

Abstract
An increase in cocaine abuse has been observed globally since the past decade. Cocaine is among the commonly abused stimulants used for recreational purposes. In this study, the SPE-UHPLC-MS/MS method was developed and validated to be applied on real specimens of 20 chronic cocaine abusers to quantify cocaine/metabolites in conventional as well as alternative biological matrices. Cocaine was extracted from biological specimens using solid-phase extraction followed by liquid chromatography tandem mass spectrometry analysis. Chromatographic separation was achieved on a Poroshell120EC-18 column (2.1 mm × 50 mm, 2.7 μm particle size) using water–acetonitrile in 0.1% formic acid as a mobile phase in gradient elution mode. The flow rate of the mobile phase was 0.5 mL/min with a gradient varying the percentage of acetonitrile linearity ranging 15–95% in 6.0 min acquisition time, and the injection volume was set at 5 μL. Positive electrospray ionization with multireaction ion monitoring mode using two ion transitions for cocaine/metabolites and one for cocaine-d3 was employed. The quantification method demonstrated good linear ranges of 0.025–250 ng/mL in blood, urine, and oral fluid (ng/mg for hair and nail) with a ≥0.991% determination coefficient. The detection limit and lower quantification limit were 0.005 and 0.025 ng/mL in all matrices, respectively. The mean extraction recovery and ionization suppression ranged from 89.3 to 99.8% and −4.6 to −14.4% in the studied matrices. Within-run and between-days precisions were 1.8–7.2% and 1.9–6.1%, respectively. This study will not only help in quantifying cocaine/metabolites in alternative specimens (hair, nail, and oral fluid) but also guide clinical and forensic toxicologists in interpretation of exhumation cases. Furthermore, multiple specimens’ analyses can be of significance in estimating the time/manner of drug exposure, in confirming the results of laboratories in cases of doubtful clinical histories, or in aiding medico-legal investigations.
1. Introduction
Globally, cocaine is among the most commonly encountered illicit central nervous system stimulants in clinical or forensic toxicology investigations and is widely abused as a recreational drug and appetite suppressant. According to the World Drug Report 2023 released by United Nations Office on Drugs and Crime (UNODC), an increase in cocaine abuse has been observed since the past decade.1 Despite the rising trends of new psychoactive substances nowadays, cocaine abuse is still a problematic concern all over the world due to its implications in human performance. It is a natural alkaloid obtained from leaves of Erythroxylum coca. The Drug Enforcement Administration has classified cocaine as a Schedule II drug due to its topical local anesthetic medicinal use. The empirical formula and molecular weight of cocaine are C17H21NO4 and 303.35, respectively.2 Cocaine is well-absorbed through all routes of administration like intranasal, intravenous, and smoking. Chemically, cocaine has two ester linkages that are subjected to spontaneous or enzymatic hydrolysis. Benzoylecgonine (BE) is a metabolite of cocaine produced by the spontaneous hydrolysis of alkyl ester linkage or action of liver methylases, whereas plasma cholinesterase and liver benzoylesterases hydrolyze the phenyl ester linkage to produce ecgoninemethylester (EME) metabolite. Cocaethylene (CE) is produced when cocaine is used in combination with ethanol. BE and EME are inactive, whereas CE is an active metabolite of cocaine.3 The toxic concentration of cocaine in blood ranges from 0.1 to 5 mg/L, whereas the reported fatal concentration is 0.9–21 mg/L in blood, 0.1–215 mg/L in urine, 0.8–13 mg/L in vitreous, 0.1–20 mg/kg in liver, 0.4–15 mg/kg in brain, 0.3–27 mg/kg in kidney, and 0.1–48 mg/kg in skeletal muscle.2,4
One of the challenges faced by toxicologists in simultaneous extraction and chromatographic separation of cocaine and its metabolites from complex biological matrices is their large polarity difference. Although the positive cutoff value for cocaine is too high, still, sensitive techniques to detect low concentrations are necessary in current practices that can improve clinical and forensic analysis in both ante-mortem and post-mortem investigations.
Immunoassays5,6 and high-performance liquid chromatography (HPLC) coupled to an ultraviolet detector7−10 for detection of cocaine/metabolites available in the literature were focused on determinations in conventional matrices like blood, plasma, or urine. The immunoassay methods possessed substantial cross-reactivity, whereas HPLC methods did not provide necessary sensitivity/specificity for detection of cocaine metabolites. Therefore, gas chromatography with mass spectrometric detection (GC-MS) was used by researchers11−16 combined with derivatization to quantitate cocaine/metabolites in complex matrices. Although GC-MS had both selectivity and sensitivity, derivatization is laborious and expensive and poses a safety risk. To overcome these deficiencies, liquid chromatography with mass spectrometry (LC-MS) and liquid chromatography tandem mass spectrometry (LC-MS/MS) supplanted the previous techniques for detection of cocaine/metabolites17−34 for routine analysis. Owing to increased sensitivity/specificity, reduced analysis cost, and shorter processing time, LC-MS/MS is still the method of choice for quantification of drugs such as cocaine. The authors reviewed the published LC-MS/MS methods and observed that they had limited applications to one or more biological matrices but were not applied simultaneously to multiple biological matrices of subjects. The study presented by the authors has wider applicability based on simultaneous assessment of cocaine and its metabolites in these matrices from the same source individuals in real time. Combined analysis of conventional and alternative matrices can give valuable information to aid medico-legal investigations particularly in exhumation cases, where specimen’s availability is limited to keratinized matrices. The limitation of the study presented by the authors is that it is not a dose–concentration relationship study; however, work can be carried out further in this direction by future researchers.
2. Materials and Methods
2.1. Reagents and Materials
Cocaine multicomponent mixture-4 containing cocaine, benzoylecgonine, ecgoninemethylester, and cocaethylene (250 μg/mL each, in acetonitrile) and deuterated cocaine-d3 stable labeled internal standard (1 mg/mL in acetonitrile) were purchased from Cerilliant (Round Rock, TX, USA). Agilent mega bond elute 6 mL solid-phase extraction columns were purchased from HA Shah and Sons, Pakistan. American Chemical Society (ACS) grade monobasic sodium phosphate, ammonium hydroxide, glacial acetic acid, hexane, dichloromethane, isopropanol, methanol, and hydrochloric acid were purchased from Merck (Germany). Liquid chromatography-mass spectrometry (LC-MS) grade solvents (methanol, acetonitrile, water, and formic acid) were purchased from Merck (Germany), and plasticware consumables were purchased from Fisher Scientific (Pittsburgh, PA).
2.2. Standard Solutions, Calibrators, and Quality Control Samples
Cocaine multicomponent mixture-4 and cocaine-d3 certified reference materials were diluted separately in acetonitrile to prepare working standard solutions at 1000 and 100 ng/mL, respectively. The working standard solution of cocaine mixture was spiked in blank matrices to prepare concentration levels of 0.005, 0.025, 0.05, 0.5, 1, 5, 10, 25, 50, 75, 100, 200, and 250 ng/mL (ng/mg for hair and nail) to be used as calibrators and quality control samples in this study.
