

Haematology is a diverse specialty embracing clinical and laboratory aspects of adult and paediatric disease, both malignant and non-malignant. We describe here some of the progress that has been made in diagnostic and therapeutic strategies. Molecular advances are continuing at a phenomenal rate (exceeding the rate of progress in therapeutics), offering highly sensitive methods for disease detection and, in some disorders, prospects of cure through gene therapy.
Methods
We used information from recent key meetings, including those of the British Society for Haematology and the American Society of Hematology; leading articles in major haematology journals; and discussion with colleagues. The choice of topics covered is largely personal, and owing to space restrictions we have not included every “advance” within the specialty.
Anaemias
Distinguishing between anaemia due to iron deficiency and anaemia of chronic disease is a difficult but common problem in medical practice. Anaemia of chronic disease is complex and involves inflammatory cytokines,1 reduced marrow response to erythropoietin, reduced red cell life span, and impaired reuse of iron.2 In typical, uncomplicated iron deficiency anaemia the haemoglobin concentration, mean cell volume, and serum ferritin and iron concentrations are reduced with raised total iron binding capacity; unfortunately cases are often not typical, and the results of these tests may seem conflicting. Furthermore, if a patient with anaemia of chronic disease is also iron deficient, the parameters for diagnosing iron deficiency are altered, making the diagnosis difficult; often a bone marrow aspirate, stained for iron, is the only method for accurately assessing iron status. This is expensive, time consuming, and unpleasant for the patient. Recently the serum transferrin receptor assay has been developed, enabling more accurate assessment of iron status in this group of patients.
Serum ferritin, a 480 kDa multisubunit protein, represents the body’s iron storage pool. A reduced serum ferritin concentration generally indicates depletion of the iron stores. Ferritin, however, is an “acute phase protein,” whose concentration is raised in inflammatory disorders; in a patient with, for example, active rheumatoid disease the ferritin concentration may be normal (or even raised) even if the patient is truly iron deficient. The transferrin receptor assay aims to distinguish clearly between simple iron deficiency and anaemia of chronic disease in most cases.
Recent advances
Iron deficiency anaemia can now be distinguished from anaemia of chronic disease by using serum transferrin receptor assays
A recently discovered human herpesvirus may have a role in development of myeloma
Additional thrombotic risk factors—for example, factor V and prothrombin gene mutations, hyperhomocysteinaemia—have been identified
Newly designed DNA probe arrays have the potential to allow rapid automated detection of mutations
Antisense therapy, ribozyme technology, and other novel treatment methods are being developed
How does the transferrin receptor assay work? Transferrin is a protein that carries plasma iron. Cells that require iron express the transferrin receptor on the cell surface.3 The receptor binds plasma transferrin with bound iron atoms, after which the transferrin receptor-transferrin complex is internalised (fig 1). After unloading the iron atoms, the transferrin receptor-transferrin complex moves back to the cell surface and dissociates, leaving transferrin free to pick up further iron atoms. In iron deficiency anaemia the number of transferrin receptors increases (is upregulated),4 whereas in anaemia of chronic disease the number of transferrin receptors remains normal. Assays for transferrin receptors may be performed on serum with an enzyme linked immunosorbent assay (ELISA), allowing large numbers of samples to be batch tested. According to available data, the test seems as reliable as testing bone marrow aspirate for diagnosing iron deficiency and should be valuable in patients with rheumatology disorders and others with inflammatory disorders.
Figure 1.
