

Abstract
Objective.
Systemic juvenile idiopathic arthritis–associated lung disease (SJIA-LD) is a life-threatening disease complication. Key questions remain regarding clinical course and optimal treatment approaches. The objectives of the study were to detail management strategies after SJIA-LD detection, characterize overall disease courses, and measure long-term outcomes.
Methods.
This was a prospective cohort study. Clinical data were abstracted from the electronic medical record, including current clinical status and changes since diagnosis. Serum biomarkers were determined and correlated with presence of LD.
Results.
We enrolled 41 patients with SJIA-LD, 85% with at least one episode of macrophage activation syndrome and 41% with adverse reactions to a biologic. Although 93% of patients were alive at last follow-up (median 2.9 years), 37% progressed to requiring chronic oxygen or other ventilator support, and 65% of patients had abnormal overnight oximetry studies, which changed over time. Eighty-four percent of patients carried the HLA-DRB1*15 haplotype, significantly more than patients without LD. Patients with SJIA-LD also showed markedly elevated serum interleukin-18 (IL-18), variable C-X-C motif chemokine ligand 9 (CXCL9), and significantly elevated matrix metalloproteinase 7. Treatment strategies showed variable use of anti–IL-1/6 biologics and addition of other immunomodulatory treatments and lung-directed therapies. We found a broad range of current clinical status independent of time from diagnosis or continued biologic treatment. Multidomain measures of change showed imaging features were the least likely to improve with time.
Conclusion.
Patients with SJIA-LD had highly varied courses, with lower mortality than previously reported but frequent hypoxia and requirement for respiratory support. Treatment strategies were highly varied, highlighting an urgent need for focused clinical trials.
INTRODUCTION
Systemic juvenile idiopathic arthritis (SJIA) is a distinct subtype of juvenile idiopathic arthritis with a prevalence of 0.4 to 0.8 per million children, now broadly considered to represent an auto-inflammatory disorder.1,2 Indeed, children with SJIA largely fail to respond to therapies such as methotrexate used in other forms of JIA3 but demonstrate dramatic response in most cases to biologic medications that target proinflammatory cytokines such as interleukin-1 (IL-1) and IL-6.4–7 In the past decade, such therapies have largely become standard of care in North America and Europe.2,8 SJIA is also distinguished from other forms of JIA by the risk for life-threatening complications. Chief among these is macrophage activation syndrome (MAS), a cytokine storm syndrome characterized by overwhelming systemic inflammation with extreme hyperferritinemia, coagulopathy, and multiorgan dysfunction.9 Recent work has highlighted the central role of interferon gamma (IFNγ) as a driver for MAS,10,11 and an early-phase clinical trial of the anti-IFNγ monoclonal antibody emapalumab has shown promise.12
Another emerging complication of SJIA is severe lung disease (SJIA-LD). In 2013, 25 patients with SJIA were reported with pulmonary artery hypertension, interstitial lung disease, and pulmonary alveolar proteinosis (PAP).13 We have recently reported our single-center experience of 18 patients with SJIA-LD14 in a study that described clinical, radiographic, and histologic features of the disease. Compared with patients with SJIA without LD, patients with SJIA-LD were generally younger at diagnosis, had higher IL-18 levels, and were more likely to have had MAS and adverse drug reactions. This study also provided the first pathophysiological description of SJIA-LD, finding high levels of IL-18 in bronchoalveolar lavage fluid and increased expression of IFNγ-associated pathways and T cell activation in lung tissue. Concurrently, an international case series of 61 patients with SJIA-LD described several patterns of lung disease, including those associated with prior biologic exposure.15 In addition, adverse reactions, including delayed-type hypersensitivity or drug reaction with eosinophilia and systemic symptoms (DRESS)–like reactions to a variety of anti–IL-1 and IL-6 biologics, have been frequently reported in patients with SJIA-LD,14,15 and recent data have associated these DRESS-like features with the presence of HLA-DRB1*15 alleles.16
Despite these descriptions of SJIA-LD, key questions remain unanswered regarding disease course, outcomes, and optimal treatment approaches. In particular, the two largest case series reported significant long-term mortality rates,13,15 but individual case reports and our previous single-center series show more positive outcomes.14,17–19 In addition, the recent description of DRESS-like features after biologic therapy in patients with SJIA highlights the critical need to understand the role of such medications in management of SJIA-LD. Here, we report on the treatments, disease course, and outcomes of our prospective cohort of children with SJIA-LD evaluated in our multidisciplinary clinic. The objectives of the study were to detail management strategies of patients after LD detection, characterize overall disease courses, and measure long-term clinical outcomes.
METHODS
Study population and definitions.