2.3. Real Sample Collection, Preservation, and Storage
For validation studies, drug-free blood, urine, oral fluid, scalp hair, and finger nails were donated by healthy volunteers on their consent. These control samples were tested for drugs and verified to be drug-free to be used as negative control matrices. Twenty male patients were included in this study that had cocaine and ethanol exposure only. The patients with history of polydrug abuse and who had reported any hair or nail treatment were excluded from this study. Whole blood, urine, scalp hair, finger nail, and oral fluid specimens of patients, aged 16–60 years with at least past three-month cocaine abuse history and ≤36 h recent cocaine exposure, were collected on their consent to be used as real samples in this study. These samples were collected from patients prior to their treatment for substance abuse disorder. Demographic details and past history of cocaine consumption were recorded.
For oral fluid collection, the patients were refrained from drinking, eating, smoking, or putting anything in their mouth 30 min prior to sample collection. Patients were requested to spit into a 50 mL sample collection cup containing a sodium fluoride preservative. Whole blood (5 mL) was collected in a gray-top vial with a sodium fluoride preservative. Urine was collected in a 50 mL collection cup without any preservative. Blood, urine, and oral fluid samples were immediately stored at refrigeration temperature. Hair samples (tuft of hair having 1/4th thickness of a pencil) from the posterior vortex region of head were cut near the scalp with root ends tied to mark the direction and were stored in a paper envelope. The first 3–5 mm segment of hair from the proximal end was used in analysis. Finger nails (2–3 mm of distal edges of all 10 fingers) were clipped and stored in a paper envelope. Hair and nail samples were stored at ambient temperature until analysis.
2.4. Sample Pretreatment
Blood, urine, and oral fluid were used as such without any sample pretreatment or dilution. Scalp hair and finger nail specimens were subjected to washing first with deionized water and then twice with 5 mL of dichloromethane at room temperature for 5 min to remove external contaminants. Absorbent paper sheets were used to dry hair and nail samples after each washing step. The dichloromethane extracts were transferred into a clean tube, evaporated to dryness in a nitrogen evaporator at 40 °C, reconstituted with 500 μL of acetonitrile, and refrigerated at 4 °C until analysis was performed to determine the presence of external contaminations.
Cocaine is unstable in strong alkaline solution; therefore, acid hydrolysis is a recommended digestion method for cocaine. Therefore, 25 mg each of washed and dried hair and nail specimens were finely cut with decontaminated scissors and incubated overnight with 1 mL of hydrochloric acid (1 N) at room temperature. Negative control hair and nail specimens were also digested by using the same procedures.
2.5. Sample Preparation
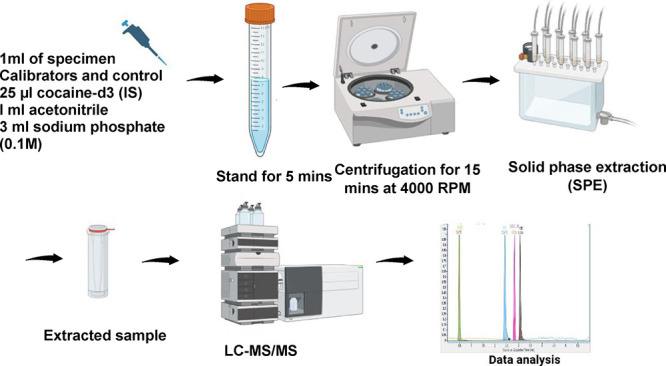
A total of 1 mL each of calibrators, controls, and real samples was transferred into labeled 15 mL polypropylene tubes followed by addition of 25 μL of cocaine-d3 working internal standard solution and 3 mL of 0.1 M phosphate buffer (pH 6). Tubes were centrifuged for 15 min at 4000 rpm, and the specimens were then decanted into the solid-phase extraction (SPE) columns (previously, conditioned with 3 mL of methanol followed by 3 mL of deionized water and 2 mL of 0.1 M phosphate buffer, pH 6). The columns were rinsed with 3 mL each of deionized water, 0.5 M acetic acid, and methanol. Eluted SPE columns with 3 mL of freshly prepared solution of methylene chloride, isopropanol, and ammonium hydroxide (78:20:2, v/v) were added to 100 μL of 0.1N hydrochloric acid to the organic extracts and subjected to dryness using a nitrogen evaporator at 40 °C. Each dried tube was reconstituted with 150 μL of acetonitrile (containing 0.1% formic acid), vortex-mixed, transferred into an autosampler vial with 250 μL insert, and capped for subsequent LC-MS/MS analysis.
2.6. Liquid Chromatography Tandem Mass Spectrometry Operational Parameters
The chromatographic separation of cocaine and its metabolites was achieved using an Agilent Poroshell120EC-18 (2.1 mm × 50 mm, 2.7 μm particle size) analytical column at 60 °C. An Agilent Infinity ultrahigh-performance liquid chromatography (1260) with tandem mass spectrometry (6470) instrument was employed for quantification. The mobile phases in gradient mode of elution used include LC-MS-grade water in 0.1% formic acid (mobile phase A) and LC-MS-grade acetonitrile in 0.1% formic acid (mobile phase B). The mobile phase flow rate was set to 0.5 mL/min with gradient varying the percentage of acetonitrile linearity: 0.0 min, 15%; 0.5 min, 15%; 3.0 min, 65%; 4.0 min, 95%; 5.0 min, 95%; and 6.0 min, 15%. The injection volume was 5 μL with 6.0 min acquisition time and 2.0 min postrun time. The electron spray ionization (ESI) source with Agilent Jet Stream (AJT) technology was utilized to enhance sensitivity in positive polarity mode (delta EMV, +200) with 375 °C sheath gas temperature, 10 L/min gas flow, 45 psi nebulizer pressure, and 3500 V capillary voltage. The compounds' specific settings used for the ionization and fragmentation are listed in Table 1, along with the selected precursor and product ions. Instrument control, sample acquisition, and data analysis were performed by Mass hunter 1.6.3 software. The spectrometric analysis parameters were optimized, and two MRM transitions were selected for each analyte and one MRM transition for the internal standard according to the mass spectrometry standards. All results were based on the ratio of peak areas of the analyte and the internal standard. Certified reference materials of analytes were used for optimization to select the best parameters of the mass spectrometer for each of them. An Agilent optimizer was used for method optimization. The positive ionization mode was found to be the best for the tested analytes. The MRM transitions, fragmention voltage, collision energy, cell acceleration voltage, and dwell time for each analyte are presented in Table 1.
