

Abstract
Immune-checkpoint inhibitors have shown modest efficacy against immunologically ‘cold’ tumours. Interleukin-12 (IL-12) — a cytokine that promotes the recruitment of immune cells into tumours as well as immune-cell activation, also in cold tumours — can cause severe immune-related adverse events in patients. Here, by exploiting the preferential overexpression of proteases in tumours, we show that fusing a domain of the IL-12 receptor to IL-12 via a linker cleavable by tumour-associated proteases largely restricts the pro-inflammatory effects of IL-12 to tumour sites. In mouse models of subcutaneous adenocarcinoma and orthotopic melanoma, masked IL-12 delivered intravenously did not cause systemic IL-12 signalling and eliminated systemic immune-related adverse events, led to potent therapeutic effects via the remodelling of the immune-suppressive microenvironment, and rendered cold tumours responsive to immune-checkpoint inhibition. We also show that masked IL-12 is activated in tumour lysates from patients. Protease-sensitive masking of potent yet toxic cytokines may facilitate their clinical translation.
Checkpoint-inhibition (CPI) therapies, such as those using anti-programmed death 1 (αPD-1) antibodies, have led to large successes in the treatment of certain cancer types; yet increasing evidence suggests that these therapies are largely ineffective in immunologically ‘cold’ tumours, owing to a lack of sufficiently primed CD8+ T cells1,2. The immunosuppressive tumour microenvironment (TME) is dominated by regulatory T (Treg) cells and pro-tumourigenic macrophages (that is, M2-like macrophages)3. Patients who respond well to CPI therapies tend to exhibit a T helper 1- (Th1)-biased TME, driven by the expression of interferon-γ (IFNγ) and IFNγ-related genes4,5. An approach that can safely transform immunologically cold tumours into inflamed tumours would both increase the proportion of patients responding to CPI therapies and offer alternative treatment options for CPI non-responders. Remodelling of such an immunosuppressive TME can be achieved by pro-inflammatory cues that induce Th1 cytokine and chemokine profiles.
Immunostimulatory agents, including pro-inflammatory cytokines, may overcome CD8+ T cell exclusion by triggering a wide array of inflammatory responses, leading to infiltration and activation of antitumour CD8+ T cells6,7. IL-12 is an attractive cytokine that stimulates both the innate and the adaptive immune system and is able to activate antigen-presenting cells (APCs)8. We have previously seen potency of an engineered IL-12 in multiple murine cancer models including complete remissions, which, in our hands, was stronger than other clinically-approved immunotherapies such as CPIs or IL-29. The antitumour effects of IL-12 are mostly mediated by IFNγ, a direct downstream molecule that is secreted by T and natural killer (NK) cells10.
Despite these strong therapeutic effects, clinical trials examining recombinant IL-12 have failed due to dose-limiting immune-related adverse events (irAEs)11, as anticancer activity was observed at or above the maximum tolerated dose12. Numerous attempts of IL-12 clinical translation have been made for over three decades, yet no IL-12 products have been approved in the clinic. Therefore, solving the challenge presented by IL-12-induced irAEs is crucial to translate this powerful antitumour cytokine to the clinic.
After IL-12 administration, cytokine release syndrome and liver damage have been observed in clinical trials11. A high concentration of circulating IFNγ mediates these side effects, as evidenced by studies conducted in mice lacking the IFNγ receptor13 and by administration of neutralising antibodies against IFNγ14,15. Yet IFNγ is indispensable for antitumour efficacy of IL-12 therapy10, signifying the importance of developing an engineered IL-12 that would localize the therapeutic effects to the tumour site while sparing healthy tissues and the circulation.
Here, we report a protein-engineering approach as a solution for IL-12 toxicity and to remodel the cold TME. We describe a strategy to block the signalling activity of IL-12 by masking the receptor binding site of IL-12 with a fused receptor domain, attached via a tumour protease-cleavable linker, so that the activity is selectively restored upon proteolytic cleavage in the TME.
Results
In vitro activation of masked IL-12 leads to fully restored IL-12 bioactivity
Latency can be conferred to cytokines upon fusion of domains that inhibit cytokine-receptor interaction16,17. We hypothesised that fusion of the first two fibronectin type-III domains of the mouse IL-12 receptor β1 (IL12Rβ1; Q20-A261, referred to herein as the mask, “M”) to IL-12 would render it inactive (Fig. 1a and Supplementary Fig. 1a). By exploiting the heterodimeric structure of IL-12, we fused the mask to the N-terminus of p35 subunit and co-transfected it with p40, with the idea that the mask would engage p4018. By varying the distance between M and p35, we determined that the optimal length of the linker that prevented aggregation/dimerization is approximately 45 amino acids (Supplementary Fig. 1b–d).
Fig. 1 |. Masked IL-12 fully regains activity upon treatment with recombinant proteases.
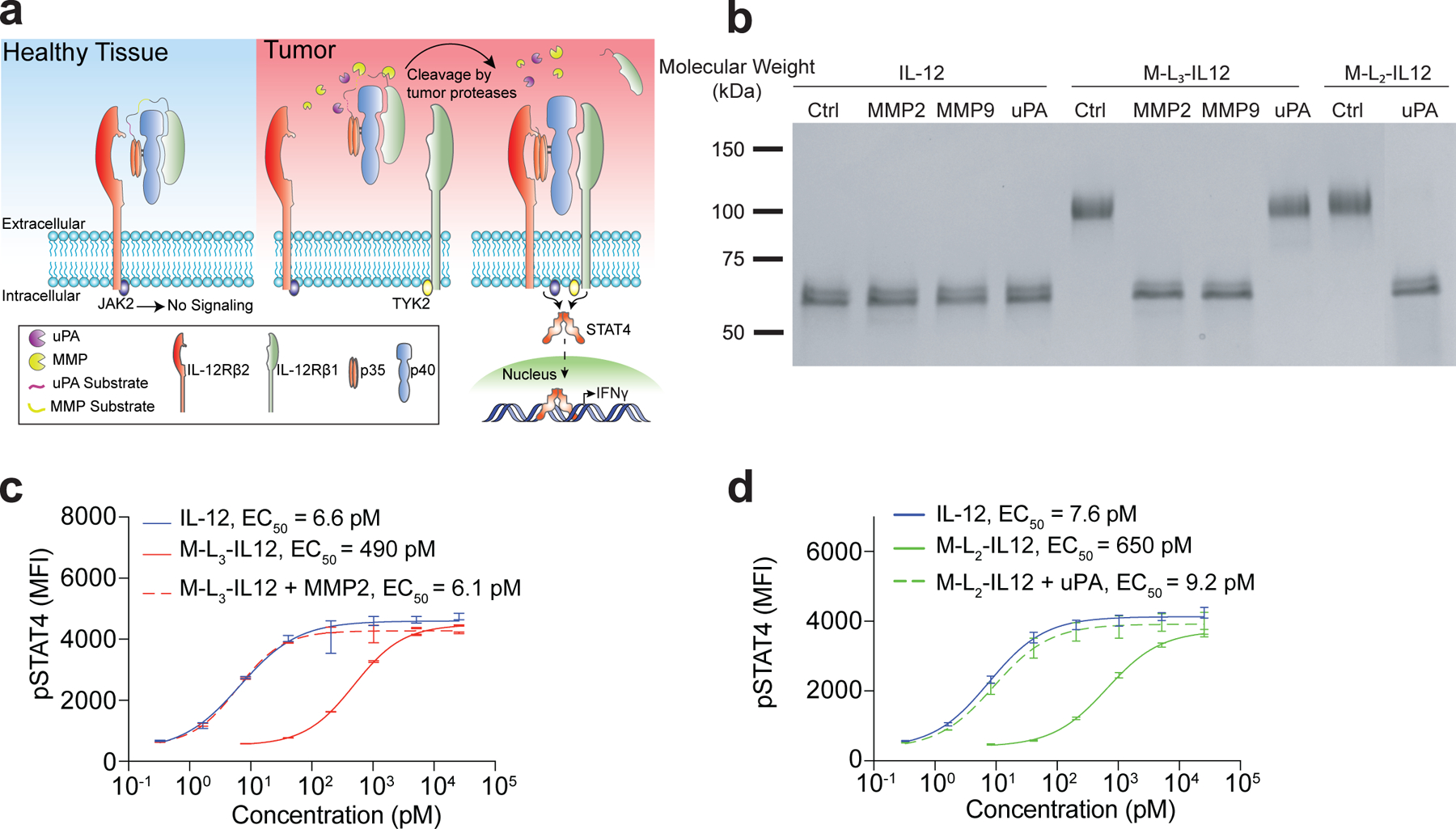
a, Schematic of masked IL-12 in healthy tissues (no signalling) and in the tumour, with the mask being cleaved by various tumour-associated proteases. b, SDS-PAGE analysis of the cleavage of masked IL-12 variants by recombinant proteases. IL-12 (50 μg/ml; 0.84 μM), M-L3-IL12 (0.84 μM) or M-L2-IL12 (0.84 μM) were incubated with activated MMP2 (2 μg/ml), MMP9 (5 μg/ml) for 30 min at 37°C or with uPA (10 μg/ml) for 2.5 hr at 37°C. c,d, Dose-response relationship of phosphorylated STAT4 (pY693) with MMP2-treated M-L3-IL12 (c) and uPA-treated M-L2-IL12 (d) in preactivated primary mouse CD8+ T cells (n=2 per condition, technical duplicates). Data are mean ± s.e.m.; EC50, half-maximum effective concentration; MFI, mean fluorescence intensity. Experiments were performed at least twice, with similar results. Representative data are shown.
We then expressed several masked IL-12 variants with various linker substrates (Ln, where n refers to linker ID) that can be cleaved by different tumour proteases (Table 1). We characterised proteolytic cleavage of masked IL-12 molecules, namely M-L3-IL12 and M-L2-IL12, by treating them with recombinant matrix metalloproteinase-2 (MMP2), MMP9 or urokinase-plasminogen activator (uPA), a serine protease (SP), and visualised the cleavage by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) (Fig. 1b). Proteolytic cleavage of MMP-sensitive M-L3-IL12 (containing three repeats of VPLSLYSG19) and SP-sensitive M-L2-IL12 (containing three repeats of LSGRSDNH20) yielded molecules corresponding to the molecular mass of IL-12. These proteases did not digest wild-type IL-12 itself. To test whether the cleaved molecules are bioactive, we stimulated preactivated mouse CD8+ T cells with either latent or activated forms of masked IL-12 (Fig. 1c,d). The intact masked IL-12 variants were ~80-fold less active than the unmodified IL-12 as measured by phosphorylation of signal transducer and activator of transcription 4 (STAT4) in the absence of proteases, whereas proteolytic activation of engineered IL-12 fully restored the activity. To confirm that the mask does not associate with IL-12 after proteolytic cleavage, we incubated the soluble, extracellular portion of IL-12Rb1 with IL-12 at 1:1 molar ratio and performed STAT4 stimulation assay (Supplementary Fig. 2). Soluble IL-12Rb1 only minimally affected IL-12 signalling, suggesting that the mask inhibits IL-12 bioactivity only when it is intact. Furthermore, the affinity of IL-12 for the full receptor complex (IL-12Rb1 + IL-12Rb2) is much higher than for either subunit alone21, suggesting that our mask’s inhibitory activity will be insignificant after cleavage.
Table 1 |.
Amino acid sequences of linkers used to generate masked IL-12 variants.
| Linker ID | Amino acid sequence | Cleaved by |
|---|---|---|
| L1 | (GGGS)2(HPVGLLAR)3(GGGS)2 | MMP2, MMP9 |
| L2 | (GGGS)2(SGLLSGRSDNH)3(GGGS)2 | uPA, matriptase, legumain |
| L3 | (GGGS)2(VPLSLYSG)3(GGGS)2 | MMP2, MMP7, MMP9 |
| L4 | (GGGS)2(VPLSLYSG)(GGGS)2(LSGRSDNH)2(GGGS)2 | MMP and Serine Proteases |
| L6 | (HPVGLLARVPLSLYSG)2(LSGRSDNH)(GGGS)2 | MMP and Serine Proteases |
| LNC | (GGGS)11 | Non-cleavable |
Linker substrates are shown in bold font.
We investigated the cleavage of several linkers by MMP2 in vitro and found that HPVGLLAR19 (substrate in M-L1-IL12) is a more reactive substrate than VPLSLYSG19 (substrate in M-L3-IL12; Supplementary Fig. 3) as visualised by SDS-PAGE analysis. Furthermore, cleavage was dependent on the number of substrate repeats, as the construct containing only one VPLSLYSG (M-L4-IL12) was processed less efficiently than the construct containing three repeats of VPLSLYSG (M-L3-IL12; Supplementary Fig. 3). SP-sensitive M-L2-IL12, containing three repeats of LSGRSDNH, was not cleaved by any of the MMPs tested (Supplementary Fig. 4). Thus, we designed one additional linker, L6, that contains two repeats each of both MMP substrates (HPVGLLARVPLSLYSG) and one repeat of SP substrate (Table 1). M-L1-IL12 and M-L6-IL12 exhibited similar reactivity against MMP-2 (Supplementary Fig. 5).
Masked IL-12 retains antitumour activity in syngeneic models and causes immunological remodelling of TME
We then sought to examine the antitumour efficacy elicited by three masked IL-12 variants in the B16F10 mouse melanoma model to determine the optimal linker. MMP/SP-reactive M-L6-IL12 induced a significantly stronger antitumour response compared to SP-only reactive M-L2-IL12 and a slightly stronger response compared to M-L4-IL12, which contained one MMP-sensitive substrate (VPLSLYSG) and two SP-sensitive substrates (Fig. 2a, Extended Data Fig. 1a,b). The superiority of L6 over L2 and L4 was further confirmed via ex vivo cleavage analysis by EMT6 tumour homogenate (Extended Data Fig. 2a). Incubation of M-L6-IL12 with EMT6 homogenate resulted in the most activation when compared to either M-L2-IL12, M-L4-IL12 or the non-cleavable M-LNC-IL12 as measured by pSTAT4 assay. This result suggests that the enzymatic sensitivity of the linker is a crucial design parameter for producing an adequate antitumour response22, and thus we selected M-L6-IL12 as our lead masked IL-12 variant.
