

Abstract
Ozone exposure induces a myriad of adverse cardiopulmonary outcomes in humans. Although advanced age and chronic disease are factors that may exacerbate a person’s negative response to ozone exposure, there are no molecular biomarkers of susceptibility. Here, we examine whether epigenetic age acceleration (EAA) is associated with responsiveness to short-term ozone exposure. Using data from a crossover-controlled exposure study (n = 17), we examined whether EAA, as measured in lung epithelial cells collected 24 h after clean air exposure, modifies the observed effect of ozone on autonomic function, cardiac electrophysiology, hemostasis, pulmonary function, and inflammation. EAA was assessed in lung epithelial cells extracted from bronchoalveolar lavage fluids, using the pan-tissue aging clock. We used two analytic approaches: (i) median regression to estimate the association between EAA and the estimated risk difference for subclinical responses to ozone and (ii) a block randomization approach to estimate EAA’s effect modification of subclinical responses. For both approaches, we calculated Fisher-exact P-values, allowing us to bypass large sample size assumptions. In median regression analyses, accelerated epigenetic age modified associations between ozone and heart rate–corrected QT interval (QTc) (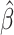 = 0.12, P-value = 0.007) and between ozone and C-reactive protein (
= 0.12, P-value = 0.007) and between ozone and C-reactive protein (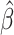 = −0.18, P = 0.069). During block randomization, the directions of association remained consistent for QTc and C-reactive protein; however, the P-values weakened. Block randomization also revealed that responsiveness of plasminogen activator inhibitor-1 (PAI-1) to ozone exposure was modified by accelerated epigenetic aging (PAI-1 difference between accelerated aging-defined block groups = −0.54, P-value = 0.039). In conclusion, EAA is a potential biomarker for individuals with increased susceptibility to ozone exposure even among young, healthy adults.
= −0.18, P = 0.069). During block randomization, the directions of association remained consistent for QTc and C-reactive protein; however, the P-values weakened. Block randomization also revealed that responsiveness of plasminogen activator inhibitor-1 (PAI-1) to ozone exposure was modified by accelerated epigenetic aging (PAI-1 difference between accelerated aging-defined block groups = −0.54, P-value = 0.039). In conclusion, EAA is a potential biomarker for individuals with increased susceptibility to ozone exposure even among young, healthy adults.
Keywords: accelerated aging, ozone, epigenetic aging, environmental susceptibility
Introduction
Research has established that exposure to ambient particulate matter and gases is associated with a wide range of adverse health outcomes that involve every major organ system [1, 2]. From years 2000 to 2019, the estimated global mortality burden associated with ground-level ozone exposure increased from 290 400 deaths to 423 100 deaths (a 46% increase), primarily due to increased exposure in urban areas [3]. Long-term ozone exposure is associated with an increased risk of mortality from cardiopulmonary disease, whereas short-term ambient ozone exposure increases the usage of short-acting beta-2 agonists (e.g. albuterol) in adults and children with asthma [4, 5]. In preclinical pulmonary studies, ozone exposure was shown to activate the Nod-like Receptor family pyrin domain containing 3–mediated inflammasome, which is responsible for activating pro-inflammatory cytokines, macrophages, and neutrophils [6–10]. The LAMARCK study, first published in 2012, highlighted the subclinical biological effects of ozone in humans by showing that a 2-h controlled exposure to 0.3 ppm ozone increases the circulating levels of interleukin (IL)-8, IL-1, and C-reactive protein (CRP), coupled with increased infiltration of neutrophils in lungs [11]. The LAMARCK study further revealed that ozone exposure modulates high-frequency (HF) domain of heart rate variability, forced expiratory volume in 1 s (FEV1), and heart rate–corrected QT interval (QTc), which indicated ozone’s effect on cardiopulmonary function and autonomic nervous system response [11].
Although air pollutants like ozone are ubiquitous, their health effects are not evenly distributed among the population. Work on environmental justice has shown that groups facing discrimination, such as racial minorities or those exposed to social stressors such as poverty, disadvantaged neighborhoods, or lack of access to resources like healthcare, often have elevated environmental health risks [12–14]. Differences in exposure concentration only account for a portion of the health risk within a given community. Additional factors such as resilience to the environment or conversely sensitivity (or susceptibility) to the environment play important roles in determining environmental health risk at the community scale [15, 16]. Individual factors also play a key role in understanding susceptibility to environmental health risks, with both age and chronic disease being associated with elevated adverse responses to air pollutants [17, 18]. Despite the growing body of research describing the factors that modify responses to the environment [19], there are few known molecular markers that are associated with altered biological and physiological responses to environmental exposure. Such markers could help to (i) identify individuals who have increased environmental health risks, (ii) facilitate the quantification of increased risk, and (iii) enable the tracking of the effectiveness of interventions to affect environmental resilience at the individual and population levels.
Chronological age is a well-established factor that may alter resilience to the environment, with aged adults often having increased environmental health risk [17]. The mechanisms accounting for the increased susceptibility of the aged to environmental exposures are not known, although organ system dysfunction and increased frailty have been proposed [20, 21]. DNA methylation–based aging biomarkers can be used to estimate the biological age of individuals, with differences between DNA methylation age and chronological age associated with mortality, cancer, and chronic disease [22, 23]. A recent study showed that accelerated DNA methylation age is associated with increased cardiovascular risk for those who are exposed to elevated traffic-related air pollution [24]. This study is one of the first to provide evidence that epigenetic aging biomarkers may be able to indicate sensitivity to environmental exposures. However, this was an observational epidemiological study and thus limited in the causal statements that could be made about the relationship between aging, air pollution exposure, and health outcomes. Further studies preferably using controlled exposure designs where exposures to pollutants are controlled by the researcher (and thus less subject to confounding) are needed to further validate the findings of this initial, observational study.
Here, we seek to use data from the LAMARCK controlled exposure study to examine whether accelerated epigenetic age is associated with increased hemostatic, cardiovascular, or pulmonary responses to short-term ozone exposure. In this study, we examine heart rate variability, the time for ventricular depolarization and repolarization, as well as markers of systemic inflammation (interleukins and CRP), hemostatic function, and lung function as outcomes. Each of these markers are related to key pathophysiologic pathways which are associated with ozone exposure [25]; however, molecular factors, such as accelerated epigenetic aging, which may exacerbate the impact of ozone, have not been examined. A previous study of LAMARCK examined associations between ozone exposure and DNA methylation age and found changes in some DNA methylation loci. However, this study did not examine epigenetic aging and did not examine if changes associated with ozone exposure were modified by factors such as biological age [26]. Understanding how aging impacts responses to ozone exposure can help to identify vulnerable individuals, which is key to advancing environmental public health for all. Therefore, for this study, we calculate the DNA methylation age of lung epithelial cells to estimate the biological age of the primary target organ of inhaled ozone exposures—the lung. In doing so, we hope to better understand how biological aging contributes to variability in responses to short-term air pollution exposure and provide further evidence that accelerated epigenetic aging may be an indicator of environmental sensitivity.