Table 1. Tandem Mass Spectrometer Settings and Retention Times for Cocaine, Its Metabolites, and the Internal Standard.
| compound | precursor ion | product ion | fragmentor voltage (V) | collision energy (V) | cell acceleration voltage (V) | dwell time (ms) |
|---|---|---|---|---|---|---|
| cocaine | 304.2 | 182.1 | 138 | 5 | 7 | 100 |
| 77.0 | 45 | |||||
| benzoylecgonine | 290.1 | 168.1 | 118 | 10 | ||
| 77.0 | 45 | |||||
| ecgonine methyl ester | 200.2 | 182.1 | 118 | 18 | ||
| 81.9 | 18 | |||||
| cocaethylene | 318.2 | 196.1 | 123 | 5 | ||
| 82.1 | 18 | |||||
| cocaine-d3 (IS) | 307.1 | 185.1 | 138 | 30 |
2.7. Analytical Method Validation for Quantification of Cocaine and Its Metabolites
This presented analytical method was validated following the standard practices for method validation in forensic toxicology by SWGTOX and Society of Hair Testing (SoHT) guidelines for drug testing in hair.42,43
A seven-point calibration model using linear regression analysis of the ratio of the peak area of analyte to the peak area of the deuterated internal standard was used. Linearity was assessed by analyzing triplicates of each calibration level (0.025, 1, 5, 25, 50, 100, and 250 ng/mL or ng/mg) for 5 days in each matrix (blood, urine, oral fluid, hair, and nail). Instrumental response from the 5-day runs was plotted against concentration levels to establish the calibration model. Matrix-matched calibrators were used to minimize matrix interferences while quantifying cocaine/metabolites in the real samples. A blank matrix and blank matrix containing only an internal standard (negative quality control) were analyzed with each calibration batch but not included in the calibration curves. For bias and precision studies, four concentrations (0.05, 10, 75, and 200 ng/mL or ng/mg) were analyzed in triplicate for 5 days. The coefficient of variance (%CV) and bias were calculated for within-run and between-run studies. For acceptance, the %CV shall not exceed 20% at each concentration.42 The limit of detection (LOD) and lower limit of quantification (LLOQ) were determined as the lowest precise concentration giving a response of at least three times the average of the baseline noise (S/N > 3) and the lowest accurate concentration with S/N ratio ≥10, respectively. Proficiency tests were used to check the reproducibility of the analytical method. The stability of the extracted compounds was evaluated. Triplicate aliquots of quality control samples (0.075 and 75 ng/mL) in each matrix were spiked with the internal standard, extracted, and analyzed at different time periods (0, 1, 4, 8, 12, 18, and 24 h). The concentration at each time period was compared to the initial concentration (at 0 h) to evaluate the stability. For freeze–thaw stability study, the above fortified samples were aliquoted into three different storage tubes for each concentration and frozen at the intended storage temperature for 24 h. This was followed by an unassisted thaw at room temperature. When completely thawed, the first set of samples was analyzed in triplicate, while the others were refrozen for 12–24 h under the same conditions. This freeze–thaw cycle followed by analysis was repeated twice. The analyte was considered as stable until the response (ratio of peak area of analyte to internal standard) compared to the time zero average signal falls outside of the method’s acceptable bias (20%). To evaluate the impact of room temperature storage of processed samples sitting on the autosampler before analysis, the authors conducted processed sample stability study, which was achieved by preparing fortified matrix samples at two concentrations, 0.075 and 75 ng/mL. Twelve aliquots of each concentration were extracted. Reconstituted extracts for each concentration were combined and vortexed to ensure adequate mixing. The concentration pool was then divided into 12 autosampler vials and placed on the autosampler. The first vial of each level was injected three times to represent the time zero (t0) sample. The remaining vials for each concentration were analyzed in triplicate every 6 h up to 72 h. Analyte signals from the triplicate analyses were averaged and compared with the t0 signal. The analyte was considered as stable until the response (ratio of peak area of analyte to internal standard) compared to the time zero average signal falls outside of the method’s acceptable bias (20%). Dilution integrity was tested for high analyte concentrations over the highest calibration range or low sample volume. This is accomplished by repeating bias and precision studies at dilution ratios (1:2 and 1:5), verifying precision and accuracy to be within 20%. Pre- and postinjections of background matrices with the highest calibration level were studied (n = 5) to evaluate the carryover effect. Ion suppression/enhancement was evaluated using the post-extraction addition approach.42
3. Results
3.1. Method Validation Results
An average of the relative response for each calibrator was plotted against the relative concentration to plot the linearity/calibration curve. The correlation coefficients (r2) obtained were used to evaluate the linearity of each curve. A correlation coefficient of 0.98 is minimally acceptable.44 The method showed good linearity up to 250 ng/mL (ng/mg for hair and nails) with determination coefficients ranging from 0.991 to 0.999 in tested matrices, as shown in Table 2.
Table 2. LC-MS/MS Method Validation Results for Cocaine and Its Metabolites in Biological Matrices.
| drug/metabolite | parameter | blood | urine | oral fluid | scalp hair | finger nail |
|---|---|---|---|---|---|---|
| cocaine | limit of detection (ng/mL or ng/mg) | 0.005 | 0.005 | 0.005 | 0.005 | 0.005 |
| lower limit of quantification (ng/mL or ng/mg) | 0.025 | 0.025 | 0.025 | 0.025 | 0.025 | |
| determination coefficient (r2) | 0.998 | 0.997 | 0.995 | 0.993 | 0.991 | |
| ion suppression/enhancement (±%) | –6.2 | –9.7 | –4.6 | –11.8 | –14.4 | |
| benzoylecgonine | limit of detection (ng/mL or ng/mg) | 0.005 | 0.005 | 0.005 | 0.005 | 0.005 |
| lower limit of quantification (ng/mL or ng/mg) | 0.025 | 0.025 | 0.025 | 0.025 | 0.025 | |
| determination coefficient (r2) | 0.997 | 0.999 | 0.996 | 0.994 | 0.993 | |
| ion suppression/enhancement (±%) | –5.7 | –7.7 | –4.6 | –10.2 | –11.2 | |
| ecgoninemethylester | limit of detection (ng/mL or ng/mg) | 0.005 | 0.005 | 0.005 | 0.005 | 0.005 |
| lower limit of quantification (ng/mL or ng/mg) | 0.025 | 0.025 | 0.025 | 0.025 | 0.025 | |
| determination coefficient (r2) | 0.995 | 0.997 | 0.999 | 0.995 | 0.996 | |
| ion suppression/enhancement (±%) | –6.4 | –8.1 | –6.5 | –7.3 | –8.2 | |
| cocaethylene | limit of detection (ng/mL or ng/mg) | 0.005 | 0.005 | 0.005 | 0.005 | 0.005 |
| lower limit of quantification (ng/mL or ng/mg) | 0.025 | 0.025 | 0.025 | 0.025 | 0.025 | |
| determination coefficient (r2) | 0.997 | 0.995 | 0.996 | 0.997 | 0.998 | |
| ion suppression/enhancement (±%) | –7.3 | –9.0 | –8.4 | –5.9 | –9.1 |
Regarding the stability studies performed at low and high positive samples (0.075 and 75 ng/mL or ng/mg), no significant change in concentration was detected in extracts up to 24 h in tested matrices. The stability of cocaine and its metabolites after four consecutive freeze–thaw cycles and sample processing up to 72 h remained within tje acceptable method bias (±20%). Using 1:2 and 1:5 aqueous dilutions, the bias (8.2–9.9%) and precision [within-run (2.9–4.0%) and between-run (3.7–4.4%)] remained within acceptable limits. No additional peaks due to endogenous substances and carryover interfering with cocaine, its metabolites, and internal standard were detected. Representative extracted ion chromatograms showing overlaid MRM transitions for the lower limit of quantitation (0.025 ng/mL or ng/mg), low positive control (10 ng/mL or ng/mg), negative quality control, and blank samples in conventional and alternative matrices are shown in Figure 1. The method validation results are summarized in Tables 2 and 3.
