

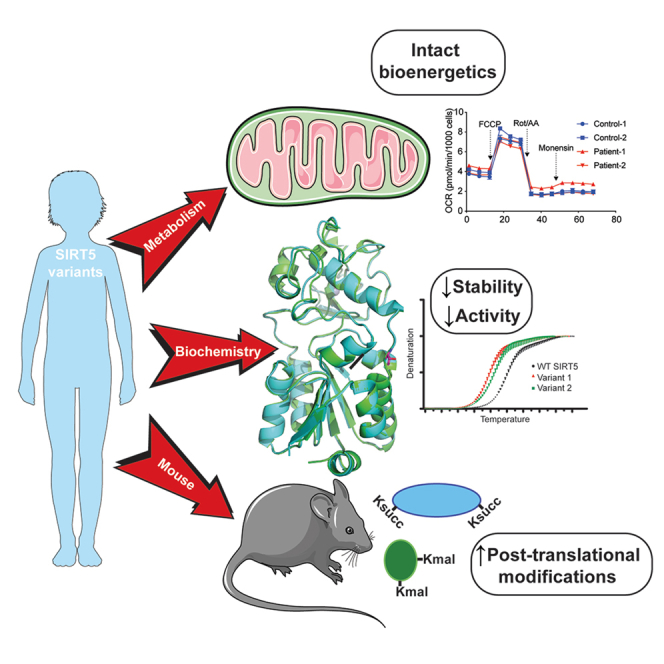
Summary
SIRT5 is a sirtuin deacylase that removes negatively charged lysine modifications, in the mitochondrial matrix and elsewhere in the cell. In benign cells and mouse models, under basal conditions, the phenotypes of SIRT5 deficiency are quite subtle. Here, we identify two homozygous SIRT5 variants in patients suspected to have mitochondrial disease. Both variants, P114T and L128V, are associated with reduced SIRT5 protein stability and impaired biochemical activity, with no evidence of neomorphic or dominant negative properties. The crystal structure of the P114T enzyme was solved and shows only subtle deviations from wild-type. Via CRISPR-Cas9, we generated a mouse model that recapitulates the human P114T mutation; homozygotes show reduced SIRT5 levels and activity, but no obvious metabolic abnormalities, neuropathology, or other gross phenotypes. We conclude that these human SIRT5 variants most likely represent severe hypomorphs, but are likely not by themselves the primary pathogenic cause of the neuropathology observed in the patients.
Subject areas: health sciences, biochemistry, physiology, molecular biology, neuroscience, structural biology
Graphical abstract
Highlights
-
•
Two SIRT5 variants were identified in patients suspected of mitochondrial disease
-
•
Both SIRT5 variants reduce SIRT5 protein level and biochemical activity
-
•
Neither SIRT5 variant affects bioenergetics in human fibroblasts or MEFs
-
•
A P114T mouse model does not recapitulate neuropathology observed in patients
Health sciences; Biochemistry; Physiology; Molecular biology; Neuroscience; Structural biology.
Introduction
Mitochondria are essential for maintaining the health of eukaryotes, through their functions as bioenergetic, biosynthetic, and signaling organelles.1 Consequently, mitochondria are vital for normal tissue function, and mitochondrial defects lead to a myriad of diseases,2,3,4 which vary tremendously in clinical impact, from relatively subtle age-associated defects in single organ systems, to devasting multi-system disorders that are fatal early in life. This latter group includes Leigh Syndrome (LS), characterized by failure to thrive, psychomotor regression, and death in childhood.5 In general, tissues with high energy requirements, such as brain, liver and heart, are more sensitive to mitochondrial defects and display severe clinical phenotypes.6 Mitochondrial disease can be caused by defects in proteins involved in oxidative phosphorylation (OXPHOS) complex assembly, in mitochondrial structure, mtDNA maintenance, and other aspects of mitochondrial function.7,8,9,10
Mammalian sirtuins are a group of seven enzymes that target specific lysine modifications to regulate the biological properties of their protein targets and promote proteome integrity.11 The sirtuin SIRT5 localizes primarily to the mitochondrial matrix, but is also present in the cytosol, peroxisomes, and nucleus. It primarily acts on the negatively charged post-translational modifications (PTMs) succinylation, malonylation, and glutarylation.12 The abundance of global succinylation, malonylation, and glutarylation levels is markedly increased upon SIRT5 depletion in tissues, and most of SIRT5’s target proteins are enzymes involved in various metabolic pathways.13,14,15,16,17,18 SIRT5 remains a somewhat enigmatic protein. SIRT5 plays major roles in specific cancer types, either as an oncogene or a tumor suppressor, including melanoma,19 pancreatic cancer,20 breast cancer,21 and AML,22 in a context-specific manner. By contrast, in mice, under basal unstressed conditions, the effects of SIRT5 deficiency tend to be quite subtle.23 SIRT5-deficient mice develop cardiac fibrosis and mildly impaired heart contractile function with age,17 and, under specific dietary conditions, hyperammonemia.24 Whether SIRT5 might play other important, as-yet undiscovered roles in whole animals remains unclear.
Identification of patients with diseases associated with defects in specific genes and pathways has frequently provided new, unexpected insights into gene function and human biology. A number of polymorphisms and mutations have been identified in sirtuin genes in humans, some of which are associated with clinical phenotypes.25,26,27,28 Prior work has linked SIRT5 polymorphisms in humans to the rate of brain aging29 and overall longevity,30 coronary artery disease and acute myocardial infarction,31 and the risk of developing gastric cancer.32 In this study, we describe two newly identified SIRT5 variants in patients with mitochondrial disease-like pathology. We show that each of the two SIRT5 variants results in reduced SIRT5 protein levels and impaired NAD+-dependent activity, driving an increase in global protein succinylation levels. We show that one of these variants, P114T, has minimal effects on the structure of SIRT5. Via CRISPR-Cas9, we generate mice with the P114T variant, and find that they have severely reduced SIRT5 protein levels and activity. However, P114T homozygous mice are grossly normal, and cells with this mutation are bioenergetically indistinguishable from wild-type controls. Moreover, the SIRT5 P114T variant was also present in a sibling who did not develop neuropathologic complications. The possible implications for SIRT5 and its role in mitochondrial disease are discussed.
Results
Whole exome sequencing identifies SIRT5 variants in two patients with mitochondrial-like disease pathology
Whole exome sequencing of DNA from two unrelated patients presenting with severe mitochondrial-like pathology resulted in the identification of two predicted-pathogenic SIRT5 variants. These patients did not have common copy number genetic variants according to CGH microarray analysis or major chromosomal aberrations associated with other mitochondrial genetic variants. In the first patient, we identified a SIRT5 homozygous missense variant (c. 340C>A, NM_012241.3) that led to a proline114 to threonine (P114T) change in the SIRT5 protein. This variant was deemed the most likely candidate for follow-up, since SIRT5 localizes to the mitochondria and regulates mitochondrial functions. Two other variants (SEMA6A and FRMPD4) were also classified as being “likely to be pathogenic” but their protein functions did not align well with the patient pathology, which was also true for two other disease-associated variants (MYH7 and KIF20A), which are associated with cardiomyopathy, but which was not observed in the patient (Table S1). In the second patient, a distinct homozygous SIRT5 missense variant was identified (c.382C>G, NM_012241.3) resulting in a leucine 128 to valine change (L128V).
Clinically, the first patient (a 7-year-old boy) presented with a profound global developmental delay, seizures, spastic quadriparesis, optic atrophy, blindness, and deafness (Figure 1A). An MRI of the brain, performed at 2 years of age, showed global cerebral and cerebellar atrophy with a marked reduction of white matter volume, as well as a delayed myelination pattern (Figure 1B, left panel). Besides these severe physical abnormalities, echocardiography indicated normal cardiac function, and plasma and urine metabolite levels were largely non-remarkable. The patient was born to first cousin parents. He has an older healthy sister, and two sisters born after him who died in early infancy at the age of 3 months with progressive hypoalbuminemia and nephrotic range proteinuria, a phenotype distinct from patient 1. Neither sister had visual tracking nor responded to sounds right from the beginning. One of the sisters was heterozygous for the SIRT5 variant, and her whole exome sequencing (WES) was negative. There was no DNA available from the second sister. The patient has a seven-month-old brother who is homozygous for the SIRT5 c.340C>A (p.P114T) variant. Thus far, this child has appropriate development and an unremarkable neurological examination, sitting unsupported, grasping, and transferring objects, and babbling.
Figure 1.

Two SIRT5 homozygous variants identified in patients with mitochondrial-like disease
(A) Clinical symptoms of the two patients.
(B) MRI images of brains of the patient 1 (at the age of 2 years) and patient 2 (at the age of 25 years).
(C) Schematic representation of the human SIRT5 protein structure. The catalytic core consists of Rossmann-fold (green box) and zinc-binding domains (cyan box), flanked by a mitochondrial target sequence (MTS, orange box) and the C-terminal region (yellow box). Three sites known important for catalytic activity, Y102, R105, and H158 are indicated. Two altered residues P114 (patient 1), and L128 (patient 2) are represented as star symbols.
(D and E) Evolutionary conservation of two mutated sites SIRT5 P114 and L128 across (D) different species, and across (E) human sirtuin family. Substrate specificity-defining residues Y102 and R105 are indicated in blue; P114 and L128 are highlighted in dark red and pink, respectively.
(F) Spatial position of the altered amino acids P114 and L128 in human SIRT5. The structure was generated based on the crystal structure of human SIRT5 (PBD: 3RIY). Affected residues P114 and L128 are indicated in red. The zinc-binding domain and Rossmann-fold domain are in blue and green, respectively. The Zinc atom is included as a yellow sphere.
The second patient (a 30-year-old female) presented clinically with neuroregression, dysarthria, and dystonia (Figure 1A). At the age of 22 years, patient 2 began to manifest ataxia and proptosis, and shortly thereafter developed tremors, that gradually resulted in loss of motor skills until she became bedridden. An MRI of the brain at age of 25 years showed bilateral symmetrical signal abnormalities in the basal ganglia, as well as in the posterior limb of the internal capsule and brain stem (Figure 1B, right panel). Similar to patient 1, these severe clinical manifestations were not associated with plasma or urine biochemical abnormalities. This patient also manifested a borderline complex IV deficiency (229 mU/UCS versus control range 228–1032 mU/UCS) in a skeletal muscle biopsy (Table S2, lower panel).
To gain insights into the impact of the two missense SIRT5 variants identified in the patients, we first investigated the characteristics of the two involved residues, P114 and L128. Both P114 and L128 are located in the catalytic core domain of SIRT5 (Figure 1C). Moreover, these two residues are highly evolutionarily conserved (Figure 1D), as well as within the human sirtuin family (Figure 1E). In addition, both P114 and L128 localize close to the well-established active site residues Y102 and R105 (Figure 1D), which are critical for substrate binding.33 Structurally, P114 lies in the flexible loop that connects the helix of the zinc-binding domain and helix α6 of the NAD+-binding Rossmann fold domain, and L128 is positioned in α6 helix of the NAD+-binding Rossmann domain (Figure 1F). The evolutionary conservation as well as nearly coinciding positions of these two altered residues, P114 and L128, suggest their potential importance for SIRT5 function in vivo. Indeed, both variants P114T and L128V are predicted to be pathogenic according to PolyPhen (http://genetics.bwh.harvard.edu/pph2/). Each of these variants has been detected at a very low frequency in the heterozygous state in the general population (10/280,762 alleles for P114T, and 5/250,928 alleles for L128V in the Genome Aggregation Database), but neither has ever been reported in the homozygous state (https://gnomad.broadinstitute.org/).
SIRT5 variants are associated with reduced SIRT5 protein levels and increased global succinylation in human fibroblasts
To understand the consequences of the SIRT5 P114T (patient 1) and L128V (patient 2) variants on SIRT5 protein and function, we generated primary skin fibroblasts from both patients and compared them to fibroblasts from healthy controls. SIRT5 protein levels in whole cell lysates were markedly reduced in patients 1 and 2 as compared to controls (Figure 2A), despite normal SIRT5 mRNA expression levels (Figure 2B). To rule out the possibility that lower SIRT5 protein levels detected in patients’ fibroblasts might be artifactual, due to reduced antibody affinity driven by amino acid substitution, we confirmed the reduction of SIRT5 protein level using two different SIRT5 antibodies generated against distinct antigens (Figure S1A). In contrast to the reduced SIRT5 levels, protein levels of SIRT3 and SIRT4 remained unchanged (Figure 2A). Moreover, complex V (ATP5A subunit) protein levels were not different between patients and controls, suggesting that total mitochondrial content was not affected in the SIRT5 variant cell lines (Figure 2A).