Methods
Controlled exposure model, human participants, and clinical diagnostics
The LAMARCK study was a randomized, single-blind, crossover study, in which research participants were exposed for 2 h to clean air or for 2 h to 0.3-ppm ozone [11]. Today, individuals in the USA are seldom exposed to 0.3-ppm ozone due to enhancements in air quality. However, peak hourly conditions in cities such as Beijing can approximate conditions of 0.3-ppm ozone [27, 28]. Moreover, a 2-h exposure to 0.3-ppm ozone is similar to what an individual would be exposed to during an 8-h exposure to the current United States Environmental Protection Agency National Ambient Air Quality Standards standard of 0.07-ppm per hour. Study participants received the ozone and clean air exposures in randomized order. During each 2-h exposure, the participants rotated between 15 min of exercise on an ergonomic bicycle and 15 min of rest to simulate increased ventilation attendant to physical labor or exercise. Exposure sessions were separated by a minimum of 14 days to limit carryover effects associated with the participant’s first exposure. Electrocardiographic (ECG) measurements, venous blood samples, respiratory performance measurements, and bronchoalveolar lavage fluids (BALFs) were collected from participants immediately before, immediately after, and 24 h after each exposure session. The following ECG estimates of heart function were measured in participants: HF (0.15 to 0.40 Hz) heart rate variability, ventricular depolarization complexity (QRS complexity), and ventricular repolarization (QTc). Venous blood samples were used to estimate biomarkers of systemic inflammation, including CRP, IL-1, IL-6, and IL-8. Venous blood was also used to estimate plasminogen activator inhibitor-1 (PAI-1), a systemic marker of hemostatic function. Lastly, the FEV1 was measured in participants via spirometry analysis. Figure 1 illustrates the randomized crossover study design.
Figure 1:
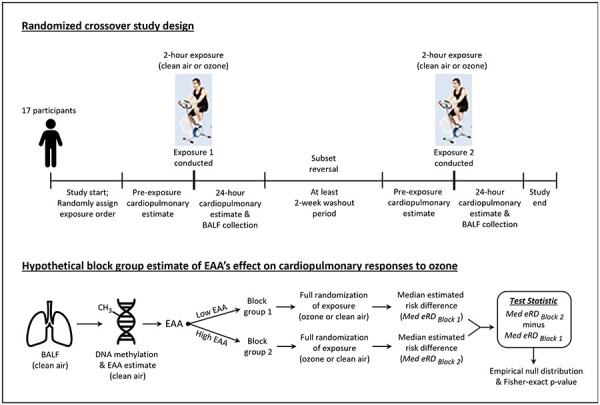
Randomized crossover study design and the hypothetical block randomization model. Abbreviation: Med, median
Measuring DNA methylation in BALF
Participants received a bronchoscopy with brush biopsy so that BALF could be collected from participants at the 24-h post-exposure (clean air or ozone) time point. The procedure for isolating epithelial cells from BALF samples, as well as the procedure for measuring DNA methylation in the epithelial cells from BALF samples using the Illumina 450K array, has been previously described [26]. Based on the percent methylation measured using the Illumina 450K array, we estimated DNA methylation age using the pan-tissue “Horvath” DNA methylation aging clock as implemented in the online age calculator (http://dnamage.genetics.ucla.edu/home) [29]. We used DNA methylation age to generate two estimates of accelerated aging. Epigenetic age acceleration difference (EAA Difference) was calculated as the difference between DNA methylation age and chronological age. Epigenetic age acceleration residual (EAA Residual) is estimated as the residual from an ordinary least squares linear regression of DNA methylation age on chronological age. pan-tissue DNA methylation age (DNAmAge) was estimated for lung epithelial cells collected 24 h after both clean air and ozone exposure; however, we used the clean air exposure DNAmAge for all analyses and estimations of EAA Difference and EAA Residual as we considered this to be more reflective of aging within lung epithelial cells absent any intervention. The minimum 2 weeks of washout period between exposures was implemented to remove any effects of prior exposure [11], so we do not expect there to be residual effects from prior ozone exposure for those study participants who received their air exposure after their ozone exposure. EAA Difference can be estimated using only the data from a single individual, namely, the individual’s chronological and epigenetic age. However, correlations between chronological age and EAA Difference can complicate interpretations of associations with this aging measure. For this reason, studies may either use a residual-based estimation of age acceleration (i.e. EAA Residual) or include chronological age as a confounder in regression models. Unlike EAA Difference, EAA Residual cannot be estimated using the data only on a single individual and thus is dependent on the population in which it is estimated. In our study, the Pearson correlation coefficient between EAA Difference and chronological age was small (R = −0.38, P = 0.13), suggesting little to no correlation between these measures. Thus, for these analyses, EAA Difference was implemented as the primary outcome of interest, while EAA Residual was used to evaluate whether our results were independent of potential confounding, although confounding was likely limited given the small correlation noted earlier.
Calculating the estimated risk difference for clinical responses
The true causal risk difference is inherently impossible to directly calculate, because, for each participant, at a given time, one can observe only one of the potential outcomes, i.e. the observed epigenetic outcome after exposure to either ozone or clean air. By using a crossover design and making the assumptions of no carryover and no time effects, we can estimate the risk difference [estimated risk difference (eRD)] for each outcome using the outcome measured under ozone exposure minus the outcome measured under clean air exposure. As done in the initial analysis of LAMARCK, outcomes (all of which were measured 24 h post-exposure) were normalized by dividing them by their pre-exposure values to reduce interindividual variability [30].
Median regression to estimate EAA’s association with the eRD
We used median regression (using R package quantreg [31]) to estimate the association between EAA and the eRD for each outcome. These regression estimates indicated the degree to which accelerated aging was associated with the estimated biological/physiological response related to ozone exposure. We used median regression because it is more robust to influential points. Outliers were identified based on regression of accelerated aging difference on each outcome using the resulting Cook’s distance and the standard residual from the regression. Outliers were defined as points with a Cook’s distance (Cook’s D [32]) greater than 3 and standardized residual > 2.5 [30, 33]. Our analyses were performed without and with outliers. Given the randomized controlled exposure and crossover study design, we did not adjust for time-invariant confounders. This approach follows previous analyses of these data [11, 26].
