

Abstract
Protein 2CATPase of picornaviruses is involved in the rearrangement of host cell organelles, viral RNA replication, and encapsidation. However, the biochemical and molecular mechanisms by which 2CATPase engages in these processes are not known. To characterize functional domains of 2CATPase, we have focused on a cysteine-rich motif near the carboxy terminus of poliovirus 2CATPase. This region, which is well conserved among enteroviruses and rhinoviruses displaying an amino acid arrangement resembling zinc finger motifs, was studied by genetic and biochemical analyses. A mutation that replaced the first cysteine residue of the motif with a serine was lethal. A mutant virus which lacked the second of four potential coordination sites for zinc was temperature sensitive. At the restrictive temperature, RNA replication was inhibited whereas translation and polyprotein processing, assayed in vitro and in vivo, appeared to be normal. An intragenomic second-site revertant which reinserted the missing coordination site for zinc and recovered RNA replication at the restrictive temperature was isolated. The cysteine-rich motif was sufficient to bind zinc in vitro, as assessed in the presence of 4-(2-pyridylazo)resorcinol by a colorimetric assay. Zinc binding, however, was not required for hydrolysis of ATP. 2CATPase as well as its precursors 2BC and P2 were found to exist in a reduced form in poliovirus-infected cells.
Picornaviridae is a family of nonenveloped, single-stranded positive-sense RNA viruses. The family is subdivided into six genera: Enterovirus, Rhinovirus, Parechovirus, Cardiovirus, Aphthovirus, and Hepatovirus. The most intensively studied picornavirus is Poliovirus, a member of the genus Enterovirus and the causative agent of poliomyelitis.
The mechanism of replication of picornavirus genomes is poorly understood. As the genome enters the host cells, it does not find enzymes specific for RNA-dependent RNA synthesis. Therefore, the genomes of picornaviruses contain sequences that code for several proteins designed to replicate the viral RNA. The genomes of picornaviruses encode one polyprotein that is cleaved by endogenous proteinases into functional proteins (63). The primer- and template-dependent RNA polymerase 3Dpol takes a central role in RNA replication. In vitro, the enzyme catalyzes three different types of reactions. First, 3Dpol synthesizes its primer by uridylylating the genome-linked protein VPg to VPg-pU(pU) (43) and uses this nucleotidyl protein to initiate chain elongation, a process that has been called protein priming (43, 49). Second, 3Dpol transcribes the RNA templates, yielding minus- and plus-strand RNA (21, 48). Third, 3Dpol unwinds double-stranded RNA during chain elongation (13).
Biochemical and genetic evidence indicate that 3Dpol is not sufficient for RNA replication and that additional viral (63) and cellular proteins are required (reviewed in reference 65). Little is known about the mechanisms by which accessory proteins participate in RNA replication. Among the virus-encoded proteins, the membrane-associated RNA-binding protein 3AB (59), the precursor of VPg (=3B) (63), stimulates the RNA chain elongation activity of 3Dpol (35, 41). Another viral polypeptide essential for RNA replication is proteinase 3CDpro, which forms RNP complexes with the 5′-terminal cloverleaf structure of the viral genome, together with the cellular protein poly(rC)-binding protein 2 (3, 40) or 3AB (27, 64). 3AB has been shown to inhibit the secretory pathway in eukaryotic cells (17), a function also exhibited by the viral protein 2B and its precursor, 2BC (5).
Protein 2CATPase (Fig. 1), the carboxy-terminal cleavage product of 2BC, contains a nucleoside triphosphate-binding motif conserved among Picornaviridae (23), Caliciviridae (31), and other small RNA and DNA viruses (24). A recent biochemical study has revealed that poliovirus 2CATPase specifically hydrolyzes ATP and that the ATPase activity is inhibited by 2 mM guanidine hydrochloride, an inhibitor of poliovirus RNA replication (44). 2BC is also capable of selectively hydrolyzing ATP (T. Pfister and E. Wimmer, unpublished results). 2CATPase as well as 2B and 2BC are associated with intracellular membranes of the host cell (7, 20). The membrane-targeting signal of 2CATPase has been mapped to an amino-terminal region (18) partially overlapping a predicted amphipathic helix (42) (Fig. 1). Poliovirus protein 2BC (2, 5, 14) as well as a fragment of 2CATPase comprising the amino-terminal 274 residues (57) are sufficient to induce in eukaryotic cells formation of vesicular structures resembling the structures observed in poliovirus-infected cells. The vesicular structures carry the viral replication complexes (RCs) attached to their surfaces (8).
FIG. 1.

General features of poliovirus protein 2CATPase. Regions of protein 2CATPase with suggested functions are indicated by brackets and amino acid positions. A, B, and C denote motifs conserved among superfamily 3 helicases and are required for the ATPase activity (44). Black diamonds indicate locations of previously published mutations that affect RNA replication; white diamonds indicate locations of hallmark mutations found in most guanidine-resistant and -dependent virus mutants (58). Arrows delineate a predicted domain organization (57).
Subcellular fractions containing the RCs are able to replicate viral RNA in vitro provided that the membranous structures are intact (8, 55, 56). It has been proposed that 2BC and/or 2CATPase play a role in the spatial organization of the RC, required for RNA replication (10). 2CATPase has been shown to bind RNA in vivo (10) and in vitro (47). Two RNA-binding domains have been mapped to the amino- and carboxy-terminal regions of 2CATPase (46) (Fig. 1). Genetic analyses, either by site-directed mutagenesis of 2CATPase (63) or by localization of mutations that confer drug resistance (45, 58, 60), have implied 2CATPase in a variety of functions during virus replication, ranging from virus uncoating and host cell rearrangement to RNA replication and encapsidation. The molecular mechanisms by which 2CATPase engages in these functions are unknown.
In an effort to learn more about the function(s) of poliovirus 2CATPase, we have focused on a cysteine-rich motif near the carboxy terminus (residues 269 through 286 [Fig. 1]). Genetic analyses revealed the involvement of the cysteine-rich motif in RNA replication and the requirement of a zinc finger-like zinc-binding motif. A biochemical approach indicated that the motif binds zinc in vitro.
MATERIALS AND METHODS
Alignments.
The alignment of enterovirus and rhinovirus polyprotein sequences was obtained online (A. C. Palmenberg, personal communication). The alignment was modified by hand in order to align the cysteine residues optimally.
Molecular cloning.
Plasmids were propagated in Escherichia coli DH5α grown in Luria-Bertani broth (Life Technologies, Gaithersburg, Md.). DNA manipulations were done by standard procedures (50) and according to manufacturers' protocols. Site-directed mutagenesis was carried out by the two-step overlapping PCR method (28).