Figure 2.

Each of the two SIRT5 variants leads to reduced SIRT5 protein level and increased global succinylation in human fibroblasts
(A) Immunoblots for SIRT5, SIRT3, and SIRT4 in whole cell lysates from control and patient fibroblasts. ATP5A and beta-actin served as loading control.
(B) SIRT5 mRNA normalized expression level in control and patient fibroblasts. Three reference genes were used for expression normalization. n = 3 independent experiments and duplicates in each experiment. Data represent mean ± SD.
(C) Immunoblots for succinyllysine and loading control beta-actin in control and patient fibroblasts.
(D) Representative immunofluorescence staining images of control and patient fibroblasts. Cells were stained with succinyllysine antibody and with Mitotracker orange. Control fibroblasts exposed to 4 mM cell-permeable dimethyl succinate-ester was used as positive control for succinyllysine staining.
(E) Quantification of immunofluorescence staining for succinyllysine in control and patients’ fibroblasts. N = 46 images were analyzed for each cell line. Data represent mean ± SD. Statistical analyses in (B) and (E) were performed using one-way ANOVA, followed by Bonferroni’s multiple comparison test. Each patient fibroblast was tested against average of controls. ∗∗ indicates p < 0.01. ∗∗∗ indicates p < 0.001. ns, not significant.
Next, we asked whether SIRT5 biochemical activity in cells was affected by the SIRT5 gene variants. Because SIRT5 possesses robust desuccinylase, demalonylase, and deglutarylase activities,33,34 we first analyzed levels of these three SIRT5-related acylation marks by immunoblot. Succinyllysine levels were increased in both patient fibroblasts as compared to controls (Figure 2C), whereas no clear difference in malonyllysine and glutaryllysine levels were detected (Figures S1B and S1C). Upon exposure to dimethyl succinate-ester, a cell permeable succinate analogue, we observed an increase in succinylation levels in both control and patient fibroblasts (Figure S1D), indicating that the succinylation machinery in the cells was not affected, and increased succinylation levels in the patient cells resulted from SIRT5 dysfunction.
We tested whether the localization of succinyllysine was altered in cells bearing the SIRT5 variants. Localization of intracellular succinyllysine was visualized using immunofluorescence staining. Exposure of fibroblasts to dimethyl succinate-ester confirmed that protein succinylation was most apparent in mitochondria (Figure 2D). Consistent with immunoblot results, significantly greater succinyllysine fluorescence signal was detected in both patient fibroblasts in comparison with succinlyllysine fluorescence in controls (Figures 2D and 2E). Intracellular localization of succinyllysine did not appear to be altered. Overlayed images of succinyllysine and MitoTracker demonstrated that succinylation was present in both mitochondria and cytosol, especially in patient cells. Together, these results show that both SIRT5 gene variants markedly reduced SIRT5 protein levels and significantly increased global succinyllysine levels in patient cells.
SIRT5 variants result in loss of SIRT5 thermal stability and desuccinylase activity
To gain additional insight into the mechanism of how the SIRT5 variants result in reduced SIRT5 protein levels and higher succinylation levels, we biochemically characterized the variants in vitro. The patient SIRT5 variants, P114T and L128V, were engineered using site-directed mutagenesis (Figure 3A) and these His-tagged SIRT5 recombinant proteins were purified over Ni2+-affinity resin purity (Figure S2A).
Figure 3.

SIRT5 P114T and SIRT5 L128V variants decrease protein thermal stability and NAD+-dependent desuccinylase activity
(A) Sanger sequencing confirmed correct amino acid substitution in recombinant SIRT5; SIRT5 c.340C>A (p.P114T) represents SIRT5 variant identified in patient 1 and c.382C>G (p. L128V) represents SIRT5 variant identified in patient 2.
(B) Thermal shift assay of recombinant SIRT5 WT and two variants. Triplicates for each sample. Representative for 3 independent experiments.
(C) Steady-state kinetics of desuccinylation was determined by varying succinyl-peptide (2–200 μM) in the presence of 1 mM NAD+ and 0.03 μg of recombinant SIRT5 protein. Duplicates for each data point.
(D) Steady-state kinetics of desuccinylation were measured by varying NAD+ (20–640 μM) in the presence of 120 μM succinyl-peptide and 0.03 μg recombinant SIRT5 protein. Duplicates for each data point.
(E) IC50 values of nicotinamide (NAM) for NAD+-dependent desuccinylase activity were assessed for recombinant WT and two SIRT5 variants. Triplicates for each data point.
(F) Steady-state desuccinylase and deacetylase activities were measured in recombinant WT and the two SIRT5 variants, using 10 μM acyl-peptide and 500 μM NAD+. Triplicates for desuccinylation and duplicates for deacetylation activity. Representative for 2 independent experiments. Data represent mean ± SD. ∗∗∗∗ indicates p < 0.0001.
We initially assessed whether the protein stability of the variants was affected. Thermal stability analysis demonstrated that both the P114T and L128V SIRT5 variants exhibited a lower denaturation temperature than WT SIRT5 (Figure 3B), implying impaired thermostability, which could contribute to the reduced steady-state SIRT5 variant protein levels at physiological temperature. Next, we investigated whether the enzymatic kinetics of the SIRT5 variants were altered. Since succinylation levels, but not glutarylation and malonylation levels, were increased in patient fibroblasts, we focused on SIRT5’s desuccinylase activity. The Michaelis-Menten constant (Km) of the desuccinylation of a fluor de lys succinyl-substrate by SIRT5 P114T was 74% higher, and catalytic efficiency (Kcat/Km) was 42% lower, compared to WT SIRT5. For the SIRT5 L128V, the Km for the succinyl-substrate was 30% higher, and catalytic efficiency was 20% lower compared to WT (Figure 3C). Additionally, the Km for NAD+ was 53% higher for SIRT5 P114T, and the catalytic efficiency was 35% lower compared to WT. No evident difference in catalytic efficiency with regards to NAD+ was observed between L128V and WT (Figure 3D). Also, no difference was found in the IC50 value for the general sirtuin inhibitor, nicotinamide (NAM), for the P114T and L128V variants as compared to WT recombinant SIRT5 (Figure 3E).
Since SIRT5 desuccinylase kinetics were only mildly decreased in the recombinant SIRT5 variants, we analyzed whether desuccinylase activity was affected to a greater extent under lower substrate conditions, which could be physiologically relevant, because locally in the mitochondria NAD+ levels can fluctuate dramatically in response to altered metabolic conditions. Under these substrate limiting conditions, with 10 μM of succinyl-substrate and a NAD+ concentration of 500 μM, desuccinylase activity was reduced by 30% in P114T and in L128V SIRT5 variant relative to WT (Figure 3F). All SIRT5 alleles had neglectable deacetylation activity (Figure 3F). In the context of limiting NAD+ to 10 μM, the desuccinylase activity of each of the SIRT5 patient variants was decreased further, to 50% of the activity of WT (Figure S2B). Together, these results indicate that the two SIRT5 variants exhibit reduced SIRT5 protein thermal stability as well as NAD+-dependent desuccinylase activity, likely contributing to the significant decrease in SIRT5 protein levels and increase in succinylation levels observed in the patient fibroblasts.
These studies were repeated independently in another laboratory using a different preparation of WT and SIRT5 P114T protein (Figures S2C and S2D), using peptide substrates whose sequence was derived from Succinate Dehydrogenase (SDHA), a SIRT5 substrate that we and others previously identified.13,35,36 Again, the SIRT5 P114T enzyme showed markedly reduced in vitro biochemical activity, in this case against succinylated, malonylated, and glutarylated peptide substrates compared to WT control (Figure S2E). To test whether P114T might exert dominant negative effects, we combined WT and P114T enzymes together in an equimolar ratio. There was no apparent inhibition of WT SIRT5 by the P114T variant, suggesting that P114T did not inhibit the biochemical activity of the WT enzyme (Figure S2F). Consistent with our other results, this preparation of SIRT5 P114T, but not a catalytic H158Y mutant,13 demonstrated reduced thermal stability compared to WT protein (Figure S2G).
The SIRT5 proline114 substitution to threonine mutation is not associated with gross changes in 3-dimensional structure
To further gain insight into the impaired biochemical activity of the SIRT5 P114T variant, we solved the crystal structures of human P114T and WT SIRT5 enzymes in parallel. Here, we report the first crystal structure of human SIRT5 without a bound co-factor or substrate mimetic in the active site. The structure of WT SIRT5 was solved to 2.25 Å resolution with two protein molecules in the asymmetric unit. The apo structure of P114T was solved to 2.7 Å resolution with two molecules in the asymmetric unit. Although the apo P114T protein has a significantly lower melting temperature than the apo WT SIRT5 protein ( Tm = −5.71 to −6.05°C), their structures are nearly identical aligning with an overall RMSD of 0.518 Å as calculated by SMS in Coot37 (Figure 4A). The cause for the lower melting temperature of the variant structure may be attributed to the fact that a proline, which is a hydrophobic residue having a very rigid structure, was mutated to a more flexible, hydrophilic threonine residue. In the WT structure, Pro114 forms ten hydrophobic interactions with the sidechains of Lys148, Ala149, His118, Leu145 and Glu113 and its carbonyl oxygen forms a hydrogen bond with Arg119 (Figures 4B and S3A). Thr114 forms only five hydrophobic interactions with the sidechains of Arg119, His118, Ala149. Although Thr114 loses the hydrogen bond between its carbonyl oxygen and the side chain of Arg119, it gains a hydrogen bond between its hydroxyl group and the carbonyl oxygen of Leu145 (Figures 4C and S3B). The formation of this new hydrogen bond could interfere with hydrogen bond between the backbone amides of Leu145 and Ala149 in the WT structure. The effects of crystal packing appear to be negligible on the positioning of residue 114 in either the WT or P114T structure (Figure S4). There are no direct symmetry contacts on the loop containing residues 114. The closest symmetry contacts involve a loop containing residues 148–150. The contact of Pro114 appears to add order to the sidechain of Lys148, where the presence of the OG1 atom of P114T causes the destabilization of the Lys148 sidechain. We conclude that the SIRT5 P114T variant shows impaired biochemical activity and thermal stability, but no dramatic change in 3-dimensional structure as assessed by crystallography.
Figure 4.

Structural alignment of the apo structures of SIRT5 WT (green) and P114T (cyan)
(A) Thr114 is shown as sticks with carbons colored magenta.
(B) Pro114 and the residues that interact with it are shown as sticks.
(C) Thr114 and the residues that interact with it are shown as sticks. Hydrogen bonds are depicted as dashed lines.
Proline114 substitution to threonine mice show reduced SIRT5 levels and elevated levels of SIRT5’s target post-translational modifications
Since the biochemical activity and thermal stability of the SIRT5 P114T variant were impaired, but the homozygosity of the P114T SIRT5 variant was also observed in an apparently healthy sibling of patient 1, we aimed to gain a more detailed understanding of the impact of the P114T SIRT5 variant in mice. Although various whole-body and floxed Sirt5 mouse mutants have been described, it remained a possibility that the P114T change might exert unexpected gain-of-function or dominant negative effects in vivo. To model the effects of P114T homozygosity in mice, we used CRISPR-Cas9 technology to generate mice with the P114T variant (Figures 5A, S5A, and S5B). We generated three distinct lines based on three mosaic founders, and confirmed the identity of the correct mutation using Sanger sequencing on genomic DNA derived from tail snips (Figure 5B). Heterozygous and homozygous P114T animals were born at the expected Mendelian ratios and were grossly unremarkable (Figures 5C and S5C). Overall body weight, as well as heart and brain weight, were indistinguishable between WT controls and P114T homozygous adults (Figure 5D).
Figure 5.

P114T variant leads to reduced SIRT5 protein levels and increased global lysine succinylation and lysine malonylation levels in mouse tissues
(A) Mouse Sirt5 exon 4 showing guide RNA target site for CRISPR/Cas9 to introduce the c.340C>A (p.P114T) change.
(B) Representative Sanger sequencing peaks of genomic DNA derived from tail snips of SIRT5 WT, P114T heterozygous and P114T homozygous mice.
(C) P114T pups were born at the expected Mendelian ratios (ns, Chi-squared test).