P-values from regression outputs are estimated based on asymptotic assumptions that are typically achieved with large sample size. Previous analyses have shown that in small sample size studies such as our study, the null randomized distribution of standard test statistics can differ greatly from the approximating asymptotic distribution (and even be multi-modal), and thus Fisher-exact P-values are preferred [34]. To calculate Fisher-exact P-values based on the test statistic obtained from the median regression, we followed the Bind and Rubin approach [34]. We assumed a Bernoulli assignment mechanism (i.e. 2N possible permutations with N being the number of LAMARCK participants). From each permutation of exposure, the test statistic was the estimated median regression T-statistic. Null randomization distributions were constructed for each test statistic, and two-sided Fisher-exact P-values were calculated.
Hypothetical block randomization analysis of the modification of the eRD by EAA
In the LAMARCK study, the “order” of each participant’s exposure (ozone versus clean air) was the only randomized factor. DNA methylation age was not randomized; therefore, we used a hypothetical block randomization design to estimate the effect modification by EAA. We divided participants into two “blocks” based on their EAA. One block was composed of study participants with above median EAA (high EAA), and the other block contained study participants with below or equal to median EAA (low EAA). These blocks were determined for EAA Difference and EAA Residual in separate analyses. We then assumed that the order of exposure was randomized for each participant within each block (Fig. 1). We calculated the median of each outcome under both ozone and clean air exposure within each block. The difference between these two median differences (ozone minus clean air) was used as our test statistic (Tdiff_in_diff). We calculated two-sided Fisher-exact P-values for each clinical outcome.
R (version 4.2.1) and R Studio were used to perform all analyses. In accordance with modern scientific approaches to hypothesis testing [35, 36], we do not rely on Fisher-exact P-values < 0.05 as the sole determiner of associations and instead consider all factors in the analysis.
Results
Cohort description
The description of this study population has been previously described in both the original analysis of the LAMARCK study [11] and the initial analysis of epigenetic responses to ozone within these study participants [34]. The study included 17 healthy, young adults (15 males, 2 females) who were exposed to both clean air and ozone with exposures occurring in randomized order and a minimum of 2 weeks between exposures [11]. The mean chronological age among the study participants was 25.3 years, while the mean epigenetic age (DNAmAge, as measured in BALF cells during clean air exposure) was 21.6 years with a mean EAA Difference of −3.7 years and a mean EAA Residual of −0.9 years (Table 1). DNA methylation was only assessed in BALF collected 24 h after either clean air or ozone exposure. In Table 1, we illustrate the mean estimate for subclinical parameters (in their natural units), according to pre-exposure measurements and 24-h post-exposure follow-up measurements, for clean air and ozone. The pre-exposure normalized values for subclinical parameters are also shown in Table 1 (arbitrary units). Supplementary Fig. S1 shows the empirical distributions of DNAmAge, EAA Difference, and EAA Residual under clean air exposure, as well as the distribution for chronological age. As stated in the Methods section, although DNAmAge was estimated from BALF collected 24 h after both clean air and ozone exposure, we used the clean air exposure for all analyses as we considered this to be more reflective of the lung epigenetic age in the absence of any intervention. As seen in multiple studies [29, 37], we observed a strong correlation between chronological age and DNAmAge in epithelial cells from BALF (R = 0.64, P = 0.005) (Supplementary Fig. S1). This is particularly noteworthy, as BALF epithelial cells were not included in the original creation of Horvath’s pan-tissue epigenetic age estimator [29]. However, Horvath’s estimator still performed as expected.
Table 1:
Descriptive statistics for clinical measurements and epigenetic aging estimates, according to environmental exposure
| Clean air | Ozone | |||||
|---|---|---|---|---|---|---|
| Estimated outcome (units; n participants) | Pre-exposure mean (SD) | 24-h follow-up mean (SD) | Normalized value | Pre-exposure mean (SD) | 24-h follow-up mean (SD) | Normalized value |
| HF (ms2; 16) | 2266 (1850) | 2988 (1138) | 2.21 (2.22) | 2808 (2245) | 2552 (1886) | 1.05 (0.58) |
| QRS (wave complexity; 16) | 0.35 (0.12) | 0.37 (0.13) | 1.07 (0.09) | 0.37 (0.15) | 0.38 (0.14) | 1.04 (0.07) |
| QTc (ms, HR corrected; 16) | 405 (22) | 403 (25) | 0.99 (0.04) | 402 (19) | 398 (23) | 0.99 (0.03) |
| CRP (ng/ml; 12) | 889 (1716) | 1010 (1866) | 1.39 (1.37) | 1500 (3797) | 1560 (2791) | 2.16 (1.81) |
| IL-1 (pg/ml; 12) | 0.23 (0.14) | 0.22 (0.17) | 0.96 (0.69) | 0.15 (0.10) | 0.27 (0.23) | 1.76 (1.01) |
| IL-6 (pg/ml; 12) | 2.35 (1.32) | 2.25 (1.14) | 0.98 (0.19) | 2.22 (1.53) | 2.07 (1.30) | 0.98 (0.23) |
| IL-8 (pg/ml; 12) | 1.25 (0.88) | 1.22 (0.57) | 1.13 (0.40) | 1.00 (0.70) | 1.02 (0.51) | 1.17 (0.44) |
| PAI-1 (ng/ml; 11) | 3.32 (2.02) | 3.42 (2.46) | 1.25 (0.94) | 3.20 (2.21) | 2.32 (2.33) | 0.76 (0.53) |
| FEV1 (l/s; 16) | 4.53 (0.61) | 4.53 (0.66) | 0.99 (0.03) | 4.48 (0.63) | 4.41 (0.63) | 0.98 (0.02) |
| DNAmAge (years; 17) | 21.59 (3.82) | 22.47 (4.75) | ||||
| EAA Difference (years; 17) | −3.70 (3.16) | −2.82 (3.25) | ||||
| EAA Residual (years; 17) | −0.86 (2.98) | 0.02 (3.28) | ||||
Abbreviations: HF = high-frequency heart rate domain; HR corrected = heart rate corrected; QRS = QRS wave complexity; SD = standard deviation.