Fig. 2 |. Masked IL-12 induces a strong antitumour response and potentiates CPI therapy.
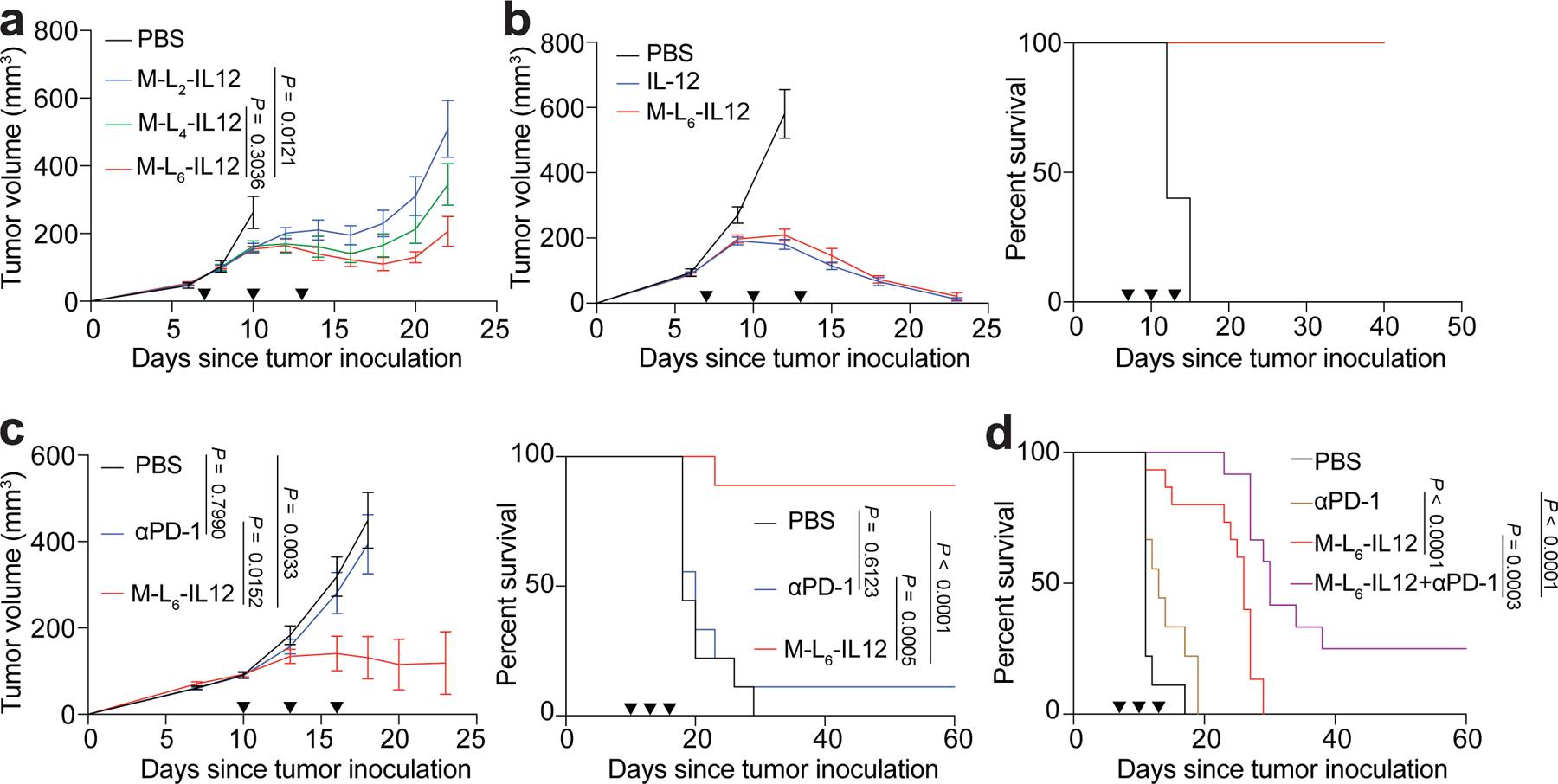
a, B16F10 melanoma-bearing mice were treated with PBS (n=6), or 250 pmol of M-L2-IL12 (n=7), M-L4-IL12 (n=8), M-L6-IL12 (n=7) i.v. on days 7, 10 and 13 post tumour inoculation. Tumour growth curves are shown. b, Subcutaneous MC38 colon adenocarcinoma-bearing mice were treated with PBS (n=5), 5 μg (83.3 pmol) IL-12 (n=7) or 250 pmol of M-L6-IL12 (n=7) i.v. on days 7, 10 and 13 post tumour inoculation. Tumour growth curves (left) and survival (right) are shown. c, Orthotopic EMT6 mammary carcinoma-bearing mice were treated with PBS (n=9), αPD-1 (n=9, 100 μg, i.p.) or M-L6-IL12 (n=9, 250 pmol, i.v.) on days 10, 13 and 16 post tumour inoculation. Tumour growth curves (left) and survival (right) are shown. d, Orthotopic B16F10 melanoma-bearing mice were treated with PBS (n=9), αPD-1 (n=9, 100 μg, i.p.), M-L6-IL12 (n=15, 250 pmol, i.v.) or M-L6-IL12 + αPD-1 (n=12) on days 7, 10 and 13 post tumour inoculation. Survival curves are shown. Data are mean ± s.e.m. Arrowheads indicate times of treatment. Experiments in a,b,c were performed twice with similar results. Data in d were pooled from two independent experiments. Statistical analyses were performed using ordinary one-way ANOVA with Tukey’s multiple comparison tests. For survival plots, Mantel-Cox test was used.
Next, we compared the ex vivo cleavage of M-L6-IL12 versus the non-cleavable M-LNC-IL12 by EMT6 tumour homogenate or serum obtained from EMT6-bearing mice via western blotting (Extended Data Fig. 2b). As expected, no cleavage was observed for M-LNC-IL12 upon incubation with EMT6 homogenate or serum. For M-L6-IL12, however, incubation with EMT6 homogenate, but not with serum, resulted in time-dependent cleavage of the mask. This demonstrates that M-L6-IL12 stays stable in vitro when incubated with serum from tumour-bearing mice for at least up to 6 hr. Finally, to show that cleavage takes place in vivo, we treated B16F10 melanoma-bearing mice with either M-L6-IL12 or M-LNC-IL12 and collected the tumours 2 hr post-injection for western blot analysis (Extended Data Fig. 2c). M-L6-IL12 was partially unmasked, whereas no cleaved IL-12 band was observed for M-LNC-IL12. Together, these data indicate that M-L6-IL12 stays intact in the periphery yet is activated selectively in the tumour.
We then performed a dose escalation study using M-L6-IL12 (16.7 pmol, 83.3 pmol, 250 pmol) in B16F10 model, and compared the antitumour efficacy to 83.3 pmol of unmodified IL-12 (Extended Data Fig. 3a). M-L6-IL12 exhibited a dose-dependent antitumour efficacy and at 250 pmol dose was similarly efficacious as 83.3 pmol of unmodified IL-12. To evaluate the blood plasma pharmacokinetics in naïve mice, we injected equimolar amounts of IL-12 and M-L6-IL12 i.v. and observed an increase in circulation time for the masked IL-12 (Extended Data Fig. 3b). We then compared the antitumour efficacy of M-L6-IL12 to that of unmodified IL-12 in subcutaneous MC38 colon adenocarcinoma. In this immunogenic model, both molecules exhibited high efficacy, achieving a 100% complete response (CR) rate (Fig. 2b and Supplementary Fig. 7). To assess the antitumour efficacy of M-L6-IL12 in the CPI-unresponsive model, we treated mice bearing established orthotopic EMT6 triple-negative breast tumours, which are characterised by transforming growth factor β signature and T cell exclusion23. Treatment with M-L6-IL12, but not αPD-1 antibody, led to significant extension of survival (8 CR out of 9 mice) when compared to saline-treated mice (Fig. 2c and Supplementary Fig. 8). We further investigated the efficacy of the combination of M-L6-IL12 and αPD-1 in mice bearing established orthotopic B16F10 melanomas, a cold model (Fig. 2d and Supplementary Fig. 9). Addition of aPD-1 to M-L6-IL12 resulted in significantly extended survival when compared to M-L6-IL12 treatment alone. aPD-1 treatment had no major effect on the survival of these mice, consistent with previous observations9.
To study the mechanism behind the therapeutic action of M-L6-IL12, we characterised immunological responses in B16F10 melanoma, a model that displays low basal inflammation. We collected the tumours from treated mice for intratumoural cytokine/chemokine profiling and lymphocyte infiltration analysis. IFNg, which is the direct downstream molecule and main mediator of antitumour activity of IL-1224, was equally upregulated in the TME in both treatment groups (Fig. 3a). Tumour necrosis factor-a (TNFa), which supports cancer cell cycle arrest25, was also equally expressed between the two groups. Significant production of granulocyte-macrophage colony-stimulating factor (GM-CSF), a maturation factor for antigen presenting cells (APCs)26, was noted as well, demonstrating that masked IL-12 treatment activates the innate immune compartment. Upregulation of IL-1β and IL-1α indicates Th1-biased TME remodelling27,28. We also observed elevated expression of CXCL9, CXCL10, CCL5, and CCL4, chemokines that are associated with increased infiltration of CD8+ T cells29. Other cytokines and chemokines (Extended Data Fig. 4a–f) were expressed at similar levels in both unmodified IL-12- and M-L6-IL12-treated groups. These results show that masked IL-12 can potently activate the type II IFN pathway, which leads to secretion of wide variety of inflammatory molecules.
Fig. 3 |. Masked IL-12 therapy elicits a wide range of inflammatory responses and causes immune cell infiltration in melanoma.
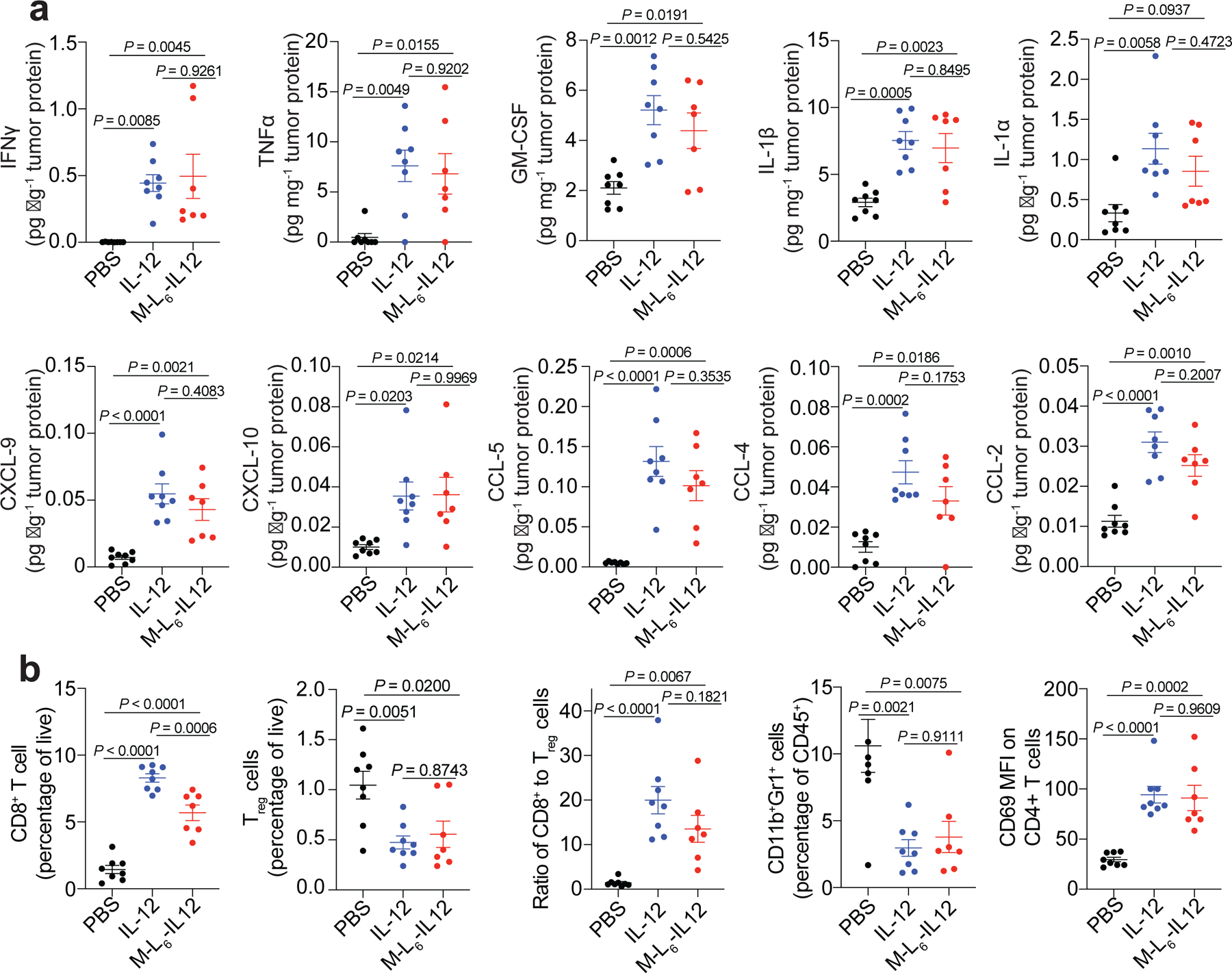
Orthotopic B16F10 melanoma-bearing mice were treated with PBS (n=8), 5 μg (83.3 pmol) IL-12 (n=8) or 250 pmol of M-L6-IL12 (n=7) i.v. on days 6 and 9 post tumour inoculation. Tumours were excised on day 11 and were homogenised for intratumoural cytokine/chemokine analysis via LEGENDPlex (a) and processed for flow cytometric analysis (b). Intratumoural cytokines were measured and normalised by total tumour protein content. Data are mean ± s.e.m. Experiments were performed twice with similar results. Statistical analyses were performed using ordinary one-way ANOVA with Tukey’s multiple comparison tests.