(D) WT and P114T homozygous mice show no difference in body weight, heart weight and brain weight. Data represent mean ± SD, ns indicates not significant.
(E–G) Immunoblots showing reduced levels of SIRT5 protein and increased Ksucc and Kmal in P114T heterozygous and P114T homozygous tissues (heart, liver, and brain) compared to WT controls.
Given the impaired thermal stability of the P114T variant, we assessed SIRT5 protein levels in tissues of mice with this mutation. SIRT5 protein levels were markedly reduced in P114T heterozygotes, and virtually undetectable in homozygotes (Figures 5E–5G). We interrogated levels of SIRT5 target PTMs in tissues from P114T heterozygotes and homozygotes. Consistent with prior findings in Sirt5 null animals, Ksucc and Kmal levels were markedly elevated in P114T variant hearts, liver, and brain (Figures 5E–5G). Kglu levels appeared to be potentially elevated as well, but the relatively poor quality of the antibody reagent available made it difficult to render firm conclusions regarding changes in levels of this mark (Figures S5D and S5E).
Similar to the results from tissues, SIRT5 protein levels were markedly reduced in P114T heterozygous mouse embryonic fibroblasts (MEFs), and virtually undetectable in homozygous MEFs (Figure S6A). To assess whether variant SIRT5 might be lost via proteasomal-mediated degradation, we incubated WT, P114T heterozygous, or variant cell lines in the presence or absence of the proteasomal inhibitor MG132 (Figure S6B). As a control, we also probed for levels of MYC, a well-known proteasomal target.38 We were unable to restore cellular SIRT5 levels in P114T cells with MG132, although the induction of MYC protein by MG132 was robust in all three genotypes (Figure S6B). To further investigate the impact of the P114T mutation on SIRT5 levels, we measured Sirt5 mRNA levels in MEF lines of all three genotypes. Unexpectedly, and discrepant with our results from patient cells, we found that Sirt5 mRNA levels were reduced by roughly half in P114T heterozygotes, and further decreased in homozygous cells (Figure S6C). The molecular basis for this reduction in Sirt5 mRNA levels in the P114T variant is unclear. We explore possible implications of this finding in the discussion section.
Proline114 substitution to threonine variant fibroblasts are bioenergetically unremarkable
SIRT5 has been shown to play roles in OXPHOS, mitochondrial intermediary metabolism, ROS detoxification, and glycolysis.12 Since biochemical analyses of OXPHOS complexes (complex I, II, III, IV and V) activities were normal in the two patients’ fibroblasts and in the muscle biopsy from patient 2 (Table S2), we reasoned that a more comprehensive assessment with various metabolic challenges would allow us to gain a better understanding of the consequence of SIRT5 variants in bioenergetics. For this, we exposed fibroblasts to diverse metabolic stress tests and quantified their bioenergetic characteristics using the Seahorse Analyzer. In these stress tests, we forced cells to strongly rely on either the glycolytic pathway, by applying CoCl2, as a HIF1A mediated inducer of glycolysis, or on oxidative phosphorylation, by applying galactose. In normal glucose media, no changes in basal and maximal respiration were observed in the SIRT5 variants’ fibroblasts (Figure 6A). The ATP-coupled oxygen consumption rate, as indicated by mitoATP production rate, and proton leak were comparable between control and patients’ fibroblasts (Figure 6B). The P114T but not L128V variant fibroblasts appeared more somewhat glycolytic in glucose media (Figure 6C) as well as glycolysis-stimulated conditions (Figure 6D). This is indicated by the higher glycolytic proton efflux rate (Figures 6C and 6D) and lower ATP rate index (Figure 6C). The ATP rate index reflects the contribution of mitochondrial OXPHOS to total ATP production rate (sum of mitochondrial and glycolytic ATP generation). As altered glycolysis was observed in only one of the patients’ fibroblasts, we asked whether this represented a consistent phenotype or biological variation. To test this, we challenged the OXPHOS system of the cells using galactose media. We hypothesized that if P114T variant induced a more glycolytic state in fibroblasts, a significant OXPHOS impairment would be expected under galactose conditions. Both variant fibroblasts responded with a similar pattern in galactose (Figures 6E–6G). However, we found no significant difference in several important aspects of bioenergetics, although proton leak rates were 2-fold higher in galactose-exposed SIRT5 variant fibroblasts (Figure 6F). Basal respiration was comparable between control and variant fibroblasts, and there was no difference in the maximal respiration (Figure 6E). Oxygen consumption rate coupled to ATP production, as shown by the mitoATP production rate, was also not altered in the SIRT5 variant cells (Figure 6F). The ATP rate index was not different between control and SIRT5 variants’ fibroblasts in galactose media (Figure 6G). Taken together, our data shows no overt adverse effects in the bioenergetic capacity in each of the variant fibroblasts, even though there was a large reduction in SIRT5 protein levels.
Figure 6.

SIRT5 variants have no apparent impact on the bioenergetics of human fibroblasts or mouse embryonic fibroblasts (MEFs)
(A–C) Seahorse metabolic flux parameters of human fibroblasts in glucose medium (measured at 72 h; For parameter OCRbasal, and PERglycol, basal, n = 14–15 technical replicates; n = 6–8 technical replicates for the other parameters in each fibroblast cell line. Data represent mean ± SEM.
(D) Seahorse metabolic flux parameters of human fibroblasts in glycolysis-challenging conditions (glucose medium supplemented with 100 μM CoCl2 for 6 h; n = 6–8 technical replicates for each fibroblasts cell line, Data represent mean ± SEM). PERglyco, glycolytic proton efflux rate.
(E–G) Seahorse metabolic flux parameters of human fibroblasts in OXPHOS-challenging conditions (5.5 mM galactose medium, measured at 72 h; n = 3 independent experiments, 6–8 technical replicates in each independent experiment). Data represent mean ± SEM, ∗ indicates p < 0.05, ns indicates not significant. Two-tailed t-test, average of three control fibroblasts was tested against average of two SIRT5 variant patients’ cell lines.
(H–K) Cellular bioenergetic phenotype of P114T MEFs is indistinguishable from WT. P114T MEFs maintain glycolytic, mitochondrial, and total ATP production rates (I), glycolytic function (J), and mitochondrial respiration (K) compared with littermate WT MEFs. Mitochondrial respiration, glycolytic stress tests, and ATP production rates were measured using a Seahorse XFe96 Analyzer. All rates are normalized to total protein content per sample. Each point represents an individual biological sample (average of n = 6). Data represent mean ± SEM, ns indicates not significant. Significance calculated by t-test. OCR, oxygen consumption rate; ECAR, extracellular acidification rate.
(L) H2DCFDA oxidation rate of human fibroblasts cultured in either glucose (5.5 mM) or galactose (5.5 mM) for 72 h. N = 4 independent experiments, 3–6 technical replicates in each experiment. Data represent mean ± SEM, ns indicates not significant. Two-tailed t-test, average of three control fibroblasts was tested against average of two SIRT5 variant patients’ cell lines.
(M) Flow cytometric analysis of H2DCFDA stained MEFs reveals no difference in ROS levels between WT and P114T MEFs. Each point represents an individual biological sample (average of n = 3). Data represent mean ± SEM, ns indicates not significant. Significance calculated by t-test.
Consistent with these findings, we found no apparent changes in bioenergetics in P114T MEFs. We subjected P114T MEFs and controls to detailed metabolic analysis using the Agilent Seahorse Analyzer. First, we performed the XF cell energy phenotypic test to measure the oxygen consumption rate (OCR), a measure of mitochondrial respiration, and the extracellular acidification rate (ECAR), a measure of glycolysis, under baseline and stressed conditions (Figure 6H). Relative to WT littermate controls, SIRT5 P114T MEFs did not show any changes in OCR and ECAR under basal conditions (baseline, in the presence of nonlimiting substrate quantity) or under induced energy demanding conditions (stressed, in the presence of stressor compounds). Furthermore, both WT and P114T MEFs exhibit similar metabolic potential (calculated as a percentage increase of stressed OCR over baseline OCR, and stressed ECAR over baseline ECAR), a measure of cellular ability to meet an energy demand via respiration and glycolysis. Likewise, both glycolytic and mitochondrial ATP production rates were unaffected by the P114T mutation (Figure 6I). We also performed the glycolysis stress test (Figure 6J), and the mitochondrial stress test (in the presence of either glucose or galactose) (Figures 6K and S6D) to measure other parameters associated with glycolysis or mitochondrial respiration. No significant differences were observed between WT and P114T MEFs. We conclude that the P114T mutation does not confer any striking respiratory or glycolytic phenotypes in MEFs under the conditions tested.
Given that proton leak was consistently increased in both SIRT5 variants’ fibroblasts, we asked whether this observation is biologically relevant. Elevated proton leak has been proposed to be a result of oxidative stress,39 we then analyzed the ROS levels in human fibroblasts using 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA). H2DCFDA is non-fluorescent under basal conditions but is oxidized by intracellular ROS to its fluorescent analogue (DCF). H2DCF-oxidation rates were similar in patients’ and control cells, both under normal glucose and galactose culture conditions (Figure 6L). Similar observations were made in P114T MEFs (Figure 6M). Therefore, the SIRT5 variants do not affect the ROS levels in the conditions tested, either in mouse or human fibroblasts.
The proline114 substitution to threonine mutation does not affect brain anatomy in mice
Owing to the neuropathology identified in the patients with biallelic SIRT5 variants, we performed detailed neuroanatomic analysis on the P114T mice. Midline sagittal brain sections of littermate WT and P114T homozygous mice at 21–50 weeks revealed no abnormalities. Brain weights were indistinguishable between WT controls and P114T homozygotes (Figure 5D). No evidence of neuronal loss in hippocampus, cerebellum, or cortex was detected by immunofluorescence staining for NeuN (Figures S7A–S7F). Similarly, no evidence of astrocytic or microglial activation was detected by staining for GFAP or IBA1, respectively (Figures 7A–7K and S7A–S7F).
Figure 7.

P114T mice do not show evidence of gliosis
(A–K) Brains from littermate WT and P114T mice were stained for GFAP (A–E and Figures S7A–S7C) and IBA1 (F–K and Figures S7D–S7F) and co-stained for NeuN. Images were taken from the hippocampus (Figures S7A and S7D), cerebellum (Figures S7B and S7E), and cortex (Figures S7C and S7F). The number of cells staining positive for GFAP and IBA1 and the intensity of GFAP (A–E) and IBA1 (F–K) staining were quantified for each image. Data are mean ± SEM. N = 4 for all groups, ns indicates not significant. Student’s paired t-test (df = 6, t = (A) 0.333, 0.72; (B) 0.03; (C) 0.21, 0.34; (D) 1.47, 1.21; (E) 0.28, 2.32; (F) 0.35, 1.09.
Discussion
In the present study, we describe two homozygous SIRT5 variants in two unrelated patients with mitochondrial disease-like pathologies. Comprehensive biochemical and molecular analyses revealed that each of the two SIRT5 loss-of-function variants led to a reduction in SIRT5 thermostability and protein levels, along with reduced biochemical activity, coupled with significantly higher succinylation levels in patient-derived fibroblasts. We solved the structure of one of these variants, P114T, and identified only minimal changes from the WT structure. P114T homozygous cells are bioenergetically intact, and mice with this change show no marked abnormalities, including in the CNS. Notably, the absence of a strong phenotype in P114T mice is highly reminiscent of the effects of germline SIRT5 knockout (KO) mice under basal conditions.12 Given the constellation of findings presented here, we believe that these SIRT5 variant alleles most likely represent severe hypomorphs, rather than dominant negatives or gain-of-function mutations in both human and mice.
One interesting finding of these studies is that Sirt5 mRNA levels are severely reduced in mouse cells bearing the P114T mutation, but not in humans. Currently, we do not know why this is the case. SIRT5 mRNA expression is positively regulated by PGC-1⍺, and inhibited by AMPK.40 It is possible that SIRT5 might regulate one or both of these factors, in an auto-regulatory loop, thereby effectively modulating its own expression, in a species-specific fashion. In this regard, multiple reports indicate that deficiency of SIRT3, the primary mitochondrial deacetylase, affects AMPK and PGC-1⍺ levels and activity.41 Whatever the underlying mechanism may be, it is likely that reduced steady-state Sirt5 mRNA levels in the P114T variant contribute to the markedly reduced SIRT5 protein levels in this background in the mouse, in addition to the inherent reduced stability of the P114T protein.