Plasmid pT7PVM (11) was the progenitor of plasmids containing full-length cDNA of wild-type or mutant poliovirus type 1, strain Mahoney (PVM). In pT7PVMΔPCS1, the codon for Cys269 of protein 2CATPase was changed to Ser, resulting in a mutant lacking potential zinc coordination site (PCS) 1 (Table 1). Mutated DNA fragments were generated by PCR with Pfu polymerase (Stratagene, La Jolla, Calif.) and primers 1, 2, 3, and 4 (Table 2). The final PCR product was cut with MluI and HpaI (New England Biolabs, Beverly, Mass.) and ligated to the corresponding sites of pT7PVM (T4 ligase; Boehringer Mannheim, Indianapolis, Ind.). In pT7PVMΔPCS2, the codons for Cys272 and His273 of 2CATPase were changed to codons for Ser and Gln, respectively (Table 1), eliminating PCS 2. Primers 1, 2, 5, and 6 (Table 2) were used for PCR-mediated mutagenesis. Plasmid pT7PVMΔPCS12 carried mutations eliminating PCS 1 and PCS 2. To generate the mutant PCR fragment, primers 1, 2, 7, and 8 were used. pT7PVMΔPCS12 was used as the template in a PCR with primers 1, 2, 9, and 10 for the generation of pT7PVMΔPCS, in which the five codons for Cys and one for His were replaced by codons for Ser and Gln, respectively.
TABLE 1.
Plasmids used in this study and mutations in the cysteine-rich motif
| Plasmid | Sequencea
|
|||
|---|---|---|---|---|
| PCS 1 | PCS 2 | PCS 3 | PCS 4 | |
| pT7PVM; pGEX-CRwt | TGT | TGT CAC | TGC TGT | TGT |
| Cys | Cys His | Cys Cys | Cys | |
| pT7PVMΔPCS1 | TCG | TGT CAC | TGC TGT | TGT |
| Ser | Cys His | Cys Cys | Cys | |
| pT7PVMΔPCS2 | TGT | AGT CAG | TGC TGT | TGT |
| Cys | Ser Gln | Cys Cys | Cys | |
| pT7PVMΔPCS12 | AGT | AGT CAG | TGC TGT | TGT |
| Ser | Ser Gln | Cys Cys | Cys | |
| pGEX-CRmut | AGT | AGT CAG | AGC TCT | TCA |
| Ser | Ser Gln | Ser Ser | Ser | |
Mutations are shown in boldface on the DNA (top row) and protein (bottom row) levels.
TABLE 2.
Oligodeoxynucleotides used as primers in PCR
| No. | Polarity | Positiona | Sequence (5′ to 3′)b |
|---|---|---|---|
| 1 | Sense | 4141 | GCCACGTGGGTGACAGTTGGTTGAAG |
| 2 | Antisense | 5420 | GGCGCCGCGGTTGTACCTTTGCTGTCCG |
| 3 | Sense | 4938 | CTGAAATGTCGAAGAACTG |
| 4 | Antisense | 4916 | CTTCGACATTTCAGTAGC |
| 5 | Sense | 4947 | GTAAGAACAGTCAGCAACC |
| 6 | Antisense | 4922 | GCTGACTGTTCTTACACATTTC |
| 7 | Sense | 4950 | ATGAGTAAGAACAGTCAGCAACCAGC |
| 8 | Antisense | 4916 | GTTCTTACTCATTTCAGTAGC |
| 9 | Sense | 4988 | GCTCTCCTTTAGTGTCAGGTAAGG |
| 10 | Antisense | 4960 | CACTAAAGGAGAGCTTCTC |
| 11 | Sense | 4897 | GTCATGAGAATTCATTCTAGAGATGGG |
| 12 | Antisense | 5002 | TCTAAGCTTGGAAGATTTGTCC |
| 13 | Sense | 4832 | CCCCCCCATATGGCACACAGTGACG |
Position of 3′-terminal nucleotide in pT7PVM; positive-strand numbering.
Underlined sequences are complementary to pT7PVM.
For the reconstruction of revertants prN271H and rS272C, the PCR products obtained from reverse transcription-PCR (RT-PCR) (see below) were cut with MluI and HpaI and inserted into pT7PVM.
Plasmids pGEX-CRwt and pGEX-CRmut were constructed to express wild-type (wt) and mutant cysteine-rich motifs of 2CATPase fused to glutathione S-transferase (GST) in E. coli (GST-CRwt and GST-CRmut, respectively). The fragments between nucleotides 4884 and 5015 of pT7PVM and pT7PVMΔPCS were amplified by PCR using primers 11 and 12 (Table 2), digested with EcoRI and HindIII, and inserted into the corresponding sites of pGEX-KG (26). The generation of pGEX-2C for the expression of full-length 2C in E. coli has been described previously (44). All PCR-originated parts of plasmid DNA were verified by sequencing (Sequenase; United States Biochemical, Cleveland, Ohio).
Cells and virus.
HeLa R19 cells were grown as monolayers in Dulbecco modified Eagle's medium (DMEM; Life Technologies) supplemented with 5% bovine calf serum (Life Technologies) unless otherwise stated.
PVM was produced by in vitro transcription of pT7PVM and transfection of HeLa cells with transcript RNA at 37°C as described previously (61). Upon exhibiting complete cytopathic effect (CPE), cell monolayers were subjected to three cycles of freezing and thawing in culture medium. Cell debris was pelleted at 2,000 × g. The supernatant was titrated in a standard plaque assay (45). HeLa cells (5 × 107 or 1.4 × 108) were infected at a multiplicity of infection (MOI) of 10 PFU/cell. The supernatant was titrated and used as virus stock.
Virus was passaged at 39.5°C by infection of 5 × 107 cells with virus stock at an MOI of 10 PFU/cell. At complete CPE, a volume of supernatant corresponding to 5 × 108 PFU of virus stock was transferred to 5 × 107 fresh cells. This was repeated until passage 6. Plaque purification of virus was done at 39.5°C similarly to plaque assays except that cells were stained as described by Shepley et al. (52). Plaques of different sizes were picked through the agar with disposable tips attached to a micropipettor. The tips were rinsed with DMEM–3% bovine calf serum in a 24-well plate containing HeLa cell monolayers. The plate was incubated at 37°C until the majority of cells showed CPE, at which point virus supernatants were harvested.
RT-PCR and sequencing of PCR products.
Virus supernatant (100 μl) was incubated at 37°C for 30 min in the presence of 10 U of RNase-free DNase (Boehringer Mannheim) to digest residual DNA from the RT reactions. RNA was prepared by proteinase K digestion (Sigma, St. Louis, Mo.), phenol-chloroform-isoamyl alcohol extraction, and ethanol precipitation (30). Four-fifths of the RNA was reverse transcribed with avian myeloblastosis virus reverse transcriptase (Boehringer Mannheim) in a total volume of 20 μl containing 50 pmol of random DNA nonamers at 42°C for 1 h; 5 μl of the RT reaction mixture was amplified by PCR using Taq polymerase (Boehringer Mannheim) and primers 2 and 13 (Table 2). One-fifth of the RNA was directly subjected to PCR to exclude contamination with plasmid DNA. The products of RT-PCR were gel purified and sequenced with a Sequitherm cycle sequencing kit (Epicentre Technologies, Madison, Wis.) and appropriate primers.
In vivo labeling of viral macromolecules.
HeLa cells (5 × 107) were infected at an MOI of 10 PFU/cell. Viral RNA was labeled by adding actinomycin D (5 μg/ml; Calbiochem, La Jolla, Calif.) at 0.5 h postinfection (p.i.) and [5,6-3H]uridine (5 μCi/ml; 42 Ci/mmol; ICN Pharmaceuticals, Costa Mesa, Calif.) at 1.75 h p.i. At different time points, cells were washed once with ice-cold phosphate-buffered saline and lysed on ice with 200 μl of lysis buffer (10 mM Tris-HCl [pH 7.4], 1 mM EDTA, 140 mM NaCl, 1% Nonidet P40 [Sigma]). Nuclei and debris were removed by centrifugation. Incorporation of [3H]uridine into RNA was measured in duplicate samples by liquid scintillation counting of trichloroacetic acid (TCA)-precipitable radioactivity.