Figure 1.

MRM transitions overlaid chromatograms of 0.025 ng/mL (LLOQ), 10 ng/mL (calibrator), and negative control samples showing cocaine (COC), cocaine-d3 (IS), benzoylecgonine (BE), ecgoninemethylester (EME), and cocaethylene (CE) in whole blood (a), urine (b), oral fluid (c), scalp hair (d), and finger nail (e) matrices.
Table 3. Accuracy and Precision Results for Cocaine and Its Metabolites in Biological Matrices by LC-MS/MSa.
|
accuracy
(% bias) |
precision (% RSD
or CV) |
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
within-run (n = 5) |
between-run (n = 15) |
within-run (n = 5) |
between-run (n = 15) |
||||||||||||||
| Conc. (ng/mL or ng/mg) |
Conc. (ng/mL or ng/mg) |
Conc. (ng/mL or ng/mg) |
Conc. (ng/mL or ng/mg) |
||||||||||||||
| DOA | BM | 0.05 | 10 | 75 | 200 | 0.05 | 10 | 75 | 200 | 0.05 | 10 | 75 | 200 | 0.05 | 10 | 75 | 200 |
| COC | WB | –1.3 | –1.8 | –2.7 | –3.2 | –2.2 | –4.7 | –4.3 | –2.9 | 2.0 | 0.9 | 1.6 | 3.3 | 1.2 | 2.3 | 1.3 | 5.7 |
| U | 2.1 | 2.4 | 3.5 | 3.2 | 2.2 | 1.9 | 2.8 | 3.4 | 2.3 | 2.7 | 1.6 | 1.8 | 2.8 | 2.4 | 3.3 | 2.6 | |
| OF | 2.3 | 2.2 | 2,7 | 3.1 | 2.0 | 2.5 | 3.2 | 2.8 | 3.4 | 3.0 | 4.1 | 2.6 | 3.2 | 3.8 | 2.9 | 3.1 | |
| SH | –3.3 | –2.6 | –3.2 | –4.5 | –2.2 | –2.1 | –2.6 | –3.3 | 2.1 | 2.0 | 2.3 | 2.5 | 3.1 | 3.6 | 3.4 | 3.2 | |
| FN | –6.1 | –4.6 | –5.2 | –5.6 | –7.2 | –8.6 | –4.2 | –9.1 | 2.9 | 2.2 | 5.1 | 4.8 | 3.2 | 3.8 | 5.6 | 6.7 | |
| BE | WB | –3.8 | –2.9 | –2.8 | –1.5 | –3.6 | –3.1 | –2.7 | –4.1 | 3.0 | 2.4 | 3.6 | 3.1 | 2.3 | 4.3 | 5.0 | 2.8 |
| U | 6.1 | 5.7 | 6.3 | 6.8 | 7.3 | 6.1 | 6.6 | 7.0 | 2.9 | 2.7 | 1.8 | 3.4 | 3.8 | 3.7 | 3.5 | 2.9 | |
| OF | 7.3 | 6.9 | 6.5 | 6.8 | 7.0 | 6.6 | 7.4 | 7.1 | 3.6 | 3.9 | 4.1 | 4.5 | 3.7 | 2.8 | 3.9 | 2.9 | |
| SH | 6.3 | 5.8 | 4.9 | 6.1 | 5.6 | 4.5 | 4.8 | 5.2 | 2.2 | 1.8 | 2.5 | 3.6 | 1.4 | 1.9 | 2.2 | 3.1 | |
| FN | 3.2 | 4.4 | 3.7 | 3.9 | 4.1 | 5.2 | 4.9 | 4.5 | 6.0 | 5.2 | 6.2 | 5.9 | 7.0 | 6.1 | 7.9 | 8.2 | |
| EME | WB | 6.2 | 4.6 | 6.6 | 6.7 | 5.2 | 4.9 | 5.7 | 6.1 | 2.5 | 3.2 | 3.5 | 3.2 | 2.6 | 4.0 | 3.7 | 4.3 |
| U | 5.3 | 4.5 | 4.4 | 4.7 | 2.5 | 3.7 | 4.6 | 4.4 | 3.8 | 3.1 | 2.8 | 2.6 | 4.4 | 3.9 | 4.0 | 4.2 | |
| OF | 4.6 | 5.2 | 4.9 | 4.7 | 5.1 | 4.8 | 5.5 | 5.8 | 5.1 | 4.2 | 4.4 | 4.7 | 4.1 | 3.7 | 3.6 | 4.0 | |
| SH | 3.7 | 3.1 | 4.0 | 3.6 | 4.2 | 5.3 | 4.9 | 4.6 | 4.0 | 3.8 | 5.3 | 6.1 | 3.3 | 2.9 | 3.5 | 2.9 | |
| FN | 5.2 | 6.0 | 5.8 | 6.2 | 5.1 | 5.3 | 4.8 | 5.0 | 5.8 | 6.3 | 6.8 | 5.6 | 5.8 | 5.4 | 4.9 | 5.1 | |
| CE | WB | 5.3 | 5.5 | 5.9 | 4.3 | 6.3 | 5.9 | 6.5 | 7.1 | 4.8 | 5.3 | 5.4 | 5.0 | 5.7 | 5.4 | 7.0 | 6.7 |
| U | 4.8 | 5.6 | 4.9 | 4,4 | 7.2 | 6.8 | 6.4 | 6.3 | 3.4 | 3.7 | 4.2 | 4.6 | 5.7 | 5.3 | 4.9 | 5.2 | |
| OF | 3.7 | 4.1 | 3.9 | 3.2 | 8.7 | 9.0 | 9.6 | 9.1 | 8.1 | 7.2 | 7.4 | 7.3 | 6.9 | 4.8 | 5.7 | 6.1 | |
| SH | 5.1 | 5.8 | 5.5 | 7.6 | 8.1 | 7.7 | 8.3 | 8.8 | 4.7 | 5.1 | 5.6 | 5.0 | 6.3 | 5.9 | 6.4 | 6.2 | |
| FN | 7.2 | 8.7 | 9.2 | 9.5 | 8.9 | 9.1 | 7.8 | 8.6 | 5.3 | 5.5 | 5.0 | 4.7 | 5.9 | 6.1 | 6.3 | 7.3 | |
Abbreviations used: DOA = drugs of abuse, BM = biological matrix, COC = cocaine, BE = benzoylecgonine, EME = ecgoninemethylester, CE = cocaethylene, WB = whole blood, U = urine, OF = oral fluid, SH = scalp hair, FN = finger nail, RSD = relative standard deviation, CV = coefficient of variance.
3.2. Results of Real Sample Analysis
The levels of cocaine and its metabolites were assessed in conventional and alternative biological specimens of 20 cocaine addicts using LC-MS/MS. The concentration ranges of cocaine and its metabolites obtained in their blood, urine, oral fluid, scalp hair, and finger nail specimens are presented in Table 4.
