumescheit C., Mirdita M., Lee J., Gilchrist C.L.M., Söding J., Steinegger M. Fast and accurate protein structure search with Foldseek. Nat. Biotechnol. 2023 doi: 10.1038/s41587-023-01773-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Richer J., Johnston S.A., Stafford P. Epitope identification from fixed-complexity random-sequence peptide microarrays. Mol. Cell. Proteomics. 2015;14:136–147. doi: 10.1074/mcp.M114.043513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Paull M.L., Johnston T., Ibsen K.N., Bozekowski J.D., Daugherty P.S. A general approach for predicting protein epitopes targeted by antibody repertoires using whole proteomes. PLoS One. 2019;14 doi: 10.1371/journal.pone.0217668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Paull M.L., Bozekowski J.D., Daugherty P.S. Mapping antibody binding using multiplexed epitope substitution analysis. J. Immunol. Methods. 2021;499 doi: 10.1016/j.jim.2021.113178. [DOI] [PubMed] [Google Scholar]
- 158.Carballo G.M., Vázquez K.G., García-González L.A., Rio G.D., Brizuela C.A. Embedded-AMP: a multi-thread computational method for the systematic identification of antimicrobial peptides embedded in proteome sequences. Antibiotics ((Basel)) 2023;12 doi: 10.3390/antibiotics12010139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Hampikian G., Andersen T. Absent sequences: nullomers and primes. Pac. Symp. Biocomput. 2007 doi: 10.1142/9789812772435_0034. [DOI] [PubMed] [Google Scholar]
- 160.Silva R.M., Pratas D., Castro L., Pinho A.J., Ferreira P.J.S.G. Three minimal sequences found in Ebola virus genomes and absent from human DNA. Bioinformatics. 2015;31:2421–2425. doi: 10.1093/bioinformatics/btv189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Goswami J., Davis M.C., Andersen T., Alileche A., Hampikian G. Safeguarding forensic DNA reference samples with nullomer barcodes. J. Forensic Leg. Med. 2013;20:513–519. doi: 10.1016/j.jflm.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 162.Koulouras G., Frith M.C. Significant non-existence of sequences in genomes and proteomes. Nucleic Acids Res. 2021;49:3139–3155. doi: 10.1093/nar/gkab139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Georgakopoulos-Soares I., Yizhar-Barnea O., Mouratidis I., Hemberg M., Ahituv N. Absent from DNA and protein: genomic characterization of nullomers and nullpeptides across functional categories and evolution. Genome Biol. 2021;22:245. doi: 10.1186/s13059-021-02459-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Vergni D., Santoni D. Nullomers and high order nullomers in genomic sequences. PLoS One. 2016;11 doi: 10.1371/journal.pone.0164540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Pinho A.J., Ferreira P.J.S.G., Garcia S.P., Rodrigues J.M.O.S. On finding minimal absent words. BMC Bioinformatics. 2009;10:137. doi: 10.1186/1471-2105-10-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Barton C., Heliou A., Mouchard L., Pissis S.P. Linear-time computation of minimal absent words using suffix array. BMC Bioinformatics. 2014;15:388. doi: 10.1186/s12859-014-0388-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Garcia S.P., Pinho A.J., Rodrigues J.M.O.S., Bastos C.A.C., Ferreira P.J.S.G. Minimal absent words in prokaryotic and eukaryotic genomes. PLoS One. 2011;6 doi: 10.1371/journal.pone.0016065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Chairungsee S., Crochemore M. Negative information for building phylogenies. Recent Pat. DNA Gene Seq. 2013;7:128–136. doi: 10.2174/1872215611307020007. [DOI] [PubMed] [Google Scholar]