Viral proteins were labeled in the presence of Trans35S-label (22 μCi/ml; ICN Pharmaceuticals); 0.5 h prior to addition of the label, cells were washed twice with DMEM without methionine (Life Technologies) containing 120 mM excess NaCl (39). The same medium was used during subsequent incubation and labeling. Cells were lysed, and incorporated radioactivity was counted as described above. Viral proteins were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (34) and autoradiography.
Western blot analysis.
Lysates of poliovirus-infected cells were separated by SDS-PAGE and electrotransferred to nitrocellulose membranes (Protran; Schleicher & Schuell, Keene, N.H.). Western blotting was done as described elsewhere (19), using 5% nonfat milk powder for blocking, anti-2C monoclonal antibody 91.10 (44), and an alkaline phosphatase-conjugated anti-mouse antibody (Biosource, Camarillo, Calif.). The blot was developed by using nitroblue tetrazolium–5-bromo-4-chloro-3-indolylphosphate tablets (Sigma) dissolved in water.
Expression of recombinant proteins in E. coli.
GST-2C was expressed and purified as described previously (44). E. coli BL21(DE3) carrying pGEX-CRwt or pGEX-CRmut was grown in 2× YT medium (50). GST-CRwt and GST-CRmut were expressed upon the addition of 0.1 mM isopropyl-β-d-thiogalactopyranoside (Sigma) at 30°C for 3 h and purified as described for GST-2C (44). Protein expression and purification were monitored by SDS-PAGE. The amount of protein in the eluates was determined by the Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, Calif.) and by measuring the absorption at 280 nm in the presence of 6 M guanidine hydrochloride (22).
Determination of metal concentration.
Protein samples were digested in a total volume of 50 μl with 50 μg of proteinase K (Sigma) per ml in HBS 200 (50 mM HEPES-KOH [pH 7.5], 0.2 M NaCl) at 56°C for 30 min. Subsequently, an identical volume of HBS 200 containing 5 mM iodoacetamide (IAM; Sigma) and 0.2 mM 4-(2-pyridylazo)resorcinol (PAR; Sigma) was added. The absorption at 490 nm was measured in a microplate reader (Dynatech Laboratories, Chantilly, Va.). HBS 200 containing 5 to 30 μM zinc acetate was used to create a standard curve. The amount of protein in the samples to be analyzed for metal content was adjusted so that the amount of metal released was in the linear range of the standard curve.
ATPase assay.
One microgram of protein was incubated with 3 mM ATP at 37°C for 30 min in a total volume of 60 μl containing 20 mM HEPES-KOH (pH 6.8), 4 mM magnesium acetate, and 5 mM dithiothreitol (DTT). The reaction was stopped on ice, and 60 μl of ice-cold 16% TCA was added. The released phosphate was quantified by a colorimetric assay (44).
RESULTS
Sequence comparison of the cysteine-rich motifs of picornavirus 2C proteins.
Poliovirus protein 2CATPase contains a cysteine-rich motif between amino acid residues 269 and 286 (Fig. 1) that is well conserved among enteroviruses and rhinoviruses (Fig. 2). The cysteine pattern resembles a zinc-binding motif with four PCSs (15). PCSs 2 and 3 are often occupied by two cysteine residues. PCS 2 of protein 2C of poliovirus and rhinovirus 14 consists of a cysteine and a histidine residue, both representing potential zinc-coordinating residues. In enterovirus 71, coxsackie A virus 16, and some rhinoviruses, PCS 2 does not contain a cysteine residue; instead, there are asparagine, threonine, or serine residues which potentially form a hydrogen bond with a water molecule that may coordinate zinc, an arrangement observed in some zinc-binding proteins (15).
FIG. 2.

Protein sequence alignment of the cysteine-rich motif in protein 2CATPase of enteroviruses and rhinoviruses. Cysteine residues are printed in bold. PCSs are shaded and numbered from left to right. Abbreviations: PV, poliovirus; EV, bovine enterovirus; CAV and CBV, coxsackie A virus and coxsackie B virus, respectively; SVDV, swine vesicular disease virus; HRV, human rhinovirus. a, numbering of amino acid residues according to the PVM sequence.
Surrounding the PCSs, conserved amino acid residues are apparent (Fig. 2). Charged residues are positioned between PCSs 1 and 2. A highly conserved asparagine residue often flanked by hydrophobic residues appears between PCSs 2 and 3. PCS 3 is preceded by one or two basic residues. PCSs 3 and 4 are separated by three hydrophobic residues, the first of which is exclusively a proline. PCS 4 is strictly followed by the sequence glycine-lysine-alanine and a hydrophobic alipathic amino acid residue. The conservation of a cysteine-rich motif in protein 2C of enteroviruses and rhinoviruses suggests a crucial and unique function of this domain.
Mutant polioviruses that lack PCS 1 or PCS 2.
Mutational analysis was used to address the significance of the cysteine-rich region in poliovirus protein 2CATPase. The codon for Cys269 was changed by site-directed mutagenesis to Ser (pT7PVMΔPCS1 [Table 1]), thereby eliminating PCS 1. The mutated plasmid pT7PVMΔPCS1 was transcribed into RNA, which was used for the transfection of HeLa cells. Despite repeated attempts, no virus was recovered as determined by plaque assays (not shown). Although the codon for Cys269 was changed by two nucleotides, the failure of rescuing any virus, either through direct reversion or a suppressor mutation, was surprising and indicated severe consequences of this mutation for viral replication.
PCS 2 was eliminated by replacing the codons for Cys272 and His273 to the codons for Ser and Gln, respectively (pT7PVMΔPCS2 [Table 1]). Upon transfection of HeLa cells with the transcript RNA, infectious virus (PVMΔPCS2) was obtained. The plaques of PVMΔPCS2 were slightly smaller than those of wt PVM (not shown).
RNAs that harbor the PCS 1 or PCS 2 mutations were translated in vitro, and the translation products were compared to those obtained with transcript RNA of pT7PVM (Fig. 3). Translation of the polypeptide appeared to be normal for both mutants, indicating an intact open reading frame. Moreover, neither mutant expressed a defect in polyprotein processing.
FIG. 3.

In vitro translation reactions in the absence of RNA (lane 1) or primed with transcript RNA of pT7PVM (lane 2), pT7PVMΔPCS1 (lane 3), or pT7PVMΔPCS2 (lane 4). An extract of poliovirus-infected HeLa cells was used as marker (lane M). Viral proteins were labeled with [35S]methionine and visualized by autoradiography. Positions of nonstructural proteins are indicated to the left.
PVMΔPCS2 is temperature sensitive.
A virus stock of PVMΔPCS2 was prepared at 37°C. To ensure that revertants did not accumulate during virus multiplication, the sequence of the mutated region was determined by sequencing of an RT-PCR product. No reversions were observed, indicating that PVMΔPCS2 was viable and relatively stable under standard growing conditions. Indeed, in a single-cycle growth experiment, the replication of PVMΔPCS2 was only slightly delayed compared to that of wt PVM and reached virus titers comparable to those of PVM (Fig. 4A). At 39.5°C, however, PVMΔPCS2 showed a delay in virus multiplication and a reduction in maximal virus titer. Thus, PVMΔPCS2 had a defect that was apparent only at elevated temperatures.