This was a prospective cohort study; patients were identified and enrolled upon lung disease detection or referral to Cincinnati Children’s Hospital Medical Center (CCHMC) for consultation or second opinion. Eighteen of these patients were previously reported by us.14 SJIA was diagnosed based on the International League of Associations for Rheumatology classification criteria and the Childhood Arthritis and Rheumatology Research Alliance operational definition.20,21 Patients were considered to have MAS based on diagnosis of the treating physician and, when data were available, satisfied 2016 MAS Classification Criteria.22 SJIA-LD was classified based on proposed definitions23 and subdivided based on certainty of diagnosis into the following categories:
Possible SJIA-LD: a patient with objective findings on clinical examination (including but not limited to tachypnea, cough, or clubbing) or diffuse abnormalities on chest imaging, not due to LD that preexisted SJIA diagnosis, infection, or other identifiable cause;
Probable SJIA-LD: a patient with both clinical findings and chest imaging findings as above or pulmonary hypertension as measured by echocardiogram; and
Definite SJIA-LD: a patient for whom there is tissue diagnosis consistent with interstitial lung disease (ILD), PAP/endogenous lipoid pneumonia, or pulmonary arterial hypertension.
This study was approved by the Institutional Review Board of CCHMC (2016–2234 and 2018–2408). Written informed consent was obtained from all patients and/or their legal guardians.
Data collection.
After informed consent was obtained, clinical data were collected from the electronic medical record via a standardized case report form. Reports from baseline chest computed tomography (CT) were reviewed by three members of the study team (YH, CT, and GSS) and scored for a prespecified list of radiographic features based on prior descriptions of SJIA-LD. Primary treating rheumatologists determined the overall Physician Global Assessment of Lung Disease Activity (PGALD) score for each patient at last follow-up on a 0 to 10 Likert scale, as well as a four-domain measure rating overall change in clinical status since LD diagnosis in four areas on a five-point scale (much better, slightly better, same, slightly worse, much worse). For PGALD, we defined 0 to 2 as low disease activity, 2 to 5 as moderate, and ≥5 as high.
HLA types.
HLA typing of patients was determined clinically or by analysis of research whole exome sequencing data when available. Briefly, HLA types were determined using bwakit 0.7.15. Mapping was performed using a GRCh38 reference downloaded and indexed using functions from bwakit. HLA types of patients with SJIA-LD were compared with those from a previously described age- and sex-matched cohort of patients with SJIA without LD.14
Serum biomarker determination.
Serum samples were obtained when possible from each patient, aliquoted, and stored at −80°C within 120 min of collection. Samples from patients with SJIA without LD were a convenience sample available from our biorepository. When performed as part of clinical standard of care, serum total IL-18 and C-X-C motif chemokine ligand 9 (CXCL9) were performed at the CCHMC Diagnostic Immunology Laboratory (DIL) and obtained from the medical record. When not performed clinically, total IL-18 and CXCL9 were performed on a research basis using enzyme-linked immunosorbent assay (ELISA) kits obtained from MBL (Woburn, MA) and R&D Systems (Minneapolis, MN), respectively. Normal ranges for total IL-18 and CXCL9 have been determined by the DIL to be <540 pg/ml and <121 pg/ml, respectively.24 Serum C-C motif chemokine ligand 11 (CCL11) and matrix metalloproteinase 7 (MMP7) were determined using specific ELISA kits obtained from R&D Systems.
Statistical analysis.
All data were analyzed using GraphPad Prism 9. For categorical variables, Fisher’s exact test was used for comparisons between groups. For continuous variables, Student’s t-test or one-way analysis of variance with Bartlett’s correction for multiple comparisons was used, with significance defined as P < 0.05. Correlations were expressed using Spearman’s rank correlation coefficient. A receiver operating characteristic (ROC) curve was used to quantify performance of MMP7 as a diagnostic test for SJIA-LD. Heatmaps and unsupervised clustering performed was using Morpheus (https://software.broadinstitute.org/morpheus).
RESULTS
Clinical features of patients with SJIA-LD.
From 2016 to 2022, we have enrolled 41 children with SJIA-LD who were evaluated in our multidisciplinary clinic in a prospective cohort study (Table 1). Collectively, patients in the cohort showed a slight female predominance (59% female), a median age of SJIA diagnosis of 1.5 years old, and median age of LD diagnosis of 3 years old. Of our patients, 6 of 41 (15%) had trisomy 21. More than 75% of patients had at least one episode of MAS, and 8% had been diagnosed with subclinical MAS; only 15% of patients had no history of MAS. All patients had exposure to anti–IL-1 biologics, and 54% had been treated with tocilizumab. More than 40% of patients had at least one clinically apparent adverse reaction to a biologic that resulted in discontinuation of that medication; the proportion with more mild adverse reactions is unknown. Although 93% of patients were alive at last follow-up (median follow-up 2.9 years, range 0.2–15.5 years), 37% progressed to requiring chronic oxygen or other ventilator support. In addition, 65% of patients had at least one abnormal baseline overnight oximetry study, defined as a mean oxygen saturation less than 97%, and 53% had at least one study below 95%. There were no significant differences between patients with probable disease (without biopsy) and definite LD (biopsy obtained) (Table 1) regarding age, sex, presence of digital clubbing, history of MAS, biologic exposure or adverse reactions before LD diagnosis, survival free of respiratory support, or overnight hypoxia. Notably, there were also no significant differences between the 18 patients first reported in our initial cohort and the 23 newly reported patients, aside from duration of follow-up (Supplementary Table 1).
Table 1.