- 169.Garcia S.P., Pinho A.J. Minimal absent words in four human genome assemblies. PLoS One. 2011;6 doi: 10.1371/journal.pone.0029344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kusalik A., Trost B., Bickis M., Fasano C., Capone G., Kanduc D. Codon number shapes peptide redundancy in the universal proteome composition. Peptides. 2009;30:1940–1944. doi: 10.1016/j.peptides.2009.06.035. [DOI] [PubMed] [Google Scholar]
- 171.Mouratidis I., Baltoumas F.A., Chantzi N., Chan C.S.Y., Montgomery A., Konnaris M.A., Georgakopoulos G.C., Das A., Chartoumpekis D., Kovac J., et al. kmerDB: a database encompassing the set of genomic and proteomic sequence information for each species. bioRxiv. 2023 doi: 10.1101/2023.11.13.566926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Mouratidis I., Chan C.S.Y., Chantzi N., Tsiatsianis G.C., Hemberg M., Ahituv N., Georgakopoulos-Soares I. Quasi-prime peptides: identification of the shortest peptide sequences unique to a species. NAR Genom Bioinform. 2023;5 doi: 10.1093/nargab/lqad039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Mouratidis I., Konnaris M.A., Chantzi N., Chan C.S.Y., Montgomery A., Baltoumas F.A., Patsakis M., Mareboina M., Pavlopoulos G.A., Chartoumpekis D.V., et al. Nucleic Quasi-Primes: Identification of the Shortest Unique Oligonucleotide Sequences in a Species. bioRxiv. 2023 doi: 10.1101/2023.12.12.571240. [DOI] [Google Scholar]
- 174.Acquisti C., Poste G., Curtiss D., Kumar S. Nullomers: really a matter of natural selection? PLoS One. 2007;2 doi: 10.1371/journal.pone.0001022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Pedone F., Santoni D. Preferential nucleosome occupancy at high values of DNA helical rise. DNA Res. 2012;19:81–90. doi: 10.1093/dnares/dsr043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Pedone F., Santoni D. Sequence-dependent DNA helical rise and nucleosome stability. BMC Mol. Biol. 2009;10:105. doi: 10.1186/1471-2199-10-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Using minimal absent words to build phylogeny. Theor. Comput. Sci. 2012;450:109–116. [Google Scholar]
- 178.Pratas D., Silva J.M. Persistent minimal sequences of SARS-CoV-2. Bioinformatics. 2021;36:5129–5132. doi: 10.1093/bioinformatics/btaa686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kanduc D. Correlating low-similarity peptide sequences and allergenic epitopes. Curr. Pharm. Des. 2008;14:289–295. doi: 10.2174/138161208783413257. [DOI] [PubMed] [Google Scholar]
- 180.Kanduc D., Tessitore L., Lucchese G., Kusalik A., Farber E., Marincola F.M. Sequence uniqueness and sequence variability as modulating factors of human anti-HCV humoral immune response. Cancer Immunol. Immunother. 2008;57:1215–1223. doi: 10.1007/s00262-008-0456-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kanduc D. Immunogenicity in peptide-immunotherapy: from self/nonself to similar/dissimilar sequences. Adv. Exp. Med. Biol. 2008;640:198–207. doi: 10.1007/978-0-387-09789-3_15. [DOI] [PubMed] [Google Scholar]
- 182.Blondelle S.E., Moya-Castro R., Osawa K., Schroder K., Wilson D.B. Immunogenically optimized peptides derived from natural mutants of HIV CTL epitopes and peptide combinatorial libraries. Biopolymers. 2008;90:683–694. doi: 10.1002/bip.21020. [DOI] [PubMed] [Google Scholar]
- 183.Vergni D., Gaudio R., Santoni D. The farther the better: Investigating how distance from human self affects the propensity of a peptide to be presented on cell surface by MHC class I molecules, the case of Trypanosoma cruzi. PLoS One. 2020;15 doi: 10.1371/journal.pone.0243285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Tuller T., Chor B., Nelson N. Forbidden penta-peptides. Protein Sci. 2007;16:2251–2259. doi: 10.1110/ps.073067607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Otaki J.M., Gotoh T., Yamamoto H. Potential implications of availability of short amino acid sequences in proteins: an old and new approach to protein decoding and design. Biotechnol. Annu. Rev. 2008;14:109–141. doi: 10.1016/S1387-2656(08)00004-5. [DOI] [PubMed] [Google Scholar]