Table 4. Concentration Ranges of Cocaine and Its Metabolites in Specimens of 20 Male Cocaine Abusers.
| history (months) | no. of addicts | drug/metabolite | blood (ng/mL) | urine (ng/mL) | oral fluid (ng/mL) | scalp hair (ng/mg) | finger nail (ng/mg) |
|---|---|---|---|---|---|---|---|
| ≥1 to <3 | 9 | cocaine | 11.70–33.02 | 39.20–57.29 | 17.21–39.34 | 1.07–8.32 | 0.09–0.20 |
| benzoyl ecgonine | 91.60–124.50 | 121.20–202.30 | 11.20–60.31 | 0.48–8.40 | 0.70–1.90 | ||
| ecgonine methyl ester | 64.50–167.90 | 153.50–214.90 | 4.50–9.36 | 0.03–0.06 | 0.10–11.2 | ||
| cocaethylene | 34.60–83.30 | 65.70–97.86 | 10.10–15.82 | 0.08–0.17 | 0.06–0.37 | ||
| ≥3 to <6 | 7 | cocaine | 53.06–87.21 | 76.09–100.20 | 34.71–58.77 | 6.82–23.21 | 0.15–0.45 |
| benzoyl ecgonine | 89.27–135.10 | 97.11–106.31 | 46.78–47.31 | 0.66–13.11 | 1.60–2.20 | ||
| ecgonine methyl ester | 75.17–116.28 | 165.72–237.80 | 1.28–3.11 | 0.05–0.21 | 0.24–0.39 | ||
| cocaethylene | 45.21–93.67 | 73.18–89.43 | 2.69–21.08 | 0.11–0.34 | 0.08–0.57 | ||
| ≥6 to <9 | 4 | cocaine | 87.33–149.62 | 59.06–77.59 | 44.07–61.31 | 2.50–53.90 | 1.06–3.25 |
| benzoyl ecgonine | 100.01–153.11 | 65.41–97.23 | 105.37–219.52 | 9.93–36.70 | 3.67–12.96 | ||
| ecgonine methyl ester | 99.07–178.67 | 38.60–63.11 | 6.90–13.65 | 0.23–1.41 | 2.31–5.62 | ||
| cocaethylene | 57.22–73.59 | 41.01–53.28 | 4.98–15.10 | 0.10–0.45 | 0.17–0.59 |
4. Discussion
The authors of the proposed study successfully validated the ESI-LC-MS/MS method for assessment of cocaine and its metabolites in the conventional and alternative matrices as presented in Tables 2 and 3 following the SWGTOX validation guidelines.42 Furthermore, the method was applied to real specimens (whole blood, urine, oral fluid, scalp hair, and finger nail) of 20 male cocaine abusers to determine the relative concentrations of cocaine and its metabolites in conventional and alternative specimens. Concerning sample preparation procedures for cocaine analysis, solid-phase extraction is the most used procedure5,7−15,23−27,36−41 due to better sample cleanup, less amounts of organic solvents used, low matrix effect, and greater recovery as compared to liquid–liquid extraction methods. Furthermore, SPE is more easily automated and provides a greater sample throughput. Solid-phase microextraction, microextraction in a packed syringe, microextraction in micropipette tips, liquid-phase microextraction, and accelerated solvent extraction are also reported as miniaturized extraction procedures in the literature, but more developments are needed before these techniques can be routinely applied in the determination of cocaine and its metabolites in biological fluids; indeed, the number of papers dealing with some of the aforementioned procedures is still scarce. Our proposed method is specific with an LOQ of 0.025 ng/mg for cocaine and its metabolites. The confirmation cutoff concentrations for cocaine and its metabolites in nail specimens are unavailable, but the Society of Hair Testing (SoHT) recommended limits of quantification ≤0.5 ng/mg for cocaine and ≤0.05 ng/mg for cocaine metabolites in hair.43 Our proposed extraction and detection method is sensitive enough as compared to microextraction methods reported in the literature to precisely quantify below the recommended cutoff values for cocaine and its metabolites. The use of UHPLC and Jet Stream Technology (AJT) electron-spray ionization in tandem mass spectrometry had further enhanced sensitivity and promoted fast analysis.
Conventional biological specimens (blood and urine) are of considerable importance in assessment of recent cocaine exposure, but the use of oral fluid and keratinized alternative specimens (hair and nail) to detect cocaine exposure is gaining significance in clinical and forensic toxicology worldwide.35−41 Although analysis of cocaine and its metabolites in blood is very common, the invasive sample collection, limited specimen volume, and shorter detection time window limit its effectiveness in medico-legal investigations. Oral fluid seems to be another promising biological matrix for detection of recent exposure to drugs and has the benefit of noninvasive sample collection. Therefore, the authors collected oral fluids by expectoration from the addicts as well to assess cocaine levels. The variation in the levels of cocaine and its metabolites in oral fluid as compared to blood was observed by the authors of this study. Based on the findings, oral fluid is suggested as a good alternative specimen to blood for assessment of recent cocaine exposure with an additional merit of noninvasive sample collection as compared to blood. Urine is the mainstay of drug monitoring, while oral fluid and blood require extensive handling prior to analysis, which includes homogenization along with a cleanup step.45 This is attributed to the fact that the drug detection window is wide in urine as compared to blood or oral fluid, but urine can be easily tampered and additional checks are required to ensure specimen validity. Further, the stability of drug/metabolite in conventional matrices is challenging, and specialized collection, preservation, storage, and transportation protocols are required. Due to these demerits, use of keratinized matrices (hair and nail) for detection of cocaine and its metabolites is gaining worldwide significance.
Drugs are capable of surviving for hundreds of years in the hair shaft under favorable conditions46−48; therefore, the window for drug detection is wider than conventional matrices and provides a retrospective history of an individual’s drug use if multisectional analysis is used. The pH gradient between plasma (pH 7.3) and melanocytes/keratinocytes (pH 3–6) favors more the basic drug incorporation into the hair as compared to acidic drugs.49 Other factors must also be taken into account in comparing concentrations among matrices. Decontamination of hair or nail samples before analysis to rule out chances of external contamination is a critical aspect and a requirement by scientific communities. Therefore, authors of this study analyzed the first hair washings using organic solvent to determine possible external contamination following the recommendation of SoHT.43 Cocaine and its metabolites were not detected in any organic solvent wash of addict’s hair, which suggests cocaine abuse rather than external contamination. Since the past decade, interest is growing in the analysis of drugs in nail specimens for chronic exposure. Drugs are incorporated into nails by deposition or incorporation into the nail bed during nail growth, which accounts for a wide retrospective window of drug detection. More published work is available on hair analysis, but reports on estimation of chronic exposure of cocaine and its metabolites in nails are scanty.
