

Abstract
Nitrosopumilus maritimus is an ammonia-oxidizing archaeon that is crucial to the global nitrogen cycle1,2. A critical step for nitrogen oxidation is the entrapment of ammonium ions from a dilute marine environment at the cell surface and their subsequent channelling to the cell membrane of N. maritimus. Here we elucidate the structure of the molecular machinery responsible for this process, comprising the surface layer (S-layer), using electron cryotomography and subtomogram averaging from cells. We supplemented our in situ structure of the ammonium-binding S-layer array with a single-particle electron cryomicroscopy structure, revealing detailed features of this immunoglobulin-rich and glycan-decorated S-layer. Biochemical analyses showed strong ammonium binding by the cell surface, which was lost after S-layer disassembly. Sensitive bioinformatic analyses identified similar S-layers in many ammonia-oxidizing archaea, with conserved sequence and structural characteristics. Moreover, molecular simulations and structure determination of ammonium-enriched specimens enabled us to examine the cation-binding properties of the S-layer, revealing how it concentrates ammonium ions on its cell-facing side, effectively acting as a multichannel sieve on the cell membrane. This in situ structural study illuminates the biogeochemically essential process of ammonium binding and channelling, common to many marine microorganisms that are fundamental to the nitrogen cycle.
Subject terms: Cryoelectron tomography, Cryoelectron microscopy, Archaeal biology
The Nitrosopumilus maritimus surface layer (S-layer) concentrates ammonium ions on its cell-facing side, acting as a multichannel sieve on the cell membrane.
Main
The ocean is our planet’s largest biome, where pelagic microbial Thaumarchaeota (syn. Nitrososphaerota) represent one of the most abundant organisms3. The numerical dominance of marine thaumarchaea suggests that they have a major role in global biogeochemical cycles1,2. N. maritimus, an intensely studied marine thaumarchaeon, grows chemolithoautotrophically by aerobically oxidizing ammonia to nitrite1. This organism has also been shown to regenerate oxygen under anoxic conditions, and to fix CO24,5, placing it in an important position in the global nitrogen and carbon dioxide biogeochemical cycles.
Owing to the low concentration of ammonia (NH3) or ammonium (NH4+) ions in the oceans, reported in the 10−8 to 10−9 M range6, marine archaea such as N. maritimus have evolved specialized molecular machinery to attract ammonium ions on their cell surface to facilitate ammonium oxidation at the cell membrane7. N. maritimus, like most archaeal cells, is encased by a paracrystalline, proteinaceous surface layer or S-layer8–11. Bulk modelling of the N. maritimus S-layer has predicted that this cellular organelle might have a key role in elevating the ammonium concentrations in the pseudoperiplasmic space between the S-layer and the cell membrane12,13; however, how this occurs on a mechanistic and molecular level is unclear. At the overall morphological scale, the N. maritimus S-layer has been reported to have a hexagonal arrangement9,12 and is postulated to consist of repeating subunits of the proteins Nmar_1547 or Nmar_1201 (two proteins with 91% sequence identity), based on transcriptomic and proteomic data14,15. Here, to understand the rules governing ammonium binding and enrichment by N. maritimus and related marine Thaumarchaeota, we investigated the molecular structure of the N. maritimus S-layer using structural, biochemical, cellular and bioinformatic methods.
Cryo-ET analysis of the N. maritimus S-layer
To gain insights into this problem, we used electron cryotomography (cryo-ET) and subtomogram averaging techniques. We have previously applied these methods to determine in situ structures of prokaryotic S-layers from in vitro purified specimens containing cellular fragments16–18. Our goal was to solve the structure of the N. maritimus S-layer directly from whole cells. Cryo-ET analysis of N. maritimus cells revealed an S-layer surrounding the approximately 300-nm-wide elongated cells, which contained a compact nucleoid and several cytosolic ribosomes (Fig. 1a). Using subtomogram averaging, we mapped the locations of the S-layer repeating units on the cell, which were arranged in a hexagonal array (Fig. 1b). We found that the S-layer hexamers coated the cells with near-perfect continuity along their lengths, while local pentameric defects were present on the cell edges, completing the S-layer (Fig. 1b), which was confirmed by quantification of hexamer and pentamer positions relative to the centre of the cell (Fig. 1c).
Fig. 1. The molecular structure and assembly of the N. maritimus S-layer in intact cells.

a, A denoised36,37 tomographic slice through a N. maritimus cell shows ultrastructural details of this marine archaeon (annotated). Inset: top and side views of the subtomogram average of the S-layer. Scale bars, 500 Å (main image) and 100 Å (inset). Cellular tomography was performed at least 27 times (Extended Data Table 1). b, Map of the subtomogram positions in the cellular S-layer showing the presence of pentameric defects at the edge of the cell. Scale bar, 500 Å. c, A histogram of subtomogram positions from all tomograms relative to the three-dimensional centre of the cell body (n = 41,303 hexamers (grey) and n = 203 pentamers (pink); one out of two biological replicates shown). d, The subtomogram averaging map enables derivation of a molecular model directly from cellular data. Amino acid residue side chains resolved are marked. See also Extended Data Figs. 1 and 2. e, A ribbon model of the cashew-shaped NmSLP monomer is shown in two orthogonal views. f,g, The structure of the S-layer hexamer displayed in two orthogonal views shows that NmSLP monomers are arranged as an array of Ig-like domains; each domain (D) is coloured differently (a schematic is shown below). The first eight Ig-like domains are resolved in the cryo-ET and subtomogram averaging map.
Next, we used state-of-the-art cryo-ET imaging and image-processing workflows, which have been shown to support high-resolution in situ structure determination from purified specimens19. As a result, we produced a high-resolution map of the N. maritimus S-layer hexamer from intact cells (Fig. 1d, Extended Data Fig. 1, Extended Data Table 1 and Supplementary Videos 1 and 2). The central region of the S-layer hexamer in the map had a resolution of 3.3 Å, with the resolution decaying to around 4.5 Å towards the periphery (Extended Data Fig. 1). The subtomogram averaging map contained sufficient details to enable us to derive an atomic model of the S-layer (Fig. 1d–g and Extended Data Fig. 2). The structure shows that the S-layer is pseudohexagonal (Fig. 1f,g) and consists of the repeated interactions of the Nmar_1547 (hereafter, NmSLP) S-layer protein. Despite the high sequence similarity of NmSLP to the other previously predicted SLP, Nmar_1201, a unique segment of NmSLP between residues 911 and 977 was clearly resolved in our map. This enabled us to identify NmSLP as the primary N. maritimus SLP on cells through direct structure determination (Extended Data Fig. 2). This observation confirms previous transcriptomic data showing high expression levels of the Nmar_1547 gene compared with Nmar_120114. However, we cannot rule out that Nmar_1201 could be present at lower copy numbers.
Extended Data Fig. 1. Subtomogram averaging (STA) of the N. maritimus S-layer.

a, Fourier shell correlation (FSC) curves of the STA reconstruction. b-c, Local resolution of two-fold symmetrised (C2) N. maritimus S-layer plotted onto the cryo-ET STA map, shown from the top and from the side. d, 3-D Fourier shell correlation (FSC) curves of the STA reconstruction. e, Local resolution of two-fold symmetrised (C2) N. maritimus S-layer shown from the bottom, from the inside of the cell. f, Local resolution of six-fold symmetrised (C6) N. maritimus S-layer map shown from the side. g, Particle distribution from the 0° projection image. h, Processing schematic from tomographic reconstructions to high resolution reconstruction of the S-layer. Scale bars for panels (b), (c), (e) and (f): 10 Å.
Extended Data Table 1.
Cryo-ET data collection, refinement, and validation statistics

Extended Data Fig. 2. Structure determination from whole-cell cryo-ET data.