- 186.Patel A., Dong J.C., Trost B., Richardson J.S., Tohme S., Babiuk S., Kusalik A., Kung S.K.P., Kobinger G.P. Pentamers not found in the universal proteome can enhance antigen specific immune responses and adjuvant vaccines. PLoS One. 2012;7 doi: 10.1371/journal.pone.0043802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Santoni D., Vergni D. In the search of potential epitopes for Wuhan seafood market pneumonia virus using high order nullomers. J. Immunol. Methods. 2020;481–482 doi: 10.1016/j.jim.2020.112787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Mehrotra N., Kharbanda S., Singh H. Peptide-based combination nanoformulations for cancer therapy. Nanomedicine. 2020;15:2201–2217. doi: 10.2217/nnm-2020-0220. [DOI] [PubMed] [Google Scholar]
- 189.Karami Fath M., Babakhaniyan K., Zokaei M., Yaghoubian A., Akbari S., Khorsandi M., Soofi A., Nabi-Afjadi M., Zalpoor H., Jalalifar F., et al. Anti-cancer peptide-based therapeutic strategies in solid tumors. Cell. Mol. Biol. Lett. 2022;27:33. doi: 10.1186/s11658-022-00332-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ali N., Wolf C., Kanchan S., Veerabhadraiah S.R., Bond L., Turner M.W., Jorcyk C.L., Hampikian G. Nullomer peptide increases immune cell infiltration and reduces tumor metabolism in triple negative breast cancer mouse model. Res Sq. 2023 doi: 10.21203/rs.3.rs-3097552/v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Falda M., Fontana P., Barzon L., Toppo S., Lavezzo E. keeSeek: searching distant non-existing words in genomes for PCR-based applications. Bioinformatics. 2014;30:2662–2664. doi: 10.1093/bioinformatics/btu312. [DOI] [PubMed] [Google Scholar]
- 192.Pierros V., Kontopodis E., Stravopodis D.J., Tsangaris G.T. Unique peptide signatures of SARS-CοV-2 virus against human proteome reveal variants’ immune escape and infectiveness. Heliyon. 2022;8 doi: 10.1016/j.heliyon.2022.e09222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kontopodis E., Pierros V., Vorgias C.E., Papassideri I.S., Stravopodis D.J., Tsangaris G.T. Uniquome: construction and decoding of a novel proteomic atlas that contains new peptide entities. bioRxiv. 2024 doi: 10.1101/2024.01.30.577925. [DOI] [Google Scholar]
- 194.Chowdhury B., Garai G. A review on multiple sequence alignment from the perspective of genetic algorithm. Genomics. 2017;109:419–431. doi: 10.1016/j.ygeno.2017.06.007. [DOI] [PubMed] [Google Scholar]
- 195.Chao J., Tang F., Xu L. Developments in algorithms for sequence alignment: a review. Biomolecules. 2022;12 doi: 10.3390/biom12040546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Li H., Durbin R. Genome assembly in the telomere-to-telomere era. Nat. Rev. Genet. 2024 doi: 10.1038/s41576-024-00718-w. [DOI] [PubMed] [Google Scholar]
- 197.Marchet C., Boucher C., Puglisi S.J., Medvedev P., Salson M., Chikhi R. Data structures based on -mers for querying large collections of sequencing data sets. Genome Res. 2021;31:1–12. doi: 10.1101/gr.260604.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Portik D.M., Brown C.T., Pierce-Ward N.T. Evaluation of taxonomic classification and profiling methods for long-read shotgun metagenomic sequencing datasets. BMC Bioinformatics. 2022;23:1–39. doi: 10.1186/s12859-022-05103-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Edgar R. Syncmers are more sensitive than minimizers for selecting conserved ‑mers in biological sequences. PeerJ. 