

Abstract
Immunogenic cell death (ICD), which results from insufficient cellular adaptation to specific stressors, occupies a central position in the development of novel anticancer therapies. Several academic groups and biotechnology companies are pursuing strategies to elicit ICD, either as standalone therapeutic approaches or as means to convert immunologically cold tumors that are insensitive to immunotherapy into hot and immunotherapy-sensitive lesions. The development of ICD-inducing treatments, however, is hindered by various obstacles. Some of these relate to the intrinsic complexity of cancer cell biology, while others arise from the use of conventional therapeutic strategies that were developed according to immune agnostic principles. Moreover, current discovery platforms for the development of novel ICD inducers suffer from limitations that must be addressed to improve bench-to-bedside translational efforts. Finally, an improved appreciation of the conceptual difference among key factors that discriminate distinct forms of cell death will assist the design of clinically viable ICD inducers.
Keywords: antigenicity, chemotherapy, damage-associated molecular patterns, dendritic cells, immune checkpoint inhibitors, radiation therapy
Introduction
To maintain organismal health, many cell types undergo a constant process of renewal and turnover, meaning that they are continuously generated by cell division and later removed by a combination of cell-intrinsic and -extrinsic mechanisms1. Specifically, aging cells that are on the verge of elimination activate cell-autonomous suicidal pathways coupled to the early emission of “come get-me” and “eat-me” signals to the attention of phagocytes, in particular macrophages2,3. By this mechanism, dying cells are engulfed and digested before they fragment and their content spills into the extracellular space. Such a silent removal of dying cells, which is referred to as “efferocytosis”, actively prevents potentially damaging inflammatory reactions and (auto)immune responses against dead cell antigens2,3.
However, if cell death is induced by infectious pathogens such as viruses and intracellular bacteria, it is in the best interest of the organism to mount innate immune responses connected to the activation of adaptive immune effectors4. Innate immunity ensures the prompt destruction of infected cells based on the recognition of rather unspecific signals of stress, such as the exposure of activatory ligands for natural killer (NK) cells5,6. Conversely, adaptive immunity provides the host with a mechanism to recall past episodes of infection by same pathogen and hence mobilize a specific response to re-infection in an accelerated fashion7. In this context, evolutionary ancient cell-autonomous pathways elicited by infection such as the so-called “integrated stress response” (ISR), encompassing endoplasmic reticulum (ER) stress and autophagy, as well as the secretion of type I interferon (IFN) upon cytosolic nucleic acid sensing are intimately linked to the stimulation of myeloid cells (in particular dendritic cells [DCs], which are the most potent antigen-presenting cells) and lymphoid cells (in particular T cells), thus orchestrating specific immune responses against pathogen-encoded antigens8.
Immune responses against malignant cells appear to follow similar rules4. Thus, cancer cells – which are antigenically different from normal cells and intrinsically stressed owing to altered metabolic functions - can alert myeloid and lymphoid effectors to clear them in the context of natural immunosurveillance9. Moreover, some clinically successful anticancer treatments including conventional chemotherapeutics, focal radiation therapy (at least when used according to specific dose and fractionation schedules) and targeted anticancer agents can stress and kill neoplastic cells in a fashion that mimics the infection by pathogens (so-called “viral mimicry”)10, hence eliciting tumor-targeting immune responses11–14. Accordingly, the demise of cancer cells succumbing to such “immunogenic cell death” (ICD) inducers can sensitize tumors to immune checkpoint inhibitors (ICIs) and possibly other forms of immunotherapy, as demonstrated in several clinical trials15–19 (Box 1). Of note, it appears that such an indirect, immune-dependent mechanism is even more important than direct debulking of the tumor mass by therapy for determining the long-term outcome of antineoplastic regimens20. In line with this notion, the density, composition and functionality of the pre-existing or therapy-induced cancer immune infiltrate (so-called “immune contexture”) predict disease outcome in a variety of oncological settings21.
Box 1. Immunotherapeutic agents benefiting from ICD induction.
During the past two decades, immunotherapy not only has become a major player in the clinical management of cancer, largely driven by the approval of immune checkpoint inhibitors (ICIs) for an ever increasing number of oncological indications, but has also diversified to encompass a number of approaches beyond ICIs20. Many of these immunotherapeutic interventions, some of which have already been licensed by various regulatory agencies worldwide for use in cancer patients, may benefit (directly or indirectly) from immunogenic cell death (ICD) induction, at least on theoretical grounds. For instance, while chimeric antigen receptor (CAR)-expressing T cells, which are currently employed in the clinical management of various hematological malignancies186, kill malignant cells based on the expression of a specific surface marker independent of antigen presentation on MHC Class I molecules187, cancer cell death as elicited by T cells is a bona fide variant of ICD47,48. This suggests that at least part of the efficacy of CAR T cells may originate from an endogenous T cell response to CAR-unrelated antigens (so-called “antigen spreading”) downstream of initial ICD induction by CAR T cells. Such a component of the response would be particularly important for patients that develop secondary resistance to CAR T cells upon the selection of malignant cell clones that no longer express the antigenic CAR target188. Along similar lines, multiple tumor-targeted monoclonal antibodies are believed to operate by blocking the delivery of trophic signaling to malignant cells and by favoring the activation of innate immune effector cells that express Fc gamma receptors like natural killer (NK) cells189. However, the fact that some of them, including epidermal growth factor receptor (EGFR)-targeting antibodies like cetuximab robustly drive ICD182 argue in favor of an at least supportive role for ICD in the therapeutic effects of these agents. Finally, an abundant preclinical literature suggests that ICD inducers represent promising combinatorial partners for a wide panel of immunomodulatory agents including (but not limited to): (1) agonists for immunostimulatory receptors such as TNF receptor superfamily member 9 (TNFRSF9, best known as 4–1BB)190 or CD40 (Ref.191); (2) immunostimulatory cytokines192; (3) immunomodulatory agents including (IDO1) inhibitors193; and (4) therapeutic cancer vaccines194. Taken together, these observations lend additional support to the critical position occupied by ICD in modern cancer (immuno)therapy.
Thus, it is conceptually and practically important to understand the peculiarities of ICD and to distinguish ICD from its non-immunogenic counterpart, be it immunologically silent (as in the setting of physiological development or adult tissue homeostasis) or merely pro-inflammatory (as in the setting of stress-induced cell death not coupled with the activation of adaptive immunity) (Fig 1). In a nutshell, the perception of cell death as immunogenic entails 4 key factors. First, cytotoxicity: cells must activate a series of (ultimately unsuccessful) stress responses before they die4. Second, antigenicity: cells must express and present antigenic determinants that can be recognized by the mature T cell repertoire, for instance carcinoembryonic antigens, post-translationally modified self antigens, or neoantigens emerging from non-synonymous mutations22. Third, adjuvanticity: premortem stress responses and cell death must be accompanied by the emission of adjuvant-like signals that are usually referred to as danger-associated molecular patterns (DAMPs)23,24. DAMPs ensure DC recruitment, the physical interaction between dying cells or their corpses and DCs, the transfer of tumor-associated antigens (TAAs) into DCs, DC maturation and migration to secondary or tertiary lymphoid organs, and finally DC-mediated antigen presentation to T lymphocytes4. Fourth, permissive microenvironmental conditions: not only the site of cell death must be (or become) accessible and hospitable to DCs for them to prime adaptive immunity, but also the site of target cells must be (or become) accessible and hospitable to T cells for them to mediate immune effector functions4,25. Thus, the activation of a potent tumor-targeting immune response downstream of ICD at a primary tumor site will have no effects at a metastatic site if the latter cannot be infiltrated by T lymphocytes or if malignant cells express high levels of co-inhibitory T cell ligands such as CD274 (best known as PD-L1)4,26. Importantly, in oncological settings, the absence of any of these 4 elements (cytotoxicity, antigenicity, adjuvanticity and permissive microenvironmental conditions) is likely to result in primary or secondary resistance to treatment4.
Figure 1. Conceptual differences between immunogenic and non-immunogenic cell death.
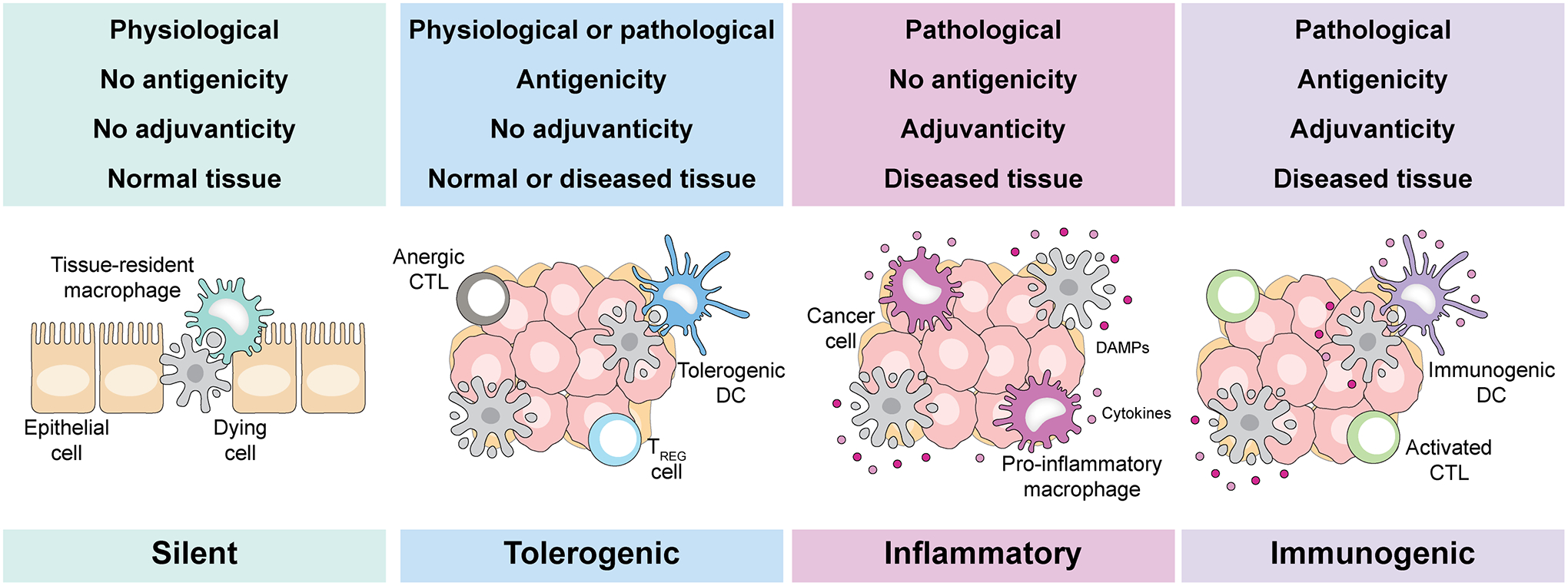
Depending on a number of parameters, dying cells can: (1) be largely ignored by the innate and adaptive immune system, a physiological scenario generally reflecting the rapid uptake of dying cells or their corpses by macrophages; (2) be actively tolerogenic, i.e., promote the establishment of antigen-specific peripheral tolerance upon the activation of regulatory T (TREG) cells that generally results from antigenicity in the absence of proper adjuvant signals; (3) activate innate (but not adaptive) immune mechanisms, notably inflammatory responses, as a result of abundant adjuvanticity in the absence of antigens that can be recognized by the mature T cell repertoire; or (4) overtly immunogenic, when cell death occurs in the context of sufficient adjuvanticity, failing adaptation to stress coupled with the emission of damage-associated molecular patterns (DAMPs) and pro-inflammatory cytokines, and under microenvironmental conditions that are permissive for dendritic cell recruitment, activation and consequent T cell priming (at cell death sites) as well as for T cell infiltration and effector functions (where the cellular targets of adaptive immunity reside). CTL, cytotoxic T lymphocyte; DC, dendritic cell.
In this Review, we summarize the core mechanisms of ICD that can be harnessed for cancer treatment, strategies through which malignant cells subvert ICD induction by therapeutic agents and appropriate countermeasures to reestablish anticancer immunity. Approaches for developing novel pharmacological agents that induce ICD or improve the perception of ICD by immune cells are also discussed.
Core mechanisms of immunogenic cell stress and death
Stress-driven regulated cell death (RCD) can occur according to different mechanisms including (but not limited to): (1) apoptosis, which is demarcated by caspase activation27; (2) ferroptosis, which involves lethal plasma membrane peroxidation28; (3) necroptosis, which is driven by the pore-forming protein mixed lineage kinase domain like pseudokinase (MLKL)29; and (4) pyroptosis, which involves pore-forming proteins of the gasdermin family30. In addition, cells can succumb in a non-regulated, accidental fashion that involves passive cellular lysis.
Although it has been tempting to reductively link the immunogenicity of cell death to specific subroutines (claiming for instance that apoptosis would be intrinsically non-inflammatory, non-immunogenic and even tolerogenic, while necrosis would invariably be pro-inflammatory and immunogenic)31, it is now clear that the biology of cell death and its perception by the host is more complex than such an oversimplistic model. In line with this notion, various purely apoptotic instances of ICD have been described32. Moreover, both accidental necrosis and ferroptosis are often unable to elicit adaptive immunity33,34.