We then analysed whether secretion of pro-inflammatory cytokines and chemokines led to infiltration of immune cells. Both treatments resulted in significant CD8+ T infiltration as compared to saline-treated mice (Fig. 3b). Furthermore, we observed equal reduction in Treg cells by both treatment groups. Importantly, the ratio of CD8+ T cell to Treg cells, which is considered as one of the main indicators of successful immunotherapy30, was similarly upregulated in IL-12- and masked IL-12-treated animals. Both molecules reduced the percentages of suppressive CD11b+Gr-1+ cells31 within the CD45+ compartment. Although no major change was detected in the numbers of conventional CD4+ T cells (Extended Data Fig. 4g), M-L6-IL12 treatment led to an increase in CD69 expression on these cells. Masked IL-12 caused dendritic cell (DC) and macrophage infiltration (Extended Data Fig. 4h,i). These macrophages may be M1-polarised, as we have seen previously9. Together, these results demonstrate that M-L6-IL12 and unmodified IL-12 exert comparable antitumour efficacy, causing profound immunological changes in the TME.
Given that cytokine/chemokine measurements and immune cell infiltration data were obtained from the same biological samples, we performed a correlation analysis to determine which cytokines/chemokines are associated with high CD8+ T cell-to-Treg ratios (Supplementary Fig. 10). In the B16F10 melanoma model, the strongest positive correlation was observed for IFNg, TNFa, and IL-1b, and chemokines CXCL-9/10, CCL-5, and CCL-4 (Supplementary Fig. 10a–g). A weaker correlation with increased CD8+ T cell-to-Treg ratios was seen with IL-10 and IL-1α, whereas no significant correlation was noticed with intratumoural IL-6 and CXCL-1 (Supplementary Fig. 10h–l). This analysis indicates that Th1-biased TME strongly correlates with enhanced CD8+ T cell-to-Treg ratios.
Treatment with protease-sensitive IL-12 minimises systemic irAEs
We next investigated whether the decreased in vitro bioactivity of masked IL-12 translates to fewer systemic irAEs. We treated healthy C3H/HeJ mice, a strain that is highly sensitive to low amounts of recombinant IL-1215, daily with either 0.5 μg IL-12 or 3-fold molar dose of M-L6-IL12 and monitored body weight change (Fig. 4a). Unmodified IL-12 induced marked body weight loss compared to saline-treated mice, whereas masked IL-12-treated mice maintained their body weight over the course of the study. In clinical trials, recombinant IL-12 administration led to significant elevation of IFNg, the main contributor to the irAEs13,15, and transaminases such as alanine aminotransferase (ALT) and aspartate aminotransferase (AST)11 activities in blood, markers of hepatotoxicity. We administered either unmodified IL-12 or masked IL-12 three times, every three days to healthy C57BL/6 mice and quantified inflammatory biomarkers in the blood. Fusion of the mask to IL-12 substantially reduced plasma IFNg, IL-6, TNFα and CCL-2 concentrations (Fig. 4b). We also observed a significantly decreased amount of circulating IL-12 in mice receiving the engineered IL-12 (Extended Data Fig. 5a). No significant upregulation of IL-10, IL-1α, IL-1β or IFNβ was observed at this time point (Extended Data Fig. 5b–e). Liver damage measured by ALT and AST activities, as well as pancreas damage measured by amylase activity in serum was also significantly decreased by fusing the mask to IL-12 (Fig. 4b). Albumin, blood urea nitrogen, and total protein levels were maintained among the groups (Extended Data Fig. 5f–h).
Fig. 4 |. Masked IL-12 eliminates side effects associated with IL-12 therapy in healthy animals.
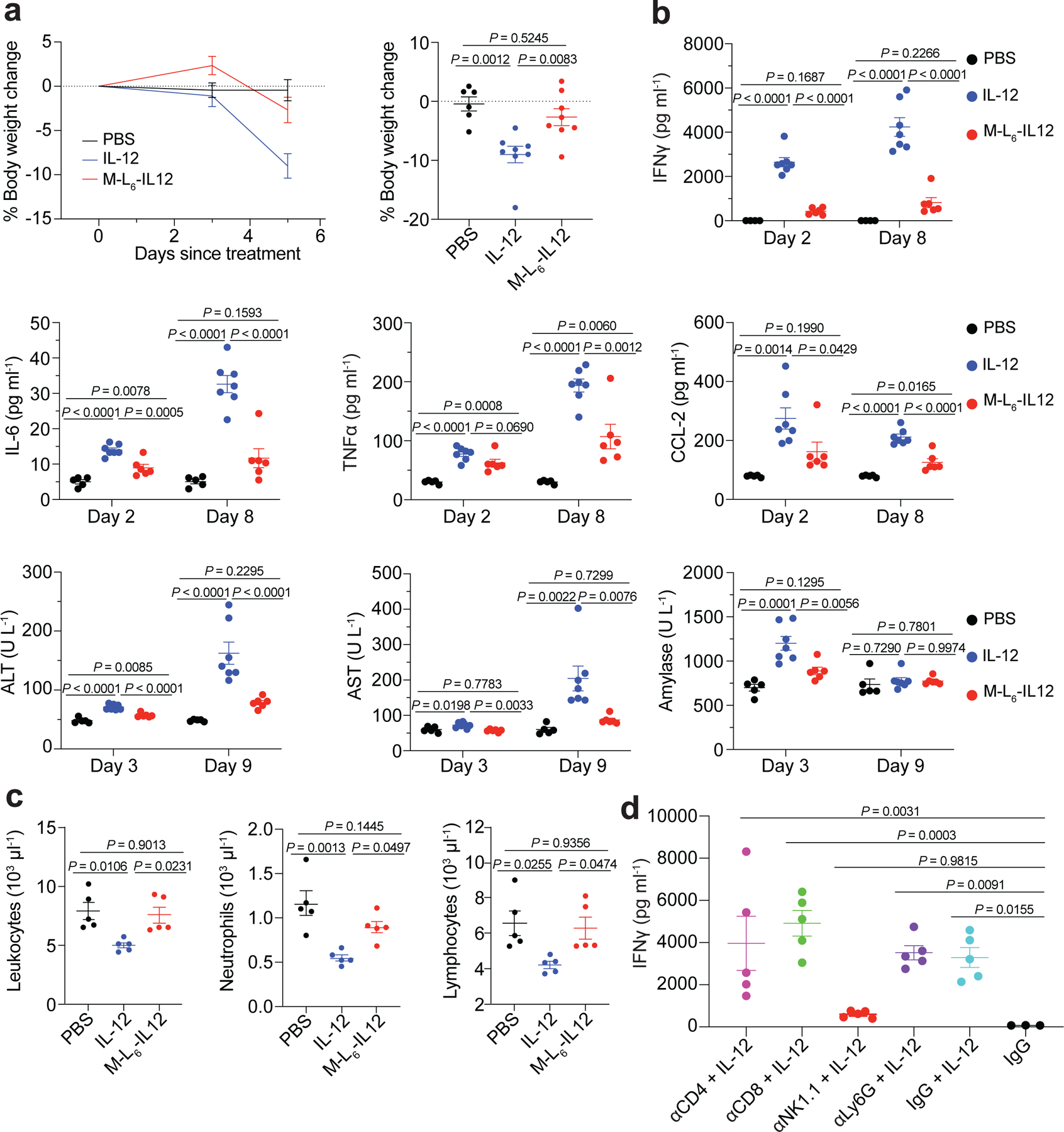
a, Healthy C3H/HeJ mice were dosed daily (starting from day 0) with PBS (n=6), 0.5 μg (8.3 pmol) of IL-12 (n=8) or 25 pmol of M-L6-IL12 (n=8) s.c. 6 times. Body weight change (left) and day 5 comparison (right) are shown. b, Healthy C57BL/6 mice were treated with PBS (n=5), 5 μg (83.3 pmol) IL-12 (n=7) or 250 pmol M-L6-IL12 (n=6) i.v. on days 0, 3 and 6. Mice were bled on days 2, 3, 8 and 9 for plasma cytokine analysis using LEGENDPlex and blood chemistry analysis. c, Healthy C57BL/6 mice were treated with PBS (n=5), 5 μg (83.3 pmol) IL-12 (n=5) or 250 pmol M-L6-IL12 (n=5) i.v. and bled 4 days later for quantification of circulating blood cells using hematology analyzer. d, Healthy C57BL/6 mice were administered neutralising antibodies (400 μg, n=5 per group) on days 0 and 3. On day 4, mice were treated i.v. with 5 μg (83.3 pmol) IL-12 and bled on day 6 for plasma IFNg measurement. Data are mean ± s.e.m. Experiments were performed twice with similar results. Statistical analyses were performed using ordinary one-way ANOVA with Tukey’s multiple comparison tests.
Another common side effect of recombinant IL-12 therapy observed in the clinic is the decrease of circulating white blood cells, such as neutrophils and lymphocytes32, which may increase the risk of infections. Treatment with unmodified IL-12, but not M-L6-IL12, induced significant leukopenia, neutropenia and lymphopenia compared to healthy controls (Fig. 4c). We next evaluated the effect of administration of neutralising antibodies on systemic IFNγ production upon IL-12 injection (Fig. 4d). We found that depletion of NK cells had a major impact on IFNγ production, whereas depletion of CD4+, CD8+ or LyG6+ cells did not reduce systemic IFNγ. This result suggests that NK cells are the main producers of IFNγ in response to IL-12, corroborating recent findings18.
We then sought to verify our findings of reduced toxicity in tumour-bearing mice (Fig. 5). As in tumour-free mice, masked IL-12 injection significantly decreased plasma IFNg concentration compared to IL-12 (Fig. 5a). The concentration of IL-12 protein in the blood, which may include some of the injected protein as well as de novo IL-12, was also decreased to the level of saline-treated mice. We did not observe significant upregulation in plasma IL-1α at the timepoint tested. IL-12 reportedly induces infiltration of myeloid cells into the spleen33 and decreases systemic hematopoiesis34. In the spleens of B16F10 melanoma-bearing mice, systemic administration of unmodified IL-12 decreased CD45+ cells, increased CD11b+Gr-1+ and CD11b+Gr-1− cells compared to saline treatment, whereas masked IL-12 did not (Fig. 5b). Significant reduction in numbers of splenic CD8+ and CD4+ T cells was noted upon treatment with IL-12, but not masked IL-12 (Fig. 5b), indicating diminished peripheral activity of M-L6-IL12. MC38-bearing mice treated with M-L6-IL12 exhibited decreased liver, kidney and pancreas damage markers when compared to mice treated with unmodified IL-12, supporting our findings in B16F10-bearing mice (Extended Data Fig. 6). Together, these data demonstrate that our masking approach significantly reduces the irAEs induced by IL-12 in both healthy and tumour-bearing mice, down to the level of saline injections.
Fig. 5 |. Treatment of melanoma-bearing mice with masked IL-12 does not generate systemic irAEs.