2021;9 doi: 10.7717/peerj.10805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Sahlin K. Effective sequence similarity detection with strobemers. Genome Res. 2021;31:2080–2094. doi: 10.1101/gr.275648.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Melsted P., Pritchard J.K. Efficient counting of k-mers in DNA sequences using a bloom filter. BMC Bioinformatics. 2011;12:333. doi: 10.1186/1471-2105-12-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Wang J., Chen S., Dong L., Wang G. CHTKC: a robust and efficient k-mer counting algorithm based on a lock-free chaining hash table. Brief. Bioinform. 2020;22:bbaa063. doi: 10.1093/bib/bbaa063. [DOI] [PubMed] [Google Scholar]
- 203.Nyström-Persson J., Keeble-Gagnère G., Zawad N. Compact and evenly distributed k-mer binning for genomic sequences. Bioinformatics. 2021;37:1338. doi: 10.1093/bioinformatics/btab323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Rizk G., Lavenier D., Chikhi R. DSK: k-mer counting with very low memory usage. Bioinformatics. 2013;29:652–653. doi: 10.1093/bioinformatics/btt020. [DOI] [PubMed] [Google Scholar]
- 205.Thomas A., Barriere S., Broseus L., Brooke J., Lorenzi C., Villemin J.-P., Beurier G., Sabatier R., Reynes C., Mancheron A., et al. GECKO is a genetic algorithm to classify and explore high throughput sequencing data. Commun Biol. 2019;2:222. doi: 10.1038/s42003-019-0456-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Erbert M., Rechner S., Müller-Hannemann M. Gerbil: a fast and memory-efficient k-mer counter with GPU-support. Algorithms Mol. Biol. 2017;12:1–12. doi: 10.1186/s13015-017-0097-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Audano P., Vannberg F. KAnalyze: a fast versatile pipelined k-mer toolkit. Bioinformatics. 2014;30:2070–2072. doi: 10.1093/bioinformatics/btu152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Mamun A.-A., Pal S., Rajasekaran S. KCMBT: a k-mer counter based on multiple burst trees. Bioinformatics. 2016;32:2783. doi: 10.1093/bioinformatics/btw345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Tang D., Li Y., Tan D., Fu J., Tang Y., Lin J., Zhao R., Du H., Zhao Z. KCOSS: an ultra-fast k-mer counter for assembled genome analysis. Bioinformatics. 2022;38:933–940. doi: 10.1093/bioinformatics/btab797. [DOI] [PubMed] [Google Scholar]
- 210.Crusoe M.R., Alameldin H.F., Awad S., Boucher E., Caldwell A., Cartwright R., Charbonneau A., Constantinides B., Edvenson G., Fay S., et al. The khmer software package: enabling efficient nucleotide sequence analysis. F1000Res. 2015;4:900. doi: 10.12688/f1000research.6924.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.KmerAnalysis.jl: K-mer counting algorithms and count-data utilities for the BioJulia framework Github.
- 212.Riquier S., Bessiere C., Guibert B., Bouge A.-L., Boureux A., Ruffle F., Audoux J., Gilbert N., Xue H., Gautheret D., et al. Kmerator Suite: design of specific k-mer signatures and automatic metadata discovery in large RNA-seq datasets. NAR Genom Bioinform. 2021;3 doi: 10.1093/nargab/lqab058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Seemann,T. kounta: Generate multi-sample k-mer count matrix from WGS Github.
- 214.Livesey,J. krust: counts k-mers, written in rust Github.
- 215.Li Y., XifengYan MSPKmerCounter: a fast and memory efficient approach for K-mer counting. arXiv [q-bio. GN] 2015 [Google Scholar]
- 216.Shen W., Le S., Li Y., Hu F. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS One. 2016;11 doi: 10.1371/journal.pone.0163962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.[No title].