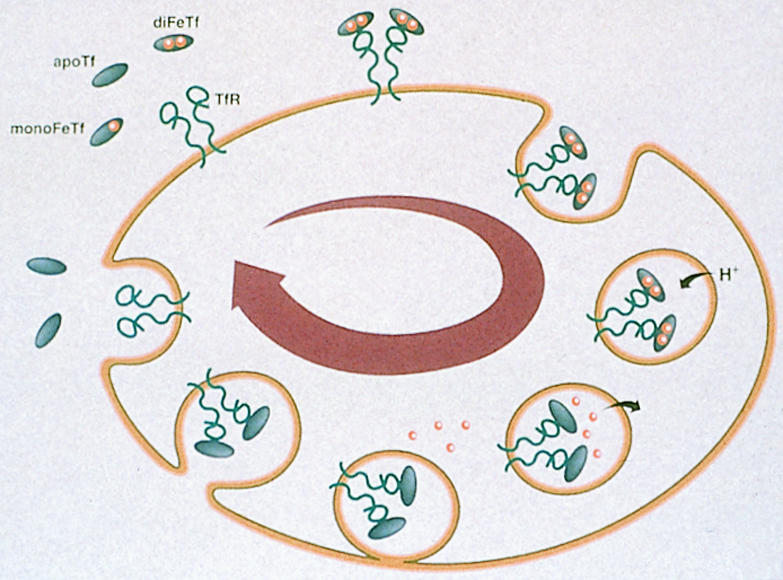
Transferrin receptor expressed on cell surface binds transferrin bound to two iron atoms. The transferrin receptor-transferrin complex is internalised; iron dissociates and moves into cytosol; and transferrin receptor-transferrin complex moves back to plasma membrane and dissociates (TfR=transferrin receptor; monoFeTf=transferrin bound to one iron atom; diFeTf=transferrin bound to two iron atoms; apoTf=transferrin lacking bound iron; H+=hydrogen ion)
Haemophilia
Haemophilias A (factor VIII deficiency) and B (factor IX deficiency, Christmas disease) are X linked bleeding disorders affecting 1:10 000 and 1:60 000 males respectively. Depending on the clotting factor concentrations, the disease is divided into mild (>5% factor), moderate (2-5%), or severe (<2%). The cornerstone of haemophilia management has been the replacement of the deficient factor with purified concentrate of factor VIII or IX as home therapy. Treatment for patients with mild haemophilia will tend to be given on demand (on the evidence of a bleed), whereas patients with severe haemophilia will receive prophylaxis from the age of 1-2 years (this artificially converts the haemophilia from severe to mild, resulting in a significant reduction in arthropathy).5 Today, although purified clotting factors are relatively safe with respect to bloodborne viruses (hepatitis and HIV), the risk of new variant Creutzfeldt-Jakob disease is indeterminable, and inhibitor development must also be considered an important issue.
Until recently, most clotting factors used for treating haemophilias A and B were obtained by the extraction and purification of clotting factor from pooled human plasma, with all the attendant risks this brings—for example, transmission of HIV and hepatitis C virus. Recombinant factor VIII concentrates were developed during the 1980s after the structures of the genes for factors VIII and IX were determined; currently available recombinant factor VIII concentrates include Recombinate (expressed in Chinese hamster ovary cells in culture), Kogenate (expressed in baby hamster kidney cells), and Refracto (second generation recombinant factor VIII lacking part of the gene, expressed in Chinese hamster ovary cells6). All patients under 16 years are now being treated with recombinant factor VIII. Recombinant factor IX will also be available in the near future.7
Patients may still be exposed to viruses, however, as the cells that express recombinant proteins (as described above) may themselves harbour viruses. In addition, the cells are grown in complex culture media containing fetal calf serum, bovine serum albumin, and other nutrients that may also harbour viruses. Finally, many factor concentrates are stabilised with human albumin, which may harbour viruses too. Third generation products, available in the near future, will have no added human or bovine protein.
Disorders associated with increased risk of thrombosis
The term thrombophilia is used to describe inherited or acquired disorders of blood coagulation that result in an increased risk of thrombosis. The number of heritable disorders has grown considerably since the discovery of antithrombin III deficiency in 1965. Recently described inherited polymorphisms include mutations of the factor V and prothrombin genes. Of major interest is the recent finding that thrombophilia is associated with recurrent fetal loss and pre-eclampsia.