Thus, the accidental release of DAMPs from permeabilized cells can mediate inflammatory responses and is likely to contribute to ICD by interacting with receptors on immune cells (notably DCs), but is not sufficient to optimally stimulate adaptive immunity4. This applies to the unregulated release: (1) nucleotides, which can mediate chemotactic and immunostimulatory functions via purinergic receptors; (2) nucleic acids, which exert immunostimulatory effects by binding Toll-like receptors (TLRs); (3) the cytosolic protein annexin A1 (ANXA1), which has a formyl peptide receptor 1 (FPR1)-dependent chemotactic activity; (4) heat shock proteins (HSPs), which are sensed by TLR2 or TLR4 to promote immune cell activation; and (5) the nuclear chromatin-binding protein high mobility group box 1 (HMGB1), which acts on TLR4 and advanced glycation end product receptor (AGER) with immunostimulatory effects. Altogether, these DAMPs are not sufficient to activate pattern recognition receptors (PRRs) in a way that culminates with T cell priming by DCs4. Similarly, the accessibility of F-actin (which is usually shielded by the plasma membrane) on cellular corpses to its DC-associated receptor C-type lectin domain containing 9A (CLEC9A, best known as DNGR-1) may be necessary for cell death to be immunogenic, but it is unlikely to be sufficient35.
Rather, adaptative premortem responses operating in stressed cells including the protracted secretion of lysosomal ATP in the context of autophagy36 and the exposure of ER components including HSPs and calreticulin (CALR) on the plasma membrane in the context of the ISR37 appear to be essential for ICD. ATP is one of the first DAMPs appearing in the microenvironment of stressed cells, where it can be sensed by DC or their precursors via purinergic receptor P2Y2 (P2RY2), which promotes chemotaxis38,39, hence attracting DCs to the proximity of dying cells40, or purinergic receptor P2X 7 (P2RX7), which promotes DC activation41. Specifically, P2RX7 signaling driven by extracellular ATP activates the NLRP3 inflammasome and hence drives caspase 1 (CASP1) maturation, culminating with the secretion of bioactive interleukin 1B (IL1b) and IL1841. Of note, extracellular ATP-derived ADP also support ICD by promoting DC micropinocytosis via purinergic receptor P2Y12 (P2RY12)42.
Surface-exposed CALR, which translocates to the plasma membrane in complex with protein disulfide isomerase family A member 3 (PDIA3), acts as a potent “eat-me” signal for DCs upon interaction with LDL receptor related protein 1 (LRP1, best known as CD91)43,44. Importantly, ICD-associated CALR surface exposure occurs before the apoptotic exposure of phosphatidylserine (PS)32, which efficiently redirects dying cells or their corpses from macrophages, mediating immunologically silent efferocytosis2, to conventional type 1 DCs (cDC1s), a key DC subset for the activation of tumor-targeting immunity45. Surface-exposed CALR also interacts with natural cytotoxicity triggering receptor 1 (NCR1, best known as NKp46) on NK cells, hence promoting their cytotoxic functions46. Interestingly, cancer cell death as mediated by CD8+ cytotoxic T lymphocytes (CTLs) is also a bona fide variant of ICD47,48, but whether the same applies to NK cell cytotoxicity remains to be formally demonstrated.
Of note, ER stress and autophagy often must be preceded by the ISR for cell death to be perceived as immunogenic. The ISR consists in the phosphorylation of eukaryotic translation initiation factor 2 subunit alpha (EIF2S1, best known as eIF2α) by a panel of kinases, namely eukaryotic translation initiation factor 2 alpha kinase 1 (EIF2AK1, best known as HRI), EIF2AK2 (best known as PKR), EIF2AK3 (best known as PERK) and EIF2AK4 (best known as GCN2), leading to a major reshuffling in protein synthesis49,50. Experimental ISR induction in cancer cells by inhibition of DNA-to-RNA transcription51 or microtubular (de)polymerization52 has been shown to promote bona fide ICD instances. Moreover, PKR, PERK and GCN2 have all been mechanistically involved in ICD as driven by various stimuli53,54. Finally, not only genetic interventions aimed at preventing eIF2α phosphorylation have been demonstrated to subvert the immunogenicity of cell death32, but eIF2α phosphorylation also constitutes a biomarker of ICD correlating with autophagy activation and CALR induction at the single cell level55 as well as with tumor immune infiltration and positive prognostic features in patients with various malignancies37.
The active secretion of pro-inflammatory cytokines by dying cells also contributes to the immunogenicity of cell death. For instance, the cell-autonomous activation of PRRs such as the cytosolic double-stranded DNA sensor cyclic GMP-AMP synthase (CGAS), its endosomal counterpart TLR9 or the endosomal RNA sensor TLR3 can stimulate the secretion of type I IFN and cytokines downstream thereof such as C-X-C motif chemokine ligand 9 (CXCL9) and CXCL10 from cancer cells succumbing to immunogenic chemotherapy, which ultimately favor T cell recruitment into the tumor bed56,57. Receptor interacting serine/threonine kinase 1 (RIPK1) signaling via the NF-κB axis in necroptotic cancer cells has also been shown to result in ICD via the secretion of IL6 (Ref. 58), similar to IL1B secretion as elicited in the context of cancer cell pyroptosis59.
The leitmotif that emerges is that cancer cells undergoing ICD condition the microenvironment through the active exposure of specific proteins (e.g., CALR, PDIA3 and HSPs), the active secretion of metabolites and cytokines (e.g., ATP, type I IFN, CXCL9, CXLC10, IL1B and IL6), the passive release of soluble macromolecules (e.g., ANXA1, HMGB1, DNA and RNA) and an increased accessibility of cytoskeleton-associated proteins (e.g., F-actin) (Fig. 2). These changes in the surfaceome and secretome of stressed or dying cancer cells occur in a way that antigen-presenting cells, in particular cDC1s and their precursors are (1) lured into the tumor microenvironment (TME), (2) attracted into the immediate vicinity of stressed or dying cancer cells, (3) instigated to take up portions of these cells; and (4) driven into maturation for optimal antigen presentation to cytotoxic T lymphocytes22. Of note, this cascade of events appears to be particularly efficient if the tumor contains tertiary lymphoid structures, which are complex, spatially organized immune cell aggregates including DCs, B lymphocytes and T cells, to enable local (as compared to nodal) antigen presentation60,61.
Figure 2. Core mechanisms of immunogenic cell stress and death.
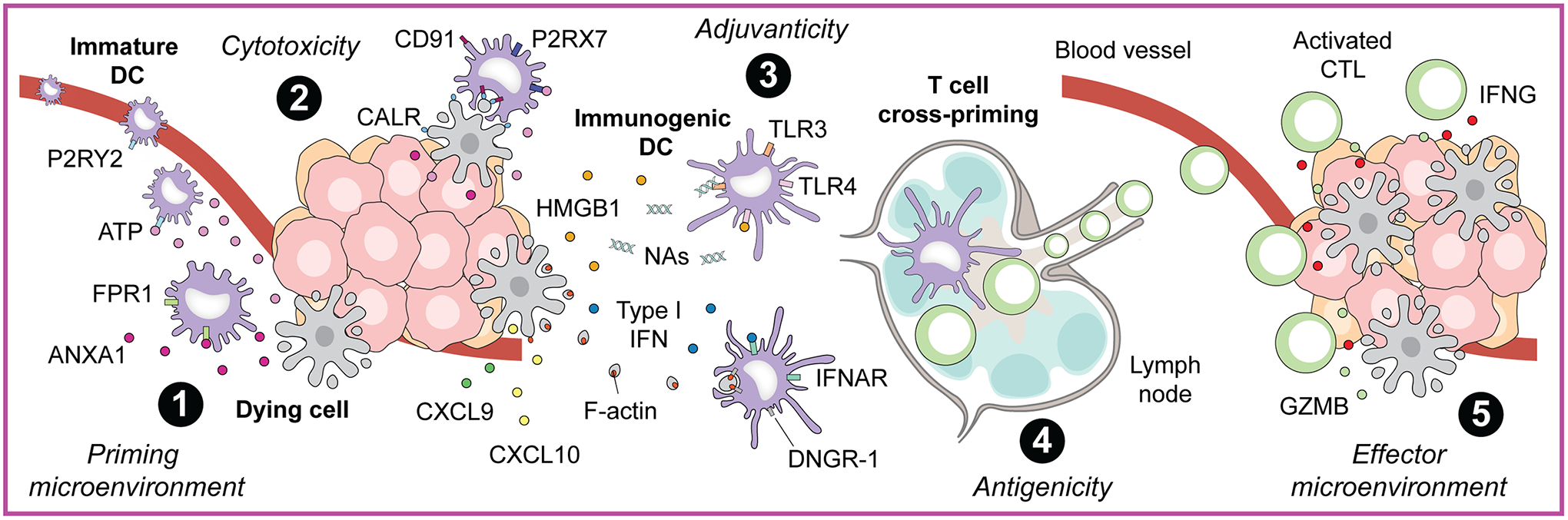
The perception of cell stress and death as overtly immunogenic (i.e., resulting in the activation of adaptive immune responses specific for dead cell-associated antigens) mechanistically relies on the following core components: (1) the site of cell death must contain or be permissive for the recruitment dendritic cells (DCs) or their precursors, and must not be dominated by immunosuppressive signals that may prevent DC activation (microenvironmental conditions for priming); (2) cell death must occur in the context of unsuccessful responses to stress (cytotoxicity) that are associated with the spatiotemporally regulated emission of damage-associated molecular patterns (DAMPs) and cytokines that: (a) unless the site of cell death is abundantly infiltrated by immune cells a priori, recruit DC precursors, (b) attract DC precursors to the close proximity of dying cells, (c) promote the phagocytic uptake of dying cells or material thereof by DC precursors, (d) provide robust immunostimulatory signals to DC precursors for their maturation and functional licensing, and (e) enable mature DCs to reach secondary or tertiary lymphoid structures and to cross-prime antigen-specific T lymphocytes (adjuvanticity); (3) dying cells must express antigenic determinants that can be recognized by the mature T cell repertoire (antigenicity); and (4) the microenvironment of cells targeted by such an adaptive immune responses is permissive for infiltration by antigen-specific T lymphocytes and the activation of their effector functions (microenvironmental conditions for effecting). ANXA1, annexin A1; CALR, calreticulin; CD91 (official name: LRP1), LDL receptor related protein 1; C-X-C motif chemokine ligand, CXCL; DNGR-1 (official name: CLEC9A), C-type lectin domain containing 9A; FPR1, formyl peptide receptor 1; GZMB, granzyme B; HMGB1, high-mobility group box 1; IFN, interferon; IFNAR, IFN interferon alpha and beta receptor; IFNG, interferon gamma; P2RX7, purinergic receptor P2X 7; P2RY2, purinergic receptor P2Y2; TLR, Toll-like receptor.
Established and innovative strategies for ICD induction
Multiple strategies that promote the demise of malignant cells efficiently drive ICD, as exemplified by drugs including conventional chemotherapeutics and targeted anticancer agents, as well as oncolytic viruses and peptides, and various physical treatment modalities, either employed as standalone regimens or as part of multimodal approaches (Table 1).
Table 1.
Major strategies for ICD induction in cancer.
| Strategy | Examples | Pros | Cons | Notes | Ref. |
|---|---|---|---|---|---|
| Chemotherapy | Doxorubicin Oxaliplatin Paclitaxel |
Widely available for clinical use | Generally associated with non-negligible toxicity | Thoroughly characterized in preclinical models | 11 |
| CTL-mediated cytotoxicity | N/A | Highly targeted to malignant cells | Difficult to control in clinical settings | May contribute to the efficacy of CAR T cells |
47
48 |
| ECP | 8-MOP-based ECP | Well-defined toxicity profile | Largely restricted to hematological cancers | Associated with robust DC maturation |
95
96 |
| High hydrostatic pressure | N/A | Broad antigenic sprectrum | Restricted to DC-based vaccine development | Under clinical evaluation | 86 |
| Microwave thermal ablation | N/A | Well-defined toxicity profile | Limited to a few oncological indications | Scarce preclinical characterization | 87 |
| Oncolytic peptides | LTX-315/VPX-315 | Minimally invasive | Restricted to injectable (skin) tumors | Promising clinical activity in BCC | 81 |
| Oncolytic virotherapy | T-vec | Minimally invasive | Restricted to injectable (skin) tumors | Approved for use against melanoma | 74 |
| PDT | Hypericin-based PDT | Not invasive and associated with limited toxicity | Restricted to skin cancers | Immunogenicity varies with photosensitizer | 53 |
| Radiotherapy | N/A | Not invasive and associated with limited toxicity | Potentially limited by conventional approaches | Thoroughly characterized in preclinical models | 84 |
| Targeted anticancer therapy | Cabozantinib Cetuximab Crizotinib |
More selective than chemotherapy | Generally associated with non-negligible toxicity | Potent synergy with immunotherapeutics |
66
67 182 |
Abbreviations: 8-MOP; 8-methoxypsoralen; BCC, basal cell carcinoma; CTL, cytotoxic T lymphocyte; DC, dendritic cell; ECP, extracorporeal photochemotherapy; ICD, immunogenic cell death; N/A, not applicable; PDT, photodynamic therapy; T-vec, talimogene laherparepvec.
Single drugs.