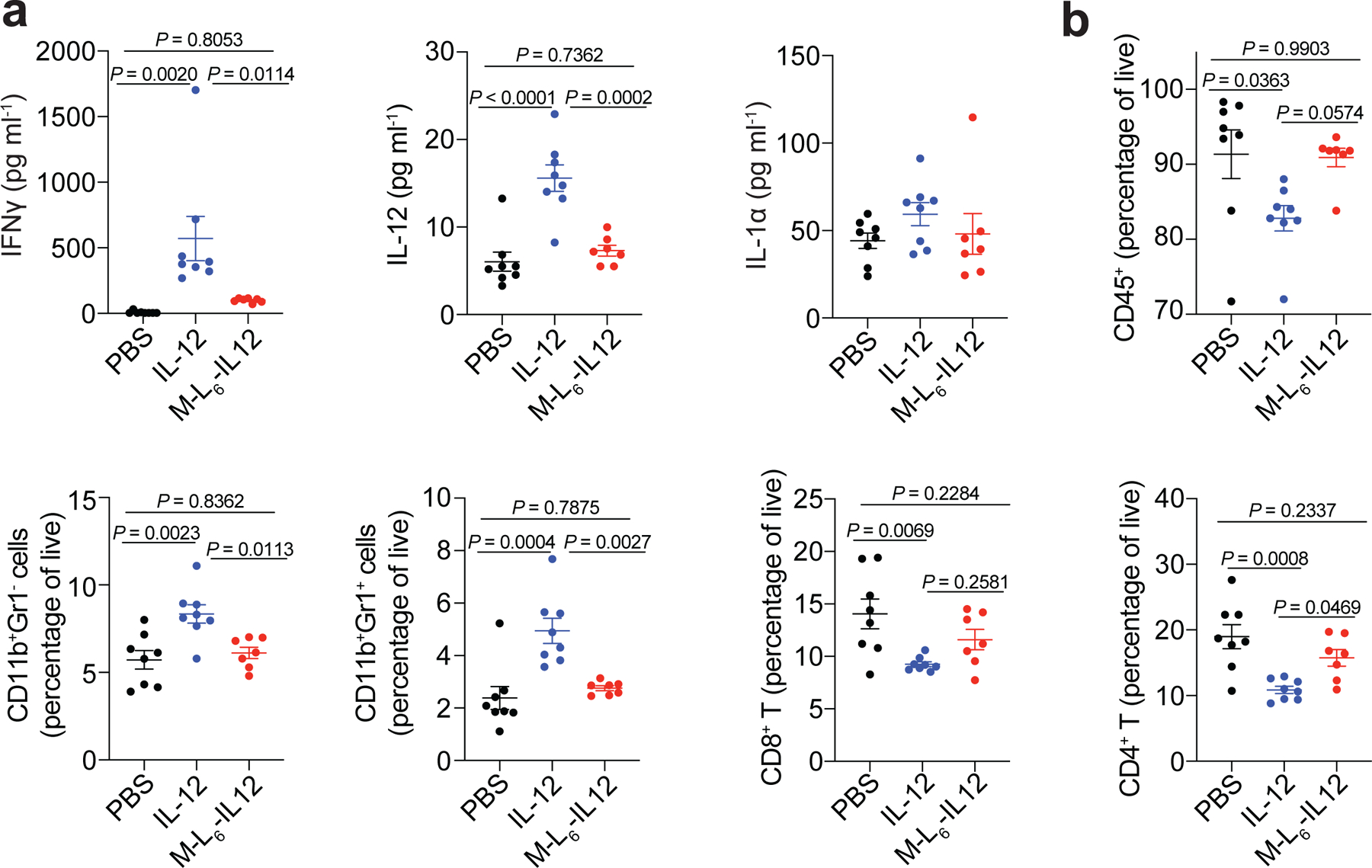
Orthotopic B16F10 melanoma-bearing mice were treated with PBS (n=8), 5 μg (83.3 pmol) IL-12 (n=8) or 250 pmol M-L6-IL12 (n=7) i.v. on days 6 and 9 post tumour inoculation. On day 11, mice were bled for plasma cytokine quantification using LEGENDPlex (a) and spleens were excised for flow cytometric analysis (b). Data are mean ± s.e.m. Experiments were performed twice with similar results. Statistical analyses were performed using ordinary one-way ANOVA with Tukey’s multiple comparison tests.
Ex vivo cleavage by human tumours activates masked IL-12
Expression of various proteases is increased in human solid tumours35, and thus we sought to investigate the stability of masked IL-12 in human tumours versus in healthy tissue or serum. To test this, we obtained a flash-frozen human melanoma biopsy and patient-matched serum. Incubation of M-L6-IL12 with human melanoma homogenate, but not matched serum, resulted in time-dependent activation of masked IL-12 as visualised by western blotting (Fig. 6a). The non-cleavable control molecule was not affected by either of the samples. To analyse the cleavage of M-L6-IL12 by tumour versus healthy tissue, we obtained human breast cancer biopsy (stage IIA infiltrating lobular carcinoma), as well as a biopsy of the adjacent normal tissue (ANT) from the same patient. We homogenised both tissues and incubated the homogenates with M-L6-IL12 and performed STAT4 phosphorylation assay (Fig. 6b). Incubation of M-L6-IL12 with the tumour homogenate, but not the ANT homogenate, resulted in activation of masked IL-12. Incubation of masked IL-12 with ANT resulted in minor cleavage as compared to masked IL-12 incubated with buffer only, which is perhaps due to cleavage of the linker by intracellular proteases. To visualise the cleavage, we also performed western blot analysis using the same breast tumour homogenate and ANT homogenate (Fig. 6c). Incubation with the tumour homogenate resulted in marked cleavage of the mask, whereas ANT led to only a slight cleavage, supporting the results of the pSTAT4 assay. Overall, these data suggest that M-L6-IL12 is cleaved ex vivo by human melanoma and breast cancer biopsies, while staying largely intact in matched serum and ANT samples.
Fig. 6 |. Cleavage of the mask by human tumours and generation of human masked IL-12.
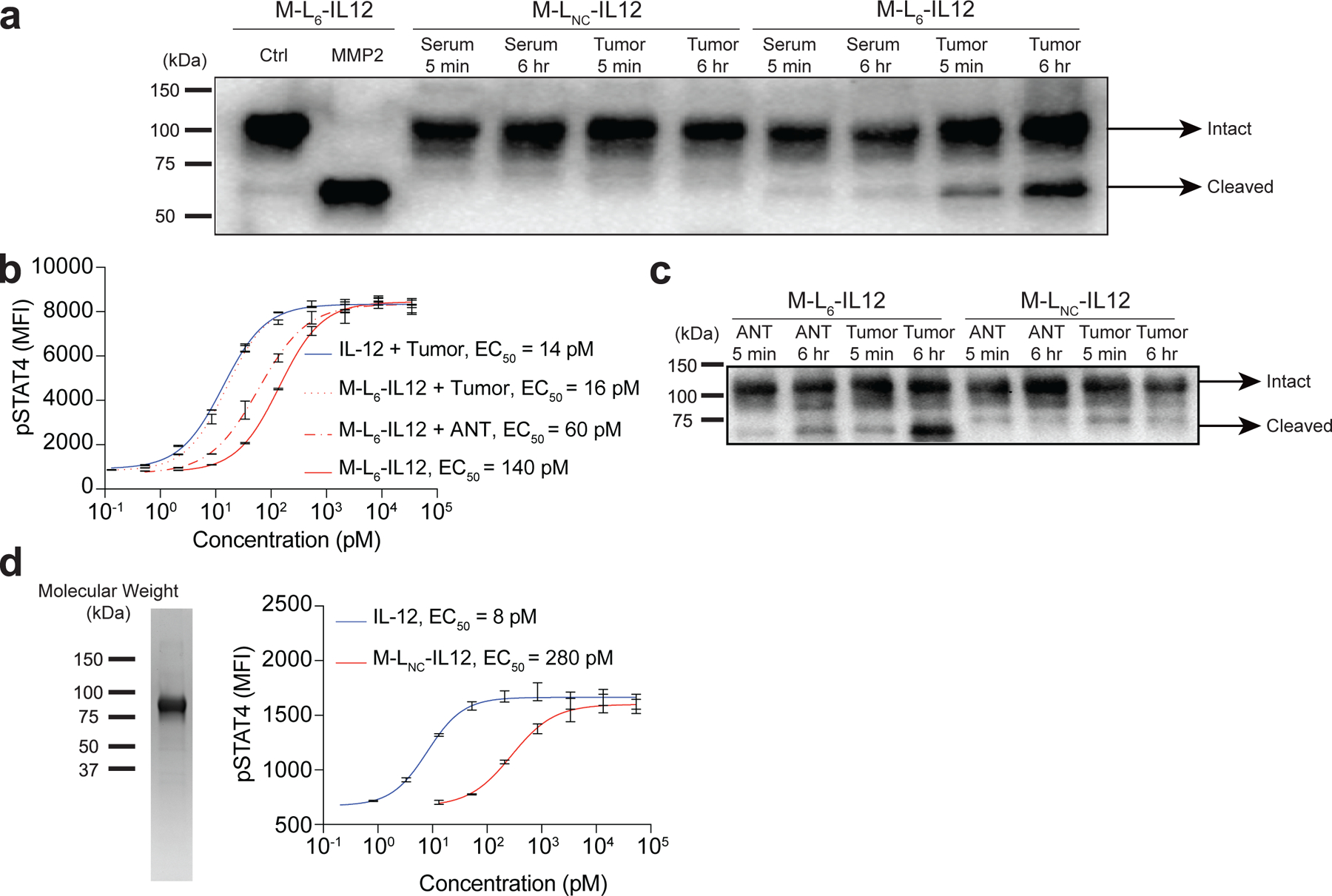
a, Western blot analysis of the cleavage of M-L6-IL12 by human melanoma and patient-matched serum. M-LNC-IL12 or M-L6-IL12 (0.84 μM) were incubated with either serum or melanoma homogenate (2 mg/mL) for indicated times at 37 °C. Untreated M-L6-IL12 and MMP2-treated M-L6-IL12 were loaded as controls. b, Cleavage of M-L6-IL12 by human breast tumour homogenate. IL-12 or M-L6-IL12 (at 0.84 μM) were mixed with either tumour homogenate or adjacent normal tissue (ANT) lysate (2 mg/mL) and incubated overnight at 37°C. Samples were then diluted and applied on pre-activated mouse CD8+ T cells and MFI of pSTAT4 was measured via flow cytometry (n=2 per condition, technical duplicates). c, Western blot analysis of the cleavage of M-L6-IL12 or M-LNC-IL12 by human breast cancer homogenate or ANT homogenate. Incubation was performed at 37°C for indicated duration. d, SDS-PAGE analysis (left) of purified human M-LNC-IL12 under nonreducing conditions and in vitro activity (right) of human M-LNC-IL12 as assessed by pSTAT4 MFI on preactivated human CD8+ T cells (n=2 per condition, technical duplicates). Data are mean ± s.e.m.; EC50, half-maximum effective concentration. ANT, adjacent normal tissue. Experiments were performed at least twice, with similar results. Representative data are shown.
To demonstrate that our approach can be applied to engineer human IL-12 as well, we generated human masked IL-12, in which the mask was derived from human IL-12Rb1C24-E234. This fusion protein was expressed at high yields and a two-step purification led to a homogenous monomer (Fig. 6d, left). Human masked IL-12 exhibited ~35-fold decreased bioactivity compared to human IL-12 (Fig. 6d, right), confirming the applicability of human IL-12Rb1 as the masking domain.
Discussion
Recent engineering strategies aimed at reducing the incidence of irAEs upon IL-12 therapy have relied on targeting IL-12 to tumour matrix9,36,37 and tumour necrosis38, attenuating IL-12 mutein18, and delivering IL-12 gene directly to tumours39–41. Despite encouraging preclinical and clinical results, the route of administration, exposure of IL-12 to circulating lymphocytes and subsequent toxicity may limit the broad use of this cytokine. Masked IL-12 produces minimal systemic side effects yet is capable of driving profound inflammation selectively within the tumour. Masked IL-12 can be administered either i.v. or subcutaneously (s.c.), both of which are highly preferred routes in the clinic.
Our data show that NK cells are the main source of systemic IFNγ, which leads to undesired effects. Avoiding the NK cell binding and activation in the blood would be critical for reducing IL-12 side effects. A recently described attenuated IL-12 mutein, which preferentially activates CD8+ T cells versus NK cells due to the difference in IL-12 receptor expression, demonstrated an enhanced therapeutic index based on this mechanism18. Thus, the reduction in toxicity of M-L6-IL12 could be partially attributed to attenuation, in addition to the selective proteolytic unmasking within the tumour. Our results reveal that the masking approach eliminates irAEs even after repeated administration of M-L6-IL12. This is encouraging given that in clinical trials, recombinant IL-12 was administered multiple times.
In contrast to very little IFNγ in the blood, masked IL-12 induced strong intratumoural IFNγ expression. IFNγ induces chemokine expression, and these chemokines likely induce effector CD8+ T cell infiltration. The activation of CD8+ T cells and reduction of Treg cells are caused by IFNγ secreted from T cells, as we previously reported9. We tested EMT6 and B16F10 melanoma, which are immunologically cold, CPI-unresponsive tumours. Our data clearly demonstrate that masked IL-12 remodels the immunosuppressive, cold TME, and bears potential to synergise with CPI therapies.
We selected the cytokine binding domain of the extracellular IL-12Rβ1 as the mask because the binding affinity (KD) of IL-12 for the full receptor (IL-12Rβ1 and β2) is in the pM range while the KD for IL-12Rβ1 alone is in the nM range21. Thus, once cleaved, the soluble mask will not inhibit IL-12 binding to the IL-12R, due to ~1000-fold difference in affinity between the full complex and a single subunit. Additionally, since IL-12Rβ1 naturally exists in the body, it is unlikely that the fusion protein will be immunogenic, and thus is more advantageous compared to other possible non-endogenous masking domains. The only possible immunogenic domain in our fusion protein is the cleavable linker connecting the mask and the p35 subunit.
Designing biologics that are sensitive to proteolytic cleavage by tumour-associated enzymes is a promising strategy to localize therapeutic effects to the disease site. Various proteases, such as MMPs and SPs, are aberrantly expressed in the TME35. These proteases are secreted by tumour cells as well as tumour-infiltrating immune cells, such as macrophages42. Yet their expression in the periphery is tightly regulated. This feature has been exploited to engineer protease-sensitive nanoparticles43,44, antibodies20,45, and certain cytokines such as IL-246,47, type I IFNs16,48 and IL-1249. Protease-sensitive masked CPI antibodies have already demonstrated encouraging early clinical trial results50.
We have previously investigated antitumour efficacies of tumour matrix-binding chemokine51, CPI52, IL-252, IL-129, doxorubicin53, and anti-CD40 antibody54. Among these important antitumour agents, tumour-targeted IL-12 showed the strongest antitumour efficacy in multiple tumour models, including complete remission of cold tumours. In this study, we show that a masking approach for IL-12, one of the most potent yet toxic cytokines, can be successfully applied, transforming it into a potential therapeutic. Whereas our matrix-binding IL-12 variant demonstrated prolonged retention in tumours, the masked IL-12 is locally activated in the tumour; indeed, it may be possible to utilise both of these technologies to form a dual-function variant. Several clinical trials utilising engineered IL-12 have been already launched with promising results38.
Collectively, our results reveal that systemic administration of masked IL-12 induces a potent antitumour effect, resulting in the eradication of established colon tumours and of immune-excluded orthotopic breast tumours; yet fusion of a masking domain abrogates IL-12-induced peripheral toxicity, as evidenced by a variety of systemic inflammatory markers. We have also shown that ex vivo patient tumour lysates effectively activate masked IL-12, emphasizing the translatability of our approach to patients with advanced oncologic diseases.