Factor V mutations
Several hereditary causes for thrombosis have been described (box), but there are still many cases of familial thrombotic disease for which the underlying cause is not known. Major advances have been made, however—notably, the discovery of the factor V (Leiden) and prothrombin gene mutations. The factor V Leiden mutation was described by Bertina and coworkers8 after observations made by Dahlback et al that in plasma samples from one family with thrombotic tendency the effect of activated protein C was reduced.9 Activated protein C is a key anticoagulant that rapidly downgrades thrombin generation by cleaving and inactivating phospholipid-bound activated forms of coagulation factors V and VIII (factors Va and VIIIa). The inactivation takes place at three sites: Arg506, Arg306, and Arg679. Resistance to activated protein C, resulting in a poor anticoagulant response, is present in 5-10% of normal subjects in Europe but is virtually absent in Japanese and in Africans.10,11 Factor V Leiden, first described in the Leiden thrombophilia study,8 is a variant factor V caused by the substitution of Gln at Arg506 and is responsible for 90% of the cases of resistance to activated protein C. Recently, a new factor V mutation associated with this resistance has been described where the amino acid substitution is at Arg306 (factor V Cambridge).12 The factor V mutations are associated with an estimated relative risk of venous thrombosis of about 7 for heterozygotes12,13 and about 80 for homozygotes.14 The phenotype for resistance to activated protein C is heterogeneous and influenced by multiple genetic and environmental factors, such as pregnancy, the contraceptive pill, and increased concentrations of factor VIII.
Inherited disorders associated with hypercoagulability
Antithrombin (previously termed antithrombin III) deficiency
Protein C deficiency
Protein S deficiency
Resistance to activated protein C and factor V Leiden mutation
Prothrombin gene mutation
Abnormalities of fibrinogen or plasminogen
Factor XII deficiency
Prothrombin gene mutation
Prothrombin is the precursor of thrombin, a key enzyme in haemostasis and thrombosis. Poort and colleagues have examined the prothrombin gene in detail using molecular techniques—for example, polymerase chain reaction with DNA sequencing—in patients with venous thromboembolism. Of these patients, 18% had a G→A switch at nucleotide 20210 in the 3′ untranslated region of the gene15; this was found in only 1% of normal control subjects.16 In a population based, case-control study the researchers found that this mutation increased the frequency of venous thromboembolism threefold, with raised plasma prothrombin concentrations in subjects with the mutant allele. Other genetic disorders with increased tendency to thromboembolic disease will be described in the near future.
Causes of hyperhomocysteinaemia
Inherited Enzyme deficiencies
Cobalamin coenzyme synthesis Methionine synthase Others
Transport defects
Transcobalamin II deficiency
Acquired
Vitamin B-12 deficiency Folate deficiency Pyridoxine deficiency Chronic renal disease Hypothyroidism Folate antagonists—for example, methotrexate Vitamin B-12 antagonists—for example, nitrous oxide
Homocysteinaemia and thrombotic disease
Homocysteine is a amino acid that contains sulphur, and the homocysteine pathway involves enzymes that require vitamin B-12, folate, and pyridoxine as cofactors. High blood concentrations of homocysteine are an independent risk factor for both venous and arterial thrombosis. Abnormalities of homocysteine metabolism may occur through inherited or nutritional disorders (box). In nutritional deficiencies, frank deficiency of vitamin B-12 or folate will lead to megaloblastic anaemia, and milder deficiencies are associated with a predisposition to thrombosis. In the inherited form the high concentrations of homocysteine directly damage the vascular system. Patients homozygous for an inborn error of metabolism resulting in homocystinuria and hyperhomocysteinaemia were first reported to be prone to precocious occlusive vascular disease by McCully in 1969.17–19 The assay of plasma homocysteine has now been added to the ever growing number of procedures in screening for thrombophilia.