- 218.Kurtz S., Narechania A., Stein J.C., Ware D. A new method to compute K-mer frequencies and its application to annotate large repetitive plant genomes. BMC Genomics. 2008;9:517. doi: 10.1186/1471-2164-9-517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Roy R.S., Bhattacharya D., Schliep A. Turtle: identifying frequent k-mers with cache-efficient algorithms. Bioinformatics. 2014;30:1950–1957. doi: 10.1093/bioinformatics/btu132. [DOI] [PubMed] [Google Scholar]
- 220.Zhang J., Guo J., Yu X., Yu X., Guo W., Zeng T., Chen L. Bioinformatics Research and Applications. Springer International Publishing,; 2017. Mining K-mers of various lengths in biological sequences; pp. 186–195. [Google Scholar]
- 221.Vurture G.W., Sedlazeck F.J., Nattestad M., Underwood C.J., Fang H., Gurtowski J., Schatz M.C. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics. 2017;33:2202–2204. doi: 10.1093/bioinformatics/btx153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Kaplinski L., Lepamets M., Remm M. GenomeTester4: a toolkit for performing basic set operations - union, intersection and complement on k-mer lists. Gigascience. 2015;4:58. doi: 10.1186/s13742-015-0097-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Mapleson D., Garcia Accinelli G., Kettleborough G., Wright J., Clavijo B.J. KAT: a K-mer analysis toolkit to quality control NGS datasets and genome assemblies. Bioinformatics. 2017;33:574–576. doi: 10.1093/bioinformatics/btw663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Cha S., Bird D.M. Optimizing k-mer size using a variant grid search to enhance de novo genome assembly. Bioinformation. 2016;12:36–40. doi: 10.6026/97320630012036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Melsted P., Halldórsson B.V. KmerStream: streaming algorithms for k-mer abundance estimation. Bioinformatics. 2014;30:3541–3547. doi: 10.1093/bioinformatics/btu713. [DOI] [PubMed] [Google Scholar]
- 226.Mohamadi H., Khan H., Birol I. ntCard: a streaming algorithm for cardinality estimation in genomics data. Bioinformatics. 2017;33:1324–1330. doi: 10.1093/bioinformatics/btw832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Rangavittal S., Stopa N., Tomaszkiewicz M., Sahlin K., Makova K.D., Medvedev P. DiscoverY: a classifier for identifying Y chromosome sequences in male assemblies. BMC Genomics. 2019;20:641. doi: 10.1186/s12864-019-5996-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Harris R.S., Medvedev P. Improved representation of sequence bloom trees. Bioinformatics. 2020;36:721–727. doi: 10.1093/bioinformatics/btz662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Wang Y., Chen Q., Deng C., Zheng Y., Sun F. KmerGO: A Tool to Identify Group-Specific Sequences With k-mers. Front. Microbiol. 2020;11:2067. doi: 10.3389/fmicb.2020.02067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Pan T., Flick P., Jain C., Liu Y., Aluru S. In Proceedings of the 7th ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics, BCB ’16. Association for Computing Machinery; New York, NY, USA: 2016. Kmerind: A Flexible Parallel Library for K-mer Indexing of Biological Sequences on Distributed Memory Systems; pp. 422–433. [DOI] [PubMed] [Google Scholar]
- 231.Pandey P., Almodaresi F., Bender M.A., Ferdman M., Johnson R., Patro R. Mantis: A Fast, Small, and Exact Large-Scale Sequence-Search Index. Cell Syst. 2018;7:201–207.e4. doi: 10.1016/j.cels.2018.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Karasikov M., Mustafa H., Danciu D., Zimmermann M., Barber C., Rätsch G., Kahles A. MetaGraph: Indexing and Analysing Nucleotide Archives at Petabase-scale. bioRxiv. 2020 doi: 10.1101/2020.10.01.322164. [DOI] [Google Scholar]