a, The STA map of the S-layer (isosurface shown) was used to build a model of NmSLP (ribbon) directly from cellular data. (See also Fig. 1(b–e)). b-i, Examples of cryo-ET density and the built model for the Ig-like domains one to eight (D1-D8) of one NmSLP subunit. The local resolution decreases from the central two-fold axis of the NmSLP hexamer (see Extended Data Fig. 1).
At the sequence level, NmSLP is arranged into ten immunoglobulin-like (Ig-like) domains (Fig. 1f,g). The first eight domains were well resolved in our 3.3–4.5-Å-resolution subtomogram averaging map. By contrast, the last two domains appeared less ordered, with the local resolution decaying towards the C terminus of NmSLP, away from the centre of the S-layer hexamer (Extended Data Figs. 1 and 2). At the N terminus, the C6 symmetry of the hexamer is broken, revealing a distinctly two-fold symmetric central pore (Extended Data Fig. 2). Each monomer of NmSLP in the S-layer adopts a rough ‘cashew’ shape, facilitating several interactions around the hexamer of NmSLP (Fig. 1e–g). Domain 4, nestled within domain 3 and linked by short connectors, is slightly raised relative to the base of the cashew-shaped Ig-array of NmSLP, projecting towards the extracellular milieu.
Although the amino acid residues (37–1499) from the first eight Ig-like domains could be unambiguously built into the subtomogram averaging map, several unexplained densities were observed, emanating from surface-exposed asparagine residues (Extended Data Fig. 2). Given that archaeal SLPs are known to be heavily glycosylated20, we hypothesized that these densities might correspond to glycans. Another notable set of unexplained densities was observed near the negatively charged amino acid residue side chains of Asp73 and Glu74 at the central C2 pore, as well as between NmSLP monomers around the hexamer (Extended Data Fig. 2). Considering that positively charged ions have been previously observed bound to S-layers21,22, we hypothesized that these additional densities on the N. maritimus S-layer could potentially correspond to bound cations, although the resolution of the map prevented us from unambiguously assigning their chemical identities.
Cryo-EM shows a porous S-layer
The N. maritimus S-layer has previously been predicted through bulk biophysical modelling to help attract ammonium ions12. Considering the tight sheath formed by the S-layer around cells (Fig. 1b,c), providing an extremely large surface area for interaction with the marine environment, we postulated that the S-layer functions as a negatively charged ammonium trap. This would facilitate the movement of cations such as ammonium towards the cell membrane, specifically to the sites of ammonium oxidation. To test these hypotheses with higher resolution structures, in which ion and other densities would be better resolved, we purified N. maritimus cell envelopes for cryo-electron microscopy (cryo-EM) analysis. We then used single-particle techniques, as applied previously to two-dimensional S-layer sheets for structure determination18,23, to solve a 2.7-Å-resolution structure of the S-layer (Fig. 2a, Extended Data Fig. 3 and Extended Data Table 2).
Fig. 2. Cryo-EM structure of isolated N. maritimus S-layer sheets.

a, In vitro cryo-EM structure (global resolution, 2.7 Å) of isolated S-layer sheets from N. maritimus. The colour scheme for ribbon diagrams is the same as in Fig. 1; domains of one NmSLP are marked. b–j, Magnified views of the pores lined with negatively charged residues, which are ubiquitous in the S-layer sheet. The location of the pores is given in the titles of the panels. k, The sharpened38 cryo-EM map shows 17 glycans decorating each NmSLP; the map shown at a lower contour level in two different orientations. A schematic of the glycan locations on the NmSLP sequence is shown below.
Extended Data Fig. 3. Single particle analysis of isolated N. maritimus S-layer sheets.

a, Cryo-EM image of isolated N. maritimus cell envelopes show repeating units of the pseudo-hexagonal (tilted) S-layer. Insets – characteristic top and side views observed in class averages. This single image is representative from a data set containing 12,557 images from three independent data collections (see Extended Data Table 2). b, FSC estimation of the resolution of the unsymmetrized (C1), two-fold (C2) and six-fold (C6) symmetrised maps. c-e, Local resolution of the C2 cryo-EM map estimated in RELION, plotted into the density, shown in from the top (c), side (d) and bottom (e). The resolution of outer domains (D8-D9) is slightly lower. f, Directional 3D-FSC between two random halves of the data57. g, Angular distribution of the particles in the data set, shown on a relative scale (purple denotes low and yellow denotes high). h, Histogram of local resolutions in voxels of the cryo-EM map (C2-symmetrised). Scale bars: (a) 500 Å, Inset 200 Å; (c-d) 20 Å.
Extended Data Table 2.
Cryo-EM data collection, refinement, and validation statistics

The single-particle structure was very similar (root mean squared deviation of 2.21 Å for the full composite model, 0.54 Å for residues 35–455 (refined in C2) and 1.39 Å for residues 466–1498 (refined in C6)) to the subtomogram averaging structure (Fig. 2, Extended Data Fig. 4 and Supplementary Video 3), enabling us to extend our structure by modelling the ninth Ig-like domain, reaching up to residue 1616 out of 1734 (Fig. 2a and Extended Data Fig. 3). The last Ig-like domain remains unresolved in our map, with only disordered, diffuse density observed beyond the ninth Ig-like domain of NmSLP in the pseudoperiplasmic space, indicating flexibility relative to the rigid part of the S-layer. The NmSLP hexamer in the single-particle structure appears to be slightly expanded compared with the subtomogram averaging structure (Supplementary Video 3), perhaps due to differences in the S-layer curvature.
Extended Data Fig. 4. Comparison of the STA and SPA N. maritimus S-layer reconstruction.

a, Close-up view of the STA map with the built NmSLP model (domain 1) showing resolved large and small side chains. b, Close-up view of the SPA map in the same view as shown in panel (a). c-d, Close-up view of the STA map (c) and (d) of domain 3 of the NmSLP with resolved small and bulky side chains. e-f, Close-up views of the N-glycan densities of the STA map (a) and SPA map (f) in domain 1. g-h, The density of the N-glycans is enhanced in the sharpened38 map (Methods) of the same region. i-l, Close-up views of some of the N-glycan densities of the STA (i,k) and SPA map (j,l) of domain 4.
The N. maritimus genome also encodes a homologue for a cell-anchoring SlaB protein (Extended Data Fig. 5a,b and Supplementary Table 1) that is known to bind the S-layer of the archaeon Sulfolobus acidocaldarius to the cell membrane24. However, proteomic data on N. maritimus indicate that this protein is considerably less abundant in the cell than NmSLP15. We speculate that the last (tenth), unresolved Ig-like domain of NmSLP may have a role in anchoring the S-layer to molecules present on the cell envelope, therefore partially reducing the need for stoichiometric anchoring by a SlaB or a SlaB-like protein in N. maritimus.
Extended Data Fig. 5. Bioinformatic analysis of Ig-like domain-containing archaeal SLPs.