ICD can be triggered by chemotherapeutics that inhibit DNA-to-RNA transcription (e.g., actinomycin D, anthracyclines, lurbinectedin, oxaliplatin) or that interfere with microtubules (e.g., taxanes, vinca alkaloids)11. In line with this notion, the clinical response to such chemotherapeutics is associated with an increased infiltration of malignant lesions by DCs and CTLs along with a relative decrease in the intratumoral amount of immunosuppressive CD4+CD25+FOXP3+ regulatory T (TREG) cells52,62. Moreover, induction therapy with ICD-inducing chemotherapeutics has been shown to sensitize malignant lesions to ICIs targeting PD-L1 or its receptor programmed cell death 1 (PDCD1, best known as PD-1)15,18,19. Bortezomib, a proteasome inhibitor used for the treatment of multiple myeloma, also induces ICD63,64. So far, however, no bortezomib-containing regimen has been shown to cooperate with ICIs in this oncological indication. Instead, efforts are being made to target myeloma cells with ICD-inducing drug-antibody conjugates such as the B cell maturation antigen (BCMA)-targeting agent belantamab mafodotin65. More than 20 clinical trials are currently testing the safety and therapeutic efficacy of this agent in patient with multiple myeloma (source www.clinicaltrials.gov).
Some targeted anticancer agents and notably tyrosine kinase inhibitors (TKIs) also act as bona fide ICD inducers, as documented for the broad specificity TKIs crizotinib66 and cabozantinib67. That said, while cabozantinib can be advantageously combined with the PD-1 blocker nivolumab and the cytotoxic T lymphocyte-associated protein 4 (CTLA4) blocker ipilimumab for the treatment of metastatic renal cancer68, likely reflecting its ICD-inducing activity, two clinical trials in which crizotinib was combined with nivolumab or another PD-1 inbhitor (pembrolizumab) for the treatment of ALK-positive non-small cell lung carcinoma (NSCLC) had to be terminated due to severe liver damage69,70. Such hepatotoxicity could be mimicked by the simultaneous administration of crizotinib and a monoclonal antibody specific for PD-1 antibody in mice, but it turned out to be avoidable in the context of preserved antitumor activity by adopting a sequential administration schedule (with crizotinib first)66. Whether such a revised administration schedule would prevent hepatotoxicity in patients remains to be investigated.
Galenic formulations.
Major efforts are underway to design novel galenic formulations for ICD-relevant drugs, usually in the form of nanoparticles (including liposomes and solid scaffolds), to pursue one or more of the following goals: (1) selective targeting of malignant lesions, for instance by harnessing tumor-specific antibodies of intrinsic features of the TME (e.g., hypoxia, reduced pH, vascular abnormalities); (2) local release of activation of the drug in response to physical triggers that can be focalized on malignant lesions (e.g., γ irradiation, light, heat, ultrasounds); and (3) the timely (simultaneous or sequential) release of drugs and immunostimulants, including (but not limited to) various PRR agonists and ICIs delivered as recombinant proteins or ICI-coding constructs71,72. More than one thousand papers developing such formulations have been published over the past few years, mostly from Chinese research groups. It will be interesting to investigate the clinical utility of the most successful galenic formulations as determined by preclinical tests. Major challenges in this respect are posed by the absence of systematic side-by-side comparisons of distinct galenic formulations in preclinical tumor models, as well as by very choice of rodents as the model of choice, as the selection and characterization of clinically viable galenic approaches may indeed demand the use of larger animals to account for scalability.
Oncolytic viruses and peptides.
Reflecting its evolutionary origin, ICD can be induced by viruses including oncolytic viruses73,74. The first oncolytic virus approved by the US Food and Drug Administration (FDA) was talimogene laherparepvec (best known as T-Vec), a genetically modified human herpes simplex virus 1 (HSV-1)75. Of note, one of these modifications consisted in the deletion of the neurovirulence factor ICP34.5, which promotes eIF2α dephosphorylation and hence blocks the ISR76,77. Thus, the ability of T-Vec to trigger ICD has been improved by genetic engineering to drive an exacerbated ISR. Several other experimental oncolytic viruses have also been shown to induce ICD, which now is invoked as a central mechanism of their antineoplastic action78. Logically, numerous attempts are underway to equip oncolytic viruses with transgenes encoding immunostimulatory factors that enhance DC- and T cell-mediated immune responses, as well as to combine oncolytic viruses with ICIs73,79. While likely, whether these next generation oncolytic virotherapies will provide a therapeutic benefit to patients with cancer remains to be investigated in properly controlled clinical studies. Finally, oncolytic peptides may also induce ICD80, as documented for LTX-315/VPX-315 (Refs. 81,82). This agent, which mediates ICD by permeabilizing plasma and mitochondrial membranes81, has been shown to efficiently synergize with CTLA (but not PD-1) blockers, as well as with other immunostimulatory agents such as radiation therapy, in multiple preclinical tumor models82,83. Recently, LTX-315/VPX-315 has demonstrated promising single agent activity against basal cell carcinoma in a Phase II clinical trial (source https://newsweb.oslobors.no/message/596578), further exemplifying the potential of ICD inducers for clinical cancer management.
Physical stimuli.
Cancer cell stress and death as elicited by a variety of physical stimuli including (but not limited to) focal radiation therapy, microwave ablation and high hydrostatic pressure has been shown to result in the activation of tumor-targeting immune responses that can synergize with ICIs or other forms of immunotherapy in a variety of preclinical cancer models84–87. However, the translatability of these findings to a broad panel of oncological indications in humans still presents multiple obstacles. For instance, while focal radiotherapy has been successfully combined with ICIs in patients with NSCLC16,88,89 and esophageal or gastroesophageal junction cancer who received preoperative (neoadjuvant) chemoradiotherapy90, the same degree of cooperativity could not be documented in patients with glioblastoma91,92 and head and neck squamous cell carcinoma93. At least in part, this reflects the use of conventional radiotherapy schedules and target volumes that are associated with considerable immunosuppression locally (e.g., when tumor-draining lymph nodes are in the radiation field) or systemically (e.g., when large volumes of blood are irradiated) and hence are intrinsically incompatible with ICI-driven immunostimulation94. However, at the frontier between pharmacology and biophysics, both photodynamic therapy (PDT) and extracorporeal photochemotherapy (ECP) have been successfully developed novel strategies for ICD induction in patients53,95,96. In both settings, photosensitizers are administered systemically and then activated by light, either in situ (for PDT, which is used for skin tumors), or within specialized apheresis devices that expose circulating lymphocytes (for ECP, which is used for cutaneous T cell lymphoma)97,98 Supporting a role for ICD induction in the therapeutic activity of these approaches, clinical responses to PDT correlate with enhanced immune infiltration of treated lesions99, while the peripheral blood mononuclear cells (PBMCs) of patients responding to ECP manifest increased secretion of pro-inflammatory cytokines including tumor necrosis factor (TNF) as compared to PBMCs from non-responders100. Whether PDT and ECP can be safely and effectively combined with ICIs or other immunotherapeutics in patients, however, has not yet been formally evaluated.
Combinatorial strategies.
ICD can be caused by the combination of agents that per se do not operate as bona fide ICD inducers. One example is provided by the combination of cisplatin, which is not an optimal ICD driver as it generally fails to elicit the ISR, with an agent capable of correcting such signaling defect, such as cardiac glycosides or crizotinib66,101. Along similar lines, gemcitabine elicits all the immunostimulatory ICD-relevant DAMPs, yet fails to stimulate adaptive anticancer immunity due to its ability to stimulate the production of prostaglandin E2 (PGE2), which has powerful tumor-repopulating and immunosuppressive effects102,103. Thus, combining gemcitabine with the prostaglandin-endoperoxide synthase 2 (PTGS2, best known as COX2) inhibitor celecoxib robustly triggers ICD in preclinical tumor models104, a possibility that is currently being investigated in at least two clinical trials enrolling patients with pancreatic carcinoma (NCT03498326) or bladder cancer (NCT02885974; source www.clinicaltrial.gov). In addition, it is possible to boost the efficacy of ICD-inducing chemotherapeutics by combination regimens. For example, bona fide ICD inducers such as mitoxantrone or oxaliplatin can be advantageously combined with autophagy-inducing agents (e.g., aspirin, hydroxycitrate or spermidine) to obtain improved tumor control and therapeutic cooperativity with PD-1 blockade105–107. Such a positive interaction likely involves the favorable effects of autophagy inducers on ATP secretion by stressed cancer cells36,62, as well as an improvement of the stem cell-like properties of memory T cells108. At least one clinical trial testing the ability of aspirin to improve the immune contexture of patients with ovarian carcinoma is currently ongoing (NCT05080946; source www.clinicaltrial.gov). To the best of our knowledge, instead, hydroxycitrate and spermidine are not being tested for the ability to promote ICD in the clinic.
ICD suppression by cancer cells
Pathogenic viruses often encode proteins that suppress: (1) cell death, for instance by inhibiting caspases or mitochondrial permeabilization; (2) the ISR, for instance by blocking PKR or promoting eIF2α dephosphorylation; (3) autophagy, for instance by interfering with the core autophagy regulator beclin 1 (BECN1); (4) type I IFN responses, for instance by inhibiting master transcription factors such as interferon regulatory factor 3 (IRF3) or limiting the expression of type I IFN receptors; (5) phagocytosis, for instance by promoting the expression of the CALR antagonist CD474. All such proteins act as virulence factors that limit ICD by interfering with cytotoxicity and adjuvanticity. In addition, viruses can limit the antigenicity of infected cells, for instance by suppressing MHC Class I expression (as documented for ICP47, which is a virulence factor expressed by HSV-1 but deleted from T-Vec)109. Finally, viruses can encode immunosuppressive factors (for instance by blocking TNF signaling) or inhibitors of T cell effector molecules such as granzyme B (GZMB) to avoid the recognition of infected cells by the immune system110,111.
Similar to viruses, malignant cells are subjected to a constant Darwinian selection in favor of the fittest, resulting in the progressive selection of cells with increased replicative potential, resistance to cell death and escape from (ICD-dependent) immune recognition112 (Table 2). Interestingly, cancer cells often exhibit one or more defects in ICD-relevant pathways before treatment with an ICD inducer (primary resistance), a fact that likely reflects the importance of spontaneous ICD for immunosurveillance. Indeed, as they acquire increasingly malignant features, cancer cells experience considerable perturbations of intracellular homeostasis that result in the activation of ICD-relevant response pathways and in a relatively high propensity for spontaneous death, resulting in their detection by immune effectors113,114. Alternatively, cancers may initially respond to therapeutic ICD inducers, but subsequently develop secondary resistance to the treatment. Fortunately, each strategy of ICD subversion by cancer cells can be addressed by appropriate countermeasures, at least at the preclinical level, as discussed below.
Table 2.
Main strategies for ICD evasion by cancer cells and potential countermeasures.
| Strategy | Consequence(s) | Mechanism(s)* | Countermeasure(s) | Ref. |
|---|---|---|---|---|
| Evading antigenicity | Limited recognition by tumor-targeting T cells | Antigen loss | Activation of antigen-independent cytotoxicity |
183
184 |
| Decreased MHC Class I expression | Restoration of MHC Class I presentation | 22 | ||
| Autophagy activation | Autophagy inhibition | 181 | ||
| Evading cytotoxicity | Limited release of antigenic material for DC uptake | Active RCD suppression | Engagement of alternative RCD pathways |
118
119 |
| Evading adjuvanticity | Limited uptake of tumor-derived material by DCs | ANXA1 downregulation | Recombinant CALR administration | 157 |
| Autophagy inhibition | Autophagy activation |
107
106 105 |
||
| B7-H4 expression | Inhibition of B7-H4 glycosylation | 137 | ||
| CALR downregulation | Recombinant CALR administration | 34 | ||
| tCALR secretion | tCALR neutralization | 141 | ||
| CD47 upregulation | CD47 blockage SIRPA blockage |
142
140 |
||
| ERO1A expression | ERO1A inhibition | 138 | ||
| STC1 expression | STC1 inhibition | 139 | ||
| Limited DC activation and antigen cross-presentation to cytotoxic T lymphocytes | Autophagy activation | Autophagy inhibition | 151 | |
| FPR1 loss | TLR3 agonism | 158 | ||
| GSN secretion | GSN neutralization | 148 | ||
| HGMB1 downregulation | TL4 agonism | 82 | ||
| Type I IFN inhibition | Accrued MOMP | 151 | ||
| CASP3 inhibition |
121
122 127 |
|||
| Conditioning the TME | Active suppression of DC and/or T cell functions | Immunosuppressive cell recruitment | MDSC and TREG cell depletion |
161
162 |
| Immunosuppressive cytokine secretion | Cytokine neutralization | 185 | ||
| PD-L1 expression | PD-1 or PD-L1 blockage | 82 |
Abbreviations: ANXA1, annexin A1; B7-H4 (official name: VTCN1), V-set domain containing T cell activation inhibitor 1; CALR, calreticulin; CASP3, caspase 3; CD39 (official name: ENTPD1), ectonucleoside triphosphate diphosphohydrolase 1; CD73 (official name: NT5E), 5′-nucleotidase ecto; DC, dendritic cell; ERO1A, endoplasmic reticulum oxidoreductase 1 alpha; GSN, gelsolin; HMGB1, high mobility group box 1; ICD, immunogenic cell death; IFN, interferon; ISR, integrated stress response; FPR1, formyl peptide receptor 1; MDSC, myeloid-derived suppressor cell; MOMP, mitochondrial outer membrane permeabilization; PD-1 (best official name: PDCD1), programmed cell death 1; PD-L1 (official name: CD274); RCD, regulated cell death; SIRPA, signal regulatory protein alpha; STC1, stanniocalcin 1; tCALR, truncated calreticulin; TME, tumor microenvironment; TLR, toll-like receptor; TREG, regulatory T.
main examples.