- 233.Marchet C., Limasset A. Scalable sequence database search using partitioned aggregated Bloom comb trees. Bioinformatics. 2023;39:i252–i259. doi: 10.1093/bioinformatics/btad225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Rangavittal S., Harris R.S., Cechova M., Tomaszkiewicz M., Chikhi R., Makova K.D., Medvedev P. RecoverY: k-mer-based read classification for Y-chromosome-specific sequencing and assembly. Bioinformatics. 2018;34:1125–1131. doi: 10.1093/bioinformatics/btx771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Solomon B., Kingsford C. Fast search of thousands of short-read sequencing experiments. Nat. Biotechnol. 2016;34:300–302. doi: 10.1038/nbt.3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Yu Y., Liu J., Liu X., Zhang Y., Magner E., Lehnert E., Qian C., Liu J. SeqOthello: querying RNA-seq experiments at scale. Genome Biol. 2018;19:167. doi: 10.1186/s13059-018-1535-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Pibiri G.E. Sparse and skew hashing of K-mers. Bioinformatics. 2022;38:i185–i194. doi: 10.1093/bioinformatics/btac245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Sun C., Medvedev P. Toward fast and accurate SNP genotyping from whole genome sequencing data for bedside diagnostics. Bioinformatics. 2018;35:415–420. doi: 10.1093/bioinformatics/bty641. [DOI] [PubMed] [Google Scholar]
- 239.Gnerre S., Maccallum I., Przybylski D., Ribeiro F.J., Burton J.N., Walker B.J., Sharpe T., Hall G., Shea T.P., Sykes S., et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl. Acad. Sci. U. S. A. 2011;108:1513–1518. doi: 10.1073/pnas.1017351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Holley G., Melsted P. Bifrost: highly parallel construction and indexing of colored and compacted de Bruijn graphs. Genome Biol. 2020;21:249. doi: 10.1186/s13059-020-02135-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Koren S., Walenz B.P., Berlin K., Miller J.R., Bergman N.H., Phillippy A.M. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722–736. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Iqbal Z., Caccamo M., Turner I., Flicek P., McVean G. De novo assembly and genotyping of variants using colored de Bruijn graphs. Nat. Genet. 2012;44:226–232. doi: 10.1038/ng.1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Guidi G., Raulet G., Rokhsar D., Oliker L., Yelick K., Buluç A. In Proceedings of the 51st International Conference on Parallel Processing. ICPP ’22. Association for Computing Machinery; New York, NY, USA: 2023. Distributed-Memory Parallel Contig Generation for De Novo Long-Read Genome Assembly; pp. 1–11. [Google Scholar]
- 244.Li D., Liu C.-M., Luo R., Sadakane K., Lam T.-W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015;31:1674–1676. doi: 10.1093/bioinformatics/btv033. [DOI] [PubMed] [Google Scholar]
- 245.Mikheenko A., Prjibelski A., Saveliev V., Antipov D., Gurevich A. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics. 2018;34:i142–i150. doi: 10.1093/bioinformatics/bty266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Souvorov A., Agarwala R., Lipman D.J. SKESA: strategic k-mer extension for scrupulous assemblies. Genome Biol. 2018;19:153. doi: 10.1186/s13059-018-1540-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Mikheenko A., Bzikadze A.V., Gurevich A., Miga K.H., Pevzner P.A. TandemMapper and TandemQUAST: mapping long reads and assessing/improving assembly quality in extra-long tandem repeats. bioRxiv. 2019 doi: 10.1101/2019.12.23.887158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Mikheenko A., Bzikadze A.V., Gurevich A., Miga K.H., Pevzner P.A. TandemTools: mapping long reads and assessing/improving assembly quality in extra-long tandem repeats. Bioinformatics. 2020;36:i75–i83. doi: 10.1093/bioinformatics/btaa440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Morgenstern B., Zhu B., Horwege S., Leimeister C.A. Estimating evolutionary distances between genomic sequences from spaced-word matches. Algorithms Mol. Biol. 2015;10:5. doi: 10.1186/s13015-015-0032-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.BBMap: A Fast, Accurate, Splice-Aware Aligner (2014).