In summary, in this study, we have identified variants in SIRT5 associated with severe mitochondrial disease, but are likely not pathogenic. These variants are associated with reduced SIRT5 levels and activity, albeit little change to SIRT5’s gross 3-dimensional structure, at least for P114T. Mice with this alteration show the accumulation of succinylated and malonylated proteins highly reminiscent of Sirt5 knockout animals. These mice lack the severe phenotypes found in the two human patients. Taken together, our cellular and mouse physiologic data on the two new SIRT5 alleles indicate that these are likely not pathogenic variants, at least on their own. However, SIRT5, like many other sirtuins, might function as a modifier protein that, in combination with other genetic or environmental factors or stressors, could influence physiology.12 Future studies might uncover more subtle phenotypes in the P114T mice similar to those observed in the Sirt5 knockout strain – for example, defects in the urea cycle,24 age-associated cardiac fibrosis17 and/or cardiac stress sensitivity.35,42
Limitations of the study
The small number of pedigrees and patients characterized in this work renders it challenging to arrive at definitive conclusions regarding the impact of hypomorphic variants in SIRT5. It is possible that more detailed biochemical analysis, for example using unbiased metabolomic studies, might uncover consistent defects conferred by SIRT5 loss-of-function, in the human or the mouse. It is also important to note that these variants could result in other changes to SIRT5 function that are not reflected in activity measurements, e.g., altered interaction(s) with binding partner(s). Likewise, specific stress conditions, such as fasting, altered diet, or even aging, might uncover phenotypes in the Sirt5 P114T strain.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-SIRT5 | Cell Signaling Technology | Cat#8782; RRID: AB_2716763 |
| Rabbit polyclonal anti-SIRT5 | Sigma-Aldrich | Cat#HPA022002; RRID: AB_1856913 |
| Rabbit polyclonal anti-SIRT3 | Cell Signaling Technology | Cat#5490s; RRID: AB_10828246 |
| Goat polyclonal anti-SIRT4 | Abcam | Cat#ab10140; RRID: AB_2188769 |
| Rabbit polyclonal pan anti-succinyllysine | PTM BioLabs | Cat#PTM-401; RRID: AB_2687628 |
| Mouse monoclonal pan anti-succinyllysine | PTM BioLabs | Cat#PTM-419; RRID: AB_2942099 |
| Mouse monoclonal anti-actin beta | Abcam | Cat#ab6276; RRID: AB_2223210 |
| Anti-Histone H3 | Cell Signaling Technology | Cat#9715; RRID: AB_331563 |
| Rabbit monoclonal mix anti-malonyllysine | Cell Signaling Technology | Cat#14942; RRID: AB_2687627 |
| Rabbit polyclonal anti-glutaryllysine | PTM BioLabs | Cat#PTM-1151 |
| Mouse monoclonal anti-total OXPHOS Human Cocktail | Abcam | Cat#ab110411; RRID: AB_2756818 |
| Rabbit IgG | Vector Laboratories | Cat#I-1000; RRID: AB_2336355 |
| Monoclonal goat anti-rabbit IgG, Alexa Fluor 488 | Thermo Fisher Scientific | Cat#A11008; RRID: AB_143165 |
| Rabbit monoclonal anti-c-MYC | Cell Signaling Technology | Cat#5605; RRID: AB_1903938 |
| Mouse monoclonal anti-β-Actin | Millipore Sigma | Cat#A5441; RRID: AB_476744 |
| Mouse monoclonal anti-α-Sarcomeric Actin | Millipore Sigma | Cat#A2172; RRID: AB_476695 |
| Mouse monoclonal anti-α-Tubulin | Santa Cruz | Cat#SC-23948; RRID: AB_628410 |
| Mouse monoclonal anti-Histone H3 | Cell Signaling Technology | Cat#14269; RRID: AB_2756816 |
| Guinea pig polyclonal anti-NeuN | Millipore Sigma | Cat#ABN90; RRID: AB_11205592 |
| Rabbit polyclonal anti-GFAP | Agilent | Cat#Z0334; RRID: AB_10013382 |
| Rabbit polyclonal Iba1 | FUJIFILM Wako Pure Chemical Corporation | Cat#019–19741; RRID: AB_839504 |
| Alexa Fluor 488 goat anti–mouse IgG (H + L) | Invitrogen | Cat#A11029; RRID: AB_2534088 |
| Alexa Fluor 594 F(ab’)2 fragment of goat anti-rabbit IgG (H + L) | Invitrogen | Cat#A11072; RRID: AB_2534116 |
| Bacterial and virus strains | ||
| TOP10 competent E. coli | Invitrogen | Cat#C4040-50 |
| BL21(DE3) E. coli | ThermoFisher Scientific | Cat#EC0114 |
| Biological samples | ||
| Patients-derived primary skin fibroblasts | Radboud University Medical Center | N/A |
| Healthy individuals-derived primary skin fibroblasts | Radboud University Medical Center | N/A |
| Patient 2-derived skeletal muscle biopsy | Radboud University Medical Center | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant SIRT5 P114T mutant protein | This paper | N/A |
| Recombinant SIRT5 L128V mutant protein | This paper | N/A |
| Critical commercial assays | ||
| fluor de lys SIRT5 fluorometric drug discovery assay kit | Enzo Life Sciences | Cat#BML-AK5130001 |
| Sypro Orange Thermal Shift | Sigma-Aldrich | Cat#S5692 |
| Deposited data | ||
| SIRT5 WT Apo crystal structure | This paper | PDBID: 8GBL |
| SIRT5 P114T Apo crystal structure | This paper | PDBID: 8GBN |
| Experimental models: Cell lines | ||
| SIRT5-WT mouse embryonic fibroblasts | This paper | N/A |
| SIRT5-P114T heterozygous mouse embryonic fibroblasts | This paper | N/A |
| SIRT5-P114T homozygous mouse embryonic fibroblasts | This paper | N/A |
| Experimental models: Organisms/strains | ||
| SIRT5-WT mice | This paper | N/A |
| SIRT5-P114T heterozygous mice | This paper | N/A |
| SIRT5-P114T homozygous mice | This paper | N/A |
| Oligonucleotides | ||
| See Table S3 for the oligonucleotides | This paper | N/A |
| See Table S3 for the primers | This paper | N/A |
| Recombinant DNA | ||
| Wild type His-SIRT5 plasmid | Addgene | Cat#25487; http://n2t.net/addgene:25487; RRID:Addgene_25487 |
| Wild type His-SIRT5 plasmid | Addgene | Cat#81030 https://www.addgene.org/81030/RRID:Addgene_81030 |
| P114T mutant His-SIRT5 plasmid | This paper | N/A |
| L128V mutant His-SIRT5 plasmid | This paper | N/A |
| Software and algorithms | ||
| GraphPad Prism version 5.04 | GraphPad | https://www.graphpad.com/guides/prism/latest/curve-fitting/index.htm |
| Seahorse Wave Desktop Software | Agilent Technology | https://www.agilent.com/en/product/cell-analysis/real-time-cell-metabolic-analysis/xf-software/seahorse-wave-desktop-software-740897 |
| ImageJ | Schneider et al.43 | https://imagej.nih.gov/ij/ |
| FlowJo 10.2 analysis software | BD Biosciences | https://www.flowjo.com/solutions/flowjo/downloads/previous-versions |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, David B. Lombard (dbl68@miami.edu).
Materials availability
SIRT5 mutant plasmids generated in this study will be shared by the corresponding authors upon request.
Data and code availability
-
•
The crystal structures of SIRT5 WT Apo and SIRT5 P114T Apo have been deposited to the PDB with accession codes 8GBL and 8GBN, respectively.
-
•
Exome sequencing data reported in this study are not publicly available due to privacy and ethical restrictions. These data and related variant call files (vcf) will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Patient ethics
Written and oral informed consent for diagnostic and research studies was obtained for both subjects in accordance with the Declaration of Helsinki and following the regulations of the local medical ethics committee.
Human participants
Patient 1 is a male from Oman. The clinical presentation and MRI of his brain was diagnosed at the age of 2 years. Patient 2 is a female from Saudi Arabia. MRI of her brain was performed at the age of 25 years. Onset of clinical manifestation was at the age of 22 years. Written and oral informed consent for diagnostic and research studies was obtained for both subjects in accordance with the Declaration of Helsinki and following the regulations of the local medical ethics committee.
Primary cell cultures
Human fibroblasts from skin biopsies of healthy subjects and two patients with SIRT5 variants were used in this study. Control fibroblasts included: #13370 (male), #MW28 (unknown sex due to lack of information), #14308 (male), and #14321 (male); patients fibroblasts harboring SIRT5 variants: #13395 (male) and #13706 (female). All cell culture media supplements were obtained from Gibco, Thermo Fisher Scientific, unless otherwise stated. Human primary skin fibroblasts were routinely cultured in fibroblasts full growth medium (medium 199 (#M3769, Sigma-Aldrich, which is formulated with 5.5 mM glucose) supplemented with 10% fetal bovine serum (#06Q3501K), 2 mM Glut-Max (#35050038), and 1x antibiotic-antimycotic mix (100 IU/mL penicillin/streptomycin, and 25 μg/mL Amphotericin B, #15240062)). Cells were incubated at 37°C and were maintained in an atmosphere containing 5% CO2. Cells were passaged once every 4–5 days when reaching 90% confluency by trypsinization (#15400054) for 2 min at 37°C. All fibroblast cell lines were tested mycoplasma-free (MycoSensor QPCR assay, #302107, Agilent Technologies). Due to the limited number of patients, the effect of sex on the results derived from human fibroblasts was not anlyased. Neverthesless, the conclusion was made based on consistent results observed in both male and female patients’ fibroblasts. Therefore, we believe the absence of sex analysis does not affect the conclusion of the study.
Vertebrate animals study approval
All mice were housed at the Biomedical Science Research Building (University of Michigan). All vertebrate animal experiments were approved by and performed in accordance with the regulations of the University Committee on Use and Care of Animals (University of Michigan).
Animals
Generation of SIRT5 P114T mice
CRISPR/Cas9 technology was used to introduce a Pro114Thr mutation in mouse Sirt5 exon 4 (Ensembl exon, exon_id = ENSMUSE00000440334.3) on chromosome 13. CRISPR.TEFOR.NET (Haeussler et al., 2016) was used to select single guide RNAs (sgRNAs). Preference for sgRNAs was given to targets that cut near Sirt5 amino acid 114. Two targets predicted to be highly specific and active were tested: sgRNA C181G1 (Table S3) with a CFD specificity score of 88 (Doench et al. 2016) and sgRNA C181G2 (Table S3) with a CFD specificity score of 89 (Doench et al. 2016). sgRNAs for Cas9 targets were obtained in the form of chemically modified synthetic sgRNA from Synthego.com (Hendel et al., 2015). Recombinant Streptomyces pyogenes enhanced specificity Cas9 endonuclease was obtained from MilliporeSigma (ESPCAS9, Slaymaker et al. 2016). C181G1 and C181G2 were assembled into sgRNA/Cas9 ribonucleoprotein (RNP) complexes and tested individually by mouse zygote microinjection to determine if they induced chromosome breaks. Briefly, zygotes were cultured in vitro to the blastocyst stage (about 64 cells per blastocyst) after RNP pronuclear microinjection. DNA was extracted from individual blastocysts and subjected to PCR and amplicon DNA sequencing to identify small insertions/deletions at the Cas9/gRNA cut sites.44 Primers used to amplify the 5′ sgRNA cut site are listed in Table S3. The amplicon size was 641 bp. Amplicons were submitted for Sanger sequencing to determine if the sgRNAs induced by chromosome breaks as shown by “peaks-on-peaks” patterns in Sanger chromatograms that indicate the presence of multiple PCR templates caused by non-homologous endjoining of chromosome breaks. sgRNA C181G1 did not induce chromosome breaks while C181G2 did induce chromosome breaks (Figure S5A). An Ultramer single-stranded oligodeoxynucleotide (ssODN, Integrated DNA Technologies) was synthesized to repair chromosome breaks in zygotes through homology directed repair.45 This to mutate Pro114 to Pro114Thr, and to block Cas9 cleavage subsequent to repair. The ssODN sequence is listed in Table S3. The Pro114Thr amino acid codon change is shown in bold. Three silent codon changes (underlined codons) were made to block Cas9 cleavage after ssODN chromosome break repair.