Effect modification by EAA Difference using median regression
We used median quantile regression to examine the association between accelerated aging and the eRD for several inflammatory and cardiopulmonary measures. We began by examining plots of accelerated aging compared to eRD along with the median regression of the association between the two, both with all observations and with outliers removed (Fig. 2 and Supplementary S2). Visual inspection of these plots suggests that an effect-modifying association might exist between EAA (Difference and Residual) and the eRD for CRP and QTc. Note that all quantities reported throughout this paper are unitless measures because of the normalization that mirrors the original LAMARCK analysis [11]. The P-values from standard median regression are based on large sample size assumptions, and those assumptions may not be met in small samples such as we have here [34]. In our results, we present P-values that are based on the empirical null distribution (i.e. Fisher-exact P-values). Our results in Tables 2 and 3 and Supplementary Tables S1–S2 indicate that the P-values generated under asymptotic, large sample size assumptions were similar to the P-values from Fisher-exact calculation. Throughout the remainder of the text, all P-values reported are Fisher-exact P-values. Median regression results showed that a 1-year increase in EAA Difference was associated with an increase in the eRD for QTc (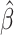 = 0.12; P = 0.006), and there was weak evidence of an attenuated eRD for CRP (
= 0.12; P = 0.006), and there was weak evidence of an attenuated eRD for CRP (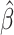 = −0.18; P = 0.095; Table 2). With outliers removed, EAA Difference’s association with QTc was maintained and the association with CRP strengthened (
= −0.18; P = 0.095; Table 2). With outliers removed, EAA Difference’s association with QTc was maintained and the association with CRP strengthened (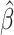 = −0.18; P = 0.069; Supplementary Table S1). Supplementary Fig. S3 shows the empirical null distribution of each clinical outcome constructed via the randomization protocol. Clear deviations from normality were seen for multiple outcomes further motivating the use of Fisher-exact P-values in small sample size scenarios.
= −0.18; P = 0.069; Supplementary Table S1). Supplementary Fig. S3 shows the empirical null distribution of each clinical outcome constructed via the randomization protocol. Clear deviations from normality were seen for multiple outcomes further motivating the use of Fisher-exact P-values in small sample size scenarios.
Figure 2:
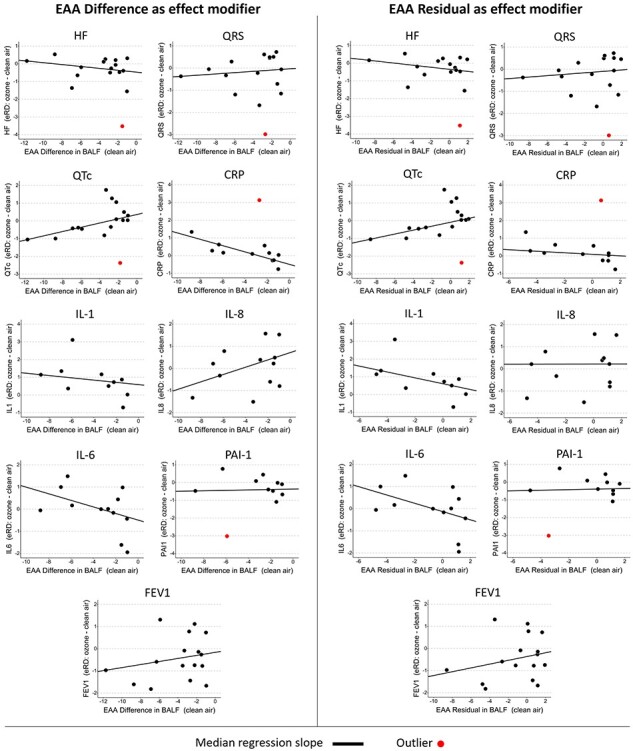
Scatterplots illustrating the median regressed relationship between the EAA Difference and the eRD of each subclinical outcome. Plots lacking a red line did not contain an outlier point. Abbreviations: HF, high-frequency heart rate domain; QRS, QRS wave complexity
Table 2:
EAA Difference median regression results
| Asymptotic | Fisher-exact | |||||
|---|---|---|---|---|---|---|
| Estimated clinical outcome | n | β | SEM | P-value | Results of P-value | |
| HF | 16 | −0.05 | 0.08 | 0.519 | 27 432/65 536 | 0.419 |
| QRS complexity | 16 | 0.03 | 0.08 | 0.719 | 44 083/65 536 | 0.673 |
| QTc | 16 | 0.12 | 0.04 | 0.006 | 421/65 536 | 0.006 |
| CRP | 12 | −0.18 | 0.09 | 0.067 | 390/4096 | 0.095 |
| IL-1 | 10 | −0.06 | 0.10 | 0.527 | 630/1024 | 0.615 |
| IL-6 | 12 | −0.15 | 0.16 | 0.380 | 824/4096 | 0.201 |
| IL-8 | 12 | 0.17 | 0.22 | 0.465 | 1904/4096 | 0.465 |
| PAI-1 | 11 | 0.01 | 0.12 | 0.925 | 1880/2048 | 0.918 |
| FEV1 | 16 | 0.07 | 0.11 | 0.545 | 45 878/65 536 | 0.700 |
Abbreviations: HF = high-frequency domain of heart rate variability; SEM = standard error; QRS = QRS wave complexity.
Table 3:
EAA Residual median regression results
| Asymptotic | Fisher-exact P-value | |||||
|---|---|---|---|---|---|---|
| Estimated clinical outcome | n | Β | SEM | P-value | Results of P-value | |
| HF | 16 | −0.06 | 0.09 | 0.523 | 22 075/65 536 | 0.337 |
| QRS complexity | 16 | 0.03 | 0.08 | 0.695 | 42 720/65 536 | 0.652 |
| QTc | 16 | 0.11 | 0.05 | 0.058 | 4927/65 536 | 0.075 |
| CRP | 12 | −0.04 | 0.09 | 0.634 | 2085/4096 | 0.509 |
| IL-1 | 10 | −0.17 | 0.15 | 0.300 | 394/1024 | 0.385 |
| IL-6 | 12 | −0.19 | 0.19 | 0.337 | 584/4096 | 0.143 |
| IL-8 | 12 | 0.001 | 0.23 | 0.997 | 4028/4096 | 0.983 |
| PAI-1 | 11 | 0.02 | 0.15 | 0.917 | 1908/2048 | 0.932 |
| FEV1 | 16 | 0.09 | 0.12 | 0.488 | 35 136/65 536 | 0.536 |
Abbreviations: HF = high-frequency domain of heart rate variability; SEM = standard error mean; QRS = QRS wave complexity.