a, Cartoon schematic of the domain architecture of SLPs in Nitrososphaerota. In addition to the main SLP, a minor SlaB homologue is encoded in the genome of most Nitrososphaerota. The SlaB protein exhibits a C-terminal transmembrane domain which potentially anchors the main S-layer canopy to the membrane. b, Cartoon schematic of the domain architecture of SLPs in Thermoproteota and Euryarchaeota. The domain architecture of AOA SLPs vary considerably from haloarchaea. c, Cluster map of Ig-like domain-containing archaeal SLPs. This map was created by collecting homologues of various representative Ig-like domain-containing SLPs and clustering them using CLANS75 based on the strength of their all-against-all pairwise BLAST P-values, with a threshold set at 1-e8. Each protein sequence in the map is depicted as a dot, and sequences within the same taxonomic class are denoted by the same colour. The intensity of the line colour indicates the significance of sequence similarities, with darker lines representing higher significance. Although comprising Ig-like domains, archaeal SLPs are extremely divergent in their sequences.
In our single-particle structure, the subunit contacts between NmSLP hexamers (Fig. 2b–j) were better resolved compared with in our subtomogram averaging map (Fig. 1). Notably, several contact sites, both between hexamers and within each hexamer, contain pores lined predominantly by rows of negatively charged amino acid residues (Fig. 2b–j). These pores are relatively small (around 5 Å) but are compatible with the size of small chemical species, fitting with the idea that the pores of the S-layer may function as cation channels. Owing to the repeating pattern of the S-layer, these pores span the entirety of the N. maritimus cell surface. Supporting the anticipated cation-binding properties of the S-layer, a bioinformatics comparison of the amino acid composition of NmSLP with that of the N. maritimus proteome and all archaeal proteins revealed a substantial increase in aspartic acid residues (10.9% in NmSLP versus 6.1% in the N. maritimus proteome versus 7.3% in all archaea). Concurrent with this, the percentages of lysine and arginine residues are reduced (1.7% and 2.2% in NmSLP versus 8.4% and 3.4% in the N. maritimus proteome versus 3.8% and 5.9% in all archaea), resulting in an S-layer that is highly negatively charged.
Overall, our structure shows that the NmSLP monomers densely populate the S-layer sheet. This arrangement is reminiscent of the S-layer in the Dead Sea archaeon Haloferax volcanii, which is composed of an SLP called csg, which also consists of tandemly repeated Ig-like domains18. Such arrays of Ig-like domains have been observed in archaeal18,25, monoderm bacterial22,26 and diderm bacterial S-layers23,27. Although these SLPs share some structural similarities, they diverge notably at the sequence level, as well as at the overall organizational level containing different number of domains (Extended Data Fig. 5c), enabling them to assemble into unique two-dimensional sheets23,24, each with distinctly different glycosylation patterns and cell anchoring mechanisms.
In total, 17 glycan densities per monomer of NmSLP were also resolved in the single-particle map (Fig. 2k and Extended Data Fig. 4). Although these densities do not support direct derivation of the chemical structure of the glycans, they project away from the cell surface at asparagine residues, which are followed by a threonine or serine residue at the +2 position (Fig. 2k). Enshrouding the outer domains of the proteinaceous NmSLP S-layer, these glycans form a thick shell, encasing the cell in a sugar-rich coat (Fig. 2k). The mesh-like arrangement of the glycans probably provides protection, potentially shielding the cell from phages28. It might also enhance the hydrophilicity of the cell surface, making it suitable for marine environments. Most of these glycans are located in the N-terminal segment of NmSLP, primarily in outermost domains 1, 2 and 4, with a single glycan present in domain 6 (Fig. 2k).
Ammonium binding of the S-layer
We confirmed that the concentration of ammonium ions in the medium strongly influences the growth of N. maritimus (Supplementary Fig. 1a), as shown previously4,9. To directly measure ammonium ion binding to the cell surface, we performed isothermal titration calorimetry (ITC), titrating a medium containing NH4Cl against whole N. maritimus cells. As expected, growing N. maritimus cells showed strong and robust ammonium binding (Fig. 3a,b and Supplementary Fig. 1b; n = 3). We subsequently performed cryo-ET analysis of the same sample after ITC measurements, revealing normal cell morphology with an intact S-layer coating the cells (Fig. 3c,d; n = 150). As ammonium is the sole energy source for these growing cells, ammonium ions must somehow reach the cell for oxidation; we therefore inferred that the measured ammonium binding occurs either directly to the S-layer or to the underlying cell after passage through the S-layer (Fig. 3c,d). We then perturbed the S-layer by pretreating the cells with ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), which is known to impair several prokaryotic S-layers16. N. maritimus cells that were pretreated with 2.5 mM EGTA showed altered ammonium binding (Fig. 3a,b and Supplementary Fig. 1b; n = 3), concurrent with observed gaps and partial S-layer disruptions seen in cryo-ET in a heterogeneous population (Fig. 3e; n = 83). Near-complete disruption of the S-layer with 5 mM EGTA entirely abolished ammonium binding (Fig. 3a,b and Supplementary Fig. 1b; n = 3), leading to rounding up of cells with exposed, uncoated membranes (Fig. 3f; n = 152). These experiments indicate that an intact S-layer is critical for ammonium binding and may also be important for cell shape maintenance in N. maritimus in a calcium-dependent manner.
Fig. 3. Ammonium ions either bind directly to the S-layer or pass through it to bind to the underlying cell.

a, ITC signal of intact N. maritimus cells titrated with ammonium chloride with different pretreatments with EGTA. b, Quantification of the ITC curves showing the total heat (Q) released in the different experiments. Data are mean ± s.d. n = 3 biologically independent replicates. Statistical analysis was performed using ordinary one-way analysis of variance (ANOVA) with correction for multiple comparisons; ****P < 0.0001, **P = 0.0075. c, Schematic of the ITC and cryo-ET experiment presented. d, Cryo-ET analysis of N. maritimus cells after titration with ammonium chloride shows complete coating with an S-layer. n = 150 from two biological replicates. e, Cryo-ET analysis of N. maritimus cells treated with 2.5 mM EGTA after titration with ammonium chloride shows gaps in the S-layer in a heterogeneous cell population. n = 83 from two biological replicates. f, Cryo-ET analysis of N. maritimus cells treated with 5 mM EGTA after titration with ammonium chloride showing round cells with a naked membrane. n = 152 from three biological replicates. For d–f, scale bars, 500 Å.
To examine the biochemically observed ammonium-binding properties of the S-layer structurally, we purified S-layer sheets in vitro, at a higher concentration of ammonium (2.5 mM compared to 1 mM NH4Cl) and resolved a 3.1-Å-resolution cryo-EM structure of the S-layer enriched in ammonium ions. Compared with our original cryo-ET structure (Supplementary Fig. 1c–f), this ammonium-enriched S-layer structure showed an increased number of unexplained densities at several S-layer pores, indicative of ammonium ion binding to the negatively charged amino acid residues lining these pores (Supplementary Fig. 1c–f).
To further investigate the ability of the lattice to bind to cations, we performed atomistic molecular dynamics (MD) simulations of the N. maritimus S-layer in the presence of ammonium (Fig. 4a, Methods, Extended Data Fig. 6 and Supplementary Fig. 2), using a framework that has recently been shown to yield strong agreement with X-ray crystallography and cryo-EM studies of cation binding to S-layers21. Analysis of residue-based ammonium occupancies over the course of three 500 ns simulations using PyLipID29 clearly identified the primary acidic residues that mediate ammonium binding (Supplementary Table 2 and Supplementary Video 4). Moreover, mapping the high-ammonium occupancy (>50%) residues onto the hexamer structure revealed multiple acidic residue clusters in excellent agreement with the pores identified in our cryo-EM and cryo-ET maps (Fig. 4b and Extended Data Fig. 7). These observations are compatible with the idea that the multiple pores in the S-layer sheet, lined with negatively charged residues, facilitate the movement of ammonium ions across the S-layer. Furthermore, in our simulations, we also found that ammonium ions could be replaced with sodium ions, suggesting that the S-layer might not discriminate between different small positively charged ions for binding (Supplementary Fig. 2).
Fig. 4. Ammonium (NH4+) binding to the negatively charged S-layer lattice.

a, MD simulations support ammonium-ion binding at the S-layer pores. Residue-based ammonium occupancies during the 0.1 M NH4+ MD simulations are plotted onto the S-layer structure on a relative scale from white to purple. b, The distribution of negatively charged residues (shown in red) in the S-layer matches well with the MD simulations showing predicted ammonium-binding residues (Extended Data Fig. 6). c, A histogram of negatively charged residues along the S-layer, overlaid onto the ammonium-ion positions in the 0.1 M NH4+ MD simulations (distance was calculated from the closest, membrane-proximal amino acid residue in the S-layer structure). For the averaged ammonium-ion residence from three independent MD simulations (averaged over the last 400 ns of each simulation), the error bars denote ±1 s.d. (Supplementary Fig. 2). d, The mean local charge of NmSLP plotted along the sequence shows a gradual but continual increase in negative charge. The z position of the NmSLP residues, derived from the S-layer structure, is indicated, with the ninth Ig-like domain forming the base of the S-layer, proximal to the cell membrane.
Extended Data Fig. 6. Molecular dynamics simulation of ammonium ion diffusion across the multi-channel, charged S-layer.

a, Unit cell design for MD simulation of the S-layer lattice. The unit cell (simulation box, outlined in yellow) was constructed to simulate an infinite two-dimensional sheet. b, Ammonium ion densities (ammonium occupancy during a single simulation shown as golden density) plotted onto the structure shown in ribbon representation. For further details on ammonium binding residues, please see Supplementary Table 2 and Supplementary Fig. 2. c-k, Pores in the S-layer (shown in Fig. 2), with pore residue side chains coloured by occupancy of ammonium ions (colour bar shown in panel k). l, Occupancy profile for the modelled NmSLP sequence; residues binding to ammonium ions (high occupancy) are shown as large letters. Residue indices corresponding to domains 1 to 9 are depicted by coloured lines below the plot.
Extended Data Fig. 7. Charge distribution across archaeal S-layers.