To generate mice carrying Pro114Thre 309 zygotes obtained from the mating of B6SJLF1 (Jackson Laboratory stock no. 100012) female and male mice were used for pronuclear microinjected as described (Behringer et al. 2014). The microinjection mixture contained sgRNA C181G2 (30 ng/ul) complexed with ESPCAS9 protein (50 ng/μL) and 10 ng/ul ssODN. Surviving zygotes were transferred to pseudopregnant females. 110 potentially gene edited G0 founder pups were screened for the Pro114Thr by PCR amplicon sequencing. Two apparently homozygous G0 founder animals carrying the mutation were mated to wild type mice for germline transmission.
Genotyping strategy
To distinguish between WT, Sirt5 P114T heterozygotes, and homozygotes, PCR was performed using DNA extracted from the tail snips of 21 days old mice. Primers used for PCR amplification are shown in Table S3. PCR products (380 bp in size) were then digested using AvaI (NEB #R0152L) and separated on agarose gel. AvaI cuts the WT amplicon after bp 85 and produces two fragments that correspond to 85 bp and 295 bp in size. AvaI does not cut P114T amplicon and thus, P114T homozygote amplicon remains as 380 bp in size. AvaI digestion of PCR product from P114T heterozygote results in three bands; 85 bp, 295 bp, and 380 bp.
Mouse embryonic fibroblasts
P114T heterozygous mice were interbred to generate mouse embryonic fibroblasts (MEFs) from day 13.5 embryos by standard methods. Genotypes were confirmed by Sanger sequencing. Wild-type, heterozygous P114T and homozygous P114T MEFs were cultured in DMEM (Gibco) containing 4.5 g/L glucose, 1 mM sodium pyruvate, 4 mM L-glutamine, 1% non-essential amino acids, 100units/mL penicillin, 100 μg/mL streptomycin, 10 mM HEPES, 115 μM 2-mercaptoethanol, and 15% heat-inactivated FBS, and were grown in a humidified chamber at 37°C containing 5% CO2 and 3% O2. Where indicated, MEFs were treated 8 μM of proteasomal inhibitor MG132 for 12 h.
Method details
Immunoblotting
After harvest by trypsinization and centrifugation (5 min, 300 × g, 4°C), the cell pellet was lysed in lysis buffer (50 mM Tris/HCl pH 7.4, 2 mM nicotinamide, 1 μM trichostatin A and protease inhibitor cocktail (#04693159001, Roche)), then sonicated on ice. Protein concentration was determined with BioRad DC assay (#5000116), and equalized to the same level with lysis buffer, after which it was incubated with NuPAGE LDS Sample Buffer (# NP0007, Invitrogen) and 50 mM dithiothreitol at 95°C for 5 min. Protein samples were separated on a 4–12% Bis-Tris Gel (#NW04125BOX, Invitrogen) and electroblotted onto nitrocellulose membrane (#88018, Invitrogen). The membrane was then probed with anti-SIRT5, anti-SIRT3, anti-SIRT4, anti-succinyllysine, anti-glutaryllysine, anti-malonyllysine, anti-complex V, anti-actin beta or anti-histone3, followed by appropriate secondary antibody (IRDye 800CW or HRP-linked IgG). Images were obtained using the Odyssey or Bio-Rad ChemiDoc XRS Systems. Actin beta and histone H3 were used as loading controls. Antibodies information is listed in key resources table. Uncropped blots are provided as supplementary information.
Immunofluorescence
Fibroblasts were seeded on sterile 24 mm cover glasses in 6-well plate. Cells were cultured in fibroblast complete growth medium as mentioned before and allowed to grow for 48 h before the assay. For succinate exposure, cells were cultured in fibroblasts full growth medium supplemented with 4 mM dimethyl succinate-ester (#112755). Mitochondria were visualized with 400 nM MitoTracker Orange (#M7510, Molecular Probes) diluted in medium 199. Cells were fixed with 4% formaldehyde solution (#252549) at 37°C for 20 min, then were permeabilized by 0.2% Triton X-100 in Tris-buffered saline (TBS). Cells underwent blocking (2% BSA in TBS, 2% goat serum, 0.1% Triton X-100, 0.05% Tween 20). Then, they were incubated with either pan anti-succinyllysine or rabbit IgG at room temperature for 90 min, washed and incubated with the secondary Alexa Fluor 488 conjugated goat anti-rabbit antibody at room temperature for 60 min. Finally, cells were counterstained with 1 μg/mL of 4′,6-diamidino-2-phenylindole (DAPI, #D9564), mounted with Fluoromount G (#0100-01, SouthernBiotech). Images were taken by Leica DM6b upright microscope and quantification was performed by ImageJ 1.52.43 46 images of each cell lines were quantified. All chemicals used in immunofluorescence experiments were from Sigma-Aldrich, unless otherwise stated.
Mutagenesis of SIRT5 plasmid
His-SIRT5 expression plasmid was a gift from Cheryl Arrowsmith (Addgene plasmid #25487). Site-directed mutagenesis of WT SIRT5 was performed using the QuickChange Lightning site-directed mutagenesis kit (#210518, Agilent Technologies), according to the manufacturer’s instruction. Plasmids were amplified in competent E. coli and purified by using an Endofree plasmid max kit (#12362, Qiagen). Plasmid DNA were sent for Sanger sequencing to confirm the introduced SIRT5 variants (BaseClear, the Netherlands). Mutagenesis primers are listed in Table S3.
Expression and Purification of Recombinant His-SIRT5
For Figure 3, His-tagged WT and variant SIRT5 proteins were expressed in the BL21(DE3) E. coli strain. Transformed BL21DE3 cultures were grown in Luria broth (LB) medium containing kanamycin (50 μg/mL) at 37°C until OD = 0.25 was reached, after which the culture was allowed to cool down to 22°C. SIRT5 expression was induced with 500 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) and the cells were harvested 18 h after the induction by centrifugation at 6000 × g for 15 min at 4°C, and snap frozen at −80°C. To purify His-SIRT5, the E. coli pellet was resuspended in 10 mM Tris/HCl pH 8.0, 500 mM NaCl supplemented with 1 mg/mL lysozyme, and sonicated on ice. After centrifugation at 12,000 × g for 15 min at 4°C, SIRT5 WT and variants were purified by Ni2+-affinity resin (#V8823, Promega), and eluted with 25 mM Tris/HCl pH 8.0, 100 mM NaCl, and 500 mM imidazole. Prior to use in further experiments, imidazole was removed from the eluted His-SIRT5 using ultra-0.5 centrifugal filter units (UFC501096, Merck Millipore). 500 μL of the recombinant SIRT5 elution fractionation was added to the ultra-0.5 centrifugal filter units and centrifuged at 14,000 × g for 15 min at room temperature. Flow-through was discarded and 450 μL of exchange buffer (25 mM Tris/HCl pH 8.0, 100 mM NaCl) was added, and the centrifugation step was repeated. Washing was repeated twice. Protein concentration of recombinant SIRT5 was subsequently determined by Bradford assay (#23200, Thermo Fisher Scientific).
SIRT5 enzymatic assay and steady-state kinetic analyses
Desuccinylase activity of recombinant His-SIRT5 WT, P114T and L128V variants was measured with the fluor de lys SIRT5 fluorometric drug discovery assay kit (#BML-AK5130001, Enzo). To assess the Michaelis-Menten constant (Km) of SIRT5 for succinyl-peptides, steady-state rates were measured by varying succinyl-substrate (2–200 μM) in the presence of 1 mM NAD+. For the determination of Km, NAD+, activity was measured under a range of NAD+ (20–320 μM) and 120 μM of succinyl-substrate. The enzymatic reaction took place at 37°C water bath with shaking (60 rpm) for 10 min. The reaction was stopped after 10 min by incubating with 1× developer and 2 mM nicotinamide at 25°C for 15 min. The fluorescence was read in half-area black 96-well plate (#3686, Corning) with excitation at 360 nm and emission at 460 nm on a Biotek Synergy HT microplate reader. Fluorescence signal of negative control (presence of succinyl-substrate and absence of NAD+ in the reaction system) was subtracted from each experimental sample. For analysis of His-SIRT5 sensitivity to NAM, fixed concentration of succinyl-substrate (10 μM) and NAD+ (500 μM) and varying concentration of NAM (0.1–1000 μM) were present in the reaction system. For analysis of His-SIRT5 desuccinylase activity under limiting conditions, succinyl-peptide was fixed at 10 μM, and NAD+ input was used as indicated in the figure legends. 0.03 μg of freshly prepared His-SIRT5 (WT, P114T, and L128V SIRT5) were used in all the experiments. For kinetics parameters calculation, data were fitted to the Michaelis-Menten equation in GraphPad Prism software (version 5.04). For IC50 of NAM for each His-SIRT5, data were fitted to the log(inhibitor) vs. response (three parameters) equation in GraphPad Prism software (version 5.04).
Protein thermal shift assay
For Figure 3B, wild-type and two variants of His-SIRT5 were freshly prepared as mentioned in “Expression and Purification of Recombinant His-SIRT5” and used in the experiment. After protein concentration determination, all His-SIRT5 were equalized to 320 μg/mL with exchange buffer (25 mM Tris/HCl pH 8.0, 100 mM NaCl) containing 20× Sypro Orange (#S5692, Sigma-Aldrich, delivered at 5000×). Samples (25 μL) were added to qPCR plate and analyzed with Bio-Rad CFX96 Real-Time System C1000 thermal cycler. The temperature was increased at a rate of 0.5 °C/min ranging from 25°C to 95°C, and fluorescence signal was monitored in the FRET channel of the thermal cycler. Fluorescence signal obtained from blank wells (absence of His-SIRT5) was subtracted from each experimental sample. Melting temperature was determined by performing non-linear fitting of normalized thermal denaturation fluorescence signal. All data points after two data points of the maximal raw fluorescence data were deleted, and a “truncated dataset” was generated. The minimal data point in the “truncated dataset” was subtracted from each data point, subsequently each data point was normalized to the maximal data point. The normalized data were then fitted to Boltzmann Sigmoidal curve in GraphPad Prism software (version 5.04).
RNA extraction and cDNA synthesis
1× ice-cold Hank’s balanced salt solution (HBSS, #14175129, Gibco) was added to the human fibroblasts immediately after culture medium was discarded. Fibroblasts were scraped off flasks by scraper, followed by centrifugation at 300 × g, 4°C for 5 min. Cells were washed twice with ice-cold 1× HBSS. Collected cell pellet was then immediately processed to RNA extraction using RNeasy mini kit (#74106, Qiagen), according to the manufacturer’s instructions. Total RNA was dissolved in DNase/RNase-free water, and extracted RNA was kept on ice onwards from now. RNA concentration and quality were measured by a Nanodrop spectrophotometer (IsoGen Life Science). RNA integrity was checked by capillary electrophoresis using Agilent D1000 ScreenTape on the TapeStation 2200 (Agilent Technologies). Subsequently, 1 μg of total RNA was reverse transcribed to cDNA by using an iScript kit (#1708891, Bio-Rad) on a Mastercycler (Eppendorf), with 5 min at 25°C, 30 min at 42°C, and 5 min at 85°C. one cDNA synthesis reaction with an RNA sample without reverse transcriptase (the minus RT reaction) was included as a negative control for further real-time RT-PCR analysis. cDNA was stored at −20°C until use. This method was applied in Figure 2B.
Real-Time RT-PCR Analysis
For Figure 2B, transcript expression was measured using iQ SYBR Green Supermix (#1725006CUST, Bio-Rad), and the fluorescence signal was monitored with a CFX96 Real-Time PCR Detection System. The PCR reaction program was 3 min at 95°C, 40 cycles of 15 s at 95°C and 45 s at 60°C, followed by the melt-curve analysis. Low-level expressed gene ribosomal protein S15 (RPS15) was preamplified with SsoAdvanced PreAmp Supermix (#1725160, Bio-Rad), with 3 min at 95°C, and 12 cycles of 15 s at 95°C and 4 min at 58°C. A negative control (a sample minus reverse transcriptase) was included in the preamplification. cDNA from all (preamplified) samples was pooled and serial dilutions were made for standard curves. Samples were diluted 100-fold for PCR reaction, and two negative controls (DNase/RNase-free water and the minus RT reaction) were also included in the real-time RT-PCR analysis. The primer sequences are listed in Table S3. SIRT5 gene expression level was normalized to expression level of three reference genes, beta-2 microglobulin (B2M), RPS15, and erbb2 interacting protein (ERBB2IP) in each fibroblast cell line. RNA of each fibroblast cell line was isolated from three independent batches of cultures, and all the sample were analyzed in the same RT-PCR run. Duplicates were used for each sample.