FIG. 4.

Single-cycle virus growth curves. HeLa cell monolayers were infected at an MOI of 10 PFU/cell. At different time points p.i., supernatants were harvested and their virus titers were determined.
To select for revertants, PVMΔPCS2 was passaged at 39.5°C. Virus obtained after passages 1 and 6 as well as unpassaged PVMΔPCS2 (i.e., passage 0) were plaque purified at 39.5°C, and their genomic regions encoding the cysteine-rich motif of 2CATPase were sequenced (Table 3). The original mutant PVMΔPCS2 was not found in any of the plaque-purified viruses sequenced. Even unpassaged PVMΔPCS2 reverted during plaque purification at 39.5°C. Two isolates that reverted Ser272 to Cys (rS272C) without changing mutation Gln273 were found. One isolate reverted both Ser272 and Gln273 to the wild-type residues Cys and His, respectively. Interestingly, the majority of viruses sequenced changed the proximal adjacent Asn271 to His (prN271H) (Table 3).
TABLE 3.
Sequences of plaque-purified PVMΔPCS2 after passaging at 39.5°C
| Construct | Sequencea
|
Passage no. | No. of isolates/no. of isolates sequenced | |
|---|---|---|---|---|
| DNA (nucleotides 4928–4945) | Protein (residues 269–274) | |||
| pT7PVM | TGT AAG AAC TGT CAC CAA | C K N C H Q | NAb | NA |
| Virus stock PVMΔPCS2 | TGT AAG AAC AGT CAG CAA | - - - S Q - | NA | NA |
| Revertant to wt | TGT AAG AAC TGT CAC CAA | - - - C H - | 6 | 1/6 |
| Revertant rS272C | TGT AAG AAC TGT CAG CAA | - - - C Q - | 0 | 1/6 |
| 6 | 1/6 | |||
| Revertant prN271H | TGT AAG CAC AGT CAG CAA | - - H S Q - | 0 | 5/6 |
| 1 | 4/4 | |||
| 6 | 4/6 | |||
Original mutations are in boldface; reversions are underlined.
NA, not applicable (plasmid and RT-PCR product of virus stock).
The regions that encode the cysteine-rich motifs of the revertants prN271H and rS272C were cloned (by segment exchange) into the full-length cDNA of wt poliovirus to study the effect of the reversions on virus phenotype. In a single-cycle growth experiment at 39.5°C, prN271H and rS272C exhibited growth characteristics indistinguishable from those of wt virus (Fig. 4B). Thus, a single amino acid change at position 271 to His or a reversion of Ser272 to wt Cys was sufficient to overcome the temperature-sensitive phenotype of PVMΔPCS2.
Translation of PVMΔPCS2 in vivo is wt-like.
In vivo labeling of viral proteins was undertaken to compare the synthesis and processing of viral proteins in cells infected with either wt PVM or mutant PVMΔPCS2. Incorporation of 35S-labeled methionine was measured between 3 and 4.5 and between 4 and 5.5 h p.i., both at 37 and 39.5°C (Fig. 5A). Ten minutes before the addition of translabel, guanidine-HCl was added to a final concentration of 2 mM to suppress viral RNA replication. The total amount of viral proteins synthesized in cells infected with the mutant was roughly half of the amount scored with PVM. Wild-type and mutant viruses showed similar, relatively small reductions of protein synthesis at the elevated temperature at both time periods during which labeling was performed. These data make it unlikely that the defect of PVMΔPCS2 at 39.5°C is due to inefficient translation.
FIG. 5.

In vivo labeling of viral proteins with [35S]methionine. (A) Viral proteins were labeled in the presence of guanidine-HCl at the time period and temperature indicated. TCA-precipitable radioactivity was measured by liquid scintillation counting. (B) Viral proteins were labeled in the absence of guanidine-HCl from 4 to 5.5 h p.i. Proteins were separated by SDS-PAGE and visualized by autoradiography. Positions of nonstructural proteins are indicated.
SDS-PAGE analysis of the viral proteins synthesized in vivo between 4 and 5.5 h p.i. in the absence of guanidine revealed no difference in the processing patterns of the wt and mutant polyproteins at 37 and 39.5°C (Fig. 5B). We conclude that temperature sensitivity of PVMΔPCS2 is not due to a defect in viral polyprotein processing in vivo.
PVMΔPCS2 is impaired in RNA replication at 39.5°C.
Viral RNA synthesis was measured by in vivo labeling of viral RNA with [3H]uridine at 37 and 39.5°C, starting at 1.75 h p.i. (Fig. 6A). At 37°C, RNA synthesis of PVMΔPCS2 was only slightly impaired compared to that of PVM. At 39.5°C, however, RNA synthesis of PVMΔPCS2 was severely (50-fold) reduced compared to that at 37°C, whereas RNA synthesis of PVM was hardly affected. This result indicated that mutant PVMΔPCS2 had a defect in RNA replication at the higher temperature. The reconstructed revertants rS272C and prN271H rescued RNA synthesis at 39.5°C to near wt levels (Fig. 6B), an observation suggesting that restoration of PCS 2 reversed the defect of PVMΔPCS2 in RNA replication.
FIG. 6.

In vivo labeling of viral RNA with [3H]uridine starting at 1.75 h p.i. Cells were lysed at different time points, and TCA-precipitable radioactivity was measured by liquid scintillation counting.
Protein 2CATPase occurs in a reduced form in the infected cell.
In poliovirus-infected cells, polypeptides 2CATPase and 2BC are predominantly located in viral RCs, which are intimately connected to membranous vesicles (7, 10). The vesicles are derivatives of intracellular compartments including the endoplasmic reticulum (5, 9, 51) and may therefore feature an oxidizing lumen. Since 2B and 2BC have been reported to permeabilize biological membranes (1, 62), the oxidizing lumen of the vesicles may determine the redox equilibrium of the RC. In an oxidizing RC, thiol groups of protein 2CATPase were likely to be oxidized, rendering them unable to coordinate metal ions. To determine whether the thiol groups of protein 2CATPase and its precursors are oxidized in vivo, poliovirus-infected cells were lysed at 5.5 h p.i. in the absence or presence of 10 mM IAM. IAM does not affect disulfide bonds but alkylates free thiol groups, preventing them from subsequent oxidation (54, 66). The lysates were analyzed by SDS-PAGE in the absence or presence of the reducing agent DTT. 2CATPase, 2BC, and P2 were visualized by Western blotting (Fig. 7). In the absence of IAM and DTT, 2CATPase and its precursor proteins migrated faster than expected and the bands appear “fuzzy,” indicating a heterogeneous protein structure (lane 1). Addition of 10 mM DTT to the gel loading buffer resulted in a sharpening of bands and a slight retardation of their migration (compare lane 3 with lane 1). Thus, the heterogeneous electrophoretic properties of 2CATPase and its precursors in the absence of DTT are due to oxidation events. No oxidation was observed if the cells were lysed in the presence of IAM (lane 2), in which case addition of DTT had no effect (lane 4). This observation indicates that the thiol groups of protein 2CATPase, 2BC, and P2 are in a reduced form in the infected cell. Moreover, the result shows that the poliovirus RC represents a reducing environment.