Clinical and demographic features of patients with SJIA-LD*
| All patients (n = 41) | Definite LD (n = 13) | Probable LD (n = 24) | P value (definite vs probable) | |
|---|---|---|---|---|
|
| ||||
| Sex, ratio | 17 M:24 F | 6 M:7 F | 8 M:16 F | 0.49 |
| Age at SJIA diagnosis (IQR), y | 1.5 (0.93–3.38) | 1.5 (1–5) | 1.3 (0.9–3) | 0.47 |
| Age at LD diagnosis (IQR), y | 3 (2–6) | 4 (2–7) | 3 (2–6.3) | 0.64 |
| Disease duration at LD diagnosis (IQR), y | 1.2 (0.7–3) | 1 (0.75–3) | 2 (0.77–3) | 0.60 |
| Clubbing present, % (n/total n) | 78 (32/41) | 85 (11/13) | 79 (19/24) | 1.0 |
| Trisomy 21, % (n/total n) | 15 (6/41) | 23 (3/13) | 8 (2/24) | 0.32 |
| Oxygen requirement ever, % (n/total n) | 37 (15/41) | 46 (6/13) | 38 (9/24) | 0.73 |
| Abnormal overnight oximetry, % (n/total n) | 65 (11/17) | 78 (7/9) | 50 (4/8) | 0.33 |
| MAS, % (n) | ||||
| Never | 15 (6) | 8 (1) | 9 (2) | 1.0 |
| Subclinical | 8 (3) | 15 (2) | 4 (1) | 0.28 |
| One episode | 32 (13) | 38 (5) | 29 (7) | 0.72 |
| >One episode | 46 (19) | 38 (5) | 58 (14) | 0.31 |
| Biologic exposure, % (n) | ||||
| IL-1 | 100 (41) | 100 (13) | 100 (24) | 1.0 |
| IL-6 | 54 (22) | 54 (7) | 50 (12) | 1.0 |
| Adverse event to biologic, % (n/) | 41 (17) | 38 (5) | 38 (9) | 1.0 |
| HLADR5/HLA-DR1*15, % (n/total n) | 84 (21/25) | 100 (11/11) | 75 (9/12) | 0.21 |
| Median follow-up (IQR), y | 2.9 (0.2–15.5) | 3.4 (0.9–6.4) | 2.1 (0.2–15.5) | 0.56 |
| Survival, % (n/total n) | 93 (38/41) | 92 (12/13) | 92 (22/24) | 1.0 |
P values are Fisher’s exact test for categorical data, t-test for continuous data. IQR, interquartile range; IL, interleukin; MAS, macrophage activation syndrome; SJIA-LD, systemic juvenile idiopathic arthritis–associated lung disease.
Our previous work has described frequent pathologic patterns found on chest CT in patients with SJIA-LD. Supplementary Table 2 shows the prevalence of specific imaging findings on the baseline CT obtained at lung disease diagnosis for the 37 patients with probable or definite LD. The most common pattern was septal thickening, which was reported in 14 of 41 cases (64%). Lymphadenopathy (44%), bronchial wall or peribronchovascular thickening (41%), and ground-glass opacities (41%) were also frequently detected. We also examined whether the presence of baseline chest CT findings was associated with progression to home oxygen requirement or respiratory support. As shown in Supplementary Table 2, ground-glass opacities were detected more commonly in patients who later required respiratory support (56%) compared with patients who did not (32%), although this association was not statistically significant (P = 0.19).
Recent data have reported a striking association of HLA-DRB1*15 with suspected DRESS reactions in patients with SJIA with LD.16 In our cohort, genetic data suitable for HLA typing were available for 25 patients. Patients with such data available had a significantly longer follow-up, as well as being more likely to have abnormal overnight oximetry results (Supplementary Table 3). Overall, 21 patients (84%) had HLA-DRB1*15 detected or were presumed to be positive because of detection of the strongly linked HLA-DRB5 allele by gene expression.25 This included 11 of 11 patients (100%) with definite LD, 9 of 12 patients (73%) with probable LD, and 1 of 2 patients (50%) with possible LD. We also compared the prevalence of this HLA haplotype in patients with SJIA-LD with our previously described cohort of age/sex-matched patients with SJIA without LD.14 As shown in Supplementary Figure 1A, patients with SJIA-LD were significantly more likely to carry HLA-DRB1*15 than patients with SJIA without LD (78% vs 30%, P = 0.004). Finally, we examined the relationship between this HLA haplotype and clinical features associated with SJIA-LD. Interestingly, we found a similar prevalence of HLA-DRB1*15 in patients both with and without clinically recognized drug reactions. In contrast, this haplotype was numerically more frequent in patients with clubbing (18 of 20 [90%] vs 3 of 5 [60%]), requiring oxygen support (12 of 12 [100%] vs 9 of 13 [67%]), and history of MAS ever (19 of 21 [90%] vs 2 of 4 [50%]) than those without these features, but none of these associations were statistically significant.
Serum biomarkers of SJIA and SJIA-LD.