Cell death resistance.
Alteration in a number of oncoproteins and tumor suppressor proteins confer cancer cells with an increased resistance to cell death induction, as exemplified by alterations in the tumor protein p53 (TP53, best known as p53) system, which are the most common somatic alterations across all cancer types115,116. However, the inactivation of one RCD variant (e.g., apoptosis) does not necessarily imply resistance to another RCD variant (e.g., ferroptosis, necroptosis), pointing to the possibility to use rather distinct drugs (or combinations thereof) to enforce the demise of malignant cells117. For this reason, efforts are underway to develop pharmacological ferroptosis inducers that would kill apoptosis- or necroptosis- resistant cells in an immunogenic fashion118,119. However, this strategy has not yet reached clinical development.
Apoptotic caspase activation.
Importantly, most cancer cells are resistant to the induction of apoptosis120, but often retain an intact apparatus that operates downstream of mitochondrial outer membrane permeabilization (MOMP) to precipitate cell death in the absence of immunostimulation, i.e., “executioner caspases” (Ref. 27). Executioner caspases and notably caspase 3 (CASP3) have indeed been shown to actively operate in support of immunological tolerance, most likely as an organismal defense from autoimmune reactions elicited by physiological cellular turnover, which largely proceeds through apoptotic mechanisms27. More specifically, CASP3 as well as its activator CASP9 actively suppress type I IFN secretion as elicited by mitochondrial DNA (mtDNA) exposed to the cytosol upon MOMP121–124, at least in part owing to the ability of CASP3 to proteolytically inactivate CGAS125. CASP3 activation also promotes immunosuppression by favoring the synthesis and secretion of PGE2 (Ref. 103), as well as by stimulating the rapid exposure of PS on the plasma membrane of dying cells126, which elicits the immunogenically silent clearance of dying cells by macrophages. Finally, caspase-independent variants of cancer cell death are more prone to elicit inflammatory NF-κB signaling in support of anticancer immunity than their caspase-dependent counterparts127. Taken together, these observations point to the inhibition of executioner caspases in the context of preserved or accrued MOMP as promising strategy to improve the immunogenicity of cancer cell death.
Reduced extracellular ATP accumulation.
Autophagy is often inactivated during early oncogenesis, hence curtailing ICD-associated autophagy-dependent ATP secretion128. In line with this notion, autophagy inhibition downstream of insulin like growth factor 1 receptor (IGF1R) hyperactivation is a poor prognostic marker coupled with signs of deficient immunosurveillance in breast cancer129. At least theoretically, this opens the possibility to reactivate autophagy by inhibitors of IGF1R or its signal transducers, notably mechanistic target of rapamycin (MTOR). The latter is indeed hyperactivated in a number of malignancies (often through IGF1R-independent mechanisms), resulting in a robust suppression of autophagic flux that may be relieved with the MTOR complex 1 (MTORC1) inhibitor rapamycin130. In addition, malignant cells as well as immunosuppressive cells populating the TME can enzymatically degrade extracellular ATP by the sequential activity of ectonucleoside triphosphate diphosphohydrolase 1 (ENTPD1, best known as CD39), which catalyzes the dephosphorylation of ATP to ADP and AMP, and 5′-nucleotidase ecto (NT5E, best known as CD73) then converts AMP into adenosine, which has potent immunosuppressive effects36,131. Elevated levels of CD39, CD73 and adenosinergic receptors have a poor prognostic impact in a number of oncological settings, suggesting that they may constitute useful pharmacological targets132. Various Phase I clinical trials have evaluated adenosine A2a receptor (ADORA2A) antagonists with encouraging results133,134. Moreover, two parallel Phase II studies reported promising activity for a monoclonal antibody neutralizing CD73 (i.e., oleclumab) in combination with the PD-L1 blocker durvalumab delivered as neoadjuvant interventions to patients with operable NSCLC135 or as part of the management of unresectable NSCLC136.
Inhibited phagocytosis.
Neoplastic cells can inactivate the ISR at the apex of the CALR exposure pathway by overexpressing V-set domain containing T cell activation inhibitor 1 (VTCN1, best known as B7-H4)137 or endoplasmic reticulum oxidoreductase 1 alpha (ERO1A)138. In addition, cancer cells can limit CALR expression37, retain CALR intracellularly by overexpressing stanniocalcin 1 (STC1)139, minimize CALR binding sites on the cell surface140, secrete a truncated version of CALR that masks CD91141, or upregulate CD47, which neutralizes the pro-phagocytic function of CALR140,142. Accordingly, reduced eIF2α phosphorylation or CALR expression, as well as high STC1 and CD47 expression, are poor prognostic markers in several distinct malignancies37,139,140. A number of experimental strategies have been developed to enforce CALR exposure on the cancer cell surface or to block CD47 functions37. In this context, promising results have been obtained in Phase I clinical trials testing CD47-blocking antibodies in patients with hematological cancers143–145. Conversely, approaches aimed at blocking the CD47 receptor signal regulatory protein alpha (SIRPA) have not reached clinical development yet146,147.
Other mechanisms.
In addition to the aforementioned mechanisms, malignant cells can avoid ICD by abundantly secreting gelsolin (GSN), which competitively inhibits the interaction between DNGR-1 and F-actin, and high GSN mRNA levels are indeed associated with poor prognosis in multiple oncological indications148. Moreover, cancer cells can interfere with type I IFN responses in multiple ways, including: (1) the downregulation of CGAS, its main signal transducer stimulator of interferon response cGAMP interactor 1 (STING1), or its main transcriptional effectors IRF3 and IRF7 (Refs. 149,150); (2) the autophagy-dependent degradation of cellular sources of CGAS-activatory DNA species, including permeabilized mitochondria151 and micronuclei152,153; and (3) defects in the signal transducers of type I IFN receptors, such as Janus kinase 1 (JAK1)154. While the clinical translation of currently available autophagy inhibitors still present multiple obstacles155, it is plausible that maximizing the release of CGAS-activating DNA from mitochondria by FDA approved agents such as the selective BCL2 apoptosis regulator (BCL2) inhibitor venetoclax (which is currently used for the management of multiple hematological tumors)120 may considerably boost the immunogenicity of cell death.
During tumor progression, neoplastic cells can also lose the expression of some DAMPs, including ANXA1 and HMGB1156,157. The immunological defect caused by HMGB1 loss can be successfully repaired – at least in preclinical tumor models – by the intratumoral injection of synthetic TLR4 agonists82. The loss of ANXA1 phenocopies loss-of-function mutations of FPR1 (which encodes the ANXA1 receptor). In preclinical models, the resistance to immunogenic chemotherapy caused by ANXA1 or FPR1 defects can be abolished by the systemic administration of TLR3 agonists158. Of note, the loss of FPR1 in mice accelerates carcinogen-induced breast and colon carcinogenesis158,159, in line with the observation that a frequent FPR1 loss-of-function polymorphism (rs867228, allelic frequency 20%) is linked to premature diagnosis of various carcinomas including luminal B breast cancer, gastrointestinal carcinomas as well as head and neck cancer158,160. It remains to be determined whether TLR3 agonists can be used to postpone the development of cancers developing in the context of FPR1 deficiency.
In sum, although ICD signaling can be perturbed at multiple levels in tumors that escape from natural or therapy-induced immunosurveillance, there are effective strategies for overcoming such perturbation, at least at preclinical level of development. We propose to refer to such strategies as “ICD correctors”.
Therapeutic obstacles against ICD-driven anticancer immunity
Although ICD-inducing drugs have demonstrated substantial clinical activity, there are several obstacles to their therapeutic efficacy. As discussed above, cancer cells can subvert ICD signaling to escape from immunosurveillance as a primary or secondary mechanism of resistance. Moreover, the clinical activity of ICD inducers can be limited by including inappropriate dosing and scheduling, inefficient combination regimens, as well as suboptimal timing of the treatment with respect to surgery.
Dosing and scheduling.
A plethora of established chemotherapeutic agents induce anticancer immunity through their capacity to kill cancer cells via ICD. Speculatively, it appears plausible that such ICD-inducing agents have been empirically selected during clinical development over other drugs that are unable to evoke anticancer immunity. Moreover, clinical optimization has likely favored doses and schedules that avoid the inhibition of immune responses, for instance as a consequence of myelosuppression or lymphodepletion. Indeed, in routine clinical praxis, established chemotherapeutics are generally used at doses that are well below the maximum tolerated dose (MTD) and are not administered in a constant fashion, but rather in cycles, most likely because such treatment schedules are compatible with antitumor immunity. Further supporting this contention, biomarkers of immunosuppression such as an elevated neutrophil-to-lymphocyte ratio are associated with poor disease outcome in the vast majority of oncological indications. Moreover, the poor cooperativity of specific ICD inducers with ICIs in at least some patient cohorts may reflect local or systemic immunosuppressive effects that are unavoidable with conventional doses and schedules94. These considerations should be taken into account for the design of clinical trials involving ICD inducers, both as standalone therapeutics and as part of combinatorial regimens involving immunotherapy.
Combinatorial regimens.
The empiric optimization of chemotherapeutic regimens achieved in the past (which was completely immune agnostic) has yielded drug cocktails that are relatively efficient such as doxorubicin plus cyclophosphamide or taxanes plus cyclophosphamide for the management of breast cancer, the FOLFOX regimen (folinic acid plus 5-fluorouracil plus oxaliplatin) for the treatment of colorectal cancer, and cisplatin plus gemcitabine for patients with NSCLC. A posteriori, it appears that the therapeutic effects of ICD induction by doxorubicin and taxanes may be amplified by cyclophosphamide due to its capacity to deplete TREG cells161. Along similar lines, oxaliplatin-induced ICD may benefit from the depletion of immunosuppressive myeloid-derived suppressor cells (MDSCs) by 5-fluoruracil162. Moreover, although imperfect at eliciting ICD, gemcitabine may enhance the effects of cisplatin, which per se fails to induce ICD104. These examples highlight the possibility to design optimal combinatorial regimens involving ICD inducers and “ICD boosters” in a data-driven fashion. Obviously, there is a growing rationale to combine ICD-stimulatory therapies with ICIs20. However, while most ICD inducers show favorable interactions with ICIs specific for PD-1 or PD-L1 in preclinical tumor models, this is not fully generalizable. For example, tumor lysis by LTX-315/VX-315 drives an anticancer immune response that is improved by CTLA4, but not PD-1, blockade82. The mechanistic underpinnings of this discrepancy remain elusive.
Adjuvancy versus neoadjuvancy.
A long-lasting paradigm of clinical cancer care has been to propose, whenever possible, the surgical removal of the tumor together with one (“sentinel”) or multiple tumor-draining lymph nodes, followed by adjuvant treatment with chemotherapy, radiotherapy, targeted therapy and/or immunotherapy. Conversely, neoadjuvant therapies have classically been reserved to patients with inoperable (e.g., highly metastatic) cancers. However, recent studies strongly suggest that, at least in some types of cancer such as microsatellite-unstable (MSI) colorectal cancer and melanoma, immunotherapy with ICIs can be administered in a neoadjuvant setting at an operable stage (as true for MSI colorectal cancer), or without lymph node dissection (as true for melanoma) with a highly favorable treatment outcome163,164. Mechanistically, this may be explained by the fact that the presence of the primary tumor or the tumor-draining lymph nodes offers superior chances to ICIs to (re)establish a therapeutically relevant immune response as compared to a post-surgical scenario in which only (mostly dormant) micrometastases are present20,165. In this context, it appears tempting to speculate that immunostimulation by chemotherapy or targeted agents would also be more efficient in the neoadjuvant than in the adjuvant setting. Indeed, patients with operable NSCLC have been advantageously treated by neoadjuvant chemoimmunotherapy166. That said, whether conventional anticancer agents inducing ICD should also be systematically used in a preoperative fashion when they must be administered alone, for instance in underdeveloped countries, still remains elusive.
ICD assays for drug discovery
Since the capacity of a given approach to elicit ICD appears to mark the therapeutic success of anticancer therapy, considerable efforts have been devoted to the discovery of novel ICD inducers. Interestingly, ICD inducers, and in particular those that inhibit transcription, appear to exhibit specific physicochemical properties that distinguish them from non-ICD inducers11. However, medium- and high-throughput screening campaigns followed by validation experiments in preclinical tumor models remain required for the identification of bona fide ICD-inducing drugs. Alongside, attempts have been launched to identify drugs that enhance the capacity of DCs to sense ICD and to cross-present tumor-associated antigens to T lymphocytes, which we refer to as “ICD enhancers”. The ability of a given molecule to act as an ICD enhancer must also be validated in vivo, in suitable preclinical tumor models (Fig. 3).
Figure 3. Potential pipeline for preclinical development of novel ICD-relevant drugs.