Irrespective of duration of chronic exposure, results presented in Table 4 depict that the concentrations of cocaine and its metabolites in scalp hair of 20 addicts are greater than the concentrations in finger nail. Kuwayama et al. reported a higher concentration of basic drugs in nail as compared to hair.50 One of the possible reasons for higher concentrations of cocaine and its metabolites in hair as compared to nail might be the absence of melanin in nails in contrast to hair. Several drugs or compounds have binding affinity with melanin, which is based on their physicochemical properties. Basic drugs had shown a positive relationship to melanin binding in studies.51,52 As the growth rate of hair is 1 cm/month,53 therefore, a proximal hair segment of 3 cm represents a three-month growth that accounts for the last three-month consumption of drugs. The growth rate of finger nail is 1 mm/month, and the 1–2 mm distal-edge nail clipping represents one- to two-month consumption of drugs. Therefore, the time taken by the nail to grow from the matrix to free edge (regeneration time) must be taken into consideration while interpreting findings. The regeneration time for finger nail is 3–5 months.54 Thus, the 1–2 mm finger nail clippings correspond to 1–2 months of growth, 3–5 months ago. As a result, the collected hair and finger nail segments did not represent the same period of time. However, the samples in this study were collected from patients with a diagnosed chronic cocaine and alcohol addiction and reported stable drug consumption patterns. This study showed good agreement between the abuse history of cocaine/metabolites and their presence in hair and nail specimens as presented in Table 4. Furthermore, with an increase in exposure duration, the cocaine/metabolite concentration increased in the hair and nail specimens. The results obtained from the analysis of conventional and alternative biological specimens of cocaine addicts were in agreement with the abuse history reported by each subject (Table 4). Based on our findings, nails are a valuable alternative to hair as positive correlations and relationships are present between them. At the same time, this study underlines the need for specific LOD and LOQ concentrations of cocaine and its metabolites for nail analysis. The study proposed preliminary cutoff values for finger nail analysis for cocaine/metabolites (i.e., 0.025 ng/mg), but further studies are encouraged in this direction.
To the best of the author’s knowledge, this is the first attempt to assess levels of cocaine and its metabolites simultaneously in both conventional and alternative biological specimens (blood, urine, oral fluid, scalp hair, and finger nail) obtained from the same subjects at the same time and can be of significant contribution in clinical and forensic toxicological investigations.
5. Conclusions
A sensitive and precise UHPLC-ESI-MS/MS method for determination of cocaine and its metabolites in conventional and alternative biological specimens had been developed, validated, and successfully applied to real specimens of cocaine addicts. This study will not only help in determination of cocaine and its metabolites in alternative specimens (hair, nail, and oral fluid) but also guide clinical and forensic toxicologists in interpretation of drug levels in alternative specimens, particularly in exhumation cases. Furthermore, the analysis of multiple specimens will be useful to better estimate the time and manner of drug exposure, to confirm laboratory results in cases of doubtful clinical histories, and to aid medico-legal investigations.
Acknowledgments
H.S.M. acknowledges the professional support extended by Prof. Dr. Akhtar Sohail Chughtai and Dr. Omar Rasheed Chughtai from Chughtai Healthcare Pakistan throughout this study.
The authors declare no competing financial interest.
References
- United Nations office on Drug and Crime (UNODC) World Drug Report 2023 2023.http://www.unodc.org.
- Baselt R. C. (2020) Cocaine. In Disposition of toxic drugs and chemicals in man, 12th ed. Biomedical Publications: Seal Beach, CA, pp 497–501. [Google Scholar]
- Daniel S. I., (2020) Cocaine. In Barry S. L.; Sarah K. (eds.), Principles of forensic toxicology, 5th ed., Chapter 23. Springer Nature: Switzerland, AG, pp 371–387. [Google Scholar]
- Molina D. K. (2019) Cocaine. In Handbook of forensic toxicology for medical examiners, 2nd ed. CRC Press, Taylor & Francis Group: Boca Raton, FL, pp 65. [Google Scholar]
- Phipps R. J.; Smith J. J.; Darwin W. D.; Cone E. J. Current methods for the separation and analysis of cocaine analytes. Handbook of Analytical Separations 2008, 6, 73–125. 10.1016/S1567-7192(06)06002-5. [DOI] [Google Scholar]
- Niedbala R. S.; Kardos K.; Fries T.; Cannon A.; Davis A. Immunoassay for detection of cocaine/metabolites in oral fluids. J. Anal. Toxicol. 2001, 25 (1), 62–68. 10.1093/jat/25.1.62. [DOI] [PubMed] [Google Scholar]
- Evans M. A.; Morarity T. Analysis of cocaine and cocaine metabolites by high pressure liquid chromatography. Journal of Analytical Toxicology 1980, 4 (1), 19–22. 10.1093/jat/4.1.19. [DOI] [PubMed] [Google Scholar]
- Fernandez P.; Lafuente N.; Bermejo A. M.; Lopez-Rivadulla M.; Cruz A. HPLC determination of cocaine and benzoylecgonine in plasma and urine from drug abusers. Journal of analytical toxicology 1996, 20 (4), 224–228. 10.1093/jat/20.4.224. [DOI] [PubMed] [Google Scholar]
- Jamdar S. C.; Pantuck C. B.; Diaz J.; Mets B. A rapid, sensitive assay for cocaine and its metabolites in biological fluids using solid-phase extraction and high-performance liquid chromatography. Journal of analytical toxicology 2000, 24 (6), 438–441. 10.1093/jat/24.6.438. [DOI] [PubMed] [Google Scholar]
- Fernandez P.; Morales L.; Vazquez C.; Bermejo A. M.; Tabernero M. J. HPLC–DAD determination of opioids, cocaine and their metabolites in plasma. Forensic science international 2006, 161 (1), 31–35. 10.1016/j.forsciint.2005.10.016. [DOI] [PubMed] [Google Scholar]
- Chinn D. M.; Crouch D. J.; Peat M. A.; Finkle B. S.; Jennison T. A. Gas chromatography-chemical ionization mass spectrometry of cocaine and its metabolites in biological fluids. Journal of Analytical Toxicology 1980, 4 (1), 37–42. 10.1093/jat/4.1.37. [DOI] [PubMed] [Google Scholar]