We also examined serum levels of inflammatory mediators and other biomarkers associated with LD. Our previous report highlighted that IL-18 levels are markedly elevated in children with SJIA-LD, with serum concentrations significantly higher than those seen in age- and sex-matched patients with SJIA without LD.14 In the present cohort, median serum IL-18 at study enrollment was 27,462 pg/ml (interquartile range [IQR] 5,753–82,963); 32 of 34 patients (94%) were above the upper limit of normal, and 31 of 34 patients (91%) were above 1,000 pg/ml (Figure 1A). Median levels of IL-18 in SJIA-LD were significantly higher than those found among a convenience cohort of children with SJIA without LD but with a range of underlying SJIA disease activity (2,443 pg/ml [IQR 644–19021], P < 0.001). Our previous work has identified the IFNγ-induced chemokine CXCL9 as a reliable marker of MAS in children with SJIA. Here, we find that patients with SJIA-LD show a broad range of serum CXCL9 concentrations with a median of 88 pg/ml (IQR 62.5–287) and were elevated above the upper limit of normal in 9 of 22 patients (41%) (Figure 1B). Median CXCL9 in children with SJIA without LD was similar (70 pg/ml [IQR 37–184], P = 0.084). There was no difference in the incidence of MAS in patients with elevated CXCL9 (data not shown) and no significant correlation between IL-18 and CXCL9 levels (R = 0.24, P = 0.30).
Figure 1.
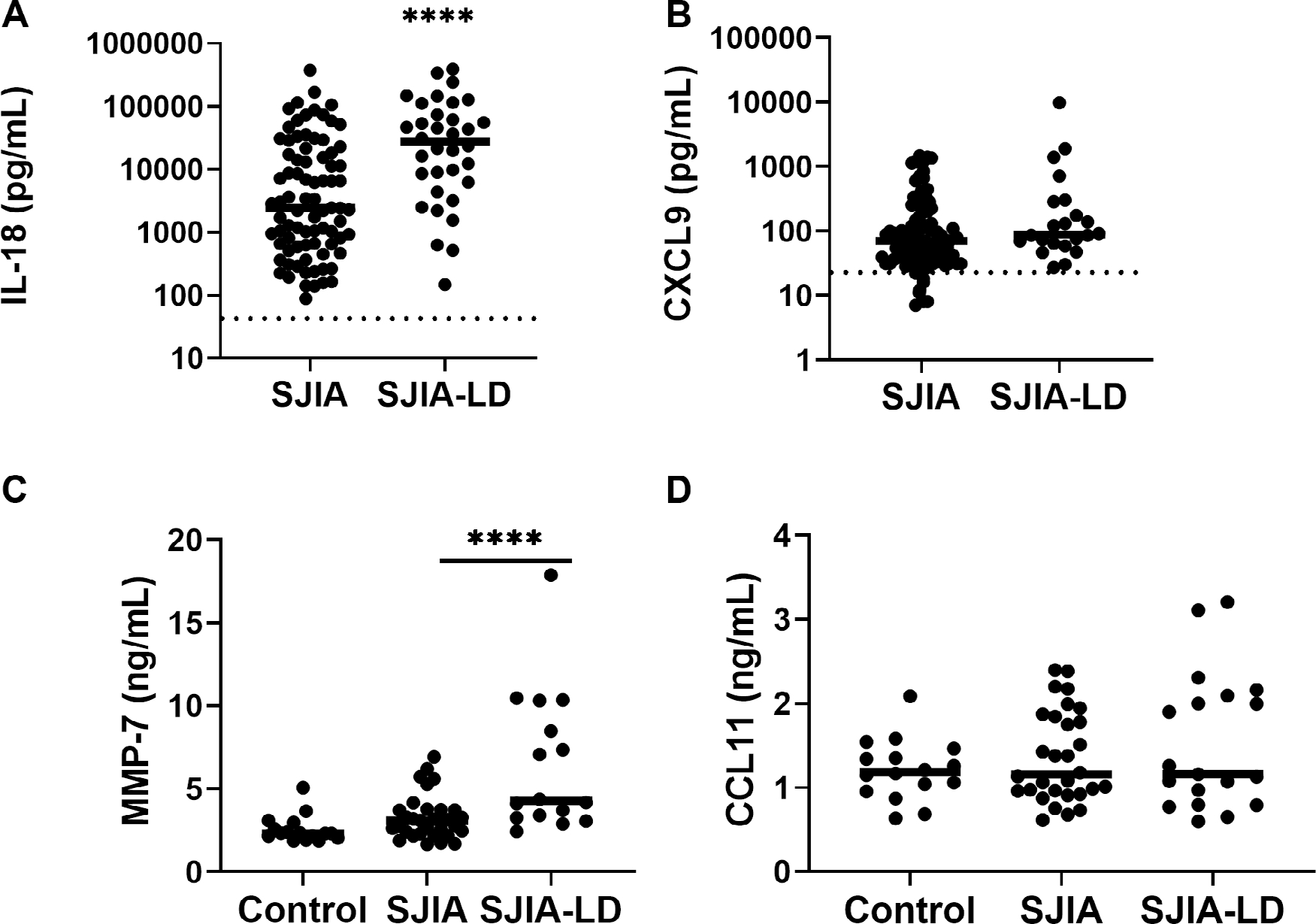
Serum biomarkers levels in patients with SJIA-LD. (A) Initial serum IL-18 level for a convenience sample of patients with SJIA with various disease activity (n = 82) compared with patients with SJIA-LD (n = 34) is shown. Upper limit of normal is 540 pg/ml. (B) Initial serum CXCL9 level for patients with SJIA (n = 93) and patients with SJIA-LD (n = 22) is shown. Upper limit of normal is 67 pg/ml. (C) Serum levels of MMP7 as determined by specific ELISA in control children (n = 15), patients with SJIA without LD (n = 30), and patients with SJIA-LD (n = 15) are shown. ****P < 0.001 vs SJIA without LD. (D) Serum levels of CCL11 (eotaxin-1) as determined by specific ELISA in control children (n = 15), patients with SJIA without LD (n = 30), and patients with SJIA-LD (n = 15) are shown. CCL11, C-C motif chemokine ligand 11; CXCL9, C-X-C motif chemokine ligand 9; ELISA, enzyme-linked immunosorbent assay; IL-18, interleukin-18; LD, lung disease; MMP7, matrix metalloproteinase 7; SJIA, systemic juvenile idiopathic arthritis.