Both immunogenic cell death (ICD)-inducing (a) and ICD-enhancing (b) drug candidates can be identified in vitro by medium-to-high screening efforts based on: (a) human cancer cell lines expressing biosensors or treated with chemical dyes that enable the assessment of autophagy activation, calreticulin (CALR) exposure on the plasma membrane, ATP secretion and high mobility group box 1 (HMGB1) loss; or (b) a conditionally immortalized mouse dendritic cell (DC) precursor cell line that can be de-immortalized ad hoc and exposed to mouse cancer cells undergoing ICD in the presence of potential ICD enhancers, followed by the assessment of ICD-related parameters including phagocytic capacity, expression of DC maturation markers, and T cell cross-priming proficiency. In the case of ICD inducers, low throughput in vitro validation with alternative technologies should be followed by in vivo assays based on prophylactic vaccination, therapeutic tests in immunocompetent vs immunodeficient hosts, and/or therapeutic assays in bilateral tumor models (when ICD induction can be achieved by local interventions). In the case of ICD enhancers, low throughput in vitro validation can involve human DCs or mouse bone-marrow derived DCs, followed by in vivo validation based on therapeutic DCs vaccination assays upon ex vivo DC exposure to candidate drugs, or ICD inducer plus enhancer treatment of mouse tumors established in syngeneic immunocompetent mice or mice lacking conventional type 1 DCs (cDC1s), such as Batf3−/− mice or mice administered with recombinant cytochrome c, somatic (CYCS). The pipeline for the discovery of agents restoring deficient ICD signaling in cancer cells, so-called ICD correctors, strictly resembles the one for ICD inducer discovery, except for the fact that combinatorial strategies are tested (i.e., suboptimal ICD inducer plus potential ICD corrector). Along similar lines, candidate ICD boosters be identified in vitro by high-throughput strategies testing regulatory T (TREG) cell or myeloid-derived suppressor cell (MDSC) viability and immunosuppressive functions, but can only be validated by low throughput in vivo assays. IFN, interferon.
In vitro assays for ICD inducers.
In a classical approach, human cancer cells of choice can be engineered with fluorescent biosensors that allow for the measurement of: (1) autophagy, based on the exogenous expression of a fluorescent variant of microtubule associated protein 1 light chain 3 beta (MAP1LC3B, best known as LC3B) that forms quantifiable puncta during the autophagic process; (2) ATP release, based on the ATP-detecting dye quinacrine staining; (3) CALR exposure on the cell surface, based on the exogenous expression of CALR fused to a fluorescent tag; (4) HMGB1 release, based on a similar principle, and (5) type I IFN signaling, based on the expression of a reporter gene under the control of a type I IFN-sensitive promoter, such as the MX1 promoter. Such biosensors are then exposed to various concentrations of each drug candidate in a robotized platform, followed by the acquisition of fluorescence signals by fluorescence microscopy or flow cytometry, and their automated analyses. Candidates eliciting all these ICD-associated processes must then undergo manual validation with alternative low-throughput approaches (ideally with genetically unmodified cancer cells), for instance the assessment of ATP release by luminescence assays, the secretion of HMGB1 and type I IFN by ELISA, and the exposure of CALR on the cell surface by flow cytometry167.
In vivo assays for ICD inducers.
Drugs that have been shortlisted as potential ICD inducers must be subjected to further validation steps in vivo. One common approach involves the treatment of mouse cancer cells with the drug of choice in vitro, followed by a washout step and their subcutaneous administration (generally in a total number of 1–3×106 cells, of which 50–70% are dying) into syngeneic, fully immunocompetent mice, in the absence of any exogenous adjuvant, with the expectation that no tumor develops at the injection site. One-two weeks later, these mice are challenged by the contralateral subcutaneous injection of healthy cancer cells of the same type (generally in a number that is 100% efficient at generating developing tumor in naïve mice). This latter “index” site is then assessed for the appearance of developing tumors, the absence of which will be interpreted as a sign of a successful vaccination downstream of bona fide ICD induction4. Another common approach involves the standard establishment of subcutaneous or orthotopic mouse tumors in syngeneic immunocompetent vs syngeneic immunodeficient (for instance upon the antibody-mediated depletion of CD4+ and CD8+ T cells or the deletion of Rag2), followed by the treatment of mice with the drug candidate. In this setting, the therapeutic activity of bona fide ICD will be reduced in immunodeficient mice. Along similar lines, immunocompetent mice bearing subcutaneous or orthotopic tumors can be treated with syngeneic DCs exposed ex vivo to cancer cells succumbing to a potential ICD inducer168–170. Of note, also in this model treatment efficacy is expected to depend in large part on CD4+ and CD8+ T cells. Finally, the immunogenicity of a drug candidate can be assessed in immunocompetent mice subcutaneously bearing two syngeneic tumors (one on each flank), only one of which will receive the drug intratumorally (or by other localized approaches, for instance with focal radiotherapy). In this case, the control of the contralateral untreated tumors will inform on the immunogenicity of treatment. While such therapeutic assays are closer to the clinical reality than prophylactic vaccination assays, they are intrinsically unable to discriminate between bona fide ICD induction versus ICD-independent immunostimulation, as demonstrated for a number of commonly employed anticancer agents171. Obviously, the switch from in vitro assays (on human cancer cells) to in vivo experiments (involving mouse cancer cells and their syngeneic hosts) constitutes a potential problem. At least theoretically, this problem might by circumvented by the use of “humanized” mice bearing a human immune system172, but this technology still requires considerable refinements.
In vitro assays for ICD enhancers.
DCs are the first-line immune effectors contacting cells that undergo ICD173. Unfortunately, DCs are terminally differentiated cells and rather rare (less than 1% of tumor-infiltrating leukocytes), rendering screening campaigns based on primary DCs impractical. However, it is possible to conditionally immortalize DC precursor cells by the transgene enforced expression of a viral oncoprotein that inactivates two cellular tumor suppressors, namely p53 and retinoblastoma 1 (RB1) under the control of a doxycycline (DOX) and dexamethasone (DEX)-inducible system174. This genetic modification enables not only the indefinite expansion of immortalized DC precursors in the presence of DOX/DEX, but also (1) the implementation of additional genetic manipulations (for instance with the CRISPR/Cas9 technology); and (2) the de-immortalization of DC precursors by DOX/DEX withdrawal, which culminates with cDC1 differentiation175. In summary, this system generates high numbers of terminally differentiated cDC1s that can be employed to assess DC functions as elicited by cancer cells potentially undergoing ICD, such as phagocytosis, expression of activation markers, and/or T cell priming175, a strategy that has been successfully employed for identifying venetoclax, navitoclax – a dual inhibitor of BCL2 and BCL2 like 1 (BCL2L1, best known as BCL-XL) – and the hexokinase 2 (HK2) inhibitor ikarugamycin as bona fide ICD enhancers176,177.
In vivo assays for ICD enhancers.
De-immortalized DCs generated as described above can be intravenously injected into wild-type mice, Batf3−/− or Wdfy4−/− mice (both of which exhibit defects in cDC1-dependent cross-presentation)45,178 to test the ability of potential ICD enhancers to boost the therapeutic activity of ICD inducers177. The capacity of these cells to migrate into tumor lesions and elicit T cell-dependent anticancer immunity can also be investigated in preclinical tumor models, adding a layer of preclinical validation to the in vitro screening assays described above. Another strategy for preclinical validation consists in the comparison of the therapeutic activity of candidate ICD enhancers in mice containing vs lacking cDC1-mediated cross-presentation due to the knockout of Batf3 or Wdfy4, or following repeated intravenous injections of recombinant cytochrome c, somatic (CYCS), which depletes cDC1s. In these two latter settings, bona fide ICD enhancers are indeed expected to mediate suboptimal anticancer effects177. On theoretical grounds, ICD enhancers should have broad therapeutic effects across multiple cancer types as they act on DCs rather than on malignant cells themselves. However, this conjecture requires further preclinical validation. Similarly, the possibility that ICD enhancers might be used as an alternative to, and perhaps in combination with, ICIs (which mostly act on T lymphocytes) awaits experimental confirmation. As a major caveat, present DC-based drug development efforts involve murine, not human DCs. Hence, it will be important to create human cDC1 precursor cell lines for high-throughput screening purposes or, at least, to design human cDC1-based systems that can be subjected to reliable low-throughput validation assays.
Concluding remarks
Progress in immunopharmacology and tumor immunology achieved over the past 2 decades suggests that the therapeutic success of nonsurgical cancer treatments including classical chemotherapies can be largely attributed to the elicitation of anticancer immune responses, often due to the induction of ICD. Spurred by this realization, considerable efforts have been made to identify new ICD-inducing drugs, some of which already been approved by the US FDA for use in patients179,180 or are under currently clinical evaluation (e.g., LTX-315/VC-315), and to combine them with immunotherapies, especially ICIs.
Notwithstanding such an undeniable progress, considerable obstacles exist in the preclinical and clinical development of ICD inducers, which can be summarized as follows:
ICD inducers, which are often cytotoxic inhibitors of DNA-to-RNA transcription or microtubular poisons, have considerable non-specific side effects, including non-negligible detrimental effects on healthy (including immune) cells. More targeted ICD inducers including TKIs or proteasome inhibitors also exhibit a similar toxicity profile. Thus, more specific strategies to induce ICD signaling events in cancer cells including the activation of the ISR or type I IFN responses are warranted. To achieve this goal, the drug development pipeline required for large-scale identification and validation of ICD inducers requires further optimization.
The conventional approach to evaluate anticancer drugs at doses close to MTD that are administered in a regular fashion to maintain drug concentrations within a stable range may cause immunosuppressive side effects that ultimately reduce therapeutic efficacy. Hence, clinical trials investigating the safety and efficacy of old and new ICD inducers should be designed to avoid non-specific immunosuppression by reducing doses and/or implementing intermittent treatment cycles. Moreover, the neoadjuvant therapy of operable tumors should be explored at a large scale, for instance in the form of window-of-opportunity trials.
By virtue of their genetic and epigenetic plasticity and heterogeneity, cancer cells can avoid efficient ICD signaling, which calls for specific countermeasures to restore ICD in a personalized fashion. This will require not only a careful molecular diagnosis of such ICD defects, but also the development of novel ICD correctors with an elevated potential to translate into clinical cancer care.
Several tumors also develop strategies to exclude, exhaust or inactivate immune effector cells and/or to recruit immunosuppressive cells, two situations that can be addressed with an arsenal of immunotherapeutic countermeasures. It remains to be seen whether novel galenic formulations that specifically enable ICD signaling at tumor sites combined with local immunotherapy will help in overcoming these hurdles or whether systemic immunotherapy with ICIs will demonstrate superiority in this respect.
The propensity of a patient to mount therapeutically relevant anticancer immune responses is not only dictated by the tumor and its microenvironment but also depends on the bodywide ecosystem (encompassing, among other factors, general metabolic status and the composition of the gut microbiome). Thus, co-morbidities such as the metabolic syndrome or intestinal dysbiosis as well as an unhealthy lifestyle may constitute additional obstacles to the successful implementation of ICD-based therapies (Box 2). These issues can only be managed by a proactive role from internal medicine physicians coupled with a holistic management of the patient.
Box 2. Emerging obstacles against anticancer immunity driven by ICD.
Tumor-targeting immune responses as elicited by immunogenic cell death (ICD) inducers can be impaired not only by issues affecting ICD signaling in malignant cells, the perception of cell death as immunogenic by the immune system or the microenvironment of dying or target cells, but also by hitherto underappreciated alterations of the bodywide ecosystem that directly or indirectly affect the immunological fitness of the patient195. Some of such alterations that (at least in preclinical tumor models) have been characterized include: (1) changes in the abundance and composition of the gut microbiome, which appear to have a direct effect on systemic immune tone196; (2) local alterations of the tumor microenvironment (TME) as imposed by the (generally unsupervised) usage of over-the-counter drugs such as aspirin, which has been associated with a potentially detrimental inhibition of cyclic GMP-AMP synthase (CGAS) signaling197; (3) detrimental alterations of immune function caused by drugs that are commonly employed for clinical cancer management but often overlooked for their (in some instances well known) immunosuppressive effects, such as glucocorticoids198,199; (4) considerably detrimental shifts of the local or systemic TME that result from inappropriate dietary habits200–202; and (5) deteriorations of intratumoral or systemic immune tone elicited by a sedentary lifestyle203 and/or psychological distress199. It is likely that multiple other over-the-counter products including common medications and nutritional supplements may impact the ability of ICD to elicit tumor-targeting immune responses. Along similar lines, it is more than plausible that additional lifestyle habits with powerful biological effects (e.g., sleep duration and quality) may influence intratumoral or systemic immune tone to alter the ultimate immunogenicity of cancer cell death. Additional work is required to assess such impact and characterize the underlying molecular and cellular mechanisms in preclinical tumor models. In the meantime, we believe that the implementation of ever more comprehensive anamnestic questionnaires by internal medicine physicians may provide an invaluable source of data for correlatively assessing the impact of the aforementioned factors on disease progression in patients with cancer treated with ICD inducers (or other drugs).
Irrespective of the obstacles enumerated above, it appears that the concept of ICD has significantly contributed to the development of anticancer drugs and will continue to do so. Indeed, multiple academic institutions and biotech companies are identifying, characterizing and optimizing new ICD inducers. We anticipate that the oncological armamentarium of ICD inducers will be completed by additional compounds including ICD correctors (to restore defective ICD signaling in malignant cells), ICD boosters (to counteract compensatory immunosuppressive mechanisms driven by ICD) and ICD enhancers (to improve the communication between malignant cells experiencing immunogenic stress and DCs), ultimately providing patients with cancer with personalized regimens that maximize the therapeutic potential of ICD across a variety of malignancies.