Effect modification by EAA Residual using median regression
When using EAA Residual as a measure of epigenetic aging, we observed some evidence for an association between EAA Residual and QTc (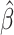 = 0.11; P = 0.075; Table 3). Otherwise, we observed no association between EAA Residual and subclinical eRD. After removing outliers, the association between EAA Residual and QTc was similar, (
= 0.11; P = 0.075; Table 3). Otherwise, we observed no association between EAA Residual and subclinical eRD. After removing outliers, the association between EAA Residual and QTc was similar, (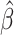 = 0.11; P = 0.063) and there was also an association observed for CRP (
= 0.11; P = 0.063) and there was also an association observed for CRP (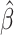 = −0.20; P = 0.032) (Supplementary Table S2). Additionally, after removal of outliers, there was weak evidence for an association between accelerated aging and the eRD for QRS complexity (
= −0.20; P = 0.032) (Supplementary Table S2). Additionally, after removal of outliers, there was weak evidence for an association between accelerated aging and the eRD for QRS complexity (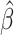 = 0.08; P = 0.093) that had not been seen with EAA Difference (Supplementary Table S2). As with the EAA Difference, we provide histograms of the empirical test statistic distribution when using EAA Residual (Supplementary Fig. S4). Finally, we also checked if there was any evidence for effect modification by chronological age on the eRD of the outcomes examined here. Chronological age was weakly associated with QRS complexity (
= 0.08; P = 0.093) that had not been seen with EAA Difference (Supplementary Table S2). As with the EAA Difference, we provide histograms of the empirical test statistic distribution when using EAA Residual (Supplementary Fig. S4). Finally, we also checked if there was any evidence for effect modification by chronological age on the eRD of the outcomes examined here. Chronological age was weakly associated with QRS complexity (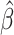 = 0.08; P = 0.083; Supplementary Table S3); otherwise, no associations were observed between chronological age and subclinical eRD. The overall evidence suggests that chronological age is not associated with the same outcomes as we observed for epigenetic age (Supplementary Table S3).
= 0.08; P = 0.083; Supplementary Table S3); otherwise, no associations were observed between chronological age and subclinical eRD. The overall evidence suggests that chronological age is not associated with the same outcomes as we observed for epigenetic age (Supplementary Table S3).
Effect modification estimated using a hypothetical block group randomized experiment
Under the design of the LAMARCK study, only the exposure to ozone versus clean air was randomized. This makes the estimation of interactions between ozone and epigenetic aging difficult. To better estimate the potential causal effect modification of accelerated epigenetic aging on responses to ozone exposure, we implemented a block-randomized approach as described in the Methods section. When including all observations, evidence for effect modification by EAA Difference was not observed across the subclinical parameters (Tables 4 and 5). After removing outliers, as observed in the median regression analyses, there was evidence of effect modification by EAA Difference for QTc (Tdiff_in_diff = 0.034; Fisher-exact P-value = 2816/32 768 = 0.086) and for CRP (Tdiff_in_diff = −0.758; Fisher-exact P-value = 192/2048 = 0.094; Supplementary Table S4; Supplementary Fig. S5). For QTc and CRP, the direction of effect modification was in the same direction as for the median regression analysis, although the statistical strength of the association was weaker based on the observed P-values. Additionally, we observed evidence of effect modification by EAA Difference for the ozone-PAI-1 association (Tdiff_in_diff= −0.544; Fisher-exact P-value = 40/1024 = 0.039; Supplementary Table S4) that was not observed in the median regression results.
Table 4:
EAA Difference block group randomization
| Sample size | Fisher-exact P-value | ||||
|---|---|---|---|---|---|
| Estimated subclinical outcome | Low EAA | High EAA | High EAA–low EAA | High EAA–low EAA | |
| HF | 8 | 8 | −0.427 | 41 286/65 536 | 0.630 |
| QRS complexity | 8 | 8 | 0.004 | 63 040/65 536 | 0.962 |
| QTc | 8 | 8 | 0.011 | 36 352/65 536 | 0.555 |
| CRP | 6 | 6 | −0.668 | 672/4096 | 0.164 |
| IL-1 | 5 | 5 | −1.530 | 144/1024 | 0.141 |
| IL-6 | 6 | 6 | −0.111 | 1536/4096 | 0.375 |
| IL-8 | 6 | 6 | 0.351 | 1420/4096 | 0.347 |
| PAI-1 | 6 | 5 | −0.115 | 1560/2048 | 0.762 |
| FEV1 | 8 | 8 | −0.022 | 20 024/65 536 | 0.306 |
Abbreviations: HF = high-frequency domain of heart rate variability; QRS = QRS wave complexity.
Table 5:
EAA Residual block group randomization results
| Sample size | Fisher-exact P-value | ||||
|---|---|---|---|---|---|
| Estimated subclinical outcome | Low EAA | High EAA | High EAA–low EAA | High EAA–low EAA | |
| HF | 8 | 8 | −0.809 | 23 132/65 536 | 0.353 |
| QRS complexity | 8 | 8 | −0.019 | 52 672/65 536 | 0.804 |
| QTc | 8 | 8 | 0.003 | 54 528/65 536 | 0.832 |
| CRP | 6 | 6 | −0.640 | 736/4096 | 0.180 |
| IL-1 | 5 | 5 | −1.530 | 144/1024 | 0.141 |
| IL-6 | 6 | 6 | −0.132 | 1088/4096 | 0.266 |
| IL-8 | 6 | 6 | 0.136 | 3102/4096 | 0.757 |
| PAI-1 | 6 | 5 | −0.115 | 1560/2048 | 0.762 |
| FEV1 | 8 | 8 | −0.020 | 25 784/65 536 | 0.393 |
Abbreviations: HF = high-frequency domain of heart rate variability; QRS = QRS wave complexity.
When using EAA Residual, all association tests failed to reject the null when outliers were included for the block group analysis, although the directions of effect modification remained the same (Table 5; Supplementary Fig. S6). After removing outliers, the effect modification test statistic and P-value were identical for CRP (Tdiff_in_diff = −0.758; Fisher-exact P-value = 192/2048 = 0.094) and for PAI-1 (Tdiff_in_diff = −0.0.544; Fisher-exact P-value = 40/4024 = 0.039) as compared to EAA Difference. However, no association was observed for QTc (Tdiff_in_diff = 0.007; Fisher-exact P-value = 16 384/32 768 = 0.500; Supplementary Table S5; Supplementary Fig. S6). This attenuated response of QTc mirrors our observation of a weaker evidence for QTc when using EAA Residual in the median regression analyses (in Table 3). Finally, all associations with subclinical parameters were null when using chronological age to assign participants into block groups (i.e. High Chronological Age versus Low Chronological Age; Supplementary Table S6).