Recent work has described biomarker patterns in children with SJIA, MAS, and SJIA-LD,26 including MMP7, a protease found in the lungs and elevated after lung injury, and CCL11 (eotaxin-1), a type II chemokine linked to hypersensitivity responses as well as pulmonary fibrosis.27 Here, we found that MMP7 was significantly elevated in SJIA-LD compared with SJIA without LD (6.4 ng/ml vs 3.3 ng/ml, P < 0.001) or SJIA-LD compared with control (6.4 ng/ml vs 2.6 ng/ml, P < 0.001) (Figure 1C). Using ROC analysis comparing levels in SJIA-LD vs SJIA without LD, we found that MMP7 has an area under the curve of 0.80, with a cutoff of 3.2 ng/ml being 81% sensitive to SJIA-LD, whereas 5.4 ng/ml was 87% specific for SJIA-LD. However, there was no significant difference found in CCL11 levels in patients with SJIA-LD compared with SJIA without LD or controls (Figure 1D). Interestingly, 17 patients with SJIA-LD had levels of both biomarkers determined, and there was a significant, moderate correlation between CCL11 and MMP7 (Pearson’s R = 0.51, P = 0.046) Together, these findings suggest that markers of lung injury may represent useful biomarkers for LD in children with SJIA.
Pharmacologic treatment patterns before and after SJIA-LD diagnosis.
As discussed above, SJIA-LD was rarely recognized before the introduction of anticytokine biologic therapy for SJIA, and this complication has been epidemiologically linked to changing patterns in medication usage.15 However, the specific medications used after LD diagnosis have not been well detailed, including continued use of biologics. Figure 2 shows the treatments used for patients with probable and definite SJIA-LD before and after LD diagnosis. Before LD diagnosis, most patients (36 of 37 [97%]) had been treated with daily corticosteroids, as well as at least one biologic including anakinra (31 of 37), canakinumab (23 of 37), and tocilizumab (20 of 37). In addition, patients were often treated with other immunomodulators frequently used for SJIA and/or MAS such as methotrexate (17 of 37) and cyclosporine A (14 of 37). After SJIA-LD diagnosis, the overall number of patients treated with anti–IL-1 or IL-6 biologics decreased (Figure 2A), although this was not significant, and most patients (26 of 27) did not permanently discontinue all biologics. In contrast, most patients added one or more immunomodulatory medications used for refractory SJIA, ILD, or MAS (Figure 2B). This includes other biologics used for JIA and other autoimmune conditions such as abatacept, adalimumab, rituximab, and intravenous immunoglobulin. Mycophenolate mofetil was used by 12 of 37 patients (32%, P = 0.005) patients with SJIA-LD after LD diagnosis. Finally, many patients (22 of 37 [59%]) started Janus Kinase (JAK) inhibitors including baricitinib (1 of 37, not significant [NS]), ruxolitinib (5 of 37, P = 0.05) and tofacitinib (19 of 37, P = 0.001). Patients in this cohort also frequently were treated with other medications directed specifically against the lung disease. This included infectious prophylaxis with trimethoprim-sulfamethoxazole (24 of 37, P = 0.0004) and pentamidine (8 of 37, P = 0.03), inhaled corticosteroids (14 of 37, P = 0.01), and short-acting (14 of 37, P = 0.01) or long-acting (4 of 37, NS) beta-agonists. Azithromycin, which is believed to have antiinflammatory effects in LD,28,29 was used in 17 of 37 patients (46%, P = 0.001), and 4 patients used airway clearance techniques (NS). Together, these findings suggest a shift in treatment strategies after LD diagnosis, with variable patterns of biologic use as well as addition of other immunomodulatory treatments and lung-directed therapies.
Figure 2.