a, The predicted net charges of archaeal SLPs at neutral pH (7.4) show that N. maritimus is highly charged, supporting its function as a cation and ammonium trap. The net charge was calculated using Isoelectric Point Calculator 2.076. b, The electrostatic charge of the N. maritimus S-layer is illustrated on the surface model. c, Residues in the N. maritimus S-layer with >50% ammonium occupancy in MD simulations from pyLipID analysis (left), compared with highlighted negatively charged residues in the S-layer (middle), show a remarkable overlap (right). d, The mean local charge of the SLPs from AOAs and other archaea, plotted along the sequence, shows a gradual but continual increase in negative charge specifically in AOAs, but not in other archaea.
Collectively, our structural data reveal that the S-layer of N. maritimus functions as a multichannel exchanger for ammonium ions, featuring negatively charged residues that line several pores in the S-layer (Fig. 2b–j). In this context, it is interesting that the negative charge of the S-layer increases when moving from the extracellular environment toward the cell membrane (Fig. 4c), specifically from the N to the C terminus of each NmSLP. This charge gradient probably facilitates the movement of ammonium ions through the S-layer towards the cell membrane. Our MD simulations support this hypothesis, showing an accumulation of ammonium ions at the cell-facing side of the S-layer (Fig. 4c, Supplementary Fig. 2 and Supplementary Video 4). To ensure that these observations did not depend on the amount of ammonium present, we conducted further MD simulations of the S-layer system in 0.05 M or 0.2 M ammonium, reproducing in both cases strong ammonium binding by the S-layer and relative accumulation of ammonium ions towards the cell-facing side of the S-layer (Supplementary Fig. 2).
The observed increase in negative charge at the structural level is also mirrored at the sequence level. There is a continuous and substantial increase in negative charge from the N to the C terminus of the NmSLP protein sequence (Fig. 4d), with the membrane-proximal C terminus being highly negatively charged. Using sensitive sequence-based homology searches and structure prediction, we found that Ig-domain-containing SLPs with a charge gradient are common across all described groups of ammonia-oxidizing archaea (AOA), suggesting similar S-layer arrangements in these species (Extended Data Fig. 7). Although the overall domain organization of such AOA and other archaeal SLPs could be similar (Extended Data Fig. 5a,b), the number of Ig-like domains and the charge distribution in each SLP vary, possibly reflecting differences in the function of these S-layers in binding positively charged molecules and ions (Extended Data Fig. 7d).
Molecular modelling of the cell surface
Together, our data enabled us to construct a molecular model of the N. maritimus cellular S-layer (Fig. 5a,b). Subtomogram position mapping of the S-layer hexamers and pentamers demonstrates that the S-layer coats the cell surface with near-perfect continuity (Fig. 5a,b), a characteristic that is also observed in other archaeal S-layers18. The continuous S-layer is heavily glycosylated, possibly protecting the cell from phages in the harsh marine environment. The dome-shaped structure of the S-layer hexamer probably supports flexibility, allowing NmSLP to coat different parts of the cell membrane with varied curvature. The S-layer is closed around cells by pentameric positions, which also appear to be composed of NmSLP (Extended Data Fig. 8). At the technical level, this study highlights the power of modern structural biology—molecular structures obtained from whole-cell cryo-ET, in conjunction with in-cell biochemistry and MD simulations, can provide key biochemical and mechanistic insights. With advancements in data collection and image-processing methodologies19,30,31, we anticipate that such in situ structural techniques will significantly enhance our molecular understanding of cells (Supplementary Fig. 3).
Fig. 5. The in situ structure of the N. maritimus S-layer shows a multichannel sieve nearly perfectly coating cells at high-copy numbers.
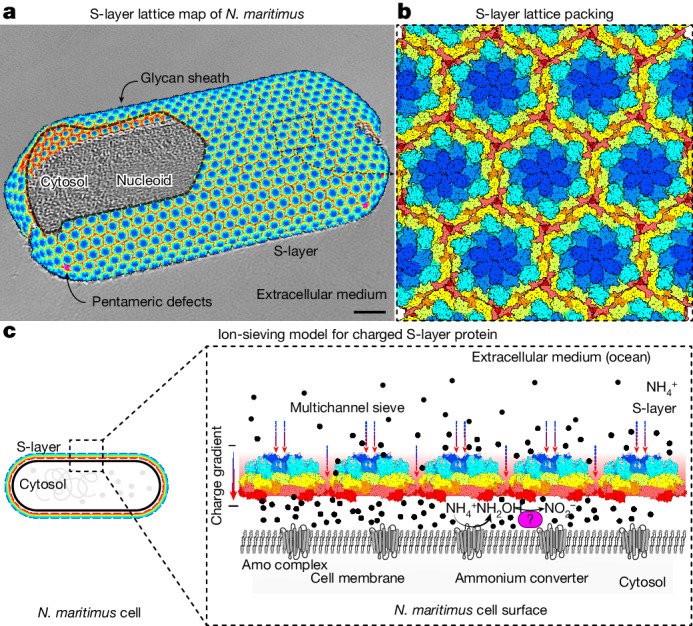
a, The S-layer lattice coats nearly the entire outer surface of N. maritimus cells. A small part of the lattice map (black dashed line) has been cut out from the top of the cell for clarity. The pseudohexagonal lattice is joined together by pentameric defects (Extended Data Fig. 8). Scale bar, 500 Å. Cellular tomography was performed at least 32 times (Extended Data Table 1). b, A model of the S-layer lattice is shown as a space-filling representation with the same colour scheme as in Fig. 1. c, The ion-exchanging model for the N. maritimus S-layer. The highly negatively charged S-layer entraps ammonium ions, and these ions may move along multiple pores in the lattice, aided by increasing negative charge along the lattice. Once beyond the S-layer, the ammonium ions can diffuse to the membrane predominantly composed of crenarchaeol39, where they are converted to nitrite through hydroxylamine (NH2OH) as an intermediate by an unknown protein complex (pink)34, setting up an ion sink and concentration gradient between the S-layer and the membrane.
Extended Data Fig. 8. Whole cell tomography of N. maritimus and S-layer lattice maps.