- Taylor R. W.; Jain N. C.; George M. P. Simultaneous identification of cocaine and benzoylecgonine using solid phase extraction and gas chromatography/mass spectrometry. Journal of analytical toxicology 1987, 11 (5), 233–234. 10.1093/jat/11.5.233. [DOI] [PubMed] [Google Scholar]
- Foltz R. L. Recent applications of mass spectrometry in forensic toxicology. International Journal of Mass Spectrometry and Ion Processes 1992, 118, 237–263. 10.1016/0168-1176(92)85064-7. [DOI] [Google Scholar]
- Virag L.; Jamdar S.; Chao C. R.; Morishima H. O. Sensitive assay for cocaine and benzoylecgonine using solid-phase extraction and gas chromatography. Journal of Chromatography B: Biomedical Sciences and Applications 1994, 658 (1), 135–141. 10.1016/0378-4347(94)00198-7. [DOI] [PubMed] [Google Scholar]
- De la Torre R.; Ortuño J.; González M. L.; Farré M.; Camí J.; Segura J. Determination of cocaine and its metabolites in human urine by gas chromatography/mass spectrometry after simultaneous use of cocaine and ethanol. J. Pharm. Biomed. Anal. 1995, 13 (3), 305–312. 10.1016/0731-7085(95)01284-R. [DOI] [PubMed] [Google Scholar]
- Cone E. J.; Oyler J.; Darwin W. D. Cocaine disposition in saliva following intravenous, intranasal, and smoked administration. Journal of analytical toxicology 1997, 21 (6), 465–475. 10.1093/jat/21.6.465. [DOI] [PubMed] [Google Scholar]
- Jeanville P. M.; Estapé E. S.; Needham S. R.; Cole M. J. Rapid confirmation/quantitation of cocaine and benzoylecgonine in urine utilizing high performance liquid chromatography and tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2000, 11 (3), 257–263. 10.1016/S1044-0305(99)00138-5. [DOI] [PubMed] [Google Scholar]
- Klingmann A.; Skopp G.; Aderjan R. Analysis of Cocaine, Benzoylecgonine Ecgonine Methyl Ester, and Ecgonine by High-Pressure Liquid Chromatography-API Mass Spectrometry and Application to a Short-Term Degradation Study of Cocaine in Plasma. Journal of analytical toxicology 2001, 25 (6), 425–430. 10.1093/jat/25.6.425. [DOI] [PubMed] [Google Scholar]
- Jeanville P. M.; Estapé E. S.; Torres-Negrón I.; Martí A. Rapid confirmation/quantitation of ecgonine methyl ester, benzoylecgonine, and cocaine in urine using on-line extraction coupled with fast HPLC and tandem mass spectrometry. Journal of analytical toxicology 2001, 25 (1), 69–75. 10.1093/jat/25.1.69. [DOI] [PubMed] [Google Scholar]
- Jagerdeo E.; Montgomery M. A.; LeBeau M. A.; Sibum M. An automated SPE/LC/MS/MS method for the analysis of cocaine and metabolites in whole blood. Journal of Chromatography B 2008, 874 (1–2), 15–20. 10.1016/j.jchromb.2008.08.026. [DOI] [PubMed] [Google Scholar]
- Langman L. J.; Bjergum M. W.; Williamson C. L.; Crow F. W. Sensitive method for detection of cocaine and associated analytes by liquid chromatography-tandem mass spectrometry in urine. Journal of analytical toxicology 2009, 33 (8), 447–455. 10.1093/jat/33.8.447. [DOI] [PubMed] [Google Scholar]
- Zancanaro I.; Limberger R. P.; Bohel P. O.; dos Santos M. K.; De Boni R. B.; Pechansky F.; Caldas E. D. Prescription and illicit psychoactive drugs in oral fluid—LC–MS/MS method development and analysis of samples from Brazilian drivers. Forensic science international 2012, 223 (1–3), 208–216. 10.1016/j.forsciint.2012.08.048. [DOI] [PubMed] [Google Scholar]
- Imbert L.; Dulaurent S.; Mercerolle M.; Morichon J.; Lachâtre G.; Gaulier J. M. Development and validation of a single LC–MS/MS assay following SPE for simultaneous hair analysis of amphetamines, opiates, cocaine and metabolites. Forensic science international 2014, 234, 132–138. 10.1016/j.forsciint.2013.11.004. [DOI] [PubMed] [Google Scholar]
- Barroso M.; Gallardo E. Assessing cocaine abuse using LC–MS/MS measurements in biological specimens. Bioanalysis 2015, 7 (12), 1497–1525. 10.4155/bio.15.72. [DOI] [PubMed] [Google Scholar]
- Schaffer M.; Cheng C. C.; Chao O.; Hill V.; Matsui P. Analysis of cocaine and metabolites in hair: validation and application of measurement of hydroxycocaine metabolites as evidence of cocaine ingestion. Anal. Bioanal. Chem. 2016, 408, 2043–2054. 10.1007/s00216-016-9354-x. [DOI] [PubMed] [Google Scholar]
- Chen X.; Zheng X.; Ding K.; Zhou Z.; Zhan C. G.; Zheng F. A quantitative LC–MS/MS method for simultaneous determination of cocaine and its metabolites in whole blood. J. Pharm. Biomed. Anal. 2017, 134, 243–251. 10.1016/j.jpba.2016.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziadosz M.; Teske J.; Henning K.; Klintschar M.; Nordmeier F. LC–MS/MS screening strategy for cannabinoids, opiates, amphetamines, cocaine, benzodiazepines and methadone in human serum, urine and post-mortem blood as an effective alternative to immunoassay based methods applied in forensic toxicology for preliminary examination. Forensic Chemistry 2018, 7, 33–37. 10.1016/j.forc.2017.12.007. [DOI] [Google Scholar]
- Mandrioli R.; Mercolini L.; Protti M. Blood and plasma volumetric absorptive microsampling (VAMS) coupled to LC-MS/MS for the forensic assessment of cocaine consumption. Molecules 2020, 25 (5), 1046. 10.3390/molecules25051046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelmaszczyk P.; Gacek E.; Wietecha-Posłuszny R. Optimized and Validated DBS/MAE/LC–MS Method for Rapid Determination of Date-Rape Drugs and Cocaine in Human Blood Samples—A New Tool in Forensic Analysis. Separations 2021, 8 (12), 249. 10.3390/separations8120249. [DOI] [Google Scholar]
- Vergne M. J. Crime Scene Investigation: Simulated Post-mortem LC–MS/MS Analysis of Cocaine and Cocaine Metabolites in Synthetic Human Serum. J. Chem. Educ. 2021, 98 (11), 3567–3571. 10.1021/acs.jchemed.1c00161. [DOI] [Google Scholar]
- Rana S.; Reichardt E.; Claire G. Cocaethylene presence in oral fluids from cocaine users. Toxicologie Analytique et Clinique 2022, 34 (3), S158. 10.1016/j.toxac.2022.06.269. [DOI] [Google Scholar]
- Whitehead H. D.; Hayes K. L.; Swartz J. A.; Prete E.; Robison-Taylor L.; Mackesy-Amiti M. E.; Lieberman M. Validated method for the analysis of 22 illicit drugs and their metabolites via liquid chromatography tandem mass spectrometry (LC–MS/MS) in illicit drug samples collected in Chicago, IL. Forensic Chem. 2023, 33, 100475 10.1016/j.forc.2023.100475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozseker P. E.; Yucel S. P.; Daglioglu N. Optimization of biochip assay for illegal substances on drug abusers’ whole blood: Randox Evidence vs LC-MS/MS. J. Immunoassay Immunochem. 2023, 44, 313–325. 10.1080/15321819.2023.2189451. [DOI] [PubMed] [Google Scholar]