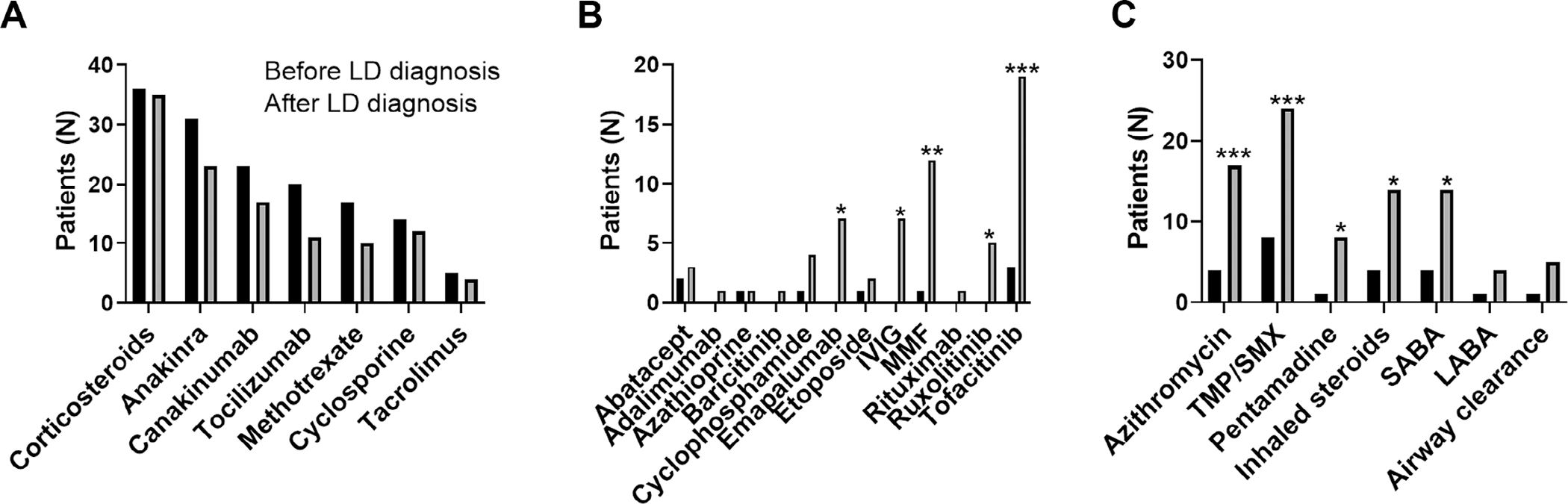
Medication use by patients with SJIA-LD before and after LD diagnosis, separated into (A) medications routinely used for SJIA, (B) other immunomodulatory therapy, and (C) treatments for pulmonary disease and prophylaxis. *P < 0.05, **P < 0.01, ***P < 0.001 compared with pre-LD diagnosis. IVIG, intravenous immunoglobulin; LABA, long-acting beta agonist; LD, lung disease; MMF, mycophenolate mofetil; SABA, short-acting beta agonist; SJIA, systemic juvenile idiopathic arthritis; TMP/SMIX, trimethoprim-sulfamethoxazole.
Clinical outcomes after LD diagnosis.
Prior published cohorts of patients with SIJA-LD show a range of mortality from 0% to 68%13–15; however, these were retrospective or cross-sectional studies and may have been limited by ascertainment and survivorship bias. In our prospective cohort, we examined both overall survival since LD diagnosis as well as a combined outcome of death or need for chronic oxygen supplementation. As shown in Table 1 and Figure 3A, 38 of 41 patients (93%) were alive at last follow-up. Two fatalities occurred within 1 year of LD diagnosis because of fulminant MAS and polymicrobial sepsis and acute renal failure and acute respiratory distress syndrome. A third patient died 3.9 years after LD diagnosis because of cytomegalovirus pneumonitis and multiorgan failure after undergoing allogenic hematopoietic stem cell transplant. Thirty-seven percent of patients with SJIA-LD had required chronic oxygen supplementation at last follow-up. As shown in Figure 3B, patients showed a progressive increase in need for respiratory support over the first 500 days after LD diagnosis before largely stabilizing. However, the predicted 5-year survival free of oxygen supplementation was 57%. As noted above, most patients who had overnight oximetry studies performed showed abnormally low mean oxygen saturation of less than 95%. In addition, several patients had serial overnight oximetry performed after LD diagnosis. Although some patients did show progressive decline in mean oximetry over time, other patients showed a gradual improvement in oxygenation in conjunction with control of their underlying SJIA (Supplementary Figure 2).
Figure 3.