Malignant disorders
There have been few recent major haematological advances in malignant disorders, and most work on acute and chronic leukaemias has focused on refining treatment protocols through ongoing clinical trials. New agents, including the purine analogues (such as fludarabine and 2-chlorodeoxyadenosine), have proved useful for treating chronic lymphoid malignancies, including chronic lymphocytic leukaemia and hairy cell leukaemia, and specialists are keen for oral preparations of these drugs to be developed for use in outpatients.
Herpes virus and multiple myeloma
The bone marrow microenvironment has been shown to be important in the pathogenesis of multiple myeloma, and non-malignant cells in the marrow may promote the growth of malignant cells.20–22 Recently a human herpesvirus, originally described in patients with Kaposi’s sarcoma and termed KS-associated herpesvirus, has been found in bone marrow dendritic cells of patients with myeloma (although not found in malignant plasma cells themselves).23 This herpesvirus has been found in 25% of patients with monoclonal gammopathy, a disorder that may transform to myeloma in a small proportion of cases.24 Through in situ hybridisation, KS-associated herpesvirus was found in 18 of 21 patients with myeloma but not in patients with other haematological malignancies or in normal subjects. In addition, the myeloma dendritic cells were found to transcribe viral interleukin 6, the human homologue of which is a growth factor for myeloma cells. No clear evidence exists yet for the direct role of KS-associated herpesvirus in the development of myeloma, but the circumstantial evidence does seem to support its importance. The results of current studies are awaited.
Other advances in haematology
Stimulation of fetal haemoglobin production to ameliorate sickling disorders
Transplantation through the umbilical cord, including bone marrow transplantation from unrelated donors
Arsenic for treatment of acute promyelocytic leukaemia (M3)
Bisphosphonates shown to be of value in myeloma bone disease
Low molecular weight heparins for treatment of thrombosis
Identification and mapping of hereditary haemochromatosis mutation
DNA vaccine trial for treatment of lymphomas and other haematological cancers
Modification of drug resistance in tumour cells
The future
Screening for specific mutations
Most molecular tests—for example, Southern blotting, polymerase chain reaction, DNA sequencing—are labour intensive, expensive, and suitable for testing relatively small numbers of samples. New, DNA based chip technology may speed up this process, allow larger numbers of samples to be tested simultaneously, and reduce the cost. The US company Affymetrix has designed GeneChip probe arrays, in which glass wafers are covered with lawns of short oligonucleotides of known sequence (fig 2).25 The chip is encased in plastic, and all reactions take place in a closed system. Test DNA binds with complementary sequences on the chip. The resultant geometric pattern of sequences defines the nature of the underlying mutation in the test sample. Manufacturing involves a photolithographic process in which a mask is used to selectively activate synthesis regions on the chip. One expression chip can analyse hundreds of different genes, while the p53 (tumour suppressor gene) chip analyses all known mutations in the p53 gene.
Figure 2.

GeneChip array (Affymetrix), a closed system containing many oligonucleotides on a lawn to which test DNA is added, has potential to screen for large numbers of mutations. Figure shows the closed chip, the lawn to which test samples hybridise, and magnified view of result for p53 analysis
Gene chip technology promises to be a powerful approach for parallel analysis of hundreds of thousands of distinct sites in the genome. The technology is currently being used in the fields of genomics, disease management, and genotyping—and perhaps eventually will be used for diagnosis. It will allow diseases to be linked to specific mutations and will enable analysis at population level. Such technology should link in well with the human genome project, the goal of which is to sequence the entire human genome and establish physical and genetic maps. Other technologies that will have a major impact over the next few years include real time polymerase chain reaction analysis (for accurate quantitation of specific DNA sequences) and spectral (colour based) karyotyping for use in accurate detection of structural chromosome abnormalities.
Molecular therapeutics
Molecular therapeutics is an exciting area, and worldwide trials of the many potential applications are under way. Major molecular strategies include antisense therapy, whereby short oligonucleotides are constructed that bind to target mRNA, preventing its processing and translation (and hence protein production).26 Molecular targets include BCR-ABL in chronic myeloid leukaemia27 and oncogenes such as c-myc, ras, and cyclin D, which are overexpressed in several haematological malignancies.28 An alternative strategy involves ribozyme technology, in which small RNA molecules are engineered that bind to target RNA species, causing their cleavage and destruction.29
Acknowledgments
We thank R&D Systems for providing figure 1 and Affymetrix (Santa Clara, CA) for figure 2, which has been adapted.