a-f, Gallery of lattice maps of the S-layer from subtomogram averaging plotted onto denoised36,37 N. maritimus cellular tomograms (only one slice shown in each case). The S-layer lattice coats nearly the entire outer surface of N. maritimus cells. A region has been cut out from each lattice map on top of the cell for clarity. The hexagonal lattice is joined together by pentameric defects, and linear lattice dislocations are also occasionally observed. Scale bars: 500 Å. Cellular tomography was performed at least 32 times (see Extended Data Table 1).
Conclusion
Our data are consistent with a scenario in which ammonium and other cations are bound and enriched at the cell surface by the negatively charged S-layer (Fig. 5c). The S-layer provides an extremely large surface area for interaction with the surrounding marine environment, where it acts as a multichannel cation exchanger and, due to its gradually increasing negative charge, leads to the accumulation of ammonium ions on the cell-facing side of the S-layer. An ‘ammonium sink’ exists at the cell membrane, where the integral membrane machinery, ammonia monooxygenase32,33, converts NH4+ to NO2− with hydroxylamine (NH2OH) as an intermediate34. This activity probably establishes an ammonium concentration gradient, extending from the membrane-proximal, ammonium-rich side of the S-layer to areas of lower concentrations near the cell membrane. A previous biophysical model has outlined how such a sink could function12, potentially initiating a chain of ammonium uptake that provides energy to the cell (Fig. 5c). This passive charge gradient of the S-layer could explain why marine AOA exhibit a 200-fold greater affinity for ammonia compared with ammonia-oxidizing bacteria7, which generally lack S-layers. This increased affinity enables AOA to thrive in low-ammonia marine environments. The energy-efficient, passive enrichment mechanism is particularly advantageous in the resource-limited, harsh conditions that are found in the ocean depths. Moreover, as S-layers similar to that of N. maritimus are found in nearly all AOA, including those in soil ecosystems (Extended Data Fig. 5a,b), this ammonium enrichment mechanism is probably conserved among these organisms35, contributing to the biogeochemically important nitrogen cycle (Supplementary Fig. 3). A comprehensive understanding of these biogeochemical processes is critical for preserving the vital ecological functions that sustain life on Earth.
Methods
Growth of N. maritimus cells
A culture of N. maritimus (SCM1) was provided by F. Elling and A. Pearson. Continuous cultures of SCM1 were grown on modified synthetic crenarchaeota medium supplemented with 1 mM NH4Cl, as previously described7,40 at 28 °C in a standing incubator that was covered to prevent excessive exposure to light. The growth of SCM1 N. maritimus cells was monitored using a NO2−-detection assay reported previously1. The same assay was used to follow SCM1 growth with differing ammonium concentrations.
Purification of N. maritimus cell envelopes
Native cell envelopes were purified from N. maritimus by adapting a previously described protocol23. A total of 12 l of N. maritimus cultures was prepared and late-log-phase cells were collected by centrifugation (10,000g, 4 °C, 30 min) and frozen and stored at −80 °C until further experimentation. The cell pellet from a 1 l culture was carefully resuspended in 3 ml lysis buffer (50 mM HEPES/NaOH pH 7.5, 500 mM NaCl, 50 mM MgCl2, 10 mM CaCl2, 1% (w/v) CHAPS, supplemented with 1× cOmplete protease inhibitor cocktail (Roche)). The cell suspension was incubated for 1 h on ice, and then lysed using sonication (10×, 5 s pulse, amplitude strength 10%). The sonicated sample was subsequently centrifuged (80,000g, 4 °C, 1 h), forming a very small white pellet at the bottom of the centrifugation tube. The pellet was resuspended into 40 µl of the same buffer and used for cryo-EM experiments. For the ammonium-enriched sample, the buffer was supplemented with 2.5 mM NH4Cl.
Cryo-EM and cryo-ET sample preparation
For cryo-EM and cryo-ET grid preparation, previously reported protocols were used18,23,41. In brief, 2.5 µl of the specimen was applied to a freshly glow discharged Quantifoil R2/2 Cu/Rh 200 mesh grid, adsorbed for 60 s, blotted for 4–5 s and plunge-frozen into liquid ethane in the Vitrobot Mark IV (Thermo Fisher Scientific), while the blotting chamber was maintained at 100% humidity at 10 °C. For tomography, the specimen was additionally supplemented with 10 nm gold conjugated with protein A. The grids were clipped and stored under liquid nitrogen until cryo-EM data collection was performed.
Cryo-ET and cryo-EM data collection
Cryo-ET data
For high-resolution in situ structure determination of the S-layer, a pipeline for high-throughput data collection was adopted42. In brief, a Titan Krios microscope was used to collect tilt-series data with a dose-symmetric tilting scheme43. Tilt series were collected at a pixel size of 1.327 Å, with a total dose of ~121 e− Å−2 applied over entire series collected between ±60° with 3° tilt increments. A total of 160 tilt series were collected with a defocus range of between −2 and −5 µm target defocus, and the samples were subjected to 0.9 s of exposure per tilt video containing 10 frames each (Extended Data Table 1). For visualization of the cellular ultrastructure, tilt-series images were acquired using the SerialEM software44 at a pixel size 3.468 Å with a defocus range of −3 to −10 μm, ±60° oscillation, 1° increments with a total dose of ~172 e− Å−2 as well as at a pixel size of 1.33 Å with a defocus range of −4 to −10 μm, ±60° oscillation, 2° increments with a total final dose of ~160 e− Å−2.
Cryo-EM single-particle data
Single-particle cryo-EM data were collected as described previously16,18,23 on the Titan Krios G3 microscope (Thermo Fisher Scientific) operating at 300 kV fitted with a Quantum energy filter (slit width 20 eV) and a K3 direct electron detector (Gatan) with a sampling pixel size of 0.546 Å running in counting super-resolution mode. For the N. maritimus purified sheets sample, a total of 12,557 videos over three sessions was collected with a dose rate of around 3.5 e− per super-resolution pixel per s on the camera level. The sample was subjected to 4.2 s of exposure, during which a total dose of around 48–51 e− Å−2 was applied, and 40 frames were recorded per video (Extended Data Table 2).
Subtomogram averaging of whole cells for structure determination
To obtain initial lattice maps, a previously described strategy was used16, in which tilt-series alignment using gold fiducials and tomogram generation was performed using IMOD45 and initial contrast transfer functions (CTFs) were estimated using CTFFIND446. Tomograms for visualization were generated using the simultaneous iterative reconstruction technique (SIRT) implemented in Tomo3D47 and denoised using Cryo-CARE36,37. Subtomogram averaging was performed using custom scripts written in MATLAB, described in detail elsewhere42,48. For initial cryo-ET structure determination, we used previously published methods17, with the major difference being the use of a recently developed 3D-CTF correction method for tomographic data49. The roughly aligned subtomogram coordinates were then imported into RELION-4 for further analysis19. We used the tilt series after video frame alignment from the initial analysis above, without additional preprocessing, along with the tilt-series alignments performed within IMOD, CTF parameters from CTFFIND446 and the Euler angle assignments and subtomogram coordinates from the original analysis to proceed with refinement. The imported parameters into RELION-4 were used for multiple cycles of pseudosubtomogram generation and realignment as described recently19. Accounting for per-particle motions with additional cycles of pseudosubtomogram improvements and realignments increased the resolution of the NmSLP hexamer to 3.4 Å in C6 symmetry. Relaxation of the symmetry50,51 led to an improved (3.3 Å) resolution overall, and 3.2 Å at the pseudohexameric axis, but decreased the resolution (~4.5 Å) at the periphery of the hexamer (Extended Data Table 1 and Extended Data Fig. 1). For spatial analysis of hexameric and pentameric S-layer positions with respect to the cell centre, the distance of each position from the cell centre was normalized by the maximally distanced hexamer/pentamer in every cell in the tomogram.
Cryo-EM single-particle analysis