Discussion
This study, utilizing data from a controlled exposure study of ozone exposures in healthy, young adults, indicates that accelerated aging of the lung epithelium is associated with responsiveness to ozone exposure. Although there may be age-related environmental effects that were captured by chronological age, these were not wholly responsible for the associations with epigenetic age. Therefore, the results indicate that epigenetic age captures aging-related environmental health risks beyond those captured by chronological age. Our study also used a hypothetical block group randomization design to estimate effect modification, which was novel in its application to a controlled exposure study and a case-crossover study design. This approach for estimating effect modification when one factor was not originally randomized may have future utility for evaluating how underlying factors alter responsiveness seen in controlled exposure studies and for conducting subgroup analyses of randomized controlled trials.
The primary observation within this study is that accelerated epigenetic aging may modify the responsiveness of biomarkers of hemostasis and thrombosis (PAI-1), inflammation (CRP), and the cardiac electrophysiologic parameters of depolarization and repolarization (QRS complexity and QTc) to short-term ozone exposure. Importantly, this is not the first study to suggest that individuals with accelerated aging might have increased environmental health risks. In a study of patients enrolled at the time of a cardiac catheterization with coronary angiography, those with accelerated aging as measured by the Horvath pan-tissue clock had increased risks of systemic hypertension and peripheral arterial disease when exposed chronically to traffic-related air pollution [24]. Our present study adds to that prior observational study by using a causal study design (controlled exposure study) with appropriate methodologies (Fisher-exact P-values and hypothetical block group randomization) to provide evidence that accelerated aging is a potential biomarker of increased environmental sensitivity to short-term ozone exposure even in young healthy individuals.
Directions of association were consistent across analyses; however, the level of statistical evidence (as indicated by the P-value) varied. Median regression analyses indicated evidence for a decreased inflammatory (CRP) and an increased cardiac repolarization responsiveness (QTc) relative to air exposure in relation to short-term ozone exposure for both the residual and difference-based measure of EAA. In the hypothetical block-randomized design approach, we observed an association for PAI-1 and weaker associations for CRP and QTc. These hypothetical block group results would be considered the strongest evidence as they utilize the case-crossover design to produce causal estimates of the effect modification of accelerated aging on responsiveness to ozone. The two-factor methodology employed here might also find applicability to other causal study designs, e.g. randomized controlled trials, seeking to evaluate how measures not accounted for within the causal study design might have modified observed effects within the study—for example, how air pollution or accelerated aging modified responses to a treatment in a randomized controlled trial.
PAI-1 is a key factor in regulating fibrinolysis and normal hemostasis [38] and is induced by activation of the transforming growth factor-β pathway. PAI-1 is a complex protein that affects physiology in a variety of ways. Complete PAI-1 deficiency causes an abnormal bleeding disorder [39], while elevated PAI-1 is associated with clinical conditions associated with aging and increased risks of thrombosis and atherosclerotic disease [40]. Our observation of attenuated PAI-1 in participants with increased age acceleration stands in conflict with published research and furthermore suggests a unique effect of ozone on PAI-1 expression. Specifically, researchers previously showed that senescence and advanced aging are associated with elevated PAI-1 [41, 42]. Additionally, a null allele in the SERPINE1 gene (where PAI-1 is encoded) is associated with decreased aging in humans [43]. More work is required to fully determine if ozone promotes the attenuation of PAI-1 in individuals with increased aging. Relative preservation of the time of ventricular repolarization, QTc, after ozone exposure was noted in those with increased EAA compared to the shortening of QTc after air exposure, a change that was concordant with greater elevations in PAI-1. Increased QTc is a risk factor for ventricular arrhythmia and sudden death [44]. Although the failure of the QT interval to adjust to rate in this case is subclinical, the fact that the rate of ventricular repolarization is slowed by epigenetic aging in response to ozone exposure suggests that epigenetic aging might serve as an additional cause of QT prolongation that could reach clinically important QT durations when superimposed on well-established risk factors for long QT syndrome such as genetic channelopathies, many pharmacological agents, electrolyte disturbances, bradycardia, left ventricular hypertrophy, heart failure, and myocardial ischemia, to name some.
CRP is an acute-phase reactant protein. Blood CRP concentrations rise gradually during aging and sharply in response to injury or inflammation [45], and exposure to ozone has been shown to increase CRP in causal study designs [11, 46]. Within this study, young adults with increased EAA had a smaller increase in CRP as compared to those with less age acceleration. While chronic elevations in inflammation are decidedly adverse for health, short-term inflammation is an important part of the body’s response to injury [47, 48]. Thus, while the directions of association for CRP appear at odds with the expected increases in inflammation from ozone exposure, one hypothesis that would resolve this discrepancy is that those with accelerated aging are having a depressed, acute inflammatory response to ozone-induced lung injury leading to worsened hemostatic and cardiac electrophysiological responses. Overall, these findings paint a picture of increased disruption of fibrinolysis and ventricular repolarization for people with accelerated aging when exposed to ozone, highlighting speculation that individuals with accelerated aging have worsened cardiovascular health risks when exposed to air pollution, and could offer a mechanism to explain increased cardiovascular risk and mortality in some population-based studies. From a public health perspective, these findings—after being reproduced in a larger study or validated using alternative study designs—may help to identify individuals who are at increased risk from environmental exposures. Further validation of these findings may also motivate testing in communities suspected of having increased environmental health risks to observe if these risks are reflected in molecular profiles, and subsequently interventions can be monitored to see if they reduce risks as reflected in molecular profiles validated to be directly tied to environmental sensitivity. In addition to further validation, the use of these findings in clinical preventive medicine may also require reductions in the cost of DNA methylation assessments facilitating their regular use in clinical preventive medicine. These findings also advance our understanding of precision environmental health whereby we understand the factors that modify an individual’s responses to the environment, e.g. EAA Difference, and use that knowledge to better understand the molecular mechanisms of environmental sensitivity.