Methods
Mice and cancer cell lines
8 to 12-week-old C57BL/6 female mice were purchased from Charles River Laboratory. 8 to 12-week-old BALB/c female mice and 8 to 12-week-old C3H/HeJ female mice were purchased from Jackson Laboratory. B16F10 melanoma, EMT6 breast and MC38 colon cancer cell lines were obtained from ATCC and cultured according to instructions. All animal experiments performed in this work were approved by the Institutional Animal Care and Use Committee of the University of Chicago. Cell lines were routinely checked for mycoplasma contamination.
Production and purification of recombinant IL-12 and masked IL-12
For the production of wild-type IL-12, optimised sequences encoding mouse p35 and p40 subunits were synthesised and subcloned into the mammalian expression vector pcDNA3.1(+) by Genscript. A sequence encoding (His)6 was added to the N-terminus of the p35 subunit to allow affinity-based protein purification. For the production of masked IL-12, a sequence encoding the mask protein (IL-12Rβ1Q20-A261) was fused to the N-terminus of the murine p35 subunit via various linkers described in the study (Supplementary Table 1). A (His)6 tag, followed by G3S, was added to the N-terminus of the Mask-p35 subunit. Sequences encoding Mask-p35 and p40 were subcloned into mammalian expression vector pcDNA3.1(+) by Genscript. Suspension-adapted HEK-293F were maintained in serum-free Free Style 293 Expression Medium (Gibco). On the day of transfection, cells were inoculated into fresh medium at a concentration of 1 × 106 cells/mL. 500 μg/L p35 (or Mask-p35) plasmid DNA, 500 μg/L p40 plasmid DNA were mixed with 2 mg/L linear 25 kDa polyethyleneimine (Polysciences) and co-transfected in OptiPRO SFM medium (4% final volume). After 4–5 days of culture, supernatants were harvested, and purification was performed as described previously9. Purified proteins were tested for endotoxin via HEK-Blue TLR4 reporter cell line, and endotoxin levels were confirmed to be less than 0.01 EU/mL. Protein purity was assessed by SDS-PAGE as described previously. Protein concentration was determined through absorbance at 280 nm using NanoDrop (Thermo Scientific). To produce recombinant human IL-12, optimised sequences encoding human p35 and p40 were synthesised and subcloned into mammalian expression vector pcDNA3.1(+) by Genscript. To produce recombinant human masked IL-12, a sequence encoding the human mask (derived from human IL-12Rβ1C24-E234) was fused to the N-terminus of human p35 via a (G3S)11 linker and was subcloned into the mammalian expression vector pcDNA3.1(+) by Genscript. Human p40 plasmid was also synthesised by and subcloned into the mammalian expression vector pcDNA3.1(+) by Genscript. Co-transfection and purification of human IL-12 (or human masked IL-12) was performed similarly to the mouse analog.
Cleavage of masked IL-12 by recombinant proteases
Recombinant mouse MMP2, MMP3, MMP8, MMP9 and recombinant human urokinase plasminogen activator (uPA) were purchased from R&D Systems. Recombinant MMPs were supplied in their inactive form and were activated using 1 mM p-aminophenylmercuric acetate (APMA, Sigma) as indicated in the product datasheet of each MMP. Following activation, MMPs and cytokines were diluted in an assay buffer containing 150 mM NaCl, 50 mM Tris, 10 mM CaCl2, 0.05% Brij-35 at pH = 7.5. Final concentrations of the proteases and cytokines are mentioned in figure legends. Samples were then analysed via gel electrophoresis. Cleavage assays using uPA, which was supplied as an active enzyme, was conducted according to manufacturer’s protocol.
Analysis of STAT4 phosphorylation by flow cytometry
Mouse CD8+ T cells were purified from spleens of C57BL/6 mice using EasySep mouse CD8+ T cell isolation kit (Stem Cell). Purified CD8+ T cells (106 cells/mL) were activated in six-well plates precoated with 5 μg/mL α-CD3 (clone 17A2, Bioxcell) and supplemented with soluble 5 μg/mL α-CD28 (clone 37.51, Biolegend) and 50 ng/mL mouse IL-2 (Peprotech) for 3 days. Culture medium was IMDM (Gibco) containing 10% heat-inactivated FBS, 1% Penicillin/Streptomycin and 50 μM 2-mercaptoethanol (Sigma Aldrich). After 3 days of culture, activated CD8+ T cells were rested for 6 hr in fresh culture medium and were transferred into 96-well plates (50,000 cells/well). Indicated amounts of IL-12 or (cleaved or intact) masked IL-12 were applied to CD8+ T cells for 15 min at 37°C to induce STAT4 phosphorylation. Cells were fixed immediately using BD Phosflow Lyse/Fix buffer for 10 min at 37°C and then permeabilised with BD Phosflow Perm Buffer III for 30 min on ice. Cells were stained with AF647-conjugated antibody against pSTAT4 (clone 38, BD) recognising phosphorylation of Tyr693. Staining was performed for 1 hr at room temperature (RT) in the dark. When assessing the inhibitory effect of extracellular domain of recombinant mouse IL-12Rβ1 (R & D), mouse IL-12 was incubated with IL-12Rβ1 at 1:1 molar ratio and applied to pre-activated mouse CD8+ T cells. For the analysis of STAT4 phosphorylation in human cells, human CD8+ T cells were magnetically sorted from frozen PBMCs using EasySep human CD8+ T Cell Isolation Kit (Stem Cell). Sorted human CD8+ T cells were activated in six-well plates precoated with 5 μg/mL α-CD3 (clone OKT3, BioLegend) and supplemented with soluble 5 μg/mL α-CD28 (clone CD28.2, BioLegend) and 50 ng/mL human IL-2 (Peprotech) for 3 days. pSTAT4 assay was performed as described above. Cells were acquired on BD LSR and data were analysed using FlowJo (Treestar). Mean Fluorescence Intensity (MFI) of pSTAT4+ population was plotted against cytokine concentration. Dose-response curve was fitted using Prism (v8, GraphPad).
Western blot analysis of masked IL-12 cleavage
Samples were separated on a 4–20% Mini-PROTEAN TGX precast gel (Bio-Rad) and transferred to a PVDF membrane (Thermo Fisher Scientific). The masked and cleaved IL-12 were detected by primary incubation with a biotinylated anti-mouse IL-12 p70 antibody (clone C17–8, Thermo Fisher Scientific) at a 1:1000 dilution, followed by secondary incubation with streptavidin-HRP (Thermo Fisher Scientific) at a 1:3000 dilution. The membrane was developed using Clarity Max Western ECL Substrate (Bio-Rad) and imaged using a ChemiDoc XRS+ System (Bio-Rad).
Cleavage of masked IL-12 by tumours, ANT and serum
Flash-frozen melanoma cancer biopsy and serum from the same patient were purchased from Cureline, Inc. (CA, USA). The patient was a 66 year-old male (Caucasian) with stage IIB mixed epithelial and spindle cell melanoma. Flash-frozen human breast cancer biopsy and the adjacent normal tissue (ANT) from the same patient were purchased from Cureline, Inc. (CA, USA). The patient was a 44-year-old female (Caucasian) and had stage IIA lobular infiltrating carcinoma. Surgery was performed in August of 2012 (for the melanoma patient) and August of 2018 (for the breast cancer patient) and samples were flash-frozen within 20 minutes of surgery. Tissues were placed in Lysing Matrix D tubes (MP Bio) along with the assay buffer containing 150 mM NaCl, 50 mM Tris, 10 mM CaCl2, 0.05% Brij-35 at pH = 7.5. Homogenization was performed using FastPrep tissue homogeniser (MP Bio). Supernatant was harvested by centrifugation at 10,000 × g for 20 min. Total protein concentration in the supernatant was determined via Pierce BCA Protein Assay kit (Thermo Fisher). Supernatants were stored in −80 °C. For the cleavage experiments, 2 mg/mL tissue lysate was incubated with 0.84 μM masked IL-12 and assay buffer containing 50 μM ZnCl2 at 37°C for the indicated times. Serum cleavage using mouse sera (undiluted) or cancer patient sera (10-fold diluted) were performed in a similar fashion. Reaction mixtures were either analysed via pSTAT4 assay or western blotting. Similar protocol was applied to cleavage experiments using mouse EMT6 tumours.
Antitumour efficacy of IL-12 and Masked IL-12
For the primary tumour model of B16F10 melanoma, 5 × 105 B16F10 cells were inoculated intradermally on the back of female C57BL/6 mice in 30 μL sterile PBS. For the EMT6 mammary carcinoma model, 5 × 105 EMT6 cells were injected into the left mammary fat pad of female BALB/c mice in 30 μL sterile PBS. For the primary tumour model of MC38 colon adenocarcinoma, 5 × 105 MC38 cells were inoculated subcutaneously on the back of the female C57BL/6 mice in 30 μL sterile PBS. Dose and schedule of the cytokines and antibodies are described in the figure legends. Cytokines were injected in 100 μL volume through tail-vein (intravenously, i.v.). For the experiments involving αPD-1 antibody (29 F.1A12, Bioxcell), antibody was administered intraperitoneally at 100 μg in PBS. The volume of the tumour was calculated using the following formula: (height) × (width) × (thickness) × (π/6). Mice were sacrificed when the tumour volume reached 1000 mm3 and/or based on humane end-point criteria.
In vivo intratumoural cleavage of masked IL-12
Mice bearing B16F10 tumours with an approximate tumour volume of 75 mm3 were treated intratumourally with 167 pmol of masked IL-12 (corresponding to 10 μg of unmodified IL-12). Mice were sacrificed 2 hr post dose. Tumours were excised and homogenised immediately in the presence of protease inhibitors and 5 mM EDTA. Homogenate supernatants were flash frozen in liquid nitrogen and stored at −80°C until assayed via western blotting.
Pharmacokinetic study of masked IL-12
Healthy C57BL/7 mice received equimolar treatment (83.3 pmol) of IL-12 or M-L6-IL12 i.v. and mice (n = 3/group) were bled 30 min, 60 min, 180 min or 480 min post injection. Plasma was separated and analysed via IL-12p70 ELISA (Invitrogen). IL-12 and M-L6-IL12 generated in our laboratory were used as standards. Plasma half-life was estimated using a one-phase decay model (v.8, GraphPad Prism).
Body weight loss in C3H/HeJ mice
8 to 12-week-old C3H/HeJ female mice were weighed on D0. After the initial weights, mice were injected with either PBS, IL-12 or M-L6-IL12 subcutaneously in 100 μL. Injections and body weight measurements were performed daily until day 5. Percent body weight change was calculated according to the following formula: [(Current body weight) – (Initial body weight)]/(Initial body weight) × 100%.
Systemic markers of toxicity
For assessment of plasma cytokines, mice were treated with either IL-12 or masked IL-12 as indicated on the figure legends. On specified days, blood was collected in EDTA-coated tubes (Eppendorf). Blood was spun at 3000 × g for 15 min to obtain plasma. For pro-inflammatory cytokine analysis, LEGENDplex Mouse Inflammation Panel (13-plex) was used (BioLegend). Plasma was diluted in half using assay buffer provided in the kit. The assay was performed according to manufacturer’s instructions. For blood chemistry analysis, mice were bled on indicated days and sera were obtained by spinning blood at 3000 × g for 15 min in in protein low-binding tubes (Eppendorf). Sera were diluted 4x in sterile water and serum ALT, AST amylase, total protein, total bilirubin, and albumin were determined using Vet Axcel blood chemistry analyzer (Alfa Wasserman). For the analysis of leukopenia, healthy C57BL/6 were injected i.v. with either IL-12 or masked IL-12 as indicated. 4 days later, blood was collected in EDTA-coated tubes and analysed using AcT 5diff CP (Beckman Coulter) hematology analyzer.
Analysis of intratumoural inflammatory markers
Mice bearing B16F10 tumours were treated with either IL-12 or M-L6-IL12 twice on days 6 and 9 post tumour inoculation. Mice were euthanised on day 11. Tumours were excised and cut approximately in half. One half was processed for flow cytometry (described below) and the other half was snap-frozen in liquid nitrogen and stored at −80 °C. Tumours were then homogenised in Tissue Protein Extraction buffer (T-PER, Thermo Fisher Scientific). Protease inhibitor tablets (Roche) were added to the T-PER buffer. Tumours were placed in Lysing Matrix D tubes (MP Bio). Homogenization was performed using FastPrep tissue homogeniser (MP Bio). Supernatant was harvested by centrifugation at 10,000 × g for 20 min. Intratumoural cytokines and chemokines were quantified using LEGENDplex Mouse Inflammation Panel (13-plex) and LEGENDplex Mouse Proinflammatory Chemokine Panel (13-plex) (both from BioLegend), respectively, and normalised by total protein content. Tumour samples were diluted in half using assay buffer provided in the kits.
Immune cell infiltrates in B16F10 melanoma
B16F10 cells (5 × 105) were inoculated intradermally on the back of the female C57BL/6 mice in 30 μL sterile PBS. Then, 6 and 9 days after tumour implantation, mice were treated with either PBS, IL-12 or M-L6-IL12. On day 11, tumours were collected and digested for 30 min at 37°C. Digestion medium was DMEM (Gibco) supplemented with 5% FBS, 2.0 mg mL−1 collagenase D (Sigma-Aldrich), 20 μg mL−1 DNase I (Worthington Biochemical), and 1.2 mM CaCl2. Single-cell suspensions were prepared using a cell strainer (70 μm; Thermo Fisher Scientific). Tumour (20 mg) was plated in each well. For antibodies against surface targets, staining was performed in PBS with 2% FBS. For intracellular targets, staining was performed according to the manufacturer’s protocols (00-5523-00, Thermo Fisher Scientific). The following anti-mouse antibodies were used: CD45 Pacific Blue (clone 30-F11, BioLegend), CD3ε BUV395 (clone 145–2C11, BD Biosciences), Foxp3 PE (clone MF23, BioLegend), CD8α BV510 (clone 53–6.7, BioLegend), CD4 BV785 (clone RM4–5, BioLegend), CD11b BUV737 (clone M1/70, BD Biosciences), CD69 AF647 (clone H1.2F3, BioLegend), CD103 BV605 (clone 2E7, BioLegend), F4/80 FITC (clone CI:A3–1, AbD Serotec), CD11c PE-Cy7 (clone N418, BioLegend), MHCII PerCP-Cy5.5 (clone, M5/114.15.2, BioLegend) and Gr-1 APC-Cy7 (clone RB6–8C5). Cell viability was determined using the fixable viability dye eFluor 455UV dye (65-0868-14, eBioscience).