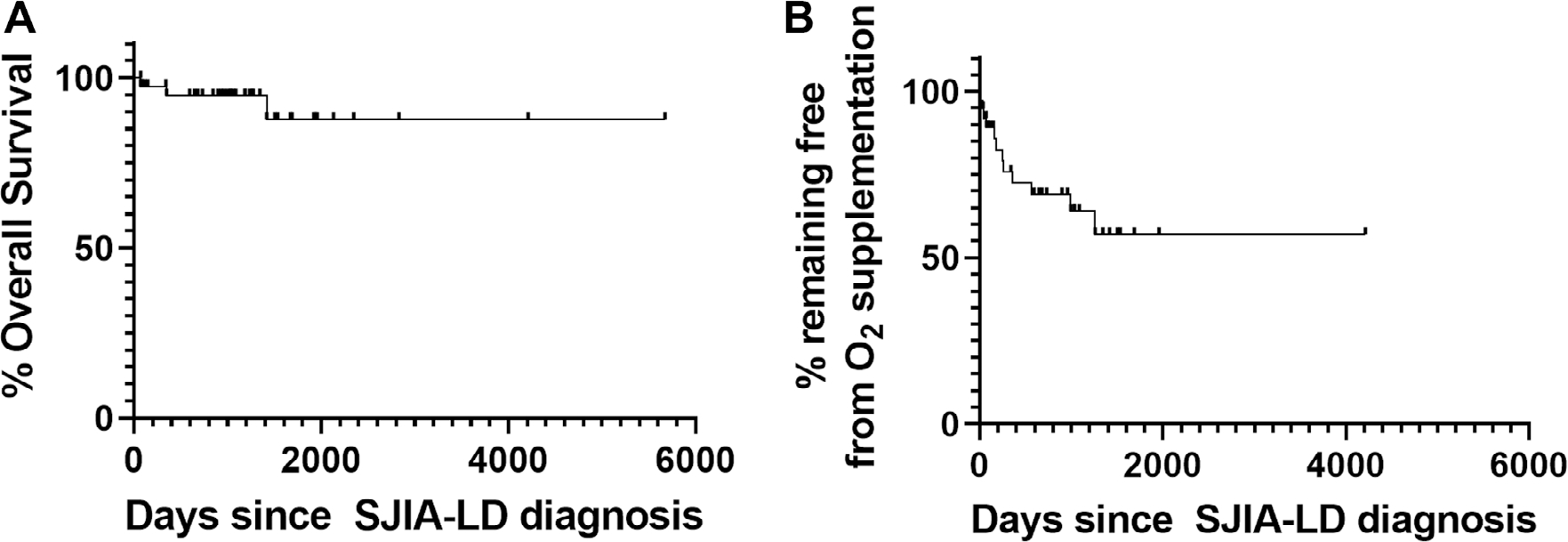
Survival curves of patients with SJIA-LD. (A) Overall survival of patients with SJIA-LD (median follow-up 2.9 years) is shown. (B) Survival free of supplemental oxygen for patients with SJIA-LD after LD diagnosis is shown. LD, lung disease; SJIA, systemic juvenile idiopathic arthritis.
To more comprehensively assess patient clinical outcomes, we defined as a new measure the PGALD, as a 0 to 10 scale measure of current lung disease severity, based upon the well-established use of the physician global assessment in rheumatology.30 As shown in Figure 4A, the PGALD at last follow-up had a median of 3 (IQR 1–5) and a range of 0 to 8. Fourteen patients (40%) had a PGALD score of 0 to 2 reflecting low disease activity, whereas 8 of 35 patients (23%) had PGALD scores ≥5 reflecting high disease activity. We also used the PGALD to examine the relationship between LD outcome and systemic inflammation, as determined by the peak measured serum IL-18. In Figure 4B, we show that the PGALD had a weak but significant positive correlation with peak IL-18 (r = 0.44, P = 0.016). The PGALD at last follow-up showed no correlation to LD duration (r = −0.13, P = 0.47). Finally, we examined whether patients who permanently discontinued anti–IL-1/6 biologic therapy had improved overall outcomes compared with those who remained on therapy. Overall, 11 patients stopped anti–IL-1/6 biologic therapy for greater than 6 months while still exhibiting active features of SJIA (2 additional patients discontinued medication after achieving clinical remission on medication). There was no significant difference in PGALD score at last follow-up between patients who discontinued such biologics (n = 11, median PGALD 3 [IQR 2–3]) and those who continued biologics (n = 24, median 3 [IQR 1–5], P = 0.68).
Figure 4.