- 251.Langmead B. Aligning short sequencing reads with Bowtie. Curr. Protoc. Bioinformatics. 2010;Chapter 11(Unit 11.7) doi: 10.1002/0471250953.bi1107s32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Li H., Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Abuín J.M., Pichel J.C., Pena T.F., Amigo J. BigBWA: approaching the Burrows-Wheeler aligner to big data technologies. Bioinformatics. 2015;31:4003–4005. doi: 10.1093/bioinformatics/btv506. [DOI] [PubMed] [Google Scholar]
- 256.Lorenzi C., Barriere S., Villemin J.-P., Dejardin Bretones L., Mancheron A., Ritchie W. iMOKA: k-mer based software to analyze large collections of sequencing data. Genome Biol. 2020;21:261. doi: 10.1186/s13059-020-02165-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Lindgreen S., Adair K.L., Gardner P.P. An evaluation of the accuracy and speed of metagenome analysis tools. Sci. Rep. 2016;6:19233. doi: 10.1038/srep19233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Ye S.H., Siddle K.J., Park D.J., Sabeti P.C. Benchmarking metagenomics tools for taxonomic classification. Cell. 2019;178:779–794. doi: 10.1016/j.cell.2019.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Koslicki D., Chatterjee S., Shahrivar D., Walker A.W., Francis S.C., Fraser L.J., Vehkaperä M., Lan Y., Corander J. ARK: Aggregation of Reads by K-Means for Estimation of Bacterial Community Composition. PLoS One. 2015;10 doi: 10.1371/journal.pone.0140644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Lu J., Breitwieser F.P., Thielen P., Salzberg S.L. Bracken: estimating species abundance in metagenomics data. PeerJ Comput. Sci. 2017;3 [Google Scholar]
- 261.Bui V.-K., Wei C. CDKAM: a taxonomic classification tool using discriminative k-mers and approximate matching strategies. BMC Bioinformatics. 2020;21:468. doi: 10.1186/s12859-020-03777-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Ounit R., Wanamaker S., Close T.J., Lonardi S. CLARK: fast and accurate classification of metagenomic and genomic sequences using discriminative k-mers. BMC Genomics. 2015;16:236. doi: 10.1186/s12864-015-1419-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Davenport C.F., Neugebauer J., Beckmann N., Friedrich B., Kameri B., Kokott S., Paetow M., Siekmann B., Wieding-Drewes M., Wienhöfer M., et al. Genometa--a fast and accurate classifier for short metagenomic shotgun reads. PLoS One. 2012;7 doi: 10.1371/journal.pone.0041224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Shen W., Xiang H., Huang T., Tang H., Peng M., Cai D., Hu P., Ren H. KMCP: accurate metagenomic profiling of both prokaryotic and viral populations by pseudo-mapping. Bioinformatics. 2023;39 doi: 10.1093/bioinformatics/btac845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Hasman H., Saputra D., Sicheritz-Ponten T., Lund O., Svendsen C.A., Frimodt-Møller N., Aarestrup F.M. Rapid whole-genome sequencing for detection and characterization of microorganisms directly from clinical samples. J. Clin. Microbiol. 2014;52:139–146. doi: 10.1128/JCM.02452-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Ames S.K., Hysom D.A., Gardner S.N., Lloyd G.S., Gokhale M.B., Allen J.E. Scalable metagenomic taxonomy classification using a reference genome database. Bioinformatics. 2013;29:2253–2260. doi: 10.1093/bioinformatics/btt389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Schmidt S., Khan S., Alanko J.N., Pibiri G.E., Tomescu A.I. Matchtigs: minimum plain text representation of k-mer sets. Genome Biol. 2023;24:136. doi: 10.1186/s13059-023-02968-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Müller A., Hundt C., Hildebrandt A., Hankeln T., Schmidt B. MetaCache: context-aware classification of metagenomic reads using minhashing. Bioinformatics. 2017;33:3740–3748. doi: 10.1093/bioinformatics/btx520. [DOI] [PubMed] [Google Scholar]
- 269.Koslicki D., Falush D. MetaPalette: a -mer painting approach for metagenomic taxonomic profiling and quantification of novel strain variation. mSystems. 2016;1 doi: 10.1128/mSystems.00020-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Chatterjee S., Koslicki D., Dong S., Innocenti N., Cheng L., Lan Y., Vehkaperä M., Skoglund M., Rasmussen L.K., Aurell E., et al. SEK: sparsity exploiting k-mer-based estimation of bacterial community composition. Bioinformatics. 2014;30:2423–2431. doi: 10.1093/bioinformatics/btu320. [DOI] [PubMed] [Google Scholar]
- 271.Roosaare M., Vaher M., Kaplinski L., Möls M., Andreson R., Lepamets M., Kõressaar T., Naaber P., Kõljalg S., Remm M. StrainSeeker: fast identification of bacterial strains from raw sequencing reads using user-provided guide trees. PeerJ. 2017;5 doi: 10.7717/peerj.3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Diaz N.N., Krause L., Goesmann A., Niehaus K., Nattkemper T.W. TACOA: taxonomic classification of environmental genomic fragments using a kernelized nearest neighbor approach. BMC Bioinformatics. 2009;10:56. doi: 10.1186/1471-2105-10-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Taxonomer: a fast and accurate metagenomics tool and its uses on clinical specimens (2016).