Footnotes
Funding: No specific funding.
Competing interests: None declared.
References
- 1.Means RT, Krantz SB. Progress in understanding the pathogenesis of the anemia in chronic disease. Blood. 1992;80:1639–1647. [PubMed] [Google Scholar]
- 2.Baer AN, Dessypris EN, Krantz SB. The pathogenesis of anemia in rheumatoid arthritis: a clinical and laboratory analysis. Semin Arthritis Rheum. 1990;19:209–223. doi: 10.1016/0049-0172(90)90001-v. [DOI] [PubMed] [Google Scholar]
- 3.Seligman PA, Schleicher RB, Allen RH. Isolation and characterization of the transferrin receptor from human placenta. J Biol Chem. 1979;254:9943–9946. [PubMed] [Google Scholar]
- 4.Kohgo Y, Niitsu Y, Nishisato T, Kato J, Kondo H, Sasaki K, et al. Quantitation and characterization of serum transferrin receptor in patients with anemias and polycythemias. Jpn J Med. 1988;27:64–70. doi: 10.2169/internalmedicine1962.27.64. [DOI] [PubMed] [Google Scholar]
- 5.Roberts HR, Ebest ME. Current management of hemophilia B. Hematol Oncol Clin North Am. 1993;7:1269–1280. [PubMed] [Google Scholar]
- 6.Berntorp E. Second generation B-domain deleted recombinant factor VIII. Thromb Haemost. 1997;78:256–260. [PubMed] [Google Scholar]
- 7.White GC, Beebe A, Nielsen B. Recombinant factor IX. Thromb Haemost. 1997;78:261–265. [PubMed] [Google Scholar]
- 8.Bertina RM, Koeleman BP, Koster T, Rosendaal FR, Dirven RJ, de Ronde H, et al. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature. 1994;369:64–67. doi: 10.1038/369064a0. [DOI] [PubMed] [Google Scholar]
- 9.Dahlback B, Carlsson M, Svensson PJ. Familial thrombophilia due to a previously unrecognized mechanism characterized by poor anticoagulant response to activated protein C: prediction of a cofactor to activated protein C. Proc Natl Acad Sci U S A. 1993;90:1004–1008. doi: 10.1073/pnas.90.3.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takiyama O, Ishida F, Kodaira H, Kitano K. APC-resistance and MnlI genotype (Gln506) of coagulation factor V are rare in Japanese population. Thromb Haemost. 1995;74:996. [PubMed] [Google Scholar]
- 11.Rees DC, Cox M, Clegg JB. World distribution of factor V Leiden. Lancet. 1995;346:1133–1134. doi: 10.1016/s0140-6736(95)91803-5. [DOI] [PubMed] [Google Scholar]
- 12.Williamson D, Brown K, Luddington R, Baglin C, Baglin T. Factor V Cambridge: a new mutation (Arg306Thr) associated with resistance to activated protein C. Blood. 1998;91:1140–1144. [PubMed] [Google Scholar]
- 13.Koster T, Rosendaal FR, de Ronde H, Briet E, Vandenbrouke JP, Bertina RM. Venous thrombosis due to poor anticoagulant response to activated protein C: Leiden thrombophilia study. Lancet. 1993;342:1503–1506. doi: 10.1016/s0140-6736(05)80081-9. [DOI] [PubMed] [Google Scholar]
- 14.Rosendaal FR, Koster T, Vandenbroucke JP, Reitsma PH. High risk of thrombosis in patients homozygous for factor V Leiden (activated protein C resistance) Blood. 1995;85:1504–1508. [PubMed] [Google Scholar]
- 15.Poort SR, Michiels JJ, Reitsma PH, Bertina RM. Homozygosity for a novel missense mutation in the prothrombin gene causing a severe bleeding disorder. Thromb Haemost. 1994;72:819–824. [PubMed] [Google Scholar]