FIG. 7.
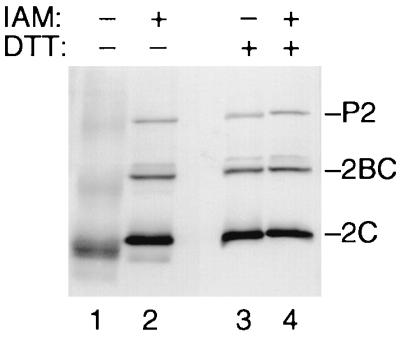
Proteins 2CATPase, 2BC, and P2 are in a reduced form in infected cells. Poliovirus-infected cells were lysed in the absence (−) or presence (+) of 10 mM IAM. Proteins were separated by SDS-PAGE in the absence (−) or presence (+) of 10 mM DTT. 2CATPase, 2BC, and P2 were visualized by Western blotting using an anti-2C monoclonal antibody.
The cysteine-rich motif of poliovirus 2CATPase binds zinc in vitro.
GST-CRwt (Fig. 8A) was immobilized on glutathione-Sepharose beads and treated with EDTA or zinc acetate according to the flow chart depicted in Fig. 8B. The eluted GST-CRwt was digested with proteinase K and treated with IAM in order to release metal ions bound to thiol groups. The metal content was determined by using PAR, which forms a Me2+-PAR2 complex (29, 38). Complex formation was monitored by an increase in absorption at 490 nm and quantified by using a standard curve plotted from known concentrations of zinc acetate (Fig. 8C). EDTA-treated GST-CRwt contained only trace amounts of metal (Fig. 8B, sample 1), whereas the zinc-treated protein (sample 2) contained almost 2 mol of metal ion per mol of protein (Fig. 8B). The same amount of metal was found if the protein has been treated first with EDTA and subsequently with zinc (sample 3). This finding indicates that GST-CRwt binds zinc in vitro. Zinc binding was reversible, since treatment with zinc followed by treatment with EDTA resulted in the loss of metal (sample 4).
FIG. 8.



The cysteine-rich motif binds zinc in vitro. (A) Amino acid sequences of peptides expressed as GST fusion proteins in E. coli. GST-CRwt contained residues 255 to 297 of poliovirus 2CATPase encompassing the cysteine-rich motif, residues 269 to 286. In GST-CRmut, the cysteine and histidine residues were replaced by serine and glutamine, respectively. GST was the product of the expression plasmid pGEX-KG. (B) Flowchart of GST-CRwt purification and treatment with zinc acetate and/or EDTA. The block diagram (bottom) shows the metal content of four protein preparations, determined by measuring the absorption at 490 nm in the presence of 0.1 mM PAR. (C) Typical standard curve showing the relationship between zinc concentration and absorption in the presence of PAR. The best-fit curve and its correlation were calculated with the program Cricket Graph (Cricket Software, Malvern, Pa.). (D) Metal content of GST-CRwt, GST-CRmut, and GST after zinc treatment and purification. The average and standard deviation of quadruplicate measurements are shown.
To determine whether the cysteine-rich motif of GST-CRwt contains the zinc-binding site, GST and GST-CRmut (Fig. 8A) were expressed in E. coli. Both fusion proteins were treated and purified as sample 2 of GST-CRwt (Fig. 8B). The metal content of the proteinase K digested eluates was determined (Fig. 8D). Both GST and GST-CRmut bound approximately one zinc ion per molecule of protein, whereas GST-CRwt bound more than two zinc ions per molecule. Thus, the cysteine-rich motif was zinc binding by virtue of its cysteine and histidine residues. In addition, the result suggested that GST itself also binds zinc.
The ATPase activity of GST-2C does not require zinc.
GST fused to full-length 2C (GST-2C) was expressed in E. coli and purified on glutathione-Sepharose as described previously (44) except that the binding reaction mixture contained either 1 mM EDTA or 0.1 mM zinc acetate. Purified GST-2C was assayed for metal content and ATPase activity. EDTA-treated GST-2C contained roughly three times less metal than the zinc-treated protein (Fig. 9A), an observation indicating that full-length 2CATPase is able to bind zinc. Since the molar ratio of zinc to protein was roughly the same for GST-2C and GST-CRwt, it is likely that the cysteine-rich motif is the only zinc-binding moiety in protein 2CATPase. We failed to express GST-2C with a mutated cysteine-rich motif, presumably due to misfolding of the mutant protein and its ultimate degradation in E. coli.
FIG. 9.

Metal content and enzymatic activity of EDTA- and zinc-treated GST-2C. Bacterially expressed GST-2C was bound to glutathione-Sepharose beads in the presence of 1 mM EDTA or 0.1 mM zinc acetate. The beads were washed four times with 100 bead volumes each and eluted. (A) Metal content of the differently treated GST-2C preparations. (B) Specific ATPase activity of the two preparations. Inorganic phosphate released from ATP was determined by a colorimetric assay (44). The average and standard deviation of triplicate measurements are shown.
Using a colorimetric ATPase assay that measures the amount of inorganic phosphate released upon hydrolysis of ATP (44), we compared the specific ATPase activities of EDTA-treated and zinc-treated GST-2C (Fig. 9B). The zinc-treated preparation was approximately 10% less active in hydrolysis of ATP than the EDTA-treated sample. This difference is considered as insignificant, since the specific ATPase activity varies in this range among different preparations of GST-2C (unpublished observation). Thus, zinc binding does not appear to be required for the ATPase activity of protein 2CATPase. This result is in agreement with an earlier study (46) in which a recombinant protein 2CATPase that lacked the carboxy-terminal 77 residues (including the cysteine-rich motif) was active in ATP hydrolysis.
DISCUSSION
We have investigated the cysteine-rich motif in poliovirus protein 2CATPase. The motif is conserved among enteroviruses and rhinoviruses. It is, however, absent in the 2C sequences of other known picornaviruses. Three lines of evidence suggest to us that the cysteine-rich motif is zinc binding. First, the arrangement of the cysteine residues follows the pattern CX2–4cX6–8CX3–4C, which is similar to the zinc-binding motif within zinc fingers of type CCCC, in which one zinc ion is coordinated by four cysteine residues (33). The apparent absence of PCS 2 in some enteroviruses and most rhinoviruses is unique among zinc fingers. In these cases, however, PCS 2 may consist of a bridging water molecule between a polar amino acid side chain and the zinc ion. This configuration is found in some metal-binding enzymes (15). Second, elimination of PCS 2 in poliovirus 2CATPase by site-directed mutagenesis resulted in a temperature-sensitive mutant that reverted to the wt sequence or to a new zinc-binding motif of type CHCC. In both cases, PCS 2 was restored. Third, the cysteine-rich motif either on its own or in the context of full-length 2CATPase was zinc binding in vitro. The possibility that the cysteine residues in protein 2CATPase are engaged in disulfide bond formation in vivo was excluded (Fig. 7), emphasizing that free thiol groups in protein 2CATPase are available for zinc binding in poliovirus-infected cells.