Immune cell infiltrates in the spleen
B16F10 cells (5 × 105) were inoculated intradermally on the back of the female C57BL/6 mice in 30 μL sterile PBS. Then, 6 and 9 days after tumour implantation, mice were treated with either PBS, IL-12 or M-L6-IL12. On day 11, spleens were collected, and single-cell suspensions were prepared by mechanically disrupting the spleen through a cell strainer (70 μm; Thermo Fisher Scientific). 2 × 106 cells were plated in each well. For antibodies against surface targets, staining was performed in PBS with 2% FBS. For intracellular targets, staining was performed according to the manufacturer’s protocols (00-5523-00, Thermo Fisher Scientific). The following anti-mouse antibodies were used: CD45 Pacific Blue (clone 30-F11, BioLegend), CD3e BUV395 (clone 145–2C11, BD Biosciences), Foxp3 PE (clone MF23, BioLegend), CD8α BV510 (clone 53–6.7, BioLegend), CD4 BV785 (clone RM4–5, BioLegend), CD11b BUV737 (clone M1/70, BD Biosciences), CD69 AF647 (clone H1.2F3, BioLegend), CD103 BV605 (clone 2E7, BioLegend), F4/80 FITC (clone CI:A3–1, AbD Serotec), CD11c PE-Cy7 (clone N418, BioLegend), MHCII PerCP-Cy5.5 (clone, M5/114.15.2, BioLegend) and Gr-1 APC-Cy7 (clone RB6–8C5, BioLegend). Cell viability was determined using the fixable viability dye eFluor 455UV dye (65-0868-14, eBioscience).
Depletion study
Depletion of cellular subsets were performed by injecting antibodies against CD4 (GK1.5, Bioxcell, 400 μg), CD8α (2.43, Bioxcell, 400 μg), NK1.1 (PK136, Bioxcell, 400 μg), Ly6G (1A8, Bioxcell, 400 μg) or rat IgG isotype control (LTF-2, Bioxcell, 400 μg) on days 0 and 3. On day 4, mice were administered 5 μg IL-12 i.v. and bled on day 6 for quantification of IFNγ via LegendPLEX assay.
Statistical analysis
Statistical analysis between groups was performed using Prism (v.8, GraphPad Prism). For multiple comparisons, one-way analysis of variance followed by Tukey post hoc test was used. For survival plots, log-rank (Mantel–Cox) tests was used.
Extended Data
Extended Data Fig. 1. Antitumor efficacy of masked IL-12 is linker-dependent.
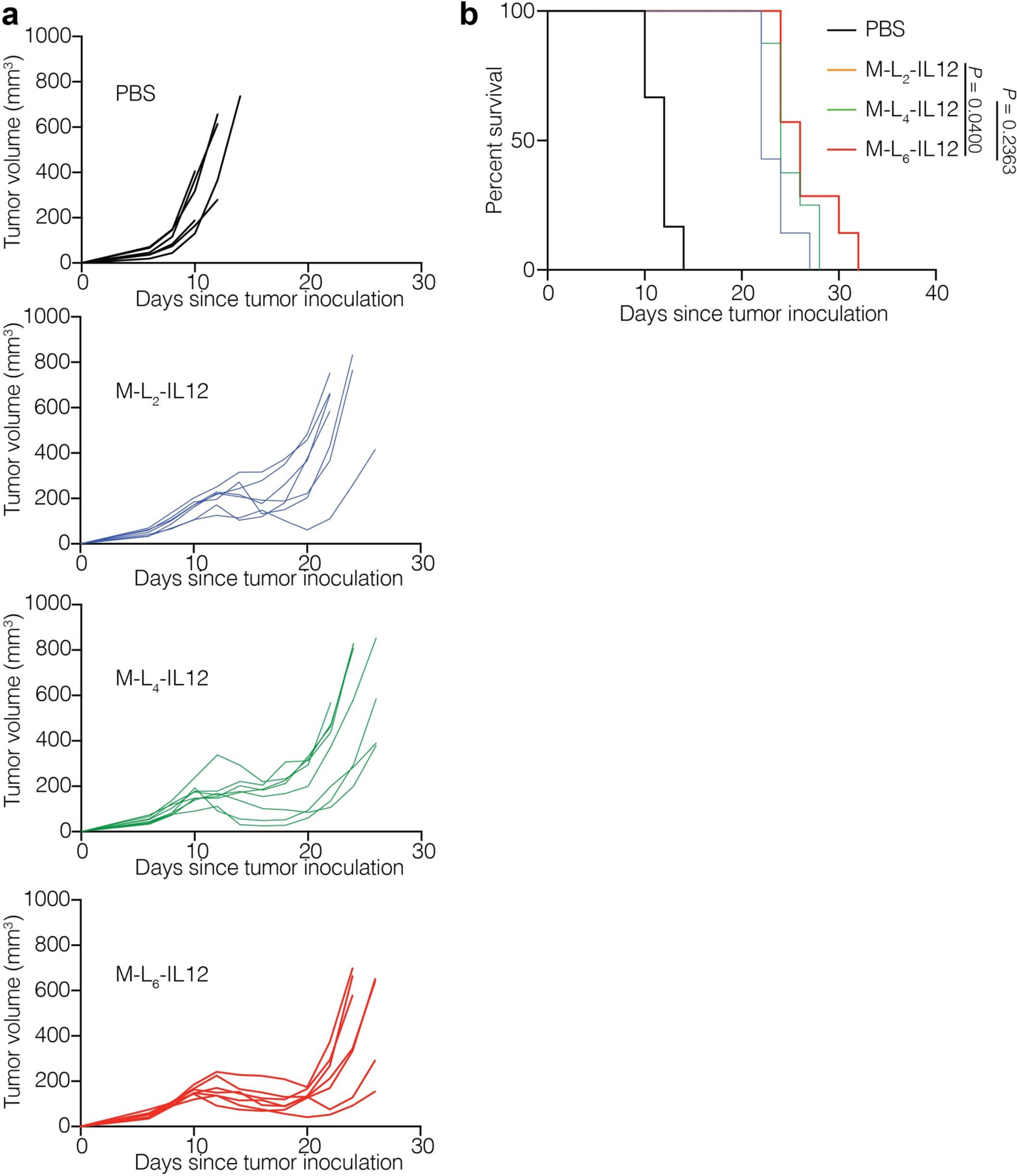
Mice were treated as described in Fig. 2a. Individual tumor growth curves (a) and survival (b) are shown. Statistical analysis in b was performed using log-rank (Mantel-Cox) test.
Extended Data Fig. 2. M-L6-IL12 is cleaved by mouse tumors ex vivo and in vivo.
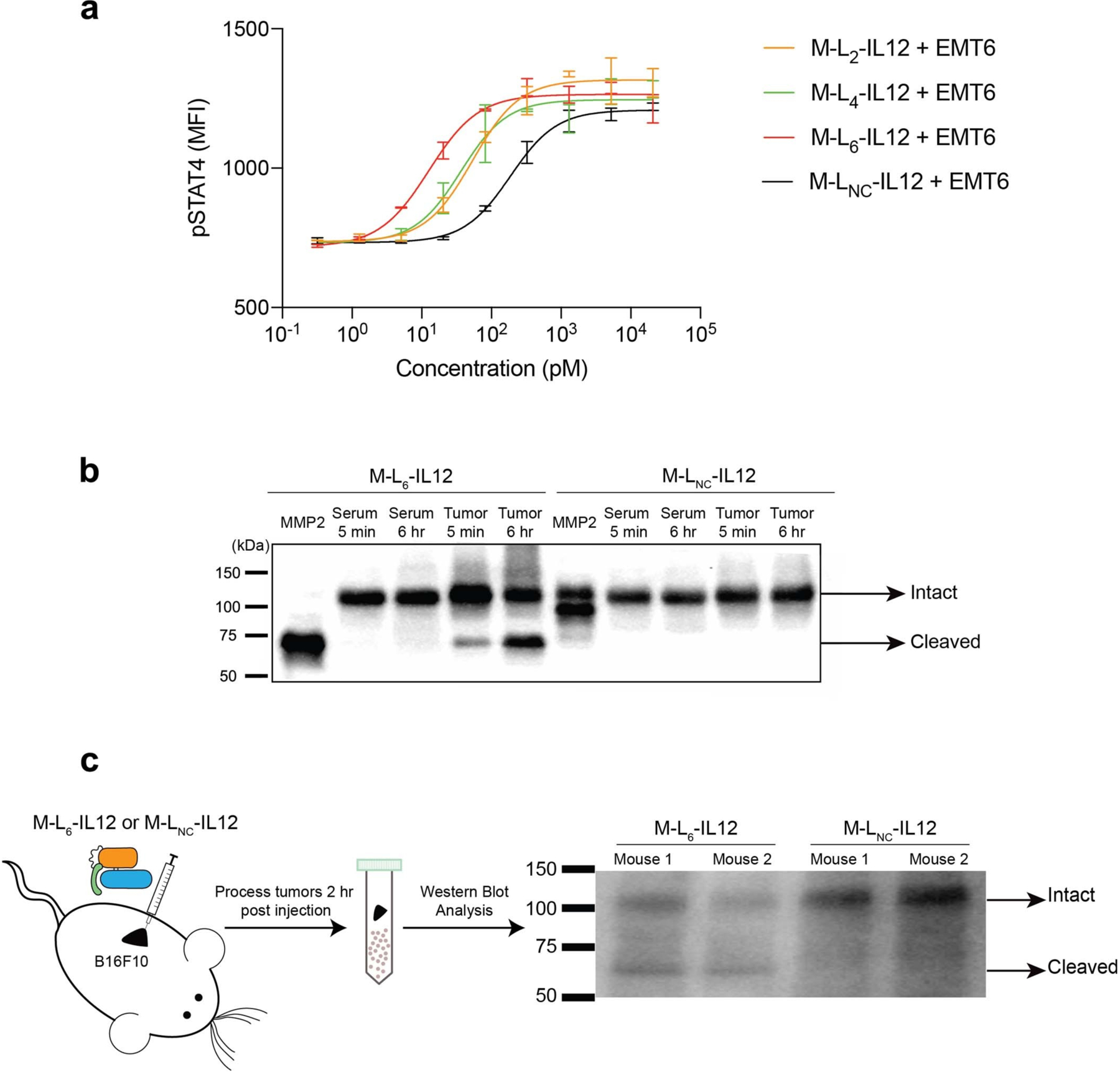
a, Masked IL-12 variants containing linkers L2, L4, L6 and the non-cleavable LNC (0.83 μM) were incubated with EMT6 homogenate (2 mg/mL) for 6 hr at 37 °C. Samples were then diluted in media and applied to pre-activated mouse CD8+ T cells and pSTAT4 MFI was measured. b, M-L6-IL12 or non-cleavable M-LNC-IL12 were incubated in tumor-bearing serum or EMT6 homogenate for indicated times at 37 °C. Reaction mixture was analyzed via western blotting. MMP2-activated M-L6-IL12 is shown as positive control. c, B16F10-bearing mice were injected intratumorally with either M-L6-IL12 or M-LNC-IL12 (167 pmol). Tumors were collected 2 hr post injection and homogenized immediately in the presence of proteases inhibitors and EDTA to stop any further degradation and analyzed via western blotting. Experiments were performed twice with similar results.
Extended Data Fig. 3. M-L6-IL12 induces a dose-dependent antitumor efficacy in B16F10 melanoma and shows extended half-life.