Acknowledgements.
LG is/has been supported (as a PI unless otherwise indicated) by one R01 grant from the NIH/NCI (#CA271915), by two Breakthrough Level 2 grants from the US DoD BCRP (#BC180476P1, #BC210945), by a grant from the STARR Cancer Consortium (#I16-0064), by a Transformative Breast Cancer Consortium Grant from the US DoD BCRP (#W81XWH2120034, PI: Formenti), by a U54 grant from NIH/NCI (#CA274291, PI: Deasy, Formenti, Weichselbaum), by the 2019 Laura Ziskin Prize in Translational Research (#ZP-6177, PI: Formenti) from the Stand Up to Cancer (SU2C), by a Mantle Cell Lymphoma Research Initiative (MCL-RI, PI: Chen-Kiang) grant from the Leukemia and Lymphoma Society (LLS), by a Rapid Response Grant from the Functional Genomics Initiative (New York, US), by a pre-SPORE grant (PI: Demaria, Formenti) and a Clinical Trials Innovation Grant from the Sandra and Edward Meyer Cancer Center (New York, US); by startup funds from the Dept. of Radiation Oncology at Weill Cornell Medicine (New York, US), by industrial collaborations with Lytix Biopharma (Oslo, Norway), Promontory (New York, US) and Onxeo (Paris, France), as well as by donations from Promontory (New York, US), the Luke Heller TECPR2 Foundation (Boston, US), Sotio a.s. (Prague, Czech Republic), Lytix Biopharma (Oslo, Norway), Onxeo (Paris, France), Ricerchiamo (Brescia, Italy), and Noxopharm (Chatswood, Australia). GK is supported by the Ligue contre le Cancer (équipe labellisée); Agence National de la Recherche (ANR) – Projets blancs; AMMICa US23/CNRS UMS3655; Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Fondation pour la Recherche Médicale (FRM); a donation by Elior; Equipex Onco-Pheno-Screen; European Joint Programme on Rare Diseases (EJPRD); European Research Council Advanced Investigator Award (ERC-2021-ADG, ICD-Cancer, Grant No. 101052444), European Union Horizon 2020 Projects Oncobiome, Prevalung (grant No. 101095604) and Crimson; Institut National du Cancer (INCa); Institut Universitaire de France; LabEx Immuno-Oncology ANR-18-IDEX-0001; a Cancer Research ASPIRE Award from the Mark Foundation; the RHU Immunolife; Seerave Foundation; SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); and SIRIC Cancer Research and Personalized Medicine (CARPEM). This study contributes to the IdEx Université de Paris ANR-18-IDEX-0001. Views and opinions expressed are those of the author(s) only and do not necessarily reflect those of the European Union, the European Research Council or any other granting authority. Neither the European Union nor any other granting authority can be held responsible for them.
Footnotes
Competing Interests. LG is/has been holding research contracts with Lytix Biopharma, Promontory and Onxeo, has received consulting/advisory honoraria from Boehringer Ingelheim, AstraZeneca, OmniSEQ, Onxeo, The Longevity Labs, Inzen, Imvax, Sotio, Promontory, Noxopharm, EduCom, and the Luke Heller TECPR2 Foundation, and holds Promontory stock options. GK has been holding research contracts with Daiichi Sankyo, Eleor, Kaleido, Lytix Pharma, PharmaMar, Osasuna Therapeutics, Samsara Therapeutics, Sanofi, Tollys, and Vascage. GK is on the Board of Directors of the Bristol Myers Squibb Foundation France. DS is a full-time employee of Violet Therapeutics. GK is a scientific co-founder of everImmune, Osasuna Therapeutics, Samsara Therapeutics and Therafast Bio. GK is in the scientific advisory boards of Hevolution, Institut Servier, Longevity Vision Funds and Rejuveron Life Sciences. GK is the inventor of patents covering therapeutic targeting of aging, cancer, cystic fibrosis and metabolic disorders. GK’s wife, Laurence Zitvogel, has held research contracts with Glaxo Smyth Kline, Incyte, Lytix, Kaleido, Innovate Pharma, Daiichi Sankyo, Pilege, Merus, Transgene, 9 m, Tusk and Roche, was on the on the Board of Directors of Transgene, is a cofounder of everImmune, and holds patents covering the treatment of cancer and the therapeutic manipulation of the microbiota. GK’s brother, Romano Kroemer, was an employee of Sanofi and now consults for Boehringer-Ingelheim. The funders had no role in the design of the study; in the writing of the manuscript, or in the decision to publish the results. FMM is a full-time employee of Sonata Therapeytics.
References
- 1.López-Otín C & Kroemer G Hallmarks of Health. Cell 184, 33–63 (2021). [DOI] [PubMed] [Google Scholar]
- 2.Mehrotra P & Ravichandran KS Drugging the efferocytosis process: concepts and opportunities. Nat Rev Drug Discov 21, 601–620 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boada-Romero E, Martinez J, Heckmann BL & Green DR The clearance of dead cells by efferocytosis. Nat Rev Mol Cell Biol 21, 398–414 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kroemer G, Galassi C, Zitvogel L & Galluzzi L Immunogenic cell stress and death. Nat Immunol 23, 487–500 (2022). [DOI] [PubMed] [Google Scholar]
- 5.Björkström NK, Strunz B & Ljunggren HG Natural killer cells in antiviral immunity. Nat Rev Immunol 22, 112–123 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kyrysyuk O & Wucherpfennig KW Designing Cancer Immunotherapies That Engage T Cells and NK Cells. Annu Rev Immunol 41, 17–38 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woodland DL & Kohlmeier JE Migration, maintenance and recall of memory T cells in peripheral tissues. Nat Rev Immunol 9, 153–161 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Galluzzi L, Yamazaki T & Kroemer G Linking cellular stress responses to systemic homeostasis. Nat Rev Mol Cell Biol 19, 731–745 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Dunn GP, Old LJ & Schreiber RD The three Es of cancer immunoediting. Annu Rev Immunol 22, 329–360 (2004). [DOI] [PubMed] [Google Scholar]
- 10.Chen R, Ishak CA & De Carvalho DD Endogenous Retroelements and the Viral Mimicry Response in Cancer Therapy and Cellular Homeostasis. Cancer Discov 11, 2707–2725 (2021). [DOI] [PubMed] [Google Scholar]
- 11.Galluzzi L, Humeau J, Buqué A, Zitvogel L & Kroemer G Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat Rev Clin Oncol 17, 725–741 (2020). [DOI] [PubMed] [Google Scholar]
- 12.McLaughlin M et al. Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat Rev Cancer 20, 203–217 (2020). [DOI] [PubMed] [Google Scholar]
- 13.Petroni G, Buqué A, Coussens LM & Galluzzi L Targeting oncogene and non-oncogene addiction to inflame the tumour microenvironment. Nat Rev Drug Discov 21, 440–462 (2022). [DOI] [PubMed] [Google Scholar]
- 14.Cytlak UM et al. Immunomodulation by radiotherapy in tumour control and normal tissue toxicity. Nat Rev Immunol 22, 124–138 (2022). [DOI] [PubMed] [Google Scholar]
- 15.Voorwerk L et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: the TONIC trial. Nat Med 25, 920–928 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Antonia SJ et al. Overall Survival with Durvalumab after Chemoradiotherapy in Stage III NSCLC. N Engl J Med 379, 2342–2350 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Campbell MT et al. Pilot study of Tremelimumab with and without cryoablation in patients with metastatic renal cell carcinoma. Nat Commun 12, 6375 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Røssevold AH et al. Atezolizumab plus anthracycline-based chemotherapy in metastatic triple-negative breast cancer: the randomized, double-blind phase 2b ALICE trial. Nat Med 28, 2573–2583 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thibaudin M et al. First-line durvalumab and tremelimumab with chemotherapy in RAS-mutated metastatic colorectal cancer: a phase 1b/2 trial. Nat Med 29, 2087–2098 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kroemer G, Chan TA, Eggermont AMM & Galluzzi L Immunosurveillance in clinical cancer management. CA Cancer J Clin (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bruni D, Angell HK & Galon J The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat Rev Cancer 20, 662–680 (2020). [DOI] [PubMed] [Google Scholar]
- 22.Jhunjhunwala S, Hammer C & Delamarre L Antigen presentation in cancer: insights into tumour immunogenicity and immune evasion. Nat Rev Cancer 21, 298–312 (2021). [DOI] [PubMed] [Google Scholar]
- 23.Krysko DV et al. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer 12, 860–875 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Zindel J & Kubes P DAMPs, PAMPs, and LAMPs in Immunity and Sterile Inflammation. Annu Rev Pathol 15, 493–518 (2020). [DOI] [PubMed] [Google Scholar]
- 25.Singleton DC, Macann A & Wilson WR Therapeutic targeting of the hypoxic tumour microenvironment. Nat Rev Clin Oncol 18, 751–772 (2021). [DOI] [PubMed] [Google Scholar]
- 26.Lucca LE & Dominguez-Villar M Modulation of regulatory T cell function and stability by co-inhibitory receptors. Nat Rev Immunol 20, 680–693 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Vitale I et al. Apoptotic cell death in disease-Current understanding of the NCCD 2023. Cell Death Differ 30, 1097–1154 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stockwell BR Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 185, 2401–2421 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weinlich R, Oberst A, Beere HM & Green DR Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol 18, 127–136 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Conrad M, Angeli JP, Vandenabeele P & Stockwell BR Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Discov 15, 348–366 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edinger AL & Thompson CB Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol 16, 663–669 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Panaretakis T et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. Embo j 28, 578–590 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wiernicki B et al. Cancer cells dying from ferroptosis impede dendritic cell-mediated anti-tumor immunity. Nat Commun 13, 3676 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]; This is the first demonstration that ferroptosis is not always immunogenic per se, and actively inhibits the immunogenicity of apoptotic cell death.
- 34.Obeid M et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med 13, 54–61 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Ahrens S et al. F-actin is an evolutionarily conserved damage-associated molecular pattern recognized by DNGR-1, a receptor for dead cells. Immunity 36, 635–645 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Kepp O et al. ATP and cancer immunosurveillance. Embo j 40, e108130 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fucikova J, Spisek R, Kroemer G & Galluzzi L Calreticulin and cancer. Cell Res 31, 5–16 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elliott MR et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 461, 282–286 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chekeni FB et al. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature 467, 863–867 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma Y et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity 38, 729–741 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Ghiringhelli F et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med 15, 1170–1178 (2009). [DOI] [PubMed] [Google Scholar]
- 42.Nicholas RA Identification of the P2Y(12) receptor: a novel member of the P2Y family of receptors activated by extracellular nucleotides. Mol Pharmacol 60, 416–420 (2001). [PubMed] [Google Scholar]
- 43.Gardai SJ et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 123, 321–334 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Gardai SJ et al. By binding SIRPalpha or calreticulin/CD91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation. Cell 115, 13–23 (2003). [DOI] [PubMed] [Google Scholar]
- 45.Hildner K et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322, 1097–1100 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sen Santara S et al. The NK cell receptor NKp46 recognizes ecto-calreticulin on ER-stressed cells. Nature 616, 348–356 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper provides an elegant demonstration that besides mediating pro-phagocytic effects, CALR can directly bind NKp46 on the surface of NK cells to promote their cytotoxicity.
- 47.Minute L et al. Cellular cytotoxicity is a form of immunogenic cell death. J Immunother Cancer 8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; These two articles were the first to show that the death of cancer cells as mediated by CD8+ CTLs is a bona fide instance of ICD.
- 48.Jaime-Sanchez P et al. Cell death induced by cytotoxic CD8(+) T cells is immunogenic and primes caspase-3-dependent spread immunity against endogenous tumor antigens. J Immunother Cancer 8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; These two articles were the first to show that the death of cancer cells as mediated by CD8+ CTLs is a bona fide instance of ICD.
- 49.Costa-Mattioli M & Walter P The integrated stress response: From mechanism to disease. Science 368 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lines CL, McGrath MJ, Dorwart T & Conn CS The integrated stress response in cancer progression: a force for plasticity and resistance. Front Oncol 13, 1206561 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Humeau J et al. Inhibition of transcription by dactinomycin reveals a new characteristic of immunogenic cell stress. EMBO Mol Med 12, e11622 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Senovilla L et al. An immunosurveillance mechanism controls cancer cell ploidy. Science 337, 1678–1684 (2012). [DOI] [PubMed] [Google Scholar]
- 53.Garg AD et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. Embo j 31, 1062–1079 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Giglio P et al. PKR and GCN2 stress kinases promote an ER stress-independent eIF2α phosphorylation responsible for calreticulin exposure in melanoma cells. Oncoimmunology 7, e1466765 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Humeau J et al. Phosphorylation of eukaryotic initiation factor-2α (eIF2α) in autophagy. Cell Death Dis 11, 433 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sistigu A et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med 20, 1301–1309 (2014). [DOI] [PubMed] [Google Scholar]
- 57.Wang Z et al. cGAS/STING axis mediates a topoisomerase II inhibitor-induced tumor immunogenicity. J Clin Invest 129, 4850–4862 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yatim N et al. RIPK1 and NF-κB signaling in dying cells determines cross-priming of CD8+ T cells. Science 350, 328–334 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zi M et al. Improved antitumor immunity of chemotherapy in OSCC treatment by Gasdermin-E mediated pyroptosis. Apoptosis 28, 348–361 (2023). [DOI] [PubMed] [Google Scholar]
- 60.Fridman WH et al. B cells and tertiary lymphoid structures as determinants of tumour immune contexture and clinical outcome. Nat Rev Clin Oncol 19, 441–457 (2022). [DOI] [PubMed] [Google Scholar]
- 61.Sarti Kinker G & da Silva Medina T Tertiary lymphoid structures as hubs of antitumour immunity. Nat Rev Cancer 23, 803 (2023). [DOI] [PubMed] [Google Scholar]
- 62.Michaud M et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 334, 1573–1577 (2011). [DOI] [PubMed] [Google Scholar]; This paper documents the importance of proficient autophagic responses in cancer cells to support optimal ICD-associated ATP release.