Several methods for the detection and quantification of zinc and other metals bound to proteins have been described. Usually, these methods require highly sophisticated equipment or high-energy radioactive isotopes. We have used a simple and inexpensive assay which can be performed without special equipment or safety precautions. Metal is detected by the chelating substance PAR, which increases the absorption of light upon formation of a Me2+-PAR2 complex (29, 38). Absorption at 490 nm appeared to be proportional to the concentration of zinc ranging from 5 to 30 μM (Fig. 8C). The limitations of the PAR assay are low sensitivity, lack of metal specificity, and the difficulty in removing the metal ions from coordinating amino acid side chains. Sufficient amounts of proteins were obtained by using a common GST fusion expression and purification system (26). Sequential treatment of immobilized GST-2C and GST-CRwt with EDTA and zinc demonstrated that the cysteine-rich motif binds zinc in vitro. Finally, to make protein-bound metal ions accessible to PAR, the proteins were digested with proteinase K and the thiol groups were alkylated with IAM. Since the purity of the protein preparations varied among different samples and the measurement of the amount of protein is imprecise, metal-to-protein ratios calculated can be only approximate. However, the good agreement between the different protein preparations, e.g., sample 2 versus sample 3 of GST-CRwt (Fig. 8B) and GST-CRmut versus GST (Fig. 8D), indicates that the PAR assay is suitable for an approximate quantification. We suggest that the cysteine-rich motif binds zinc in a 1:1 molar ratio. In an ongoing study, the metal-binding capability of the cysteine-rich motif is being addressed by synchrotron radiation-induced X-ray emission (32). Preliminary results confirm that the motif binds zinc (T. Pfister, K. W. Jones, and E. Wimmer, unpublished results).
In vitro binding of zinc appears to be indicative for a role of zinc in protein function (6). Interestingly, the zinc-requiring function of 2CATPase is not strictly dependent on an invariant zinc-binding motif, since the motif can be of type CCCC (wt and rS272C) or CHCC (prN271H). This is in contrast to the zinc finger motif of Moloney murine leukemia virus nucleocapsid protein, which was defective in cDNA synthesis upon changing the zinc-binding motif from CCHC to CCHH or CCCC (25). In that study, however, the exchange of zinc-coordinating residues did not alter the spacing between them. In prN271H, the spacing between the zinc-coordinating residues is changed (Table 3), which may have compensated for the Cys-to-His change in PCS 2. This may suggest that the spacing requirements of Cys and His residues are different for zinc binding.
The function of 2CATPase that requires zinc binding has not been determined but appears to be related to viral RNA replication. The involvement of protein 2CATPase in RNA replication is well documented (63) although its precise role is unknown. Apparently, the ATPase activity of protein 2CATPase does not require zinc bound to the cysteine-rich motif. We suggest that the zinc-binding motif is a zinc finger. Zinc fingers are recognized as motifs that mediate specific protein-protein and protein-nucleic acid interactions (33). Indeed, poliovirus 2CATPase has been shown to oligomerize and to bind to proteins 2B and 2BC (16, 58). In addition, 2CATPase has been reported to bind specifically to an RNA structure near the 3′ end of poliovirus negative-strand RNA (4). The significance of zinc binding in these interactions is under investigation.
Zinc binding of protein 2CATPase may have structural consequences. 2CATPase has been predicted to consist of three domains (Fig. 1) (57). The zinc-binding motif is located between domains 2 (α/β) and 3 (α/α). The coordination of zinc may bring domain 3 close to domain 2 in a configuration crucial for an as yet unknown function. In support of this model, a poliovirus with a temperature-sensitive replication phenotype (2C-31) (37) carrying an insertion upstream of the zinc-binding motif resulted in a pseudo-revertant virus with additional mutations downstream of the zinc-binding motif (36). One plausible explanation for the emergence of the pseudo-revertant virus suggests that the reversion restored an interaction between regions upstream and regions downstream of the zinc-binding motif, that is, between domain 2 and domain 3. The zinc-binding motif may serve as a hinge between domains 2 and 3. We have recently proposed that ATP binding and hydrolysis by protein 2CATPase may regulate assembly and function of a complex required during virus replication (44). As a zinc finger, the cysteine-rich motif may mediate the specific interaction of 2CATPase with another constituent of the complex. Specific interaction may change the position of domain 3 relative to domain 2, which may affect a function of 2CATPase.
Other picornavirus proteins have been shown to bind zinc as well. The viral proteinases 2Apro of enteroviruses and rhinoviruses share a conserved pattern of cysteine and histidine residues, which have been proposed to chelate zinc (66). Indeed, 2Apro of human rhinovirus 2 has been shown to require zinc for proteinase function (53). It was suggested that zinc is a structural component of 2Apro (53). The leader protein of cardioviruses have a zinc finger-like CHCC motif. In vitro zinc binding of the leader protein of Theiler's murine encephalomyelitis virus, a cardiovirus, has been demonstrated (12).
The discovery of a zinc-binding motif, a new genetic element involved in RNA replication, provides a new handle for the investigation of poliovirus RNA replication in general and the role of 2CATPase in this process in particular.
ACKNOWLEDGMENTS
We thank Todd Miller for help in designing the mutations and Aniko Paul for carefully reading the manuscript.
This work was supported by grants AI15122 and AI32100 of the National Institute of Allergy and Infectious Diseases.
REFERENCES
- 1.Aldabe R, Barco A, Carrasco L. Membrane permeabilization by poliovirus proteins 2B and 2BC. J Biol Chem. 1996;271:23134–23137. doi: 10.1074/jbc.271.38.23134. [DOI] [PubMed] [Google Scholar]
- 2.Aldabe R, Carrasco L. Induction of membrane proliferation by poliovirus proteins 2C and 2BC. Biochem Biophys Res Commun. 1995;206:64–76. doi: 10.1006/bbrc.1995.1010. [DOI] [PubMed] [Google Scholar]
- 3.Andino R, Rieckhof G E, Achacoso P L, Baltimore D. Poliovirus RNA synthesis utilizes an RNP complex formed around the 5′-end of viral RNA. EMBO J. 1993;12:3587–3598. doi: 10.1002/j.1460-2075.1993.tb06032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Banerjee R, Echeverri A, Dasgupta A. Poliovirus-encoded 2C polypeptide specifically binds to the 3′-terminal sequences of viral negative-strand RNA. J Virol. 1997;71:9570–9578. doi: 10.1128/jvi.71.12.9570-9578.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barco A, Carrasco L. A human virus protein, poliovirus protein 2BC, induces membrane proliferation and blocks the exocytic pathway in the yeast Saccharomyces cerevisiae. EMBO J. 1995;14:3349–3364. doi: 10.1002/j.1460-2075.1995.tb07341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berg J M, Shi Y. The galvanization of biology: a growing appreciation for the roles of zinc. Science. 1996;271:1081–1085. doi: 10.1126/science.271.5252.1081. [DOI] [PubMed] [Google Scholar]