- 16.Poort SR, Rosendaal FR, Reitsma PH, Bertina RM. A common genetic variation in the 3′-untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increased incidence of venous thrombosis. Blood. 1996;88:3698–3703. [PubMed] [Google Scholar]
- 17.McCully KS. Vascular pathology of homocysteinemia. Implications for the pathogenesis of arteriosclerosis. Am J Pathol. 1969;56:111–128. [PMC free article] [PubMed] [Google Scholar]
- 18.D’Angelo A, Selhub J. Homocysteine and thrombotic disease. Blood. 1997;90:1–11. [PubMed] [Google Scholar]
- 19.Mayer EL, Jacobsen DW, Robinson K. Homocysteine and coronary atherosclerosis. J Am Coll Cardiol. 1996;27:517–527. doi: 10.1016/0735-1097(95)00508-0. [DOI] [PubMed] [Google Scholar]
- 20.Lichtenstein A, Tu Y, Fady C, Vescio R, Berenson J. Interleukin-6 inhibits apoptosis of malignant plasma cells. Cell Immunol. 1995;162:248–255. doi: 10.1006/cimm.1995.1076. [DOI] [PubMed] [Google Scholar]
- 21.Bataille R, Harousseau J-L. Multiple myeloma. N Engl J Med. 1997;336:1657–1664. doi: 10.1056/NEJM199706053362307. [DOI] [PubMed] [Google Scholar]
- 22.Uchiyama H, Barut BA, Mohrbacher AF, Chauhan D, Anderson KC. Adhesion of human myeloma-derived cell lines to bone marrow stromal cells stimulates interleukin-6 secretion. Blood. 1993;82:3712–3720. [PubMed] [Google Scholar]
- 23.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science. 1994;266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 24.Rettig MB, Ma HJ, Vescio RA, Pold M, Schiller G, Belson D, et al. Kaposi’s sarcoma-associated herpesvirus infection of bone marrow dendritic cells from multiple myeloma patients. Science. 1997;276:1851–1854. doi: 10.1126/science.276.5320.1851. [DOI] [PubMed] [Google Scholar]
- 25.Chee M, Yang R, Hubbell E, Berno A, Huang XC, Stern D, et al. Accessing genetic diversity with high density DNA arrays. Science. 1996;274:610–614. doi: 10.1126/science.274.5287.610. [DOI] [PubMed] [Google Scholar]
- 26.Bayever E, Iversen PL, Bishop MR, Sharp JG, Tewary HK, Arneson MA, et al. Systemic administration of a phosphorothioate oligonucleotide with a sequence complementary to p53 for acute myelogenous leukemia and myelodysplastic syndrome: initial results of a phase I trial. Antisense Res Devel. 1993;3:383–390. doi: 10.1089/ard.1993.3.383. [DOI] [PubMed] [Google Scholar]
- 27.De Fabritiis P, Petti MC, Montefusco E, De Propris MS, Sala R, Belluci R, et al. BCR-ABL antisense oligodeoxynucleotide in vitro purging and autologous bone marrow transplantation for patients with chronic myelogenous leukemia in advanced phase. Blood. 1998;91:3156–3162. [PubMed] [Google Scholar]
- 28.Schrump DS, Chen A, Consoli U. Inhibition of lung cancer proliferation by antisense cyclin D. Cancer Gene Ther. 1996;3:131–135. [PubMed] [Google Scholar]
- 29.Irie A, Kijima H, Ohkawa T, Bouffard DY, Suzuki T, Curcio LD, et al. Anti-oncogene ribozymes for cancer gene therapy. Adv Pharmacol. 1997;40:207–257. doi: 10.1016/s1054-3589(08)60141-6. [DOI] [PubMed] [Google Scholar]