- Chen H. W.; Liu H. T.; Kuo Y. N.; Yang D. P.; Ting T. T.; Chen J. H.; Chiu J. Y.; Jair Y. C.; Li H. C.; Chiang P. J.; Chen W. R.; Lin M. C.; Hsu Y. H.; Chen P. S. Rapid and sensitive dilute-and-shoot analysis using LC-MS-MS for identification of multi-class psychoactive substances in human urine. J. Pharm. Biomed. Anal. 2023, 233, 115443 10.1016/j.jpba.2023.115443. [DOI] [PubMed] [Google Scholar]
- Da Cunha K. F.; Oliveira K. D.; Huestis M. A.; Costa J. L. Screening of 104 new psychoactive substances (NPS) and other drugs of abuse in oral fluid by LC–MS-MS. Journal of analytical toxicology 2020, 44 (7), 697–707. 10.1093/jat/bkaa089. [DOI] [PubMed] [Google Scholar]
- Müller V. V.; Hahn R. Z.; Lizot L. d. L. F.; Schneider A.; da Silva C. P.; Gerbase F. E.; Pereira D.; Linden R.; Antunes M. V. Validation of an analytical method for the simultaneous determination of 16 drugs and metabolites in hair in the context of driving license granting. Forensic Sci. Int. 2020, 315, 110428 10.1016/j.forsciint.2020.110428. [DOI] [PubMed] [Google Scholar]
- Cappelle D.; De Keukeleire S.; Neels H.; Been F.; De Doncker M.; Dom G.; Crunelle C. L.; Covaci A.; van Nuijs A. L. N. Keratinous matrices for the assessment of drugs of abuse consumption: A correlation study between hair and nails. Drug Test. Anal. 2018, 10 (7), 1110–1118. 10.1002/dta.2356. [DOI] [PubMed] [Google Scholar]
- Mannocchi G.; Di Trana A.; Tini A.; Zaami S.; Gottardi M.; Pichini S.; Busardò F. P. Development and validation of fast UHPLC-MS/MS screening method for 87 NPS and 32 other drugs of abuse in hair and nails: application to real cases. Anal. Bioanal. Chem. 2020, 412, 5125–5145. 10.1007/s00216-020-02462-6. [DOI] [PubMed] [Google Scholar]
- Liu P.; Liu W.; Qiao H.; Jiang S.; Wang Y.; Chen J.; Su M.; Di B. Simultaneous quantification of 106 drugs or their metabolites in nail samples by UPLC-MS/MS with high-throughput sample preparation: Application to 294 real cases. Anal. Chim. Acta 2022, 1226, 340170 10.1016/j.aca.2022.340170. [DOI] [PubMed] [Google Scholar]
- Ji J. J.; Xu D.; Yan H.; Xiang P.; Shen M. LC–MS-MS Determination of 88 Psychotropic Drugs in 1,865 Hair Samples from Addicts in Drug Abstinence. Journal of Analytical Toxicology 2023, 47 (1), 52–58. 10.1093/jat/bkac024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai W.; Qiao Z.; Xiang P.; Dang Y.; Shi Y. A UPLC-MS/MS methodological approach for the analysis of 75 phenethylamines and their derivatives in hair. J. Pharm. Biomed. Anal. 2023, 229, 115367 10.1016/j.jpba.2023.115367. [DOI] [PubMed] [Google Scholar]
- Scientific Working Group for Forensic Toxicology (SWGTOX) standard practices for method validation in forensic toxicology. In J. Anal. Toxicol. Oxford University Press, 201337 (7), , 452–474. 10.1093/jat/bkt054 [DOI] [PubMed] [Google Scholar]
- Favretto D.; Cooper G.; Andraus M.; Sporkert F.; Agius R.; Appenzeller B.; Baumgartner M.; Binz T.; Cirimele V.; Kronstrand R.; del Mar Ramirez M.; Strano-Rossi S.; Uhl M.; Vincenti M.; Yegles M. The Society of Hair Testing consensus on general recommendations for hair testing and drugs of abuse testing in hair. Drug Test. Anal. 2023, 15 (9), 1042–1046. 10.1002/dta.3526. [DOI] [PubMed] [Google Scholar]
- Society of Forensic Toxicologists/American Academy of Forensic Sciences (SOFT/AAFS). Forensic Toxicology Laboratory Guidelines 2006. version. [Google Scholar]
- Pizzolato T. M.; de Alda M. J. L.; Barceló D. LC-based analysis of drugs of abuse and their metabolites in urine. TrAC Trends Anal. Chem. 2007, 26 (6), 609–624. 10.1016/j.trac.2007.04.005. [DOI] [Google Scholar]
- López P.; Martello S.; Bermejo A. M.; De Vincenzi E.; Tabernero M. J.; Chiarotti M. Validation of ELISA screening and LC–MS/MS confirmation methods for cocaine in hair after simple extraction. Anal. Bioanal. Chem. 2010, 397, 1539–1548. 10.1007/s00216-010-3684-x. [DOI] [PubMed] [Google Scholar]
- Segura J.; Stramesi C.; Redón A.; Ventura M.; Sanchez C. J.; GonzálezG San L; Montagna M. Immunological screening of drugsof abuse and gas chromatographic-mass spectrometric confirmationof opiates and cocaine in hair. J. Chromatogr. B: Biomed. Sci. Appl. 1999, 724 (1), 9–21. 10.1016/S0378-4347(98)00531-3. [DOI] [PubMed] [Google Scholar]
- Recommendations for hair testing in forensic cases. Forensic Sci. Int. 2004, 145 (2–3), 83–84. 10.1016/j.forsciint.2004.04.022. [DOI] [PubMed] [Google Scholar]
- Pragst F.; Balikova M. A. State of the art in hair analysis for detection of drug and alcohol abuse. Clinicachimica acta 2006, 370, 17–49. 10.1016/j.cca.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Kuwayama K.; Miyaguchi H.; Iwata Y.; Kanamori T.; Tsujikawa K.; Yamamuro T.; Segawa H.; Inoue H. Time-course measurements of drug concentrations in hair and toenails after single administrations of pharmaceutical products: Time-course measurements of drug concentrations in hair and toenails. Drug Testing and Analysis 2017, 9 (4), 571–577. 10.1002/dta.1991. [DOI] [PubMed] [Google Scholar]
- Kronstrand R.; Andersson M. C.; Ahlner J.; Larson G. Incorporation of selegiline metabolites into hair after oral selegiline intake. Journal of Analytical Toxicology 2001, 25 (7), 594–601. 10.1093/jat/25.7.594. [DOI] [PubMed] [Google Scholar]
- Kronstrand R.; Förstberg-Peterson S.; Kagedal B.; Ahlner J.; Larson G. Codeine concentration in hair after oral administration is dependent on melanin content. Clinical Chemistry 1999, 45 (9), 1485–1494. 10.1093/clinchem/45.9.1485. [DOI] [PubMed] [Google Scholar]
- Barbosa J.; Faria J.; Carvalho F.; Pedro M.; Queirós O.; Moreira R.; Dinis-Oliveira R. J. Hair as an alternative matrix in bioanalysis. Bioanalysis 2013, 5 (8), 895–914. 10.4155/bio.13.50. [DOI] [PubMed] [Google Scholar]
- Palmeri A.; Pichini S.; Pacifici R.; Zuccaro P.; Lopez A. Drugs in nails: physiology, pharmacokinetics and forensic toxicology. Clinical pharmacokinetics 2000, 38, 95–110. 10.2165/00003088-200038020-00001. [DOI] [PubMed] [Google Scholar]