- 7.Bienz K, Egger D, Pasamontes L. Association of polioviral proteins of the P2 genomic region with the viral replication complex and virus-induced membrane synthesis as visualized by electron microscopic immunocytochemistry and autoradiography. Virology. 1987;160:220–226. doi: 10.1016/0042-6822(87)90063-8. [DOI] [PubMed] [Google Scholar]
- 8.Bienz K, Egger D, Pfister T, Troxler M. Structural and functional characterization of the poliovirus replication complex. J Virol. 1992;66:2740–2747. doi: 10.1128/jvi.66.5.2740-2747.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bienz K, Egger D, Rasser Y, Bossart W. Intracellular distribution of poliovirus proteins and the induction of virus-specific cytoplasmic structures. Virology. 1983;131:39–48. doi: 10.1016/0042-6822(83)90531-7. [DOI] [PubMed] [Google Scholar]
- 10.Bienz K, Egger D, Troxler M, Pasamontes I. Structural organization of poliovirus RNA replication is mediated by viral proteins of the P2 genomic region. J Virol. 1990;64:1156–1163. doi: 10.1128/jvi.64.3.1156-1163.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao X, Kuhn R J, Wimmer E. Replication of poliovirus RNA containing two VPg coding sequences leads to a specific deletion event. J Virol. 1993;67:5572–5578. doi: 10.1128/jvi.67.9.5572-5578.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen H-H, Kong W-P, Roos R P. The leader peptide of Theiler's murine encephalomyelitis virus is a zinc-binding protein. J Virol. 1995;69:8076–8078. doi: 10.1128/jvi.69.12.8076-8078.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cho M W, Richards O C, Dmitrieva T M, Agol A, Ehrenfeld E. RNA duplex unwinding activity of poliovirus RNA-dependent RNA polymerase 3Dpol. J Virol. 1993;67:3010–3018. doi: 10.1128/jvi.67.6.3010-3018.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cho M W, Tererina N, Egger D, Bienz K, Ehrenfeld E. Membrane rearrangement and vesicle induction by recombinant poliovirus 2C and 2BC in human cells. Virology. 1994;202:129–145. doi: 10.1006/viro.1994.1329. [DOI] [PubMed] [Google Scholar]
- 15.Coleman J E. Zinc proteins: enzymes, storage proteins, transcription factors, and replication proteins. Annu Rev Biochem. 1992;61:897–946. doi: 10.1146/annurev.bi.61.070192.004341. [DOI] [PubMed] [Google Scholar]
- 16.Cuconati A, Xiang W, Lahser F, Pfister T, Wimmer E. A protein linkage map of the P2 nonstructural proteins of poliovirus. J Virol. 1998;72:1297–1307. doi: 10.1128/jvi.72.2.1297-1307.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doedens J R, Kirkegaard K. Inhibition of cellular protein secretion by poliovirus proteins 2B and 3A. EMBO J. 1995;14:894–907. doi: 10.1002/j.1460-2075.1995.tb07071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Echeverri A C, Dasgupta A. Amino terminal regions of poliovirus 2C protein mediate membrane binding. Virology. 1995;208:540–553. doi: 10.1006/viro.1995.1185. [DOI] [PubMed] [Google Scholar]
- 19.Egger D, Bienz K. Protein (Western) blotting. Mol Biotechnol. 1994;1:289–305. doi: 10.1007/BF02921696. [DOI] [PubMed] [Google Scholar]
- 20.Egger D, Pasamontes L, Bolten R, Boyko V, Bienz K. Reversible dissociation of the poliovirus replication complex: functions and interactions of its components in viral RNA synthesis. J Virol. 1996;70:8675–8683. doi: 10.1128/jvi.70.12.8675-8683.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flanegan J B, Baltimore D. Poliovirus-specific primer-dependent RNA polymerase able to copy poly(A) Proc Natl Acad Sci USA. 1977;74:3677–3680. doi: 10.1073/pnas.74.9.3677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gill S C, Hippel P H. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 23.Gorbalenya A E, Blinov V M, Donchenko A P, Koonin E. An NTP binding motif is the most conserved sequence in a highly diverged monophyletic group of proteins involved in positive strand RNA viral replication. J Mol Evol. 1989;28:256–268. doi: 10.1007/BF02102483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gorbalenya A E, Koonin E V, Wolf Y I. A new superfamily of putative NTP-binding domains encoded by genomes of small DNA and RNA viruses. FEBS Lett. 1990;262:145–148. doi: 10.1016/0014-5793(90)80175-i. [DOI] [PubMed] [Google Scholar]
- 25.Gorelick R J, Chabot D J, Ott D E, Gagliardi T D, Rein A, Henderson L E, Arthur L O. Genetic analysis of the zinc finger in the Moloney murine leukemia virus nucleocapsid domain: replacement of zinc-coordinating residues with other zinc-coordinating residues yields noninfectious particles containing genomic RNA. J Virol. 1996;70:2593–2597. doi: 10.1128/jvi.70.4.2593-2597.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guan K L, Dixon J E. Eukaryotic proteins expressed in Escherichia coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathione S-transferase. Anal Biochem. 1991;192:262–267. doi: 10.1016/0003-2697(91)90534-z. [DOI] [PubMed] [Google Scholar]
- 27.Harris K S, Xiang W, Alexander L, Lane W S, Paul A V, Wimmer E. Interaction of the poliovirus polypeptide 3CDpro with the 5′ and 3′ termini of the poliovirus genome: identification of viral and cellular cofactors needed for efficient binding. J Biol Chem. 1994;269:27004–27014. [PubMed] [Google Scholar]
- 28.Ho S N, Hunt H D, Horton R M, Pullen J K, Pease L R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 29.Hunt J B, Neece S H, Schachman H K, Ginsburg A. Mercurial-promoted Zn2+ release from Escherichia coli aspartate transcarbamoylase. J Biol Chem. 1984;259:14793–14803. [PubMed] [Google Scholar]
- 30.Hyypiä T, Auvinen P, Maaronen M. Polymerase chain reaction for human picornaviruses. J Gen Virol. 1989;70:3261–3268. doi: 10.1099/0022-1317-70-12-3261. [DOI] [PubMed] [Google Scholar]
- 31.Jiang X, Wang M, Wang K, Estes M K. Sequence and genomic organization of Norwalk virus. Virology. 1993;195:51–61. doi: 10.1006/viro.1993.1345. [DOI] [PubMed] [Google Scholar]
- 32.Jones K W, Bockman R S, Bronner F. Microdistribution of lead in bone: a new approach. Neurotoxicology. 1992;13:835–841. [PubMed] [Google Scholar]
- 33.Klug A, Schwabe J W R. Zinc fingers. FASEB J. 1995;9:597–604. [PubMed] [Google Scholar]
- 34.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacterial T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 35.Lama J, Paul A V, Harris K S, Wimmer E. Properties of purified recombinant poliovirus protein 3AB as substrate for viral proteinases and as co-factor for viral polymerase 3Dpol. J Biol Chem. 1994;269:66–70. [PubMed] [Google Scholar]