- 274.Teeling H., Waldmann J., Lombardot T., Bauer M., Glöckner F.O. TETRA: a web-service and a stand-alone program for the analysis and comparison of tetranucleotide usage patterns in DNA sequences. BMC Bioinformatics. 2004;5:163. doi: 10.1186/1471-2105-5-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Koslicki D., Foucart S., Rosen G. WGSQuikr: fast whole-genome shotgun metagenomic classification. PLoS One. 2014;9 doi: 10.1371/journal.pone.0091784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Fan H., Ives A.R., Surget-Groba Y. Reconstructing phylogeny from reduced-representation genome sequencing data without assembly or alignment. Mol. Ecol. Resour. 2018;18:1482–1491. doi: 10.1111/1755-0998.12921. [DOI] [PubMed] [Google Scholar]
- 277.Leimeister C.-A., Sohrabi-Jahromi S., Morgenstern B. Fast and accurate phylogeny reconstruction using filtered spaced-word matches. Bioinformatics. 2017;33:971–979. doi: 10.1093/bioinformatics/btw776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.McHardy A.C., Martín H.G., Tsirigos A., Hugenholtz P., Rigoutsos I. Accurate phylogenetic classification of variable-length DNA fragments. Nat. Methods. 2007;4:63–72. doi: 10.1038/nmeth976. [DOI] [PubMed] [Google Scholar]
- 279.Sarmashghi S., Bohmann K., P Gilbert M.T., Bafna V., Mirarab S. Skmer: assembly-free and alignment-free sample identification using genome skims. Genome Biol. 2019;20:34. doi: 10.1186/s13059-019-1632-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Röhling S., Linne A., Schellhorn J., Hosseini M., Dencker T., Morgenstern B. The number of k-mer matches between two DNA sequences as a function of k and applications to estimate phylogenetic distances. PLoS One. 2020;15 doi: 10.1371/journal.pone.0228070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Bromberg R., Grishin N.V., Otwinowski Z. Phylogeny reconstruction with alignment-free method that corrects for horizontal gene transfer. PLoS Comput. Biol. 2016;12 doi: 10.1371/journal.pcbi.1004985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Gish W., States D.J. Identification of protein coding regions by database similarity search. Nat. Genet. 1993;3:266–272. doi: 10.1038/ng0393-266. [DOI] [PubMed] [Google Scholar]
- 283.Buchfink B., Xie C., Huson D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 2015;12:59–60. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- 284.Steinegger M., Söding J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017;35:1026–1028. doi: 10.1038/nbt.3988. [DOI] [PubMed] [Google Scholar]
- 285.Huson D.H., Xie C. A poor man’s BLASTX--high-throughput metagenomic protein database search using PAUDA. Bioinformatics. 2014;30:38–39. doi: 10.1093/bioinformatics/btt254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Zhao Y., Tang H., Ye Y. RAPSearch2: a fast and memory-efficient protein similarity search tool for next-generation sequencing data. Bioinformatics. 2012;28:125–126. doi: 10.1093/bioinformatics/btr595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Edgar R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material