Whole exome sequencing
Whole exome sequencing (WES) and data analysis were performed as described previously.46,47 In short, exome enrichment was performed using the SureSelect Human All Exon 50 Mb Kit V5 (Agilent). Sequencing was done on a HiSeq4000 (Illumina) with a minimum median coverage of 80×. Read alignment to the human reference genome (GrCH37/hg19) and variant calling was performed at BGI (Copenhagen) using BWA and GATK software, respectively. Variant annotation was performed using a custom designed in-house annotation. Intronic variants (except for splice sites), synonymous changes, and common variants were filtered and excluded from the initial datasets. Patient data were first analyzed using a custom-made virtual gene panel containing mitochondrial disease genes (as described in OMIM) as well all other genes known to encode mitochondrial proteins. As no disease-causing variants were detected, the entire exome was investigated for rare, protein damaging variants. For this, variants were selected for homozygosity, exonic or canonical splice site variants, frequency <0.5% in gnomAD and in-house database, and missense/nonsense variants. Variants were classified using the ACMG criteria.48
Mutagenesis of SIRT5 plasmid
His tagged SIRT5 expression plasmid (SIRT5-pET15b)49 was used to generate His tagged SIRT5-P114T expression plasmid (SIRT5P114T-pET15b). Site-directed mutagenesis of WT SIRT5 was performed using the QuickChange II-directed mutagenesis kit (#200523, Agilent Technologies), according to the manufacturer’s instruction. The introduction of the mutation (c.340C>A, p.P114T) was confirmed by Sanger sequencing. Mutagenesis primers used are listed in Table S3. This method was applied in Figure S2C–S2G.
Purification of recombinant proteins
Recombinant His tagged WT SIRT5 and P114T variant SIRT5 proteins were purified using Ni-NTA resin as described previously.49 Briefly, His-tagged WT and variant SIRT5 proteins were expressed in the BL21(DE3) E. coli strain. Transformed BL21(DE3) cultures were grown in Luria broth (LB) medium containing carbenicillin (50 μg/mL) at 37°C until OD = ∼0.5 was reached. SIRT5 expression was induced with 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) and the cells were harvested 16 h after the induction by centrifugation at 10,000 × g for 10 min at 4°C. To purify His-tagged SIRT5, the E. coli pellet was resuspended in 50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH 8.0, supplemented with 1 mg/mL lysozyme, 10 μg/mL RNase A, and 5 μg/mL DNase I and incubated on ice for 30 min. Following incubation, resuspended pellets were sonicated on ice and centrifuged at 10,000 × g for 30 min at 4°C. SIRT5 WT and P114T proteins were purified by Ni2+-affinity resin (#30230, Qiagen), and eluted with 50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, 5% glycerol, pH 8.0. Protein concentration of recombinant SIRT5 proteins was determined by Bio-Rad DC protein assay kit (#500-0112). This method was applied in Figure S2C–S2G.
SIRT5 activity assay
All reactions were performed in 96-well black microplate (Corning No. 3603) with a reaction volume of 50 μL per well. The assay buffer contained 50 mM Tris, 137 mM NaCl, and 2.7 mM KCl at pH 8.0. Reaction wells contained SIRT5 proteins (wild-type or variant, 0.2 μM), fluorescent substrate (Ac-K(Suc)-AMC or Ac-K(Glu)-AMC or Ac-K(Mal)-AMC, 10 μM), NAD+ (200 μM), and inhibitors (if applicable) in assay buffer.50 Control wells without enzyme were incubated in each plate with aim of excluding the interference of fluorescent compounds. The reactions were incubated for 2 h at 37°C and 140 rpm. Subsequently a solution (50 μL) containing 3–4 U/μL trypsin (Sigma-Aldrich No. T8003) and 8 mM nicotinamide was added, followed by further incubation for 20 min at 37°C and 140 rpm. Fluorescence intensity was then measured using a microplate reader (BioTek Synergy H1, λex = 360 nm, λem = 460 nm, Optics: Top). All experiments were run at least three times independently. This method was used in Figure S2E and S2F.
Purification of recombinant proteins for crystallization and thermal shift assay
The wild-type SIRT5 (WT) gene (residues 32–302) and a variant form (P114T) were each cloned into a His6-TEV (Tobacco Etch Virus) expression vector (pMCSG7) and purified by the following protocol. The constructs were transformed into Rosetta2 cells, then grown in Terrific Broth and induced with 0.4 mM isopropyl β-D-1-thiogalactopyranoside overnight at 20°C. The pelleted cells were lysed by sonication in 25 mM Tris-HCl pH 7.0, 200 mM NaCl, and 0.1% 2-mercaptoethanol with protease inhibitors. The supernatant was cleared by centrifugation and incubated with Ni-NTA resin (Qiagen) pre-equilibrated with lysis buffer. The resin was washed with 25 mM Tris-HCl pH 7.0, 200 mM NaCl and 10 mM imidazole, then the protein eluted with 25 mM Tris pH 7.0, 200 mM NaCl, and 200 mM imidazole. The His-tag was removed with TEV protease during dialysis against 25 mM Tris-HCl pH 7.5, 150 mM NaCl, and 1 mM DTT overnight at 4°C, followed by reapplication to the Ni-NTA resin. The cleaved protein was concentrated and applied to a Superdex 75 (GE Healthcare) pre-equilibrated with 20 mM Tris-HCl pH 7.8 and 150 mM NaCl. The purified proteins were concentrated to 12 mg/mL and stored at −80°C for crystallization studies.
Crystallization and Structure Determination of WT and P114T SIRT5
Apo SIRT5 WT and apo P114T proteins were crystallized by hanging drop vapor diffusion at 20°C. Crystals of apo SIRT5 WT grew in drops containing 0.75 μL of protein and 0.75 μL of well solution (0.1 M MES 6.5, 25% PEG 4K). Crystals of apo P114T grew in drops containing 0.5 μL protein and 0.55 μL well solution (0.1 M MES pH 6.5, 20% PEG 10K, 10 mM praseodymium (III) acetate hydrate). SIRT5 WT crystals were cryoprotected in well solution containing an additional 20% ethylene glycol and flash cooled in liquid nitrogen, whereas Apo P144T crystals were cryoprotected in well solution containing 20% glycerol.
Diffraction data were collected on Advanced Photon Source LS-CAT beamlines 21-ID-G (WT and P114T) and then processed using HKL2000.51 Both structures were solved by molecular replacement in Phaser52 using a deposited structure of human SIRT5 (PDB: 2B4Y) sans ligand as the search model for the WT structure, and an in-house WT structure of SIRT5 missing residue Pro114 as the search model for P114T structure. Iterative rounds of model building and refinement were performed using COOT37 and Buster,53 respectively. Data collection and structural refinement statistics are shown in Table S4.
The structure of the apo form of WT SIRT5 was solved to 2.25 Å resolution in space group P21 with two protein chains per asymmetric unit. Residues 38–302 were modeled in both chains, except for a disordered loop region from 278 to 284 in both chains and 253 to 256 in chain B. The structure of the apo form of P114T was solved to 2.70 Å resolution in space group P21with two protein chains per asymmetric unit. Residues 39–302 were modeled in chain A, except for a disordered loop region from 278 to 284. Residues 37–302 were modeled in chain B, except for two disordered loop regions (278–284 and 251 to 253).
Thermal Shift Assay
To determine the melting temperatures for SIRT5 proteins (WT, P114T, and H158Y), 5 μL of buffer (25 mM HEPES 7.0, 200 mM NaCl) containing 2× PTS dye (Applied Biosystems) and 5 μL of 2× protein (5 mg/mL in 25 mM HEPES 7.0, 200 mM NaCl) were added to a 384-well plate (Thermofisher). The plate was centrifuged at 1000 × g for 1 min. The differential scanning fluorimetry experiment was performed in a Quantstudio7 (Thermofisher) using continuous ramping mode from 25°C to 95°C with a scanning rate of 0.03°C/s and results analyzed using Protein Thermal Shift software version 1.3 (Applied Biosystems). All assays were performed in quadruplicate. This method was applied in Figure S2G.
Immunoblotting
Mice were anesthetized with isoflurane and immediately sacrificed by cervical dislocation. Tissue samples were flash frozen, pulverized, and resuspended in RIPA buffer (50 mM Tris-HCL pH7.4, 150 mM NaCl, 1% NP-40, 0.5% Na-deoxycholate, 0.1% SDS, 2 mM EDTA, 50 mM NaF) supplemented with protease and phosphatase inhibitors, 10 mM NAM, and 1 μM TSA. Samples were sonicated for 60 s on ice (where cells were used sonication was performed for 30 s), then spun down at 4°C for 10 min at 15,000xg, and the supernatant was collected for protein quantification using a DC Protein Assay (Bio-Rad #5000112). Samples were mixed with Laemmli buffer (62.5 mM Tris pH6.8, 2% SDS, 10% glycerol, 5% β-mercaptoethanol, 1% bromophenol blue) and boiled for 10′, resolved by SDS-PAGE, and transferred to PVDF membrane overnight at 4°C using a Bio-Rad Criterion system. Membranes were stained with Ponceau S to assess protein loading, and then blocked using 5% milk in TBST (TBS containing 0.1% Tween 20) at room temperature. Primary antibodies were diluted in 5% BSA in TBST and incubated with the blot overnight at 4°C on a rocker. Secondary incubation was performed at room temperature for 1 h using either mouse or rabbit secondary antibodies (Jackson ImmunoResearch 115-035-062 or 111-035-045) diluted 1:10000 in 5% milk in TBST. For detection, blots were immersed in a western chemiluminescent HRP substrate (Millipore P90720) and imaged using the GE ImageQuant LAS 4000. Primary antibodies used in this study are listed in the key resources table.
Isolation of total RNA
Total RNA was extracted from cultured MEFs using Trizol reagent (Life Technologies #15596018). To remove any contaminating genomic DNA, extracted RNA was treated with RNase-free DNase I (Roche #04716728001) for 1 h at 37°C, according to manufacturer’s instructions. DNase I treated RNA was cleaned up by phenol:chloroform extraction method.
qRT-PCR
cDNA was generated from DNase I treated total RNA using MultiScribe Reverse Transcriptase (Applied Biosystems High-Capacity cDNA Reverse Transcription Kit with RNase inhibitor # 4374966) per the manufacturer’s instructions. qPCR was performed with PowerUp SYBR Green Master Mix (Applied Biosystems # A25742), in an Applied Biosystems Step One Plus Real-Time PCR Machine. Data acquisition was performed using the manufacturer’s software and data analysis was performed using a ΔΔCT approach. The primers used for qPCR are listed in Table S3.
Agilent/Seahorse assay
Seahorse metabolic flux analyses in human fibroblasts
Fibroblasts were seeded in XFe96 V3 PS cell culture microplates (#101085-004, Agilent Technologies), and cultured in fibroblast growth medium overnight. Sixteen hours post-seeding, medium was switched to exposure medium as detailed in Table S5. On the day of the Seahorse assay, pH of freshly prepared assay medium (detailed compositions are described in Table S5) and all injection solutions were checked and adjusted to 7.4 if needed; subsequently, they were filtered through 0.2 μm cellulose acetate membranes (#28145477, VWR) and kept at 37°C until use. Prior to the Seahorse run, exposure medium was replaced with Seahorse assay medium, first for 1 h in a 37°C non-CO2 incubator with normal air. Then, the fresh Seahorse assay medium was applied to cells again just prior to the Seahorse run. For bioenergetic determinations, two different injection strategies were applied separately within one assay after basal measurement. Final concentration of the compounds used in strategy 1: i) 1 μM oligomycin (#O4876, Sigma-Aldrich), ii) 2.5 μM antimycin (#A8674, Sigma-Aldrich) plus 1.25 μM rotenone (#R8875, Sigma-Aldrich), iii) 100 mM 2-deoxyglucose (2-DG, #D8375, Sigma-Aldrich). For injection strategy 2: i) 1 μM carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP, #C2920, Sigma-Aldrich), ii) 2.5 μM antimycin plus 1.25 μM rotenone, iii) 20 μM monensin (#M5273, Sigma-Aldrich). The Seahorse analyzer was maintained at 37°C for all Seahorse assays. The assay procedure consisted of a 12 min equilibration period followed by 3 cycles to measure the basal rate, comprising of a 2 min mix, and a 3 min measurement period. Injection of compounds was followed by 3 cycles, except for 2-DG injection, which was followed by 5 cycles. After Seahorse assays, cells in plate were stained with 4 μM of 2'-(4-ethoxyphenyl)-5-(4-methyl-1-piperazinyl)-2,5′-bi-1H-benzimidazole trihydrochloride trihydrate (Hoechst 33342, #B2261, Sigma-Aldrich) dissolved in assay medium, for 1 h at 37°C non-CO2 incubator. Images were taken by Cytation 1 Imaging Reader (BioTek) and cell number was determined by ImageJ 1.52. Seahorse data were normalized to cell number in each well. For determining buffer factor capacity of Seahorse assay medium, either Seahorse assay medium or 5.0 mM HCl in assay medium was injected at port A, B and C, followed by 3 measurement cycles after each. Dulbecco’s Modified Eagle’s Medium (DMEM, #A1443001, Gibco) and L-glutamine (L-Gln, #25030024, Gibco) were used in all exposure media throughout this study. Galactose (#G0625), and CoCl2 (#C8661) were from Sigma-Aldrich. XF DMEM (pH 7.4, #27518004), XF L-Gln (#103579-100), and XF glucose (#103577-100) were all from Agilent Technologies.