- 63.Spisek R et al. Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: therapeutic implications. Blood 109, 4839–4845 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gulla A et al. Bortezomib induces anti-multiple myeloma immune response mediated by cGAS/STING pathway activation. Blood Cancer Discov 2, 468–483 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Montes de Oca R et al. Belantamab Mafodotin (GSK2857916) Drives Immunogenic Cell Death and Immune-mediated Antitumor Responses In Vivo. Mol Cancer Ther 20, 1941–1955 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu P et al. Crizotinib-induced immunogenic cell death in non-small cell lung cancer. Nat Commun 10, 1486 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Scirocchi F et al. Immunogenic Cell Death and Immunomodulatory Effects of Cabozantinib. Front Oncol 11, 755433 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Choueiri TK et al. Cabozantinib plus Nivolumab and Ipilimumab in Renal-Cell Carcinoma. N Engl J Med 388, 1767–1778 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Spigel DR et al. Phase 1/2 Study of the Safety and Tolerability of Nivolumab Plus Crizotinib for the First-Line Treatment of Anaplastic Lymphoma Kinase Translocation - Positive Advanced Non-Small Cell Lung Cancer (CheckMate 370). J Thorac Oncol 13, 682–688 (2018). [DOI] [PubMed] [Google Scholar]
- 70.Patel SP et al. Phase Ib Study of Crizotinib plus Pembrolizumab in Patients with Previously Untreated Advanced Non-Small Cell Lung Cancer with ALK Translocation. Oncologist 25, 562–e1012 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xu W et al. Stimuli-responsive nanodelivery systems for amplifying immunogenic cell death in cancer immunotherapy. Immunol Rev (2023). [DOI] [PubMed] [Google Scholar]
- 72.Zhang J et al. Nanoparticle-based drug delivery systems to enhance cancer immunotherapy in solid tumors. Front Immunol 14, 1230893 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shalhout SZ, Miller DM, Emerick KS & Kaufman HL Therapy with oncolytic viruses: progress and challenges. Nat Rev Clin Oncol 20, 160–177 (2023). [DOI] [PubMed] [Google Scholar]
- 74.Bommareddy PK, Shettigar M & Kaufman HL Integrating oncolytic viruses in combination cancer immunotherapy. Nat Rev Immunol 18, 498–513 (2018). [DOI] [PubMed] [Google Scholar]
- 75.Poh A First Oncolytic Viral Therapy for Melanoma. Cancer Discov 6, 6 (2016). [DOI] [PubMed] [Google Scholar]
- 76.He B, Gross M & Roizman B The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc Natl Acad Sci U S A 94, 843–848 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Grigg C et al. Talimogene laherparepvec (T-Vec) for the treatment of melanoma and other cancers. Semin Oncol 43, 638–646 (2016). [DOI] [PubMed] [Google Scholar]
- 78.Palanivelu L, Liu CH & Lin LT Immunogenic cell death: The cornerstone of oncolytic viro-immunotherapy. Front Immunol 13, 1038226 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pol J et al. Trial Watch-Oncolytic viruses and cancer therapy. Oncoimmunology 5, e1117740 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vitale I et al. Targeting Cancer Heterogeneity with Immune Responses Driven by Oncolytic Peptides. Trends Cancer 7, 557–572 (2021). [DOI] [PubMed] [Google Scholar]
- 81.Zhou H et al. The oncolytic peptide LTX-315 triggers immunogenic cell death. Cell Death Dis 7, e2134 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yamazaki T et al. The oncolytic peptide LTX-315 overcomes resistance of cancers to immunotherapy with CTLA4 checkpoint blockade. Cell Death Differ 23, 1004–1015 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yamazaki T et al. LTX-315-enabled, radiotherapy-boosted immunotherapeutic control of breast cancer by NK cells. Oncoimmunology 10, 1962592 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rodriguez-Ruiz ME, Vitale I, Harrington KJ, Melero I & Galluzzi L Immunological impact of cell death signaling driven by radiation on the tumor microenvironment. Nat Immunol 21, 120–134 (2020). [DOI] [PubMed] [Google Scholar]
- 85.Kepp O, Marabelle A, Zitvogel L & Kroemer G Oncolysis without viruses - inducing systemic anticancer immune responses with local therapies. Nat Rev Clin Oncol 17, 49–64 (2020). [DOI] [PubMed] [Google Scholar]
- 86.Fucikova J et al. High hydrostatic pressure induces immunogenic cell death in human tumor cells. Int J Cancer 135, 1165–1177 (2014). [DOI] [PubMed] [Google Scholar]
- 87.Yu Z et al. Treatment of osteosarcoma with microwave thermal ablation to induce immunogenic cell death. Oncotarget 5, 6526–6539 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Altorki NK et al. Neoadjuvant durvalumab with or without stereotactic body radiotherapy in patients with early-stage non-small-cell lung cancer: a single-centre, randomised phase 2 trial. Lancet Oncol 22, 824–835 (2021). [DOI] [PubMed] [Google Scholar]
- 89.Zhou Q et al. Sugemalimab versus placebo after concurrent or sequential chemoradiotherapy in patients with locally advanced, unresectable, stage III non-small-cell lung cancer in China (GEMSTONE-301): interim results of a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol 23, 209–219 (2022). [DOI] [PubMed] [Google Scholar]
- 90.Kelly RJ et al. Adjuvant Nivolumab in Resected Esophageal or Gastroesophageal Junction Cancer. N Engl J Med 384, 1191–1203 (2021). [DOI] [PubMed] [Google Scholar]
- 91.Lim M et al. Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro Oncol 24, 1935–1949 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Omuro A et al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: An international randomized phase III trial. Neuro Oncol 25, 123–134 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee NY et al. Avelumab plus standard-of-care chemoradiotherapy versus chemoradiotherapy alone in patients with locally advanced squamous cell carcinoma of the head and neck: a randomised, double-blind, placebo-controlled, multicentre, phase 3 trial. Lancet Oncol 22, 450–462 (2021). [DOI] [PubMed] [Google Scholar]
- 94.Galluzzi L, Aryankalayil MJ, Coleman CN & Formenti SC Emerging evidence for adapting radiotherapy to immunotherapy. Nat Rev Clin Oncol 20, 543–557 (2023). [DOI] [PubMed] [Google Scholar]
- 95.Tatsuno K et al. Extracorporeal photochemotherapy induces bona fide immunogenic cell death. Cell Death Dis 10, 578 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ventura A et al. Extracorporeal Photochemotherapy Drives Monocyte-to-Dendritic Cell Maturation to Induce Anticancer Immunity. Cancer Res 78, 4045–4058 (2018). [DOI] [PubMed] [Google Scholar]
- 97.Li X, Lovell JF, Yoon J & Chen X Clinical development and potential of photothermal and photodynamic therapies for cancer. Nat Rev Clin Oncol 17, 657–674 (2020). [DOI] [PubMed] [Google Scholar]
- 98.Wei BM et al. Extracorporeal Photochemotherapy: Mechanistic Insights Driving Recent Advances and Future Directions. Yale J Biol Med 93, 145–159 (2020). [PMC free article] [PubMed] [Google Scholar]
- 99.Wu A et al. The effects of 5-aminolevulinic acid photodynamic therapy on the local immune response of women with cervical intraepithelial neoplasia grade 2. Front Immunol 14, 1211114 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tsai YC et al. Boost of innate immunity cytokines as biomarkers of response to extracorporeal photopheresis in patients with leukaemic cutaneous T-cell lymphoma. Br J Dermatol 189, 603–611 (2023). [DOI] [PubMed] [Google Scholar]
- 101.Menger L et al. Cardiac glycosides exert anticancer effects by inducing immunogenic cell death. Sci Transl Med 4, 143ra199 (2012). [DOI] [PubMed] [Google Scholar]
- 102.Zelenay S et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 162, 1257–1270 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Huang Q et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat Med 17, 860–866 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hayashi K et al. Tipping the immunostimulatory and inhibitory DAMP balance to harness immunogenic cell death. Nat Commun 11, 6299 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pietrocola F et al. Caloric Restriction Mimetics Enhance Anticancer Immunosurveillance. Cancer Cell 30, 147–160 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lévesque S et al. A synergistic triad of chemotherapy, immune checkpoint inhibitors, and caloric restriction mimetics eradicates tumors in mice. Oncoimmunology 8, e1657375 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Castoldi F et al. Autophagy-mediated metabolic effects of aspirin. Cell Death Discov 6, 129 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vodnala SK et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science 363 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cheng JT et al. Novel transcription regulatory sequences and factors of the immune evasion protein ICP47 (US12) of herpes simplex viruses. Virol J 17, 101 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Keckler MS Dodging the CTL response: viral evasion of Fas and granzyme induced apoptosis. Front Biosci 12, 725–732 (2007). [DOI] [PubMed] [Google Scholar]
- 111.Franz S et al. Mumps Virus SH Protein Inhibits NF-κB Activation by Interacting with Tumor Necrosis Factor Receptor 1, Interleukin-1 Receptor 1, and Toll-Like Receptor 3 Complexes. J Virol 91 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Vitale I, Shema E, Loi S & Galluzzi L Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat Med 27, 212–224 (2021). [DOI] [PubMed] [Google Scholar]
- 113.Solimini NL, Luo J & Elledge SJ Non-oncogene addiction and the stress phenotype of cancer cells. Cell 130, 986–988 (2007). [DOI] [PubMed] [Google Scholar]
- 114.Nagel R, Semenova EA & Berns A Drugging the addict: non-oncogene addiction as a target for cancer therapy. EMBO Rep 17, 1516–1531 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Levine AJ p53: 800 million years of evolution and 40 years of discovery. Nat Rev Cancer 20, 471–480 (2020). [DOI] [PubMed] [Google Scholar]
- 116.Wang M & Attardi LD A Balancing Act: p53 Activity from Tumor Suppression to Pathology and Therapeutic Implications. Annu Rev Pathol 17, 205–226 (2022). [DOI] [PubMed] [Google Scholar]
- 117.Hadian K & Stockwell BR The therapeutic potential of targeting regulated non-apoptotic cell death. Nat Rev Drug Discov 22, 723–742 (2023). [DOI] [PubMed] [Google Scholar]
- 118.Efimova I et al. Vaccination with early ferroptotic cancer cells induces efficient antitumor immunity. J Immunother Cancer 8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Li J et al. Tumor-specific GPX4 degradation enhances ferroptosis-initiated antitumor immune response in mouse models of pancreatic cancer. Sci Transl Med 15, eadg3049 (2023). [DOI] [PubMed] [Google Scholar]
- 120.Diepstraten ST et al. The manipulation of apoptosis for cancer therapy using BH3-mimetic drugs. Nat Rev Cancer 22, 45–64 (2022). [DOI] [PubMed] [Google Scholar]
- 121.Rongvaux A et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 159, 1563–1577 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; These back-to-back articles were the first to demonstrate that the activation of executioner caspases robustly suppresses type I IFN responses elicited by MOMP.
- 122.White MJ et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 159, 1549–1562 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; These back-to-back articles were the first to demonstrate that the activation of executioner caspases robustly suppresses type I IFN responses elicited by MOMP.
- 123.Rodriguez-Ruiz ME et al. Apoptotic caspases inhibit abscopal responses to radiation and identify a new prognostic biomarker for breast cancer patients. Oncoimmunology 8, e1655964 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Han C et al. Tumor cells suppress radiation-induced immunity by hijacking caspase 9 signaling. Nat Immunol 21, 546–554 (2020). [DOI] [PubMed] [Google Scholar]
- 125.Ning X et al. Apoptotic Caspases Suppress Type I Interferon Production via the Cleavage of cGAS, MAVS, and IRF3. Mol Cell 74, 19–31 e17 (2019). [DOI] [PubMed] [Google Scholar]
- 126.Suzuki J, Denning DP, Imanishi E, Horvitz HR & Nagata S Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science 341, 403–406 (2013). [DOI] [PubMed] [Google Scholar]
- 127.Giampazolias E et al. Mitochondrial permeabilization engages NF-kappaB-dependent anti-tumour activity under caspase deficiency. Nat Cell Biol 19, 1116–1129 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This original report provided compelling evidence in support of the notion that caspase-independent cancer cell death is more immunogenic than its caspase-dependent counterpart.