For S-layer structure from two-dimensional sheets, cryo-EM data processing was performed as described previously for S-layers in our laboratory18,23. Videos collected at the scope were clustered into optics groups based on the XML metadata of the data-collection software EPU (Thermo Fisher Scientific) using a k-means algorithm implemented in EPU_group_AFIS (https://github.com/DustinMorado/EPU_group_AFIS). Imported videos were motion-corrected, dose-weighted and Fourier cropped (2×) with MotionCor252 implemented in RELION-3.153. CTFs of the resulting motion-corrected micrographs were estimated using CTFFIND446. Initially, side views of S-layer sheets were first manually picked along the edge of the lattice using the helical picking tab in RELION while setting the helical rise to 60 Å. Top and tilted views were manually picked at the central hexameric axis. Manually picked particles were extracted in 4× downsampled 128 × 128 px2 boxes and classified using reference-free 2D classification inside RELION-3.1. Class averages centred at a hexameric axis were used to automatically pick particles inside RELION-3.1. Automatically picked particles were extracted in 4× downsampled 128 × 128 px2 boxes and classified using reference-free 2D classification. Particle coordinates belonging to class averages centred at the hexameric axis were used to train TOPAZ54 in 5× downsampled micrographs using the neural network architecture conv127. For the final reconstruction, particles were picked using TOPAZ and the previously trained neural network above. Furthermore, top, bottom and side views were picked using the reference-based autopicker inside RELION-3.1, which TOPAZ did not readily identify. Particles were extracted in 4× downsampled 128 px × 128 px boxes and classified using reference-free 2D classification inside RELION-3.1. Particles belonging to class averages centred at the pseudohexameric axis were combined, and particles within 30 Å were removed to prevent duplication after alignment. All of the resulting particles were then re-extracted in 4× downsampled 128 × 128 px2 boxes. All of the side views and a subset of the top and bottom views were used for initial model generation in RELION-3.1. The scaled and low-pass filtered output was then used as a starting model for 3D auto refinement in a 512 × 512 px2 box. Per-particle defocus, anisotropy magnification and higher-order aberrations55 were refined inside RELION-3.1, followed by three rounds of focused 3D autorefinement. Bayesian particle polishing was performed subsequently in a 640 px × 640 px box55 followed by autorefinement and symmetry relaxation50,51. The final map was obtained from 354,860 particles and post-processed using a soft mask focused on the central hexamer, yielding a global resolution of 2.7 Å according to the Fourier shell correlation criterion between two independently refined half-maps at a threshold value at 0.143 (ref. 56) and a local resolution of up to 2.5 Å (Extended Data Fig. 3 and Extended Data Table 2). The two-dimensional sheet-like arrangement led to anisotropy in resolution, with lower resolution perpendicular to the plane as estimated by directional FSCs57. Further details are provided in Extended Data Table 2 and Extended Data Fig. 3.
Data visualization, analysis and model building
For model building, a previously described strategy was used18,23. For the single-particle cryo-EM map, the original 640 × 640 × 640 voxel box was cropped into a 320 × 320 × 320 voxel box. In both the cryo-ET and cryo-EM maps, and the protein backbone of NmSLP was manually traced as a poly-alanine model through a single NmSLP subunit using Coot58. Side chains were assigned at clearly identifiable positions which allowed deduction of the protein sequence register. The model was then placed into the hexameric map as six copies and subjected to several rounds of refinement using refmac559 inside the CCP-EM software suite60 and PHENIX61, followed by manually rebuilding in Coot58. At the N terminus, the C2 maps were better resolved compared to the C6 maps at the C termini of NmSLPs; therefore, multimap atomic model refinement was performed in servalcat62. Model validation was performed in PHENIX and CCP-EM, and data visualization was performed in UCSF Chimera63, UCSF ChimeraX64 and PyMOL65. To analyse lattice interfaces, multiple copies of the hexameric structure were placed in the cryo-EM map prepared with a larger box size. Figure panels containing cryo-EM or cryo-ET images were prepared using IMOD and Fiji66. Lattice maps of S-layers for visual inspection were plotted inside UCSF Chimera63 with the PlaceObject plugin67 and model coordinates were plotted inside UCSF ChimeraX64 with the sym function and the BIOMATRIX PDB file header or directly using the ArtiaX plugin68. The SPA and STA maps were postprocessed using deepEMhancer38 for visualization of the N-glycan densities (Fig. 2k and Extended Data Fig. 4g–h,k–l). Composite maps from focused refinements of the two-fold (C2) and six-fold (C6) symmetrized maps were generated using refmac559 and PHENIX61 and then converted using mtz2mrc implemented in PHENIX61.
Bioinformatic analysis
A previously described strategy for detection and analysis of SLPs was used18,23. All sequence similarity searches were performed in the MPI Bioinformatics Toolkit69 using BLAST70 and HHpred71. BLAST searches were performed against the nr_arc database, a specialized subset of the NCBI non-redundant protein sequence database filtered specifically for archaeal sequences, using the default settings to identify homologues of NmSLP in archaea. The searches were seeded with the protein sequence of N. maritimus SLP. The domain organization of several obtained matches and many experimentally characterized SLPs (Supplementary Table 1) were analysed using HHpred searches with the default settings over the PDB70 and ECOD70 databases, which are versions of the PDB and ECOD databases filtered for a maximum pairwise identity of 70%, and using structural models built using AlphaFold (v.2.2.0)72. Signal peptides were predicted using SignalP (v.6.0)73. The mean local charge of the protein sequences was calculated using the EMBOSS charge tool74, using a window length of 7.
ITC analysis
ITC measurements were made using Malvern Panalytical ITC200 instruments at 25 °C in SCM buffer without ammonium chloride. Experiments were performed at a reference power of 10 µcal s−1 and with injections at 300 s intervals to capture the large exothermic heats and broad peak profiles. The ITC cell contained N. maritimus at an optical density at 600 nm (OD600) of 1.0 and the syringe contained 10 mM ammonium chloride in the SCM buffer. In total, ten injections, with the first injection corresponding to 0.5 µl, followed by nine injections of 1 µl were performed, resulting in a final ammonium chloride concentration of 0.475 mM in the ITC cell. N. maritimus cells were pretreated with 0, 2.5 or 5 mM EGTA for 30 min, and were then centrifuged at 16,000g for 15 min and resuspended in SCM medium lacking ammonium chloride to recover before adjusting to an OD600 of 1.0. Control measurements of injections of ammonium chloride into buffer were performed and these heats were small and close to the values seen for buffer into buffer control experiments. This control heat was subtracted from the N. maritimus experiments before peak integration using Malvern Panalytical PEAQ software. Experiments were performed three times with different batches of N. maritimus prepared from cells in log-growth phase in SCM medium with 1 mM ammonium chloride as nutrient source. These cultures were centrifuged and resuspended in SCM buffer lacking ammonium chloride before adjusting to an OD600 of 1 and loading into the ITC cell.
MD simulations
The NmSLP hexamer structure was prepared for atomistic MD simulation using VMD (v.1.94)67. The system was first solvated with TIP3P water molecules and 0.5 M NaCl to mimic the salinity of sea water. Next, 312 ammonium ions (0.1 M NH4+) were randomly distributed throughout the solvent, along with an equal number of chloride counter ions to maintain a charge neutral system. Simulation parameters for NH4+ were derived through analogy with existing CHARMM parameters for methylammonium. Note that, to better help identify specific ion binding sites, no structural ions apparent from the NmSLP cryo-EM and cryo-ET structures were included. The resulting system contained 566,371 atoms, including 136,236 protein atoms, 141,657 water molecules, 2,246 sodium ions, 1,358 chloride ions and 312 ammonium ions, within a hexagonal box of dimensions x = y = 217 Å, z = 150 Å and axial angles α = β = 90°, γ = 60°. The geometry of the simulation box was chosen so that the molecular interfaces observed between neighbouring hexamers in our tomography data would be reproduced through the interactions of the NmSLP hexamer with its periodic images in the x–y plane. The system was then subjected to a series of conjugant gradient energy minimizations followed by three 500 ns MD simulations. To prevent potential distortions in the NmSLP hexamer due to the absence of structural ions offsetting its highly negative charge, protein atoms (excluding hydrogens) were harmonically restrained during simulation. Unless otherwise indicated, analyses were performed after disregarding the first 100 ns of each simulation to ensure equilibrium sampling. To assess the robustness of the observed ammonium-binding pattern, we further constructed hexamer systems containing ammonium at concentrations of 0.05 M (156 NH4+ ions) and 0.2 M (624 NH4+ ions) using an identical procedure, and these systems were subjected to a single 500 ns production simulation. Note that lower concentrations of ammonium ions could not be used due to few total ammonium ions in the box in every simulation. All simulations were conducted using NAMD (v.2.14)68 and the CHARMM36 force field38. Production simulations used the NPT ensemble with conditions maintained at 1 atm and 310 K using the Nosé–Hoover Langevin piston and Langevin thermostat, respectively. The r-RESPA integrator scheme was used with an integration time step of 2 fs and SHAKE constraints applied to all hydrogen atoms. Short-range, non-bonded interactions were calculated every 2 fs with a cut-off of 12 Å; long-range electrostatics were evaluated every 6 fs using the particle-mesh-Ewald method. Further details are provided in Supplementary Table 3.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41586-024-07462-5.
Supplementary information
Supplementary Figs. 1–3 and Supplementary Tables 1–3.
Cryo-electron tomography of N. maritimus and NmSLP S-layer structure. The cryo-ET STA map and structure of the N. maritimus S-layer show how NmSLP monomers form the lattice. Different views of the S-layer are shown with text annotations.
Cryo-electron tomography of N. maritimus. Slices through the tomogram of the same N. maritimus cell shown in Supplementary Video 1 without segmentation to enable direct assessment of the cryo-ET density.
The cryo-EM structure of isolated NmSLP and structural comparison to the cryo-ET structure. The cryo-EM map and structure of the N. maritimus S-layer compared with the in situ STA structure.