Seahorse metabolic flux analyses in mouse embryonic fibroblasts
Cellular energy phenotypes, Real-Time ATP production rate, glycolytic capacity, and mitochondrial respiration parameters were measured using the XFe96 Extracellular Flux Analyzer (Seahorse Bioscience, Agilent Technologies, Santa Clara, CA). The Seahorse Cell Energy Phenotype Test was performed to measure cellular baseline energy phenotype, stressed energy phenotype, and the metabolic potentials. The assay was performed using Seahorse XF Cell Energy Phenotype Test Kit (Agilent) as per manufacturer’s instructions. Briefly, 2 × 104 SIRT5 WT or P114T MEFs (littermates and sex matched; 2 pairs males and 2 pairs females) were plated in complete MEF growth media into each well of a 96- well Seahorse microplate. Cells were then incubated in 5% CO2 at 37°C for overnight. Following incubation, cells were washed twice, incubated in a non-CO2 incubator at 37°C, and analyzed in XF assay media (non-buffered DMEM containing 10 mM glucose, 2 mM L-glutamine, and 1 mM sodium pyruvate, pH 7.4) at 37°C, under basal conditions and in response to a mixture of 1.5 μM oligomycin + 1 μM fluoro-carbonyl cyanide phenylhydrazone (FCCP). Data were analyzed by the Seahorse XF Cell Energy Phenotype Report Generator. The data were normalized to protein content.
The Seahorse XF Real-Time ATP Rate Assay Kit (Agilent) was used to simultaneously measure the basal ATP production rates from mitochondrial respiration and glycolysis. The assay was performed per manufacturer’s instructions. Briefly, 2 × 104 SIRT5 WT or P114T MEFs (littermates and sex matched; 2 pairs males and 2 pairs females) were plated in complete MEF growth media into each well of a 96-well Seahorse microplate. Cells were then incubated in 5% CO2 at 37°C for overnight. Following incubation, cells were washed twice, incubated in a non-CO2 incubator at 37°C, and analyzed in XF assay media (non-buffered DMEM containing 10 mM glucose, 2 mM L-glutamine, and 1 mM sodium pyruvate, pH 7.4) at 37°C, under basal conditions and in response to 1.5 μM oligomycin, and 0.5 μM rotenone/0.5 μM antimycin A. Data were analyzed by the Seahorse XF Real-Time ATP Rate Assay Report Generator. ATP production rates were normalized to protein content.
To measure glucose- or galactose-dependent mitochondrial respiration, mitochondrial stress tests were performed per manufacturer’s instructions. Briefly, 2 × 104 SIRT5 WT or P114T MEFs (littermates and sex matched; 2 pairs males and 2 pairs females) were plated in complete MEF growth media into each well of a 96- well Seahorse microplate. Cells were then incubated in 5% CO2 at 37°C for overnight. Following incubation, cells were washed twice, incubated in non-CO2 incubator at 37°C, and analyzed in XF assay media (non-buffered DMEM containing 25 mM glucose or 25 mM galactose, 2 mM L-glutamine, and 1 mM sodium pyruvate, pH 7.4) at 37°C, under basal conditions and in response to 1.5 μM oligomycin (Sigma #O4876), 1 μM fluoro-carbonyl cyanide phenylhydrazone (FCCP) (Sigma #C2920) and 0.5 μM rotenone (Sigma #R8875)/0.5 μM antimycin A (Sigma # A8674). Data were analyzed by the Seahorse XF Cell Mito Stress Test Report Generator. Oxygen consumption rate (OCR) (pmol O2/min) values were normalized to the protein content.
Glycolytic parameters were measured by performing glycolysis stress tests according to manufacturer’s instructions. Briefly, 2 × 104 SIRT5 WT or P114T MEFs (littermates and sex matched; 2 pairs males and 2 pairs females) were plated in complete MEF growth media into each well of a 96- well Seahorse microplate. Cells were then incubated in 5% CO2 at 37°C for overnight. Following incubation, cells were washed twice, incubated in a non-CO2 incubator at 37°C, and analyzed in XF assay media (non-buffered DMEM containing 2 mM L-glutamine, pH 7.4) at 37°C, under basal conditions and in response to 10 mM glucose (Sigma #G7528), 1.5 μM oligomycin (Sigma #O4876), and 50 mM 2-deoxy-D-glucose (Sigma #D8375). Data were analyzed by the Seahorse XF Cell Glycolysis Stress Test Report Generator. Extracellular acidification rate (ECAR) (mpH/min) values were normalized to protein content.
Measurement of intracellular ROS levels
Measurement of intracellular ROS level in Human fibroblasts
Intracellular ROS generation was determined using the cell-permeable probe 2′,7′-dichlorofluorescin diacetate (H2DCFDA) (#D6883, Sigma-Aldrich). 7000 human fibroblasts were seeded in black 96-well plates with clear bottom (#CLS3603, Sigma-Aldrich) in fibroblast growth medium. After 18 h, cells were switched to either glucose or galactose exposure medium. After 72 h of exposure, cells were washed once with 1× DPBS and protected from light. Cells were incubated in HEPES/Tris buffer (HT, 132 mM NaCl, 4.2 mM KCl, 1 mM CaCl2, 1 mM MgCl2, either glucose (5.5 mM) or galactose (5.5 mM), and 10 mM HEPES, pH 7.4) containing 10 μM of H2DCFDA in the dark for 15 min at 37°C. Next, cells were washed twice with 1× DPBS, and HT buffer containing either glucose (5.5 mM) or galactose (5.5 mM) was added. Fluorescence was detected with excitation at 485 nm and emission at 530 nm on a Biotek Synergy HT microplate reader. Cells in HT buffer without H2DCFDA dye were used for background correction. After the assay, cells were stained with 4 μM Hoechst at 37°C for 1 h. Images were made with Cytation 1. Cell number was counted using ImageJ 1.52. ROS levels were normalized to cell number.
Measurement of intracellular ROS level in mouse embryonic fibroblasts
SIRT5 WT or SIRT5 P114T MEFs (littermates and sex matched; 2 pairs males and 2 pairs females) were plated at a density of 5 × 105 per well in quadruplicates in six-well plates and incubated overnight. After incubation, medium was removed, and cells were washed once with prewarmed PBS. CM-H2DCFDA (ThermoFisher) in prewarmed PBS was added to the cells to a final concentration of 10 μM and incubated for 30 min in CO2 incubator at 37°C. After incubation, CM-H2DCFDA containing PBS was replaced by complete growth medium and incubated for 30 min at 37°C. Cells were collected by trypsinization and analyzed by flow cytometry. Results were analyzed using FlowJo 10.2 analysis software.
Brain immunofluorescence
To prepare brain tissues for immunofluorescence, littermates and sex matched mice were perfused with saline and 4% PFA and tissue was removed. Brains were immersed in 30% sucrose for 24 h and then fixed in 4% PFA overnight at 4°C. Fixed tissues were embedded in OCT and frozen. Frozen tissues were cut on a Leica CM1900 cryostat into 10 μm sections and placed on Thermo Scientific Superfrost Plus microscope slides. For staining, slides were incubated in a solution with 0.1% Triton, 10% goat serum, and 1% BSA in PBS for 20 min. Slides were placed in blocking solution (10% goat serum, 1% BSA in PBS) for 40 min before incubating in primary antibody (1:500 NeuN, 1:500 GFAP, 1:250 Iba1) diluted in blocking solution overnight at 4°C. Slides were washed 3 times in PBS and incubated with secondary antibody (1:500) diluted in blocking solution for 1 h at room temperature. Slides were then washed 3 times in PBS and mounted with Vectashield plus DAPI (Vector Laboratories). 40× images of midline sagittal sections were captured on a Nikon A-1 confocal microscope. Images were captured from 3 different brain regions: cerebellum (lobules 3 and 5), hippocampus (CA2), and cortex. 2 Images were captured/brain region/mouse. Staining intensity for each image was measured using ImageJ. Counts of GFAP or Iba1 positive cells were done using CellProfiler. Intensity and count values from the 2 images from each brain region were averaged for each sample. Significance was determined using GraphPad Prism.
The primary and secondary antibodies used in this study are listed in the key resources table.
Quantification and statistical analysis
Statistical analyses were conducted using Graphpad Prism. Multiple-group comparisons were analyzed using two-way ANOVA followed by Sidak’s correction for multiple comparisons. For analysis of two groups, Welch’s t-test was used. A p-value of less than 0.05 was considered statistically significant. Scatterplots include median and interquartile range markers. The statistical details of each experiment can be found in the figure legends. Significance markers: (∗) p < 0.05, (∗∗) p < 0.01, (∗∗∗) p < 0.001, (∗∗∗∗) p < 0.0001.
Acknowledgments
The authors would like to thank the University of Michigan Transgenic Animal Model Core for the generation of the SIRT5 P114T mouse strain, and members of the Lombard Lab for useful feedback. This project was supported by R01GM101171 (DL) and R01CA253986 (DL/NN). TY has received financial support from the China Scholarship Council (Grant no. 201606350170). Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor for the support of this research program (Grant 085P1000817). Some images in the graphical abstract were obtained and modified from Servier Medical Art from Servier (http://www.servier.com/Powerpoint-image-bank). The Lombard lab gratefully acknowledges support through P30CA240139, and the Sylvester Comprehensive Cancer Center.
Author contributions
Project conception: TY, SK, APL, JS, NN, RR, JNS, VDB, DBL; performed experiments: TY, SK, MES, RVJ, MA, JZ, TJK, EMP, TS, JS; contributed reagents and/or patient data: FA, AM, RR; project management: JK, VDB, DL.
Declaration of interests
The authors declare no competing interests.
Published: May 15, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.109991.
Contributor Information
Vincent C.J. de Boer, Email: vincent.deboer@wur.nl.
David B. Lombard, Email: dbl68@miami.edu.