- 36.Li J-P, Baltimore D. An intragenic revertant of a poliovirus 2C mutant has an uncoating defect. J Virol. 1990;64:1102–1107. doi: 10.1128/jvi.64.3.1102-1107.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li J P, Baltimore D. Isolation of poliovirus 2C mutants defective in viral RNA synthesis. J Virol. 1988;62:4016–4021. doi: 10.1128/jvi.62.11.4016-4021.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu J, Gagnon Y, Gauthier J, Furenlid L, L'Heureux P-J, Auger M, Nureki O, Yokoyama S, Lapointe J. The zinc-binding site of Escherichia coli glutamyl-tRNA synthetase is located in the acceptor-binding domain. J Biol Chem. 1995;270:15162–15169. doi: 10.1074/jbc.270.25.15162. [DOI] [PubMed] [Google Scholar]
- 39.Nuss D, Oppermann H, Koch J. Selective blockage of initiation of host protein synthesis in RNA-virus-infected cells. Proc Natl Acad Sci USA. 1975;72:1258–1262. doi: 10.1073/pnas.72.4.1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parsley T B, Towner J S, Blyn L B, Ehrenfeld E, Semler B L. Poly (rC) binding protein 2 forms a ternary complex with the 5′-terminal sequences of poliovirus RNA and the viral 3CD proteinase. RNA. 1997;3:1124–1134. [PMC free article] [PubMed] [Google Scholar]
- 41.Paul A, Cao X, Harris K S, Lama J, Wimmer E. Stimulation of poly(U) synthesis in vitro by purified poliovirus protein 3AB. J Biol Chem. 1994;269:29173–29181. [PubMed] [Google Scholar]
- 42.Paul A V, Molla A, Wimmer E. Studies of a putative amphipathic helix in the N-terminus of poliovirus protein 2C. Virology. 1994;199:188–199. doi: 10.1006/viro.1994.1111. [DOI] [PubMed] [Google Scholar]
- 43.Paul A V, van Boom J H, Filippov D, Wimmer E. Protein-primed RNA synthesis by purified poliovirus RNA polymerase. Nature. 1998;393:280–284. doi: 10.1038/30529. [DOI] [PubMed] [Google Scholar]
- 44.Pfister T, Wimmer E. Characterization of the nucleoside triphosphatase activity of poliovirus protein 2C reveals a mechanism by which guanidine inhibits poliovirus replication. J Biol Chem. 1999;274:6992–7001. doi: 10.1074/jbc.274.11.6992. [DOI] [PubMed] [Google Scholar]
- 45.Pincus S E, Diamond D C, Emini E A, Wimmer E. Guanidine-selected mutants of poliovirus: mapping of point mutations to polypeptide 2C. J Virol. 1986;57:638–646. doi: 10.1128/jvi.57.2.638-646.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rodríguez P L, Carrasco L. Poliovirus protein 2C contains two regions involved in RNA binding activity. J Biol Chem. 1995;270:10105–10112. doi: 10.1074/jbc.270.17.10105. [DOI] [PubMed] [Google Scholar]
- 47.Rodríguez P L, Carrasco L. Poliovirus protein 2C has ATPase and GTPase activities. J Biol Chem. 1993;268:8105–8110. [PubMed] [Google Scholar]
- 48.Rothstein M A, Richards O C, Amin C, Ehrenfeld E. Enzymatic activity of poliovirus RNA polymerase synthesized in Escherichia coli from viral cDNA. Virology. 1988;164:301–308. doi: 10.1016/0042-6822(88)90542-9. [DOI] [PubMed] [Google Scholar]
- 49.Salas M. Protein-priming of DNA replication. Annu Rev Biochem. 1991;60:39–71. doi: 10.1146/annurev.bi.60.070191.000351. [DOI] [PubMed] [Google Scholar]
- 50.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- 51.Schlegel A, Giddings T H, Ladinsky M S, Kirkegaard K. Cellular origin and ultrastructure of membranes induced during poliovirus infection. J Virol. 1996;70:6576–6588. doi: 10.1128/jvi.70.10.6576-6588.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shepley M P, Sherry B, Weiner H L. Monoclonal antibody identification of a 100-kDa membrane protein in HeLa cells and human spinal cord involved in poliovirus attachment. Proc Natl Acad Sci USA. 1988;85:7743–7747. doi: 10.1073/pnas.85.20.7743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sommergruber W, Casari G, Fessl F, Seipelt J, Skern T. The 2A proteinase of human rhinovirus is a zinc containing enzyme. Virology. 1994;204:815–818. doi: 10.1006/viro.1994.1599. [DOI] [PubMed] [Google Scholar]
- 54.Takahashi N, Hirose M. Determination of sulfhydryl groups and disulfide bonds in a protein by polyacrylamide gel electrophoresis. Anal Biochem. 1990;188:359–365. doi: 10.1016/0003-2697(90)90621-f. [DOI] [PubMed] [Google Scholar]
- 55.Takeda N, Kuhn R J, Yang C F, Takegami T, Wimmer E. Initiation of poliovirus plus-strand RNA synthesis in a membrane complex of infected HeLa cells. J Virol. 1986;60:43–53. doi: 10.1128/jvi.60.1.43-53.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Takegami T, Semler B L, Anderson C W, Wimmer E. Membrane fractions active in poliovirus RNA replication contain VPg precursor polypeptides. Virology. 1983;128:33–47. doi: 10.1016/0042-6822(83)90316-1. [DOI] [PubMed] [Google Scholar]
- 57.Teterina N L, Gorbalenya A E, Egger D, Bienz K, Ehrenfeld E. Poliovirus 2C protein determinants of membrane binding and rearrangements in mammalian cells. J Virol. 1997;71:8962–8972. doi: 10.1128/jvi.71.12.8962-8972.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tolskaya E A, Romanova L I, Kolesnikova M S, Gmyl A P, Gorbalenya A E, Agol V I. Genetic studies on the poliovirus 2C protein, an NTPase: a plausible mechanism of guanidine effect on the 2C function and evidence for the importance of 2C oligomerization. J Mol Biol. 1994;236:1310–1323. doi: 10.1016/0022-2836(94)90060-4. [DOI] [PubMed] [Google Scholar]
- 59.Towner J S, Ho T V, Semler B L. Determinants of membrane association for poliovirus protein 3AB. J Biol Chem. 1996;271:26810–26818. doi: 10.1074/jbc.271.43.26810. [DOI] [PubMed] [Google Scholar]
- 60.Vance L M, Moscufo N, Chow M, Heinz B A. Poliovirus 2C region functions during encapsidation of viral RNA. J Virol. 1997;71:8759–8765. doi: 10.1128/jvi.71.11.8759-8765.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van der Werf S, Bradley J, Wimmer E, Studier F W, Dunn J J. Synthesis of infectious poliovirus RNA by purified T7 RNA polymerase. Proc Natl Acad Sci USA. 1986;78:2330–2334. doi: 10.1073/pnas.83.8.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.van Kuppeveld F J, Hoenderop J G, Smeets R L, Willems P H, Dijkman H B, Galama J M, Melchers W J. Coxsackievirus protein 2B modifies endoplasmic reticulum membrane and plasma membrane permeability and facilitates virus release. EMBO J. 1997;16:3519–3532. doi: 10.1093/emboj/16.12.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wimmer E, Hellen C U, Cao X. Genetics of poliovirus. Annu Rev Genet. 1993;27:353–436. doi: 10.1146/annurev.ge.27.120193.002033. [DOI] [PubMed] [Google Scholar]
- 64.Xiang W, Harris K S, Alexander L, Wimmer E. Interaction between the 5′-terminal cloverleaf and 3AB/3CDpro of poliovirus is essential for RNA replication. J Virol. 1995;69:3658–3667. doi: 10.1128/jvi.69.6.3658-3667.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xiang W, Paul A V, Wimmer E. RNA signals in entero- and rhinovirus genome replication. Semin Virol. 1997;8:256–273. [Google Scholar]
- 66.Yu S F, Lloyd R E. Characterization of the roles of conserved cysteine and histidine residues in poliovirus 2A protease. Virology. 1992;186:725–735. doi: 10.1016/0042-6822(92)90039-r. [DOI] [PubMed] [Google Scholar]