- 128.Klionsky DJ et al. Autophagy in major human diseases. Embo j 40, e108863 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wu Q et al. IGF1 receptor inhibition amplifies the effects of cancer drugs by autophagy and immune-dependent mechanisms. J Immunother Cancer 9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Debnath J, Gammoh N & Ryan KM Autophagy and autophagy-related pathways in cancer. Nat Rev Mol Cell Biol 24, 560–575 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Thompson EA & Powell JD Inhibition of the Adenosine Pathway to Potentiate Cancer Immunotherapy: Potential for Combinatorial Approaches. Annu Rev Med 72, 331–348 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Azambuja JH, Ludwig N, Braganhol E & Whiteside TL Inhibition of the Adenosinergic Pathway in Cancer Rejuvenates Innate and Adaptive Immunity. Int J Mol Sci 20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chiappori AA et al. Phase I Study of Taminadenant (PBF509/NIR178), an Adenosine 2A Receptor Antagonist, with or without Spartalizumab (PDR001), in Patients with Advanced Non-Small Cell Lung Cancer. Clin Cancer Res 28, 2313–2320 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lim EA et al. Phase Ia/b, Open-Label, Multicenter Study of AZD4635 (an Adenosine A2A Receptor Antagonist) as Monotherapy or Combined with Durvalumab, in Patients with Solid Tumors. Clin Cancer Res 28, 4871–4884 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Cascone T et al. Neoadjuvant Durvalumab Alone or Combined with Novel Immuno-Oncology Agents in Resectable Lung Cancer: The Phase II NeoCOAST Platform Trial. Cancer Discov 13, 2394–2411 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Herbst RS et al. COAST: An Open-Label, Phase II, Multidrug Platform Study of Durvalumab Alone or in Combination With Oleclumab or Monalizumab in Patients With Unresectable, Stage III Non-Small-Cell Lung Cancer. J Clin Oncol 40, 3383–3393 (2022). [DOI] [PubMed] [Google Scholar]
- 137.Song X et al. Pharmacologic Suppression of B7-H4 Glycosylation Restores Antitumor Immunity in Immune-Cold Breast Cancers. Cancer Discov 10, 1872–1893 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Liu L et al. Ablation of ERO1A induces lethal endoplasmic reticulum stress responses and immunogenic cell death to activate anti-tumor immunity. Cell Rep Med 4, 101206 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lin H et al. Stanniocalcin 1 is a phagocytosis checkpoint driving tumor immune resistance. Cancer Cell 39, 480–493.e486 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Feng M et al. Programmed cell removal by calreticulin in tissue homeostasis and cancer. Nat Commun 9, 3194 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Liu P et al. Immunosuppression by Mutated Calreticulin Released from Malignant Cells. Mol Cell 77, 748–760.e749 (2020). [DOI] [PubMed] [Google Scholar]
- 142.Chao MP et al. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci Transl Med 2, 63ra94 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Advani R et al. CD47 Blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin’s Lymphoma. N Engl J Med 379, 1711–1721 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ansell SM et al. Phase I Study of the CD47 Blocker TTI-621 in Patients with Relapsed or Refractory Hematologic Malignancies. Clin Cancer Res 27, 2190–2199 (2021). [DOI] [PubMed] [Google Scholar]
- 145.Querfeld C et al. Intralesional TTI-621, a novel biologic targeting the innate immune checkpoint CD47, in patients with relapsed or refractory mycosis fungoides or Sézary syndrome: a multicentre, phase 1 study. Lancet Haematol 8, e808–e817 (2021). [DOI] [PubMed] [Google Scholar]
- 146.van Helden MJ et al. BYON4228 is a pan-allelic antagonistic SIRPα antibody that potentiates destruction of antibody-opsonized tumor cells and lacks binding to SIRPγ on T cells. J Immunother Cancer 11 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Bahri M et al. SIRPα-specific monoclonal antibody enables antibody-dependent phagocytosis of neuroblastoma cells. Cancer Immunol Immunother 71, 71–83 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Giampazolias E et al. Secreted gelsolin inhibits DNGR-1-dependent cross-presentation and cancer immunity. Cell 184, 4016–4031.e4022 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article elegantly demonstrates that secreted GSN inhibits the binding of F-actin to DNGR-1 hence suppressing antigen cross-presentation by cDC1s.
- 149.Kim Y, Cho NY, Jin L, Jin HY & Kang GH Prognostic significance of STING expression in solid tumor: a systematic review and meta-analysis. Front Oncol 13, 1244962 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Chen C, Wang J, Dong C, Lim D & Feng Z Development of a risk model to predict prognosis in breast cancer based on cGAS-STING-related genes. Front Genet 14, 1121018 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Yamazaki T et al. Mitochondrial DNA drives abscopal responses to radiation that are inhibited by autophagy. Nat Immunol 21, 1160–1171 (2020). [DOI] [PubMed] [Google Scholar]; This report was the first to connect MOMP-dependent mtDNA-driven CGAS signaling in malignant cells with the ability of radiotherapy to elicit anticancer immune responses.
- 152.Harding SM et al. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bartsch K et al. Absence of RNase H2 triggers generation of immunogenic micronuclei removed by autophagy. Hum Mol Genet 26, 3960–3972 (2017). [DOI] [PubMed] [Google Scholar]
- 154.Govindan R et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell 150, 1121–1134 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Levy JMM, Towers CG & Thorburn A Targeting autophagy in cancer. Nat Rev Cancer 17, 528–542 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ladoire S et al. The presence of LC3B puncta and HMGB1 expression in malignant cells correlate with the immune infiltrate in breast cancer. Autophagy 12, 864–875 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Baracco EE et al. Contribution of annexin A1 to anticancer immunosurveillance. Oncoimmunology 8, e1647760 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Le Naour J et al. A TLR3 Ligand Reestablishes Chemotherapeutic Responses in the Context of FPR1 Deficiency. Cancer Discov 11, 408–423 (2021). [DOI] [PubMed] [Google Scholar]
- 159.Le Naour J et al. Formyl peptide receptor-1 (FPR1) represses intestinal oncogenesis. Oncoimmunology 12, 2237354 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Carbonnier V et al. Rs867228 in FPR1 accelerates the manifestation of luminal B breast cancer. Oncoimmunology 12, 2189823 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ghiringhelli F et al. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother 56, 641–648 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Vincent J et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res 70, 3052–3061 (2010). [DOI] [PubMed] [Google Scholar]
- 163.Reijers ILM et al. Personalized response-directed surgery and adjuvant therapy after neoadjuvant ipilimumab and nivolumab in high-risk stage III melanoma: the PRADO trial. Nat Med 28, 1178–1188 (2022). [DOI] [PubMed] [Google Scholar]
- 164.Chalabi M et al. Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat Med 26, 566–576 (2020). [DOI] [PubMed] [Google Scholar]
- 165.Blank CU et al. Neoadjuvant versus adjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma. Nat Med 24, 1655–1661 (2018). [DOI] [PubMed] [Google Scholar]
- 166.Forde PM et al. Neoadjuvant Nivolumab plus Chemotherapy in Resectable Lung Cancer. N Engl J Med 386, 1973–1985 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kepp O et al. A fluorescent biosensor-based platform for the discovery of immunogenic cancer cell death inducers. Oncoimmunology 8, 1606665 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Cultrara C et al. A biologic-device combination product delivering tumor-derived antigens elicits immunogenic cell death-associated immune responses against glioblastoma. J Immunother Cancer 11 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Garg AD et al. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma. Sci Transl Med 8, 328ra327 (2016). [DOI] [PubMed] [Google Scholar]
- 170.Vedunova M et al. DC vaccines loaded with glioma cells killed by photodynamic therapy induce Th17 anti-tumor immunity and provide a four-gene signature for glioma prognosis. Cell Death Dis 13, 1062 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Petroni G, Formenti SC, Chen-Kiang S & Galluzzi L Immunomodulation by anticancer cell cycle inhibitors. Nat Rev Immunol 20, 669–679 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Chuprin J et al. Humanized mouse models for immuno-oncology research. Nat Rev Clin Oncol 20, 192–206 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Wculek SK et al. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol 20, 7–24 (2020). [DOI] [PubMed] [Google Scholar]
- 174.Richter C et al. Generation of inducible immortalized dendritic cells with proper immune function in vitro and in vivo. PLoS One 8, e62621 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zhao L et al. A genotype-phenotype screening system using conditionally immortalized immature dendritic cells. STAR Protoc 2, 100732 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Zhang S et al. Anticancer effects of ikarugamycin and astemizole identified in a screen for stimulators of cellular immune responses. J Immunother Cancer 11 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Zhao L et al. BCL2 Inhibition Reveals a Dendritic Cell-Specific Immune Checkpoint That Controls Tumor Immunosurveillance. Cancer Discov 13, 2448–2469 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article provides abundant evidence supporting the notion that BCL2 inhibit antigen cross-presentation by cDC1s by preventing mtDNA-driven CGAS signaling.
- 178.Theisen DJ et al. WDFY4 is required for cross-presentation in response to viral and tumor antigens. Science 362, 694–699 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Li L et al. Lurbinectedin for the treatment of small cell lung cancer. Drugs Today (Barc) 57, 377–385 (2021). [DOI] [PubMed] [Google Scholar]
- 180.Baines AC et al. FDA Approval Summary: Belantamab Mafodotin for Patients with Relapsed or Refractory Multiple Myeloma. Clin Cancer Res 28, 4629–4633 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Yamamoto K et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 581, 100–105 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper elegantly demonstrates that efficient autophagic responses in malignant cells limit anticancer immunity by degrading MHC Class I molecules.
- 182.Pozzi C et al. The EGFR-specific antibody cetuximab combined with chemotherapy triggers immunogenic cell death. Nat Med 22, 624–631 (2016). [DOI] [PubMed] [Google Scholar]
- 183.DeSelm C et al. Low-Dose Radiation Conditioning Enables CAR T Cells to Mitigate Antigen Escape. Mol Ther 26, 2542–2552 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Sugita M et al. Radiation therapy improves CAR T cell activity in acute lymphoblastic leukemia. Cell Death Dis 14, 305 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Vanpouille-Box C et al. TGFbeta Is a Master Regulator of Radiation Therapy-Induced Antitumor Immunity. Cancer Res 75, 2232–2242 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Cappell KM & Kochenderfer JN Long-term outcomes following CAR T cell therapy: what we know so far. Nat Rev Clin Oncol 20, 359–371 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Larson RC & Maus MV Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat Rev Cancer 21, 145–161 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Yang J, Chen Y, Jing Y, Green MR & Han L Advancing CAR T cell therapy through the use of multidimensional omics data. Nat Rev Clin Oncol 20, 211–228 (2023). [DOI] [PubMed] [Google Scholar]
- 189.Pahl JH et al. Anti-EGFR antibody cetuximab enhances the cytolytic activity of natural killer cells toward osteosarcoma. Clin Cancer Res 18, 432–441 (2012). [DOI] [PubMed] [Google Scholar]
- 190.Hinterberger M et al. Intratumoral virotherapy with 4–1BBL armed modified vaccinia Ankara eradicates solid tumors and promotes protective immune memory. J Immunother Cancer 9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Charpentier M, Formenti S & Demaria S CD40 agonism improves anti-tumor T cell priming induced by the combination of radiation therapy plus CTLA4 inhibition and enhances tumor response. Oncoimmunology 12, 2258011 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Huang FY et al. A recombinant oncolytic Newcastle virus expressing MIP-3alpha promotes systemic antitumor immunity. J Immunother Cancer 8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Lu J et al. Nano-enabled pancreas cancer immunotherapy using immunogenic cell death and reversing immunosuppression. Nat Commun 8, 1811 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Bausart M et al. Combination of local immunogenic cell death-inducing chemotherapy and DNA vaccine increases the survival of glioblastoma-bearing mice. Nanomedicine 50, 102681 (2023). [DOI] [PubMed] [Google Scholar]
- 195.Kroemer G, McQuade JL, Merad M, Andre F & Zitvogel L Bodywide ecological interventions on cancer. Nat Med 29, 59–74 (2023). [DOI] [PubMed] [Google Scholar]
- 196.Blake SJ, Wolf Y, Boursi B & Lynn DJ Role of the microbiota in response to and recovery from cancer therapy. Nat Rev Immunol (2023). [DOI] [PubMed] [Google Scholar]
- 197.Dai J et al. Acetylation Blocks cGAS Activity and Inhibits Self-DNA-Induced Autoimmunity. Cell 176, 1447–1460 e1414 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Obradovic MMS et al. Glucocorticoids promote breast cancer metastasis. Nature 567, 540–544 (2019). [DOI] [PubMed] [Google Scholar]
- 199.Yang H et al. Stress-glucocorticoid-TSC22D3 axis compromises therapy-induced antitumor immunity. Nat Med 25, 1428–1441 (2019). [DOI] [PubMed] [Google Scholar]
- 200.Nunez-Ruiz A, Sanchez-Brena F, Lopez-Pacheco C, Acevedo-Dominguez NA & Soldevila G Obesity modulates the immune macroenvironment associated with breast cancer development. PLoS One 17, e0266827 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Dyck L et al. Suppressive effects of the obese tumor microenvironment on CD8 T cell infiltration and effector function. J Exp Med 219 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Boi SK et al. Obesity diminishes response to PD-1-based immunotherapies in renal cancer. J Immunother Cancer 8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Gomes-Santos IL et al. Exercise Training Improves Tumor Control by Increasing CD8(+) T-cell Infiltration via CXCR3 Signaling and Sensitizes Breast Cancer to Immune Checkpoint Blockade. Cancer Immunol Res 9, 765–778 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]