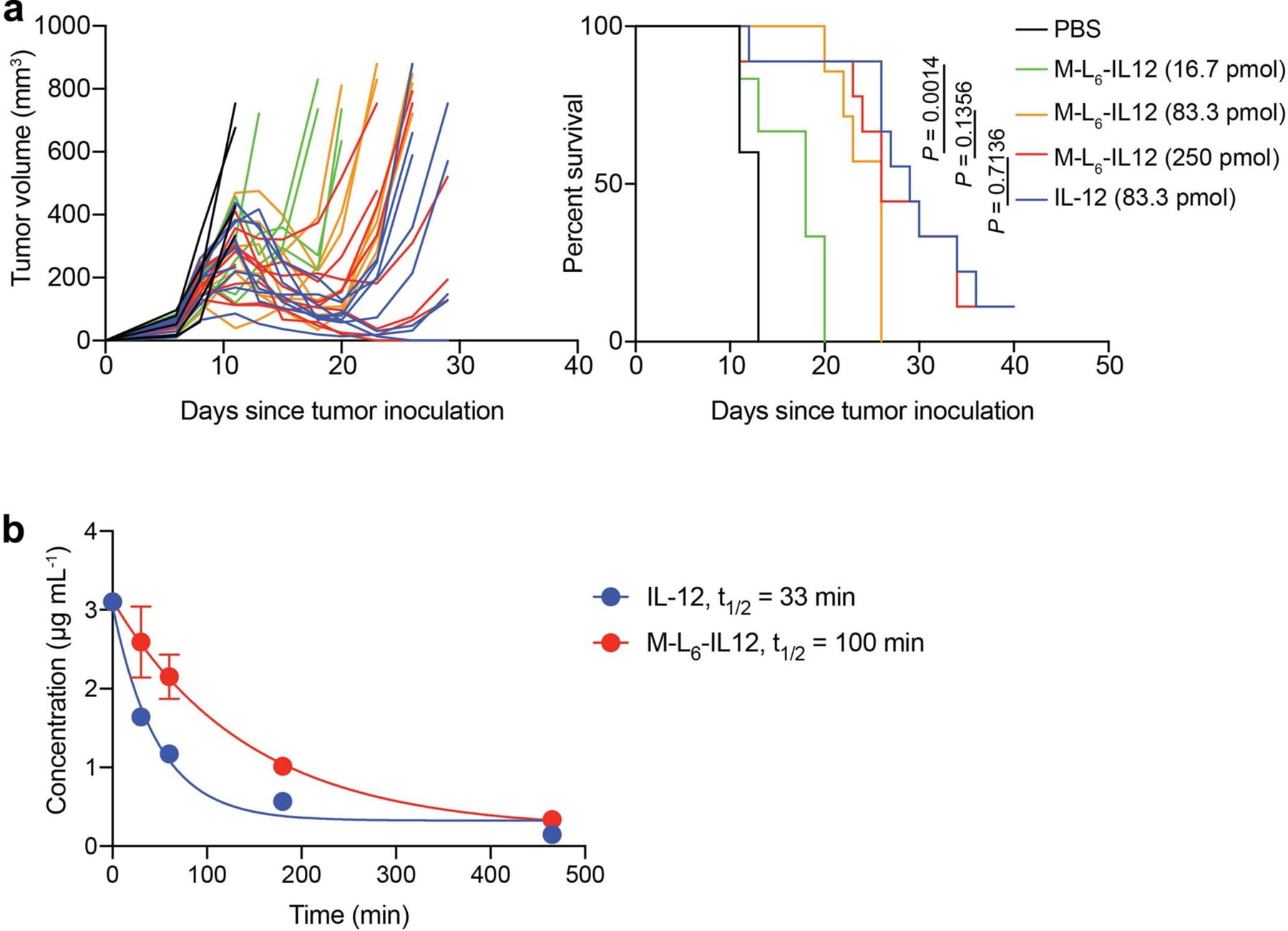
a, Mice bearing B16F10 tumors were treated i.v. with either PBS (n = 5), 16.7 pmol M-L6-IL12 (n = 6), 83.3 pmol M-L6-IL12 (n = 7), 250 pmol M-L6-IL12 (n = 9), or 83.3 pmol IL-12 (n = 9) on days 7, 10 and 13 post tumor inoculation. Individual tumor growth curves (left) and survival curve (right) are shown. Statistics was performed using Mantel-Cox test. b, Healthy C57BL/6 mice were treated i.v. with 83.3 pmol of IL-12 or M-L6-IL12 (n = 3/group) and bled at the indicated time points. Plasma was analyzed for IL-12 concentration via ELISA. Experiments were performed twice with similar results.
Extended Data Fig. 4. M-L6-IL12 and unmodified IL-12 induce similar expression of proinflammatory markers and cell infiltration in B16F10 melanoma.
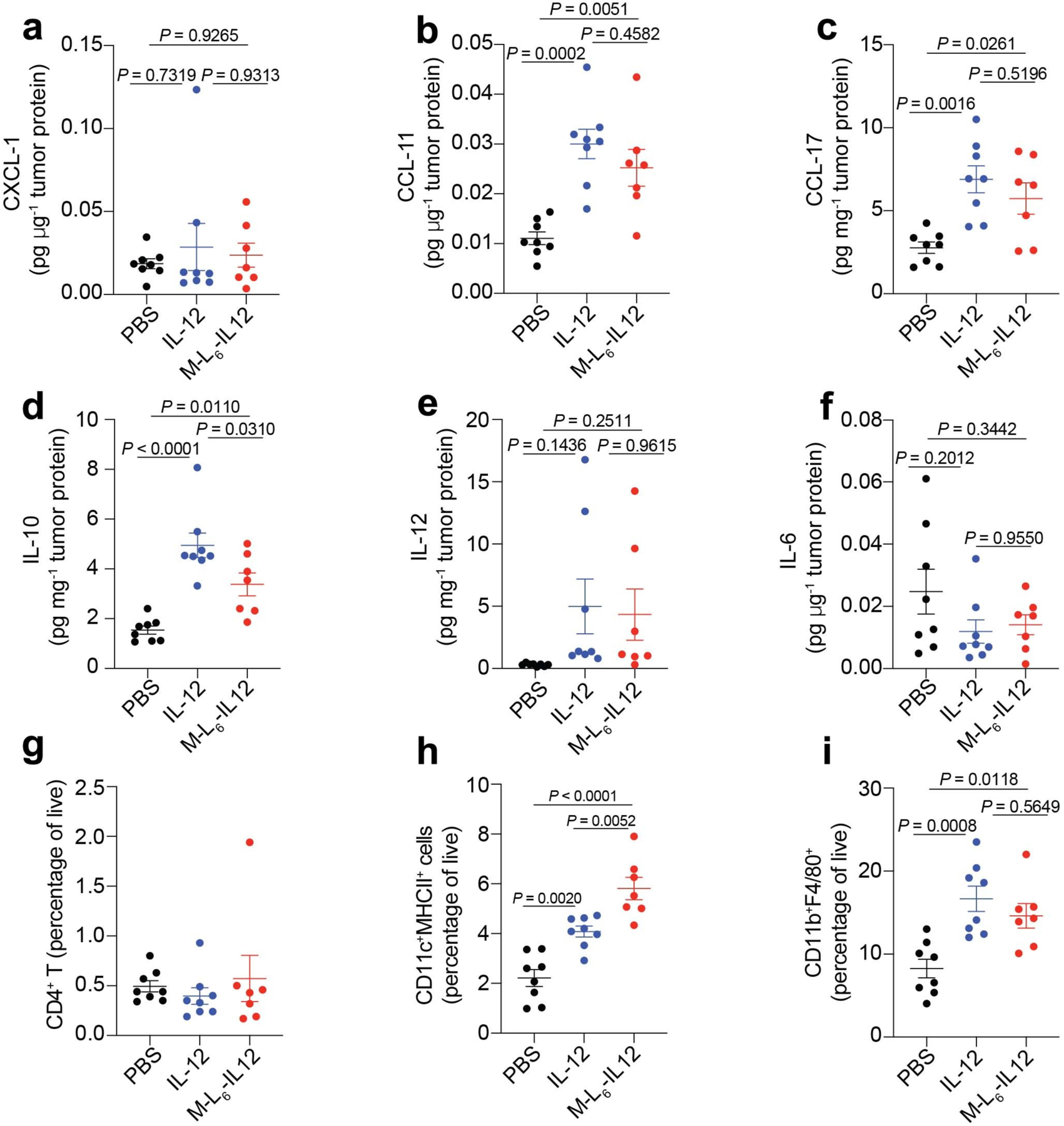
Mice were treated as described in Fig. 3. Intratumoral levels of CXCL-1 (a), CCL-11 (b), CCL-17 (c), IL-10 (d), IL-12 (e), IL-6 (f) were quantified using a LEGENDPlex assay and normalized by total tumor protein content. Frequency of CD3+CD4+Foxp3− T cells (g), CD11c+MHCII+dendritic cells (h), and CD11b+F4/80+ macrophages (i) as percentage of live cells are shown. PBS, n = 8; IL-12, n = 8; M-L6-IL12, n = 7. Data are mean ± s.e.m. Statistical analyses were performed using ordinary one-way ANOVA with Tukey’s multiple comparison test. Experiment was performed twice with similar results.
Extended Data Fig. 5. Masked IL-12 minimizes systemic inflammatory response in healthy mice.
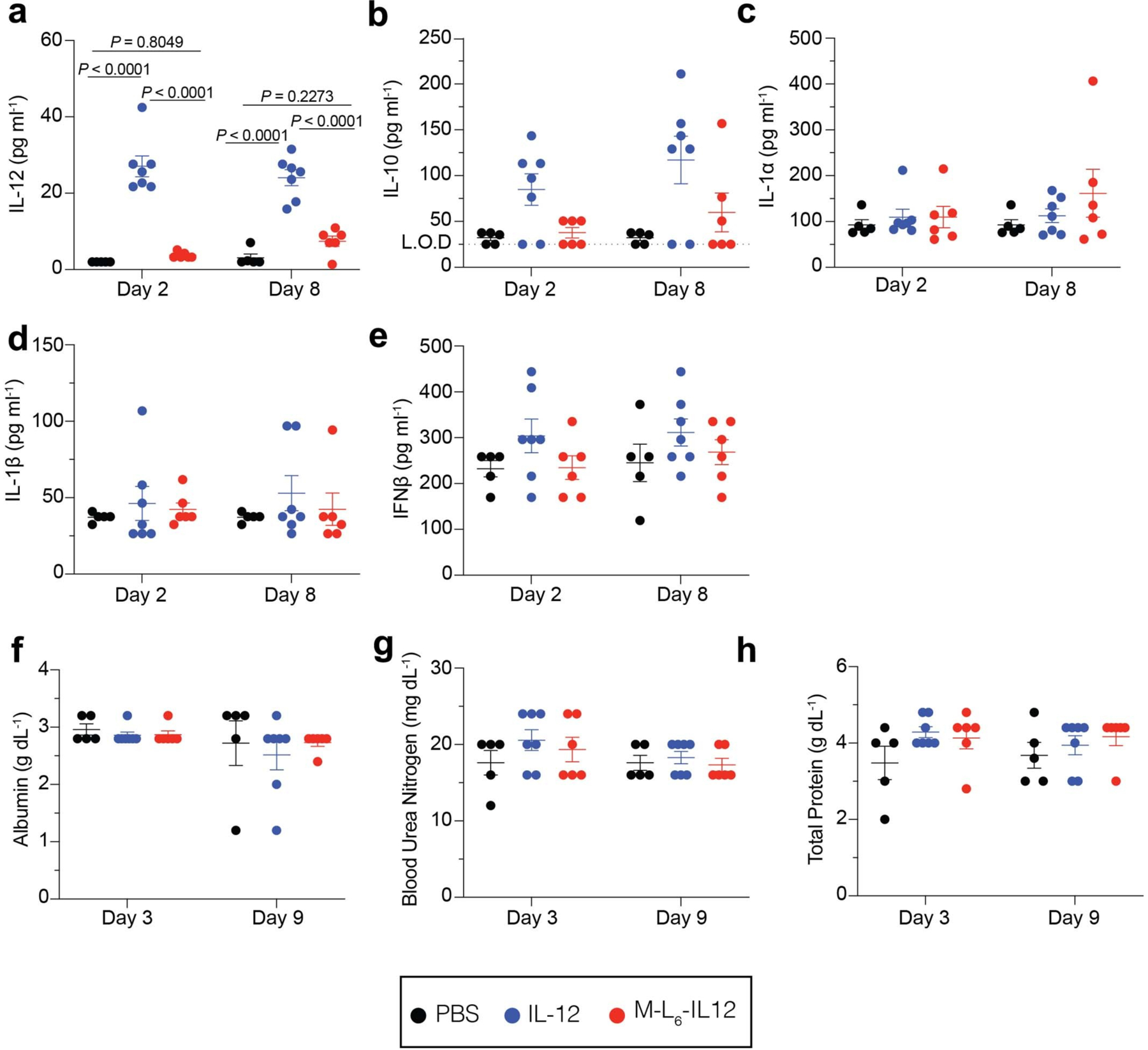
Mice were treated and analyzed as described in Fig. 4b–h. Plasma levels of IL-12 (a), IL-10 (b), IL-1a (c), IL-1b (d), and IFNb (e) were quantified using a LEGENDPlex assay. Serum levels of albumin (f), blood urea nitrogen (g), and total protein (h) were quantified using a blood chemistry analyzer. PBS, n = 5; IL-12, n = 7; M-L6-IL12, n = 6. Data are mean ± s.e.m. Statistical analyses were performed using ordinary one-way ANOVA with Tukey’s multiple comparison test. Experiments were performed twice with similar results. L.O.D = limit of detection.
Extended Data Fig. 6. Masked IL-12 reduces organ damage in MC38-bearing mice.