Ammonium binding in MD simulations. Residues identified by PyLipID with average ammonium occupancy >50% for three 0.1 M NH4+ NmSLP hexamer simulations. Each residue is coloured by its occupancy value, which was averaged over the six NmSLP monomers, and mapped onto the hexamer structure. Residue numbers and occupancies with s.d. values are given in Supplementary Table 2. This video was produced using the VDM software.
Source data
Source Data Figs. 1 and 3–5 and Source Data Extended Data Figs. 3, 5, 6, 7 and 8
Acknowledgements
This work was supported by the Medical Research Council, as part of UK Research and Innovation (programme MC_UP_1201/31 to T.A.M.B., U105184326 to J.L.). T.A.M.B. thanks the Human Frontier Science Program (grant RGY0074/2021), the Vallee Research Foundation, the European Molecular Biology Organization, the Leverhulme Trust and the Lister Institute for Preventative Medicine for support; V.A. thanks A. Lupas for continued support and the Human Frontier Science Program (grant RGY0074/2021); C.K.C. thanks P. Zhang and M. S. P. Sansom for their support as well as funding through the ERC AdG Program (grant 101021133) and a faculty start-up package from the University of Missouri-Columbia Department of Physics. We thank F. Elling and A. Pearson for the gift of a running N. maritimus cell culture; R. Rachel, S. H. W. Scheres and J. Zivanov for advice; and T. Darling, J. Grimmett, I. Clayson and J. J. E. Caesar for help with high-performance computing. One dataset for cryo-ET was acquired at the cryo-electron microscopy platform of the European Molecular Biology Laboratory (EMBL) in Heidelberg. This work was partly supported by institutional funds of the Max Planck Society; iNEXT, project number 653706, funded by the Horizon 2020 program of the European Union; and the MRC Laboratory of Molecular Biology Electron Microscopy Facility and Central Oxford Structural Molecular Imaging Centre (COSMIC). Simulations were performed on computational resources provided by HECBioSim, the UK High End Computing Consortium for Biomolecular Simulation, which is supported by the EPSRC (EP/L000253/1), as well as by the Research Computing Support Services division at the University of Missouri-Columbia, which is supported in part by the National Science Foundation (grant CNS-14229294). For the purpose of open access, the MRC Laboratory of Molecular Biology has applied a CC BY public copyright license to any Author Accepted Manuscript version arising.
Extended data figures and tables
Author contributions
A.v.K., J.L. and T.A.M.B. designed research. A.v.K, S.v.D., L.L.P., Z.F. and T.A.M.B. performed cryo-EM and cryo-ET experiments. C.K.C. and P.J.S. performed MD simulations. A.v.K. and C.B. performed ITC measurements. V.A. performed bioinformatics analyses. A.v.K., C.K.C., S.v.D., V.A., P.J.S. and T.A.M.B. analysed data. A.v.K. and T.A.M.B. wrote the manuscript with the support of all of the authors.
Peer review
Peer review information
Nature thanks Wojciech Kopec, Henry van den Bedem and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Data availability
Maps have been deposited at the Electron Microscopy Data Bank under accession codes EMDB-16482, EMDB-16483, EMDB-16484, EMDB-16486, EMDB-16487, EMDB-16489 and EMDB-16492. Model coordinates have been deposited at the Protein Data Bank under accession codes 8C8L, 8C8K, 8C8M, 8C8N, 8C8O and 8C8R. Further details are provided in Extended Data Tables 1 and 2. Source data are provided with this paper.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
is available for this paper at 10.1038/s41586-024-07462-5.
Supplementary information
The online version contains supplementary material available at 10.1038/s41586-024-07462-5.
References
- 1.Könneke M, et al. Isolation of an autotrophic ammonia-oxidizing marine archaeon. Nature. 2005;437:543–546. doi: 10.1038/nature03911. [DOI] [PubMed] [Google Scholar]
- 2.Hutchins DA, Capone DG. The marine nitrogen cycle: new developments and global change. Nat. Rev. Microbiol. 2022;20:401–414. doi: 10.1038/s41579-022-00687-z. [DOI] [PubMed] [Google Scholar]
- 3.Karner MB, DeLong EF, Karl DM. Archaeal dominance in the mesopelagic zone of the Pacific Ocean. Nature. 2001;409:507–510. doi: 10.1038/35054051. [DOI] [PubMed] [Google Scholar]
- 4.Könneke M, et al. Ammonia-oxidizing archaea use the most energy-efficient aerobic pathway for CO2 fixation. Proc. Natl Acad. Sci. USA. 2014;111:8239–8244. doi: 10.1073/pnas.1402028111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kraft B, et al. Oxygen and nitrogen production by an ammonia-oxidizing archaeon. Science. 2022;375:97–100. doi: 10.1126/science.abe6733. [DOI] [PubMed] [Google Scholar]
- 6.Wan XS, et al. Ambient nitrate switches the ammonium consumption pathway in the euphotic ocean. Nat. Commun. 2018;9:915. doi: 10.1038/s41467-018-03363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martens-Habbena W, Berube PM, Urakawa H, de la Torre JR, Stahl DA. Ammonia oxidation kinetics determine niche separation of nitrifying Archaea and Bacteria. Nature. 2009;461:976–979. doi: 10.1038/nature08465. [DOI] [PubMed] [Google Scholar]
- 8.Urakawa, H., Martens-Habbena, W. & Stahl, D. A. in Nitrification 115–155 (John Wiley & Sons, 2011).
- 9.Qin W, et al. Nitrosopumilus maritimus gen. nov., sp. nov., Nitrosopumilus cobalaminigenes sp. nov., Nitrosopumilus oxyclinae sp. nov., and Nitrosopumilus ureiphilus sp. nov., four marine ammonia-oxidizing archaea of the phylum Thaumarchaeota. Int. J. Syst. Evol. Microbiol. 2017;67:5067–5079. doi: 10.1099/ijsem.0.002416. [DOI] [PubMed] [Google Scholar]
- 10.Bharat TAM, von Kügelgen A, Alva V. Molecular logic of prokaryotic surface layer structures. Trends Microbiol. 2021;29:405–415. doi: 10.1016/j.tim.2020.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Albers SV, Meyer BH. The archaeal cell envelope. Nat. Rev. Microbiol. 2011;9:414–426. doi: 10.1038/nrmicro2576. [DOI] [PubMed] [Google Scholar]
- 12.Li PN, et al. Nutrient transport suggests an evolutionary basis for charged archaeal surface layer proteins. ISME J. 2018;12:2389–2402. doi: 10.1038/s41396-018-0191-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li PN, Herrmann J, Wakatsuki S, van den Bedem H. Transport properties of nanoporous, chemically forced biological lattices. J. Phys. Chem. B. 2019;123:10331–10342. doi: 10.1021/acs.jpcb.9b05882. [DOI] [PubMed] [Google Scholar]
- 14.Nakagawa T, Stahl DA. Transcriptional response of the archaeal ammonia oxidizer Nitrosopumilus maritimus to low and environmentally relevant ammonia concentrations. Appl. Environ. Microbiol. 2013;79:6911–6916. doi: 10.1128/AEM.02028-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qin W, et al. Stress response of a marine ammonia-oxidizing archaeon informs physiological status of environmental populations. ISME J. 2018;12:508–519. doi: 10.1038/ismej.2017.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.von Kügelgen A, et al. In Situ structure of an intact lipopolysaccharide-bound bacterial surface layer. Cell. 2020;180:348–358. doi: 10.1016/j.cell.2019.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bharat TAM, et al. Structure of the hexagonal surface layer on Caulobacter crescentus cells. Nat. Microbiol. 2017;2:17059. doi: 10.1038/nmicrobiol.2017.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.von Kügelgen A, Alva V, Bharat TAM. Complete atomic structure of a native archaeal cell surface. Cell Rep. 2021;37:110052. doi: 10.1016/j.celrep.2021.110052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zivanov J, et al. A Bayesian approach to single-particle electron cryo-tomography in RELION-4.0. eLife. 2022;11:e83724. doi: 10.7554/eLife.83724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jarrell KF, et al. N-linked glycosylation in Archaea: a structural, functional, and genetic analysis. Microbiol. Mol. Biol. Rev. 2014;78:304–341. doi: 10.1128/MMBR.00052-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herdman M, et al. High-resolution mapping of metal ions reveals principles of surface layer assembly in Caulobacter crescentus cells. Structure. 2022;30:215–228. doi: 10.1016/j.str.2021.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baranova E, et al. SbsB structure and lattice reconstruction unveil Ca2+ triggered S-layer assembly. Nature. 2012;487:119–122. doi: 10.1038/nature11155. [DOI] [PubMed] [Google Scholar]
- 23.von Kügelgen A, et al. Interdigitated immunoglobulin arrays form the hyperstable surface layer of the extremophilic bacterium Deinococcus radiodurans. Proc. Natl Acad. Sci. USA. 2023;120:e2215808120. doi: 10.1073/pnas.2215808120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gambelli L, et al. Structure of the two-component S-layer of the archaeon Sulfolobus acidocaldarius. eLife. 2024;13:e84617. doi: 10.7554/eLife.84617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gambelli L, et al. Architecture and modular assembly of Sulfolobus S-layers revealed by electron cryotomography. Proc. Natl Acad. Sci. USA. 2019;116:25278–25286. doi: 10.1073/pnas.1911262116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fioravanti A, et al. Structure of S-layer protein Sap reveals a mechanism for therapeutic intervention in anthrax. Nat. Microbiol. 2019;4:1805–1814. doi: 10.1038/s41564-019-0499-1. [DOI] [PubMed] [Google Scholar]