Strengths and limitations
The primary strength of this study is that it leverages a causal study design to provide estimates of the degree to which accelerated epigenetic aging modifies responses to short-term ozone exposure in a small group of healthy young adults. The novel application of the hypothetical block-randomized analysis may have further utility in understanding how non-randomized environmental factors or other factors impact responses seen in similar study designs. In addition, the fact that this study was done in young, healthy adults suggests that these effects are not just present in those with pre-existing disease, as seen in the previous examination of this topic [24]. Given that environmental health risks are generally greater in aged adults or in individuals with pre-existing disease, the effect modification by accelerated aging may be even greater within these groups. Limitations of this study include the small sample size and the fact that we only evaluated young adults. Due to the small sample size, we decided to report Fisher-exact P-values which provide improved P-value estimation in a small sample size setting [34]. However, we still suggest caution in interpreting these results until they have been replicated in larger sample sizes. These results are still consistent with those from the aforementioned larger, observational study. Along with the small sample size comes a lack of diversity among the study participants. This study was overwhelmingly male, with only one female participant. Another limitation is that since the clean air exposure was used for all participants, half of the participants used the DNAmAge from a clean air exposure which occurred 2 weeks (slightly longer for some due to scheduling constraints at the time of study) after their ozone exposure. However, as DNAmAge changes only slowly (2 weeks is ∼0.03 year of DNAmAge for study participants), any error introduced by this would be very small. Future studies explicitly designed to evaluate DNAmAge prior to the onset of any exposures will be able to address this limitation. The generalizability of these findings across sex, race, and other relevant sociodemographic traits remains to be addressed. The fact that the DNAmAge of these individuals was lower than their chronological age (mean difference = −3.7 years) may also present generalizability limitations with respect to populations that are epigenetically older. However, a previous study of individuals who were epigenetically older than their chronological age also observed that individuals with increased age acceleration may have increased responses to air pollution [24], suggesting that these findings may be applicable to epigenetically younger and epigenetically older populations, alike. Finally, the nature of the study means that only a limited exposure window could be examined. Whether the modification of environmental health risks by accelerated aging is similar for medium- and long-term exposures or for pollutants beyond ozone remains to be explored.
Supplementary Material
Contributor Information
William C Weston, Center for Public Health and Environmental Assessment, US Environmental Protection Agency, Chapel Hill, NC 27514, USA; US Department of Energy, Oak Ridge Institute for Science and Education, Oak Ridge, TN 37830, USA.
Marie Abèle Bind, Biostatistics Center, Massachusetts General Hospital, Boston, MA 02114, USA; Department of Medicine, Harvard Medical School, Boston, MA 02115, USA.
Wayne E Cascio, Center for Public Health and Environmental Assessment, US Environmental Protection Agency, Chapel Hill, NC 27514, USA.
Robert B Devlin, Center for Public Health and Environmental Assessment, US Environmental Protection Agency, Chapel Hill, NC 27514, USA.
David Diaz-Sanchez, Center for Public Health and Environmental Assessment, US Environmental Protection Agency, Chapel Hill, NC 27514, USA.
Cavin K Ward-Caviness, Center for Public Health and Environmental Assessment, US Environmental Protection Agency, Chapel Hill, NC 27514, USA.
Author contributions
Conceptualization C.K.W.-C., D.D.-S., M.A.-B., and R.B.D.; Data curation C.K.W.-C., D.D.-S. W.C.W., and R.B.D.; Formal Analysis W.C.W; Funding acquisition C.K.W.-C., D.D.-S., and W.E.C; Investigation C.K.W.-C. and W.C.W; Methodology C.K.W.-C, M.A.B., and W.C.W; Supervision C.K.W.-C.; Writing—original draft C.K.W.-C., M.A.-B., and W.C.W.; Writing—review & editing C.K.W.-C., M.A.-B., W.C.W., and W.E.C.
Supplementary data
Supplementary data is available at EnvEpig online.
Conflict of interest statement
At the time of this work, Dr Cavin Ward-Caviness was a scientific adviser for the Clock Foundation. The Clock Foundation had no role in any aspect of this work. This work does not necessarily represent the views or policies of the US Environmental Protection Agency.
Funding
Funding for this work came from the US Environmental Protection Agency.
Data availability
Data are available upon request from the study authors. An appropriate, approved Institutional Review Board application is required for data access.
References
- 1. Manisalidis I, Stavropoulou E, Stavropoulos A et al. Environmental and health impacts of air pollution: a review. Front Public Health 2020;8:505570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mannucci PM, Harari S, Martinelli I et al. Effects on health of air pollution: a narrative review. Int Emerg Med 2015;10:657–62. [DOI] [PubMed] [Google Scholar]
- 3. Malashock DA, Delang MN, Becker JS et al. Global trends in ozone concentration and attributable mortality for urban, peri-urban, and rural areas between 2000 and 2019: a modelling study. Lancet Planet Health 2022;6:e958–67. [DOI] [PubMed] [Google Scholar]
- 4. Pepper JR, Barrett MA, Su JG et al. Geospatial-temporal analysis of the impact of ozone on asthma rescue inhaler use. Environ Int 2020;136:105331. [DOI] [PubMed] [Google Scholar]
- 5. Lim CC, Hayes RB, Ahn J et al. Long-term exposure to ozone and cause-specific mortality risk in the United States. Am J Respir Crit Care Med 2019;200:1022–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang J, Zhao Y, Shao F. Non-canonical activation of inflammatory caspases by cytosolic LPS in innate immunity. Curr Opin Immunol 2015;32:78–83. [DOI] [PubMed] [Google Scholar]
- 7. Shi J, Zhao Y, Wang Y et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014;514:187–92. [DOI] [PubMed] [Google Scholar]
- 8. Jaspers I, Flescher E, Chen LC. Ozone-induced IL-8 expression and transcription factor binding in respiratory epithelial cells. A J Physiol 1997;272:L504–511. [DOI] [PubMed] [Google Scholar]
- 9. Michaudel C, Couturier-Maillard A, Chenuet P et al. Inflammasome, IL-1 and inflammation in ozone-induced lung injury. Am J Clin Exp Immunol 2016;5:33–40. [PMC free article] [PubMed] [Google Scholar]
- 10. Li F, Xu M, Wang M et al. Roles of mitochondrial ROS and NLRP3 inflammasome in multiple ozone-induced lung inflammation and emphysema. Respir Res 2018;19:230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Devlin RB, Duncan KE, Jardim M et al. Controlled exposure of healthy young volunteers to ozone causes cardiovascular effects. Circulation 2012;126:104–11. [DOI] [PubMed] [Google Scholar]