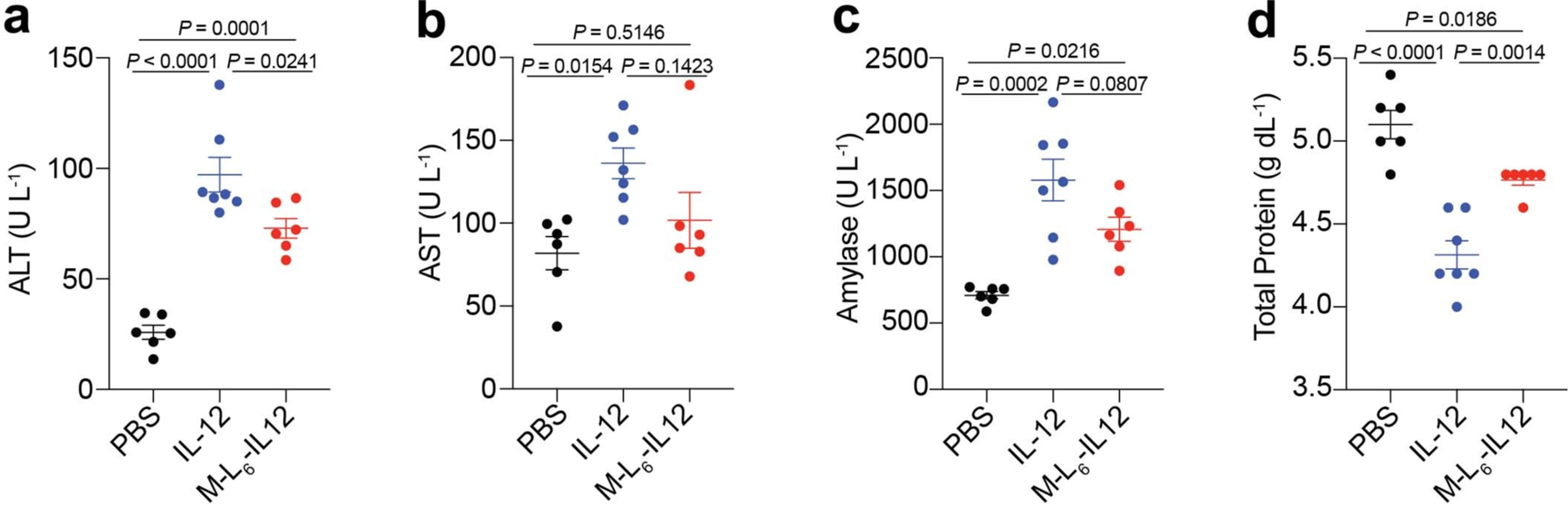
Mice bearing day 7 MC38 tumors were treated i.v. with either PBS (n = 6), IL-12 (83.3 pmol, n = 7) or M-L6-IL12 (250 pmol, n = 6) on days 7, 10 and 13. Plasma was collected on day 16 and levels of ALT (a), AST (b), amylase (c) and total protein (d) were quantified using a blood chemistry analyzer. Data are mean ± s.e.m. Statistical analyses were performed using ordinary one-way ANOVA with Tukey’s multiple comparison test. Experiments were performed twice with similar results.
Supplementary Material
Acknowledgements
This work was funded by the Chicago Immunoengineering Innovation Center of the University of Chicago and NIH R01 CA219304 (to M.A.S.). We would like to thank University of Chicago’s Cytometry and Antibody Technology facility, particularly D. Leclerc.
Footnotes
Reporting Summary. Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Competing interests
A.M., J.I., J.L.M., and J.A.H are inventors on a patent application filed by the University of Chicago covering the technology described in this work. They and M.A.S. hold equity in Arrow Immune, Inc., which is developing the technology, and J.A.H. is an officer of that company. The other authors declare no competing interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41551-01X-XXXX-X.
Data availability
The main data supporting the results in this study are available within the paper and its Supplementary Information. Source Data are provided with this paper. Additional data supporting the results of the study are also available from the corresponding authors on reasonable request.
References
- 1.Zou W, Wolchok JD & Chen L PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med 8, 328rv324, doi: 10.1126/scitranslmed.aad7118 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kalbasi A & Ribas A Tumour-intrinsic resistance to immune checkpoint blockade. Nature Reviews Immunology 20, 25–39, doi: 10.1038/s41577-019-0218-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vitale I, Shema E, Loi S & Galluzzi L Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nature medicine 27, 212–224 (2021). [DOI] [PubMed] [Google Scholar]
- 4.Gocher AM, Workman CJ & Vignali DAA Interferon-γ: teammate or opponent in the tumour microenvironment? Nature Reviews Immunology, doi: 10.1038/s41577-021-00566-3 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ayers M et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 127, 2930–2940, doi: 10.1172/jci91190 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ackerman SE et al. Immune-stimulating antibody conjugates elicit robust myeloid activation and durable antitumor immunity. Nature Cancer 2, 18–33, doi: 10.1038/s43018-020-00136-x (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mansurov A et al. Immunoengineering approaches for cytokine therapy. Am J Physiol Cell Physiol 321, C369–c383, doi: 10.1152/ajpcell.00515.2020 (2021). [DOI] [PubMed] [Google Scholar]
- 8.Langrish CL et al. IL-12 and IL-23: master regulators of innate and adaptive immunity. Immunol Rev 202, 96–105, doi: 10.1111/j.0105-2896.2004.00214.x (2004). [DOI] [PubMed] [Google Scholar]
- 9.Mansurov A et al. Collagen-binding IL-12 enhances tumour inflammation and drives the complete remission of established immunologically cold mouse tumours. Nature Biomedical Engineering 4, 531–543, doi: 10.1038/s41551-020-0549-2 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nguyen KG et al. Localized Interleukin-12 for Cancer Immunotherapy. Frontiers in Immunology 11, doi: 10.3389/fimmu.2020.575597 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Atkins MB et al. Phase I evaluation of intravenous recombinant human interleukin 12 in patients with advanced malignancies. Clin Cancer Res 3, 409–417 (1997). [PubMed] [Google Scholar]
- 12.Gollob JA et al. Phase I trial of concurrent twice-weekly recombinant human interleukin-12 plus low-dose IL-2 in patients with melanoma or renal cell carcinoma. Journal of Clinical Oncology 21, 2564–2573 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Ryffel B Interleukin-12: role of interferon-gamma in IL-12 adverse effects. Clin Immunol Immunopathol 83, 18–20, doi: 10.1006/clin.1996.4306 (1997). [DOI] [PubMed] [Google Scholar]
- 14.Sacco S et al. Protective effect of a single interleukin-12 (IL-12) predose against the toxicity of subsequent chronic IL-12 in mice: role of cytokines and glucocorticoids. Blood 90, 4473–4479 (1997). [PubMed] [Google Scholar]
- 15.Leonard JP et al. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood 90, 2541–2548 (1997). [PubMed] [Google Scholar]
- 16.Adams G, Vessillier S, Dreja H & Chernajovsky Y Targeting cytokines to inflammation sites. Nat Biotechnol 21, 1314–1320, doi: 10.1038/nbt888 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Gerspach J et al. Restoration of membrane TNF-like activity by cell surface targeting and matrix metalloproteinase-mediated processing of a TNF prodrug. Cell Death Differ 13, 273–284, doi: 10.1038/sj.cdd.4401735 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Glassman CR et al. Structural basis for IL-12 and IL-23 receptor sharing reveals a gateway for shaping actions on T versus NK cells. Cell 184, 983–999. e924 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turk BE, Huang LL, Piro ET & Cantley LC Determination of protease cleavage site motifs using mixture-based oriented peptide libraries. Nat Biotechnol 19, 661–667, doi: 10.1038/90273 (2001). [DOI] [PubMed] [Google Scholar]
- 20.Desnoyers LR et al. Tumor-Specific Activation of an EGFR-Targeting Probody Enhances Therapeutic Index. Science Translational Medicine 5, 207ra144–207ra144, doi: 10.1126/scitranslmed.3006682 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Presky DH et al. Analysis of the multiple interactions between IL-12 and the high affinity IL-12 receptor complex. J Immunol 160, 2174–2179 (1998). [PubMed] [Google Scholar]
- 22.Vasiljeva O, Menendez E, Nguyen M, Craik CS & Michael Kavanaugh W Monitoring protease activity in biological tissues using antibody prodrugs as sensing probes. Sci Rep 10, 5894, doi: 10.1038/s41598-020-62339-7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mariathasan S et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548, doi: 10.1038/nature25501 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tugues S et al. New insights into IL-12-mediated tumor suppression. Cell Death & Differentiation 22, 237–246, doi: 10.1038/cdd.2014.134 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu X et al. TNF-α sensitizes chemotherapy and radiotherapy against breast cancer cells. Cancer Cell International 17, 13, doi: 10.1186/s12935-017-0382-1 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim K-J et al. Antitumor effects of IL-12 and GM-CSF co-expressed in an engineered oncolytic HSV-1. Gene Therapy 28, 186–198, doi: 10.1038/s41434-020-00205-x (2021). [DOI] [PubMed] [Google Scholar]
- 27.Pan J et al. Interferon-gamma is an autocrine mediator for dendritic cell maturation. Immunol Lett 94, 141–151, doi: 10.1016/j.imlet.2004.05.003 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Von Stebut E et al. Interleukin 1alpha promotes Th1 differentiation and inhibits disease progression in Leishmania major-susceptible BALB/c mice. J Exp Med 198, 191–199, doi: 10.1084/jem.20030159 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dangaj D et al. Cooperation between Constitutive and Inducible Chemokines Enables T Cell Engraftment and Immune Attack in Solid Tumors. Cancer Cell 35, 885–900.e810, doi: 10.1016/j.ccell.2019.05.004 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Waldman AD, Fritz JM & Lenardo MJ A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nature Reviews Immunology 20, 651–668, doi: 10.1038/s41577-020-0306-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang L, Edwards CM & Mundy GR Gr-1+CD11b+ myeloid-derived suppressor cells: formidable partners in tumor metastasis. J Bone Miner Res 25, 1701–1706, doi: 10.1002/jbmr.154 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Portielje JE et al. Phase I study of subcutaneously administered recombinant human interleukin 12 in patients with advanced renal cell cancer. Clinical cancer research 5, 3983–3989 (1999). [PubMed] [Google Scholar]
- 33.Car BD et al. Role of interferon-gamma in interleukin 12-induced pathology in mice. Am J Pathol 147, 1693–1707 (1995). [PMC free article] [PubMed] [Google Scholar]
- 34.Eng VM et al. The stimulatory effects of interleukin (IL)-12 on hematopoiesis are antagonized by IL-12-induced interferon gamma in vivo. J Exp Med 181, 1893–1898, doi: 10.1084/jem.181.5.1893 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Winkler J, Abisoye-Ogunniyan A, Metcalf KJ & Werb Z Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat Commun 11, 5120, doi: 10.1038/s41467-020-18794-x (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Puca E et al. The antibody-based delivery of interleukin-12 to solid tumors boosts NK and CD8+ T cell activity and synergizes with immune checkpoint inhibitors. International Journal of Cancer 146, 2518–2530, doi: 10.1002/ijc.32603 (2020). [DOI] [PubMed] [Google Scholar]
- 37.Momin N et al. Anchoring of intratumorally administered cytokines to collagen safely potentiates systemic cancer immunotherapy. Science Translational Medicine 11, eaaw2614, doi: 10.1126/scitranslmed.aaw2614 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Strauss J et al. First-in-Human Phase I Trial of a Tumor-Targeted Cytokine (NHS-IL12) in Subjects with Metastatic Solid Tumors. Clinical Cancer Research 25, 99–109, doi: 10.1158/1078-0432.Ccr-18-1512 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hewitt SL et al. Intratumoral IL12 mRNA Therapy Promotes TH1 Transformation of the Tumor Microenvironment. Clinical Cancer Research 26, 6284–6298 (2020). [DOI] [PubMed] [Google Scholar]
- 40.Algazi AP et al. Phase II Trial of IL-12 Plasmid Transfection and PD-1 Blockade in Immunologically Quiescent Melanoma. Clinical Cancer Research 26, 2827–2837, doi: 10.1158/1078-0432.Ccr-19-2217 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Y et al. Multifunctional oncolytic nanoparticles deliver self-replicating IL-12 RNA to eliminate established tumors and prime systemic immunity. Nature Cancer 1, 882–893 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mason SD & Joyce JA Proteolytic networks in cancer. Trends Cell Biol 21, 228–237, doi: 10.1016/j.tcb.2010.12.002 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yao Q, Kou L, Tu Y & Zhu L MMP-Responsive ‘Smart’ Drug Delivery and Tumor Targeting. Trends Pharmacol Sci 39, 766–781, doi: 10.1016/j.tips.2018.06.003 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Kwon EJ, Dudani JS & Bhatia SN Ultrasensitive tumour-penetrating nanosensors of protease activity. Nat Biomed Eng 1, doi: 10.1038/s41551-017-0054 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Trang VH et al. A coiled-coil masking domain for selective activation of therapeutic antibodies. Nature Biotechnology 37, 761–765, doi: 10.1038/s41587-019-0135-x (2019). [DOI] [PubMed] [Google Scholar]
- 46.Puskas J et al. Development of an attenuated interleukin-2 fusion protein that can be activated by tumour-expressed proteases. Immunology 133, 206–220, doi: 10.1111/j.1365-2567.2011.03428.x (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hsu EJ et al. A cytokine receptor-masked IL2 prodrug selectively activates tumor-infiltrating lymphocytes for potent antitumor therapy. Nat Commun 12, 2768, doi: 10.1038/s41467-021-22980-w (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cao X et al. Next generation of tumor-activating type I IFN enhances anti-tumor immune responses to overcome therapy resistance. Nat Commun 12, 5866, doi: 10.1038/s41467-021-26112-2 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Skrombolas D, Sullivan M & Frelinger JG Development of an Interleukin-12 Fusion Protein That Is Activated by Cleavage with Matrix Metalloproteinase 9. J Interferon Cytokine Res 39, 233–245, doi: 10.1089/jir.2018.0129 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Autio KA, Boni V, Humphrey RW & Naing A Probody Therapeutics: An Emerging Class of Therapies Designed to Enhance On-Target Effects with Reduced Off-Tumor Toxicity for Use in Immuno-Oncology. Clin Cancer Res 26, 984–989, doi: 10.1158/1078-0432.Ccr-19-1457 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Williford J-M et al. Recruitment of CD103+ dendritic cells via tumor-targeted chemokine delivery enhances efficacy of checkpoint inhibitor immunotherapy. Science advances 5, eaay1357 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ishihara J et al. Targeted antibody and cytokine cancer immunotherapies through collagen affinity. Science translational medicine 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sasaki K et al. Engineered collagen-binding serum albumin as a drug conjugate carrier for cancer therapy. Science advances 5, eaaw6081 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ishihara J et al. Improving efficacy and safety of agonistic anti-CD40 antibody through extracellular matrix affinity. Molecular cancer therapeutics 17, 2399–2411 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The main data supporting the results in this study are available within the paper and its Supplementary Information. Source Data are provided with this paper. Additional data supporting the results of the study are also available from the corresponding authors on reasonable request.