Supplemental information
References
- 1.Chandel N.S. Evolution of Mitochondria as Signaling Organelles. Cell Metab. 2015;22:204–206. doi: 10.1016/j.cmet.2015.05.013. [DOI] [PubMed] [Google Scholar]
- 2.Koopman W.J.H., Willems P.H.G.M., Smeitink J.A.M. Monogenic mitochondrial disorders. N. Engl. J. Med. 2012;366:1132–1141. doi: 10.1056/NEJMra1012478. [DOI] [PubMed] [Google Scholar]
- 3.Breuer M.E., Koopman W.J., Koene S., Nooteboom M., Rodenburg R.J., Willems P.H., Smeitink J.A.M. The role of mitochondrial OXPHOS dysfunction in the development of neurologic diseases. Neurobiol. Dis. 2013;51:27–34. doi: 10.1016/j.nbd.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 4.Coskun P., Wyrembak J., Schriner S.E., Chen H.W., Marciniack C., Laferla F., Wallace D.C. A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim. Biophys. Acta. 2012;1820:553–564. doi: 10.1016/j.bbagen.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bakare A.B., Lesnefsky E.J., Iyer S. Leigh Syndrome: A Tale of Two Genomes. Front. Physiol. 2021;12 doi: 10.3389/fphys.2021.693734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wallace D.C. Mitochondrial diseases in man and mouse. Science. 1999;283:1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- 7.Schapira A.H.V. Mitochondrial disease. Lancet. 2006;368:70–82. doi: 10.1016/S0140-6736(06)68970-8. [DOI] [PubMed] [Google Scholar]
- 8.Scaglia F. Nuclear gene defects in mitochondrial disorders. Methods Mol. Biol. 2012;837:17–34. doi: 10.1007/978-1-61779-504-6_2. [DOI] [PubMed] [Google Scholar]
- 9.Gorman G.S., Schaefer A.M., Ng Y., Gomez N., Blakely E.L., Alston C.L., Feeney C., Horvath R., Yu-Wai-Man P., Chinnery P.F., et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015;77:753–759. doi: 10.1002/ana.24362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smeitink J., van den Heuvel L., DiMauro S. The genetics and pathology of oxidative phosphorylation. Nat. Rev. Genet. 2001;2:342–352. doi: 10.1038/35072063. [DOI] [PubMed] [Google Scholar]
- 11.Kumar S., Giblin W., Lombard D.B. In: Handbook of the Biology of Aging. Musi N., Hornsby P.J., editors. Elsevier; 2021. Sirtuins, healthspan, and longevity in mammals; pp. 77–149. [Google Scholar]
- 12.Kumar S., Lombard D.B. Functions of the sirtuin deacylase SIRT5 in normal physiology and pathobiology. Crit. Rev. Biochem. Mol. Biol. 2018;53:311–334. doi: 10.1080/10409238.2018.1458071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park J., Chen Y., Tishkoff D.X., Peng C., Tan M., Dai L., Xie Z., Zhang Y., Zwaans B.M.M., Skinner M.E., et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell. 2013;50:919–930. doi: 10.1016/j.molcel.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rardin M.J., He W., Nishida Y., Newman J.C., Carrico C., Danielson S.R., Guo A., Gut P., Sahu A.K., Li B., et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 2013;18:920–933. doi: 10.1016/j.cmet.2013.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y., Bharathi S.S., Rardin M.J., Lu J., Maringer K.V., Sims-Lucas S., Prochownik E.V., Gibson B.W., Goetzman E.S. Lysine desuccinylase SIRT5 binds to cardiolipin and regulates the electron transport chain. J. Biol. Chem. 2017;292:10239–10249. doi: 10.1074/jbc.M117.785022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nishida Y., Rardin M.J., Carrico C., He W., Sahu A.K., Gut P., Najjar R., Fitch M., Hellerstein M., Gibson B.W., Verdin E. SIRT5 Regulates both Cytosolic and Mitochondrial Protein Malonylation with Glycolysis as a Major Target. Mol. Cell. 2015;59:321–332. doi: 10.1016/j.molcel.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sadhukhan S., Liu X., Ryu D., Nelson O.D., Stupinski J.A., Li Z., Chen W., Zhang S., Weiss R.S., Locasale J.W., et al. Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc. Natl. Acad. Sci. USA. 2016;113:4320–4325. doi: 10.1073/pnas.1519858113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang G., Meyer J.G., Cai W., Softic S., Li M.E., Verdin E., Newgard C., Schilling B., Kahn C.R. Regulation of UCP1 and Mitochondrial Metabolism in Brown Adipose Tissue by Reversible Succinylation. Mol. Cell. 2019;74:844–857.e7. doi: 10.1016/j.molcel.2019.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giblin W., Bringman-Rodenbarger L., Guo A.H., Kumar S., Monovich A.C., Mostafa A.M., Skinner M.E., Azar M., Mady A.S., Chung C.H., et al. The deacylase SIRT5 supports melanoma viability by influencing chromatin dynamics. J. Clin. Invest. 2021;131 doi: 10.1172/JCI138926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu T., Shukla S.K., Vernucci E., He C., Wang D., King R.J., Jha K., Siddhanta K., Mullen N.J., Attri K.S., et al. Metabolic Rewiring by Loss of Sirt5 Promotes Kras-Induced Pancreatic Cancer Progression. Gastroenterology. 2021;161:1584–1600. doi: 10.1053/j.gastro.2021.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abril Y.L.N., Fernandez I.R., Hong J.Y., Chiang Y.L., Kutateladze D.A., Zhao Q., Yang M., Hu J., Sadhukhan S., Li B., et al. Pharmacological and genetic perturbation establish SIRT5 as a promising target in breast cancer. Oncogene. 2021;40:1644–1658. doi: 10.1038/s41388-020-01637-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yan D., Franzini A., Pomicter A.D., Halverson B.J., Antelope O., Mason C.C., Ahmann J.M., Senina A.V., Vellore N.A., Jones C.L., et al. Sirt5 Is a Druggable Metabolic Vulnerability in Acute Myeloid Leukemia. Blood Cancer Discov. 2021;2:266–287. doi: 10.1158/2643-3230.bcd-20-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lombard D.B., Alt F.W., Cheng H.L., Bunkenborg J., Streeper R.S., Mostoslavsky R., Kim J., Yancopoulos G., Valenzuela D., Murphy A., et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell Biol. 2007;27:8807–8814. doi: 10.1128/MCB.01636-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakagawa T., Lomb D.J., Haigis M.C., Guarente L. SIRT5 Deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell. 2009;137:560–570. doi: 10.1016/j.cell.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biason-Lauber A., Böni-Schnetzler M., Hubbard B.P., Bouzakri K., Brunner A., Cavelti-Weder C., Keller C., Meyer-Böni M., Meier D.T., Brorsson C., et al. Identification of a SIRT1 mutation in a family with type 1 diabetes. Cell Metab. 2013;17:448–455. doi: 10.1016/j.cmet.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hirschey M.D., Shimazu T., Jing E., Grueter C.A., Collins A.M., Aouizerat B., Stančáková A., Goetzman E., Lam M.M., Schwer B., et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol. Cell. 2011;44:177–190. doi: 10.1016/j.molcel.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aury-Landas J., Bougeard G., Castel H., Hernandez-Vargas H., Drouet A., Latouche J.B., Schouft M.T., Férec C., Leroux D., Lasset C., et al. Germline copy number variation of genes involved in chromatin remodelling in families suggestive of Li-Fraumeni syndrome with brain tumours. Eur. J. Hum. Genet. 2013;21:1369–1376. doi: 10.1038/ejhg.2013.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferrer C.M., Alders M., Postma A.V., Park S., Klein M.A., Cetinbas M., Pajkrt E., Glas A., van Koningsbruggen S., Christoffels V.M., et al. An inactivating mutation in the histone deacetylase SIRT6 causes human perinatal lethality. Genes Dev. 2018;32:373–388. doi: 10.1101/gad.307330.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Glorioso C., Oh S., Douillard G.G., Sibille E. Brain molecular aging, promotion of neurological disease and modulation by Sirtuin5 longevity gene polymorphism. Neurobiol. Dis. 2010;41:279–290. doi: 10.1016/j.nbd.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Donlon T.A., Morris B.J., Chen R., Masaki K.H., Allsopp R.C., Willcox D.C., Tiirikainen M., Willcox B.J. Analysis of Polymorphisms in 59 Potential Candidate Genes for Association With Human Longevity. J. Gerontol. A Biol. Sci. Med. Sci. 2018;73:1459–1464. doi: 10.1093/gerona/glx247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen L., Wang H., Gao F., Zhang J., Zhang Y., Ma R., Pang S., Cui Y., Yang J., Yan B. Functional genetic variants in the SIRT5 gene promoter in acute myocardial infarction. Gene. 2018;675:233–239. doi: 10.1016/j.gene.2018.07.010. [DOI] [PubMed] [Google Scholar]
- 32.Mahjabeen I., Rizwan M., Fareen G., Waqar Ahmed M., Farooq Khan A., Akhtar Kayani M. Mitochondrial sirtuins genetic variations and gastric cancer risk: Evidence from retrospective observational study. Gene. 2022;807 doi: 10.1016/j.gene.2021.145951. [DOI] [PubMed] [Google Scholar]
- 33.Du J., Zhou Y., Su X., Yu J.J., Khan S., Jiang H., Kim J., Woo J., Kim J.H., Choi B.H., et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science. 2011;334:806–809. doi: 10.1126/science.1207861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tan M., Peng C., Anderson K.A., Chhoy P., Xie Z., Dai L., Park J., Chen Y., Huang H., Zhang Y., et al. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 2014;19:605–617. doi: 10.1016/j.cmet.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boylston J.A., Sun J., Chen Y., Gucek M., Sack M.N., Murphy E. Characterization of the cardiac succinylome and its role in ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 2015;88:73–81. doi: 10.1016/j.yjmcc.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guetschow E.D., Kumar S., Lombard D.B., Kennedy R.T. Identification of sirtuin 5 inhibitors by ultrafast microchip electrophoresis using nanoliter volume samples. Anal. Bioanal. Chem. 2016;408:721–731. doi: 10.1007/s00216-015-9206-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Farrell A.S., Sears R.C. MYC degradation. Cold Spring Harb. Perspect. Med. 2014;4 doi: 10.1101/cshperspect.a014365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Divakaruni A.S., Brand M.D. The regulation and physiology of mitochondrial proton leak. Physiology. 2011;26:192–205. doi: 10.1152/physiol.00046.2010. [DOI] [PubMed] [Google Scholar]
- 40.Buler M., Aatsinki S.M., Izzi V., Uusimaa J., Hakkola J. SIRT5 is under the control of PGC-1alpha and AMPK and is involved in regulation of mitochondrial energy metabolism. FASEB J. 2014;28:3225–3237. doi: 10.1096/fj.13-245241. [DOI] [PubMed] [Google Scholar]
- 41.Lombard D.B., Zwaans B.M.M. SIRT3: As Simple As It Seems? Gerontology. 2014;60:56–64. doi: 10.1159/000354382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hershberger K.A., Abraham D.M., Martin A.S., Mao L., Liu J., Gu H., Locasale J.W., Hirschey M.D. Sirtuin 5 is required for mouse survival in response to cardiac pressure overload. J. Biol. Chem. 2017;292:19767–19781. doi: 10.1074/jbc.M117.809897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sakurai T., Watanabe S., Kamiyoshi A., Sato M., Shindo T. A single blastocyst assay optimized for detecting CRISPR/Cas9 system-induced indel mutations in mice. BMC Biotechnol. 2014;14:69. doi: 10.1186/1472-6750-14-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jasin M., Haber J.E. The democratization of gene editing: Insights from site-specific cleavage and double-strand break repair. DNA Repair (Amst) 2016;44:6–16. doi: 10.1016/j.dnarep.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wortmann S.B., Koolen D.A., Smeitink J.A., van den Heuvel L., Rodenburg R.J. Whole exome sequencing of suspected mitochondrial patients in clinical practice. J. Inherit. Metab. Dis. 2015;38:437–443. doi: 10.1007/s10545-015-9823-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Neveling K., Feenstra I., Gilissen C., Hoefsloot L.H., Kamsteeg E.J., Mensenkamp A.R., Rodenburg R.J.T., Yntema H.G., Spruijt L., Vermeer S., et al. A post-hoc comparison of the utility of sanger sequencing and exome sequencing for the diagnosis of heterogeneous diseases. Hum. Mutat. 2013;34:1721–1726. doi: 10.1002/humu.22450. [DOI] [PubMed] [Google Scholar]
- 48.Wallis Y., Payne S., McAnulty C., Bodmer D., Sistermans E., Robertson K. Association for Clinical Genetic Science & Dutch Society of Clinical Genetic Laboratory Specialists; 2013. Practice Guidelines for the Evaluation of Pathogenicity and the Reporting of Sequence Variants in Clinical Molecular Genetics; pp. 1–16. [Google Scholar]
- 49.Kumar S., Lombard D.B. Generation and Purification of Catalytically Active Recombinant Sirtuin5 (SIRT5) Protein. Methods Mol. Biol. 2016;1436:241–257. doi: 10.1007/978-1-4939-3667-0_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu Y., Debnath B., Kumar S., Lombard D.B., Neamati N. Identification of 2-hydroxybenzoic acid derivatives as selective SIRT5 inhibitors. Eur. J. Med. Chem. 2022;241 doi: 10.1016/j.ejmech.2022.114623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Otwinowski Z., Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 52.McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roversi P., Sharff A., Smart O.S., Vonrhein C., Womack T.O. Global Phasing Ltd; 2011. BUSTER Version 2.11.2. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The crystal structures of SIRT5 WT Apo and SIRT5 P114T Apo have been deposited to the PDB with accession codes 8GBL and 8GBN, respectively.
-
•
Exome sequencing data reported in this study are not publicly available due to privacy and ethical restrictions. These data and related variant call files (vcf) will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.