- 12. Paul M, Robin S. Which came first, people or pollution? Assessing the disparate siting and post-siting demographic change hypotheses of environmental injustice. Environ Res Lett 2015;10:115008. [Google Scholar]
- 13. Landrigan PJ, Rauh VA, Galvez MP. Environmental justice and the health of children. Mt Sinai J Med 2010;77:178–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brazil N. Environmental inequality in the neighborhood networks of urban mobility in US cities. Proc Natl Acad Sci 2022;119:e2117776119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stewart JA, Mitchell MA, Edgerton VS et al. Environmental justice and health effects of urban air pollution. J Natl Med Assoc 2015;107:50–8. [DOI] [PubMed] [Google Scholar]
- 16. Josey KP, Delaney SW, Wu X et al. Air pollution and mortality at the intersection of race and social class. N Engl J Med 2023;388:1396–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sacks JD, Stanek LW, Luben TJ et al. Particulate matter–induced health effects: who is susceptible? Environ Health Perspect 2011;119:446–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bell ML, Zanobetti A, Dominici F. Who is more affected by ozone pollution? A systematic review and meta-analysis. Am J Epidemiol 2014;180:15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Connor JA, Kamath R, Walker KL et al. Review of quantitative surveys of the length and stability of MTBE, TBA, and benzene plumes in groundwater at UST sites. Groundwater 2015;53:195–206. [DOI] [PubMed] [Google Scholar]
- 20. Geller AM, Zenick H. Aging and the environment: a research framework. Environ Health Perspect 2005;113:1257–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. García-Esquinas E, Rodríguez-Artalejo F. Environmental pollutants, limitations in physical functioning, and frailty in older adults. Curr Environ Health Rep 2017;4:12–20. [DOI] [PubMed] [Google Scholar]
- 22. Dhingra R, Nwanaji-Enwerem JC, Samet M et al. DNA methylation age-environmental influences, health impacts, and its role in environmental epidemiology. Curr Environ Health Rep 2018;5:317–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ryan J, Wrigglesworth J, Loong J et al. A systematic review and meta-analysis of environmental, lifestyle, and health factors associated with DNA methylation age. J Gerontol Ser A 2020;75:481–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ward-Caviness CK, Russell AG, Weaver AM et al. Accelerated epigenetic age as a biomarker of cardiovascular sensitivity to traffic-related air pollution. Aging 2020;12:24141–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang J, Wei Y, Fang Z. Ozone pollution: a major health hazard worldwide. Front Immunol 2019;10:2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bind MC, Rubin DB, Cardenas A et al. Heterogeneous ozone effects on the DNA methylome of bronchial cells observed in a crossover study. Sci Rep 2020;10:15739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kahle JJ, Neas LM, Devlin RB et al. Interaction effects of temperature and ozone on lung function and markers of systemic inflammation, coagulation, and fibrinolysis: a crossover study of healthy young volunteers. Environ Health Perspect 2015;123:310–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jun X, Yuanhang Z, Joshua SF et al. Process analysis of typical summertime ozone episodes over the Beijing area. Sci Total Environ 2008;399:147–57. [DOI] [PubMed] [Google Scholar]
- 29. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol 2013;14:3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Forthofer RN, Lee ES, Hernandez M. 13—Linear regression. In: Forthofer RN, Lee ES, Hernandez M (eds.), Biostatistics, 2nd edn. San Diego, CA: Academic Press, 2007, 349–86. [Google Scholar]
- 31. Koenker R. Quantile Regression, Econometric Society Monographs. Cambridge: Cambridge University Press, 2005. [Google Scholar]
- 32. Cook RD. Detection of influential observation in linear regression. Technometrics 1977;19:15–8. [Google Scholar]
- 33. Morano P, de Mare G, Tajani F. LMS for outliers detection in the analysis of a real estate segment of bari. Lect Notes Comput Sci 2013;7974:457–72. [Google Scholar]
- 34. Bind M-AC, Rubin DB. When possible, report a Fisher-exact P value and display its underlying null randomization distribution. Proc Natl Acad Sci 2020;117:19151–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wasserstein RL, Lazar NA. The ASA statement on p-values: context, process, and purpose. Am Stat 2016;70:129–33. [Google Scholar]
- 36. Wasserstein RL, Schirm AL, Lazar NA. Moving to a world beyond “p < 0.05”. Am Stat 2019;73:1–19. [Google Scholar]
- 37. Hannum G, Guinney J, Zhao L et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Molecular Cell 2013;49:359–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Van De Craen B, Declerck PJ, Gils A. The biochemistry, physiology and pathological roles of PAI-1 and the requirements for PAI-1 inhibition in vivo. Thromb Res 2012;130:576–85. [DOI] [PubMed] [Google Scholar]
- 39. Mehta R, Shapiro AD. Plasminogen activator inhibitor type 1 deficiency. Haemophilia 2008;14:1255–60. [DOI] [PubMed] [Google Scholar]
- 40. Vaughan DE. PAI‐1 and atherothrombosis. J Thromb Haemost 2005;3:1879–83. [DOI] [PubMed] [Google Scholar]
- 41. Ana Luisa C, Fernandes A, Aguilar-Pimentel JA et al. Towards frailty biomarkers: candidates from genes and pathways regulated in aging and age-related diseases. Ageing Res Rev 2018;47:214–77. [DOI] [PubMed] [Google Scholar]
- 42. Vaughan DE, Rai R, Khan SS et al. Plasminogen activator inhibitor-1 is a marker and a mediator of senescence. Arterioscler Thromb Vasc Biol 2017;37:1446–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Khan SS, Shah SJ, Klyachko E et al. A null mutation in SERPINE1 protects against biological aging in humans. Sci Adv 2017;3:eaao1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Robison LB, Brady WJ, Robison RA et al. QT interval prolongation and the risk of malignant ventricular dysrhythmia and/or cardiac arrest: systematic search and narrative review of risk related to the magnitude of QT interval length. Am J Emergency Med 2021;49:40–7. [DOI] [PubMed] [Google Scholar]
- 45. Pepys MB, Hirschfield GM. C-reactive protein: a critical update. J Clin Invest 2003;111:1805–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Arjomandi M, Wong H, Donde A et al. Exposure to medium and high ambient levels of ozone causes adverse systemic inflammatory and cardiac autonomic effects. Am J Physiol Heart Circ Physiol 2015;308:H1499–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Davis C, Fischer J, Ley K et al. The role of inflammation in vascular injury and repair. J Thromb Haemost 2003;1:1699–709. [DOI] [PubMed] [Google Scholar]
- 48. Levy BD, Serhan CN. Resolution of acute inflammation in the lung. Annu Rev Physiol 2014;76:467–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available upon request from the study authors. An appropriate, approved Institutional Review Board application is required for data access.