- 27.Bharat TAM, Tocheva EI, Alva V. The cell envelope architecture of Deinococcus: HPI forms the S-layer and SlpA tethers the outer membrane to peptidoglycan. Proc. Natl Acad. Sci. USA. 2023;120:e2305338120. doi: 10.1073/pnas.2305338120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim J-G, et al. Spindle-shaped viruses infect marine ammonia-oxidizing thaumarchaea. Proc. Natl Acad. Sci. USA. 2019;116:15645–15650. doi: 10.1073/pnas.1905682116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song W, et al. PyLipID: a Python package for analysis of protein-lipid interactions from molecular dynamics simulations. J. Chem. Theory Comput. 2022;18:1188–1201. doi: 10.1021/acs.jctc.1c00708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xue L, et al. Visualizing translation dynamics at atomic detail inside a bacterial cell. Nature. 2022;610:205–211. doi: 10.1038/s41586-022-05255-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tegunov D, Xue L, Dienemann C, Cramer P, Mahamid J. Multi-particle cryo-EM refinement with M visualizes ribosome-antibiotic complex at 3.5 Å in cells. Nat. Methods. 2021;18:186–193. doi: 10.1038/s41592-020-01054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hollocher TC, Tate ME, Nicholas DJ. Oxidation of ammonia by Nitrosomonas europaea. Definite 18O-tracer evidence that hydroxylamine formation involves a monooxygenase. J. Biol. Chem. 1981;256:10834–10836. doi: 10.1016/S0021-9258(19)68518-2. [DOI] [PubMed] [Google Scholar]
- 33.Hodgskiss LH, et al. Unexpected complexity of the ammonia monooxygenase in archaea. ISME J. 2023;17:588–599. doi: 10.1038/s41396-023-01367-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vajrala N, et al. Hydroxylamine as an intermediate in ammonia oxidation by globally abundant marine archaea. Proc. Natl Acad. Sci. USA. 2013;110:1006–1011. doi: 10.1073/pnas.1214272110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leininger S, et al. Archaea predominate among ammonia-oxidizing prokaryotes in soils. Nature. 2006;442:806–809. doi: 10.1038/nature04983. [DOI] [PubMed] [Google Scholar]
- 36.Buchholz, T.-O. et al. in Methods in Cell Biology Vol. 152 (eds Müller-Reichert, T. & Pigino, G.) 277–289 (Academic, 2019). [DOI] [PubMed]
- 37.Buchholz, T.-O., Jordan, M., Pigino, G. & Jug, F. Cryo-CARE: Content-aware image restoration for cryo-transmission electron microscopy data. In Proc. 2019 IEEE 16th International Symposium on Biomedical Imaging(ISBI 2019) 502–506 (IEEE, 2019).
- 38.Sanchez-Garcia R, et al. DeepEMhancer: a deep learning solution for cryo-EM volume post-processing. Commun. Biol. 2021;4:874. doi: 10.1038/s42003-021-02399-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schouten S, et al. Intact membrane lipids of “Candidatus Nitrosopumilus maritimus,” a cultivated representative of the cosmopolitan mesophilic group I Crenarchaeota. Appl. Environ. Microbiol. 2008;74:2433–2440. doi: 10.1128/AEM.01709-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leavitt, W. et al. Archaeal lipid hydrogen isotopes in a marine thaumarchaeon. Earth Space Sci. Open Arch.10.1002/essoar.10512169.1 (2022).
- 41.von Kügelgen A, van Dorst S, Alva V, Bharat TAM. A multidomain connector links the outer membrane and cell wall in phylogenetically deep-branching bacteria. Proc. Natl Acad. Sci. USA. 2022;119:e2203156119. doi: 10.1073/pnas.2203156119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wan W, et al. Structure and assembly of the Ebola virus nucleocapsid. Nature. 2017;551:394–397. doi: 10.1038/nature24490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hagen WJH, Wan W, Briggs JAG. Implementation of a cryo-electron tomography tilt-scheme optimized for high resolution subtomogram averaging. J. Struct. Biol. 2017;197:191–198. doi: 10.1016/j.jsb.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mastronarde DN. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 2005;152:36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 45.Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- 46.Rohou A, Grigorieff N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 2015;192:216–221. doi: 10.1016/j.jsb.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Agulleiro JI, Fernandez JJ. Tomo3D 2.0—exploitation of advanced vector extensions (AVX) for 3D reconstruction. J. Struct. Biol. 2015;189:147–152. doi: 10.1016/j.jsb.2014.11.009. [DOI] [PubMed] [Google Scholar]
- 48.Bharat TAM, et al. Cryo-electron tomography of Marburg virus particles and their morphogenesis within infected cells. PLoS Biol. 2011;9:e1001196. doi: 10.1371/journal.pbio.1001196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Turoňová B, Schur FKM, Wan W, Briggs JAG. Efficient 3D-CTF correction for cryo-electron tomography using NovaCTF improves subtomogram averaging resolution to 3.4 Å. J. Struct. Biol. 2017;199:187–195. doi: 10.1016/j.jsb.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ilca SL, et al. Multiple liquid crystalline geometries of highly compacted nucleic acid in a dsRNA virus. Nature. 2019;570:252–256. doi: 10.1038/s41586-019-1229-9. [DOI] [PubMed] [Google Scholar]
- 51.Abrishami V, et al. Localized reconstruction in Scipion expedites the analysis of symmetry mismatches in cryo-EM data. Prog. Biophys. Mol. Biol. 2021;160:43–52. doi: 10.1016/j.pbiomolbio.2020.05.004. [DOI] [PubMed] [Google Scholar]
- 52.Zheng SQ, et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods. 2017;14:331–332. doi: 10.1038/nmeth.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zivanov J, et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife. 2018;7:e42166. doi: 10.7554/eLife.42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bepler T, et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods. 2019;16:1153–1160. doi: 10.1038/s41592-019-0575-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zivanov J, Nakane T, Scheres SHW. Estimation of high-order aberrations and anisotropic magnification from cryo-EM data sets in RELION-3.1. IUCrJ. 2020;7:253–267. doi: 10.1107/S2052252520000081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tan YZ, et al. Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Nat. Methods. 2017;14:793–796. doi: 10.1038/nmeth.4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr. D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Murshudov GN, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Burnley T, Palmer CM, Winn M. Recent developments in the CCP-EM software suite. Acta Crystallogr. D. 2017;73:469–477. doi: 10.1107/S2059798317007859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liebschner D, et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D. 2019;75:861–877. doi: 10.1107/S2059798319011471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yamashita K, Palmer CM, Burnley T, Murshudov GN. Cryo-EM single-particle structure refinement and map calculation using Servalcat. Acta Crystallogr. D. 2021;77:1282–1291. doi: 10.1107/S2059798321009475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pettersen EF, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 64.Pettersen EF, et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 2021;30:70–82. doi: 10.1002/pro.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.The PyMOL molecular graphics system v.2.0 (Schrödinger, 2015).
- 66.Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Qu K, et al. Structure and architecture of immature and mature murine leukemia virus capsids. Proc. Natl Acad. Sci. USA. 2018;115:E11751–E11760. doi: 10.1073/pnas.1811580115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ermel UH, Arghittu SM, Frangakis AS. ArtiaX: an electron tomography toolbox for the interactive handling of sub-tomograms in UCSF ChimeraX. Protein Sci. 2022;31:e4472. doi: 10.1002/pro.4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zimmermann L, et al. A completely reimplemented MPI bioinformatics Toolkit with a new HHpred server at its core. J. Mol. Biol. 2018;430:2237–2243. doi: 10.1016/j.jmb.2017.12.007. [DOI] [PubMed] [Google Scholar]
- 70.Camacho C, et al. BLAST+: architecture and applications. BMC Bioinform. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Steinegger M, et al. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinform. 2019;20:473. doi: 10.1186/s12859-019-3019-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jumper J, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Teufel F, et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 2022;40:1023–1025. doi: 10.1038/s41587-021-01156-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 75.Frickey T, Lupas A. CLANS: a Java application for visualizing protein families based on pairwise similarity. Bioinformatics. 2004;20:3702–3704. doi: 10.1093/bioinformatics/bth444. [DOI] [PubMed] [Google Scholar]
- 76.Kozlowski LP. IPC 2.0: prediction of isoelectric point and pKa dissociation constants. Nucleic Acids Res. 2021;49:W285–W292. doi: 10.1093/nar/gkab295. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figs. 1–3 and Supplementary Tables 1–3.
Cryo-electron tomography of N. maritimus and NmSLP S-layer structure. The cryo-ET STA map and structure of the N. maritimus S-layer show how NmSLP monomers form the lattice. Different views of the S-layer are shown with text annotations.
Cryo-electron tomography of N. maritimus. Slices through the tomogram of the same N. maritimus cell shown in Supplementary Video 1 without segmentation to enable direct assessment of the cryo-ET density.
The cryo-EM structure of isolated NmSLP and structural comparison to the cryo-ET structure. The cryo-EM map and structure of the N. maritimus S-layer compared with the in situ STA structure.
Ammonium binding in MD simulations. Residues identified by PyLipID with average ammonium occupancy >50% for three 0.1 M NH4+ NmSLP hexamer simulations. Each residue is coloured by its occupancy value, which was averaged over the six NmSLP monomers, and mapped onto the hexamer structure. Residue numbers and occupancies with s.d. values are given in Supplementary Table 2. This video was produced using the VDM software.
Source Data Figs. 1 and 3–5 and Source Data Extended Data Figs. 3, 5, 6, 7 and 8
Data Availability Statement
Maps have been deposited at the Electron Microscopy Data Bank under accession codes EMDB-16482, EMDB-16483, EMDB-16484, EMDB-16486, EMDB-16487, EMDB-16489 and EMDB-16492. Model coordinates have been deposited at the Protein Data Bank under accession codes 8C8L, 8C8K, 8C8M, 8C8N, 8C8O and 8C8R. Further details are provided in Extended Data Tables 1 and 2. Source data are provided with this paper.