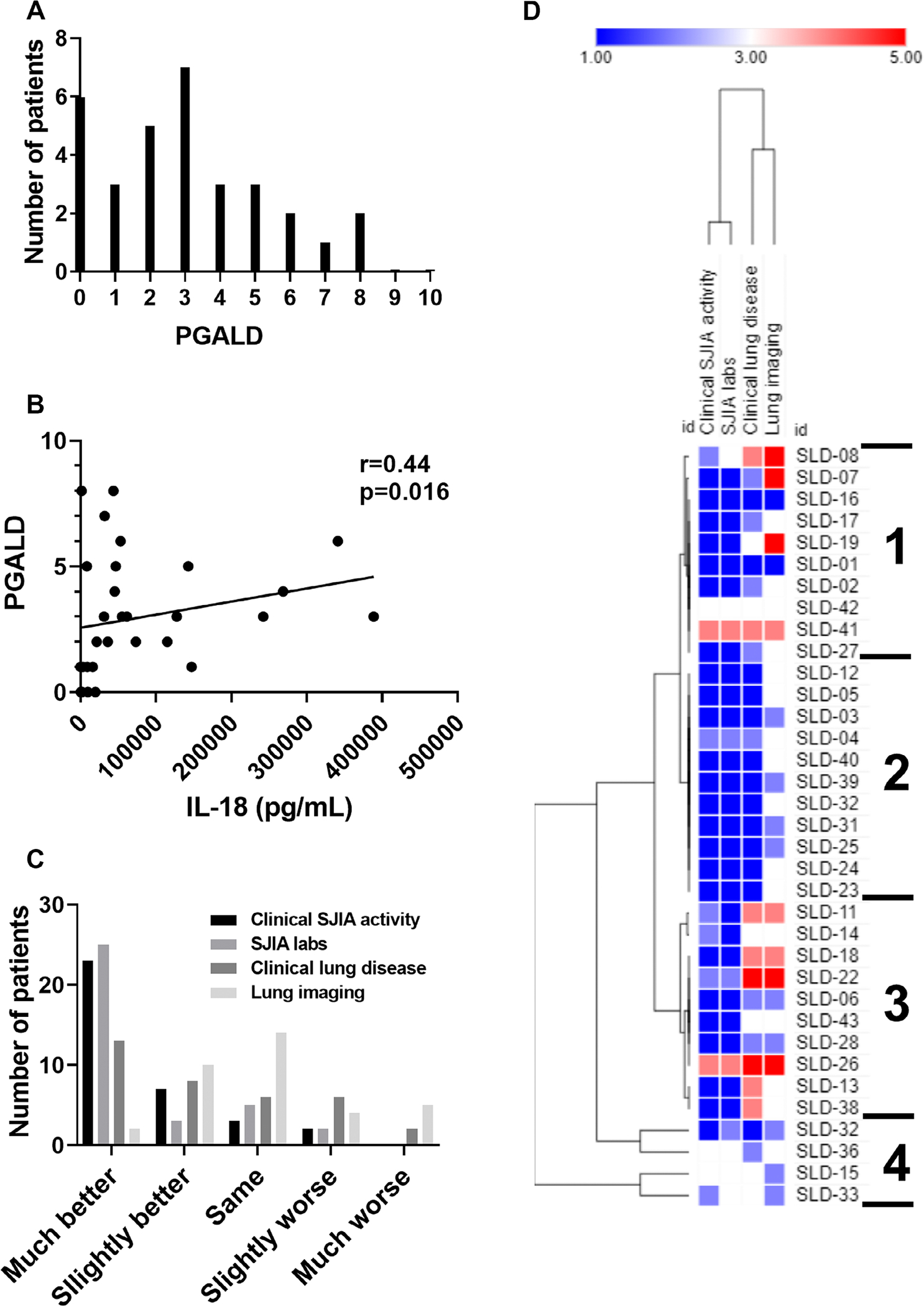
Current clinical status of patients with SJIA-LD. (A) Distribution of PGALD scores at last follow-up for patients with SJIA-LD is shown. (B) Correlation between PGALD score at last follow-up and peak serum IL-18 level is shown. (C) Change in clinical status from LD diagnosis to last follow-up in four cardinal clinical domains (chi-square, P = 0.001) is shown. (D) Hierarchical clustering of patterns of clinical change for patients with SJIA-LD is shown. IL-18, interleukin-18; LD, lung disease; PGALD, Physician Global Assessment of Lung Disease; SJIA, systemic juvenile idiopathic arthritis; SLD, SJIA Lung Disease.
We also assessed for overall change since diagnosis in using a four-domain measure: clinical SJIA activity, inflammatory or SJIA laboratory features, clinical LD activity, and lung imaging. As shown in Figure 4C, although overall outcomes were variable, most patients showed improvement in most domains over time. A majority of patients were rated as “much better” regarding clinical SJIA activity (23 of 35 [66%]) and SJIA/inflammatory laboratories (25 of 35 [71%]), and a plurality regarding clinical LD (13 of 35 [37%]). In contrast, lung imaging changes were more variable, with the largest number of patients rated as “unchanged” (14 of 35 [40%]). Performing hierarchical clustering of individual patterns of improvement (Figure 4D) showed four clusters of patient phenotypes, which were independent of disease duration. Patients in cluster 1 mostly showed improvement in clinical SJIA activity and SJIA laboratories but with more variability in clinical LD and lung imaging. Cluster 2 represented patients with improvement in all domains, although lung imaging was improved or unchanged. Cluster 3 included several patients with overall improvement in SJIA activity and laboratories but worsening in clinical LD and imaging, whereas cluster 4 included patients with little overall change since diagnosis. Both the PGALD and four-domain measure showed similar results when restricted to patients with SJIA-LD with at least 1 year of follow-up (Supplementary Figure 3, n = 30). Together, these findings suggest a range of disease trajectories in SJIA-LD.
Finally, we examined radiographic progression in several patients with serial chest CT. As shown in Figure 5, radiographic changes over time were highly variable and ranged from stabilization to slow improvement to progressive worsening. After LD diagnosis, patient 1 stopped anticytokine biologics and was treated with tofacitinib, cyclosporine A, and corticosteroids but was not able to achieve steroid-free control of underlying SJIA or improvement in radiographic findings. In contrast, other patients demonstrated progressive radiographic worsening of LD despite numerous biologic and nonbiologic treatments. Patient 2 at LD diagnosis demonstrated scattered parenchymal abnormalities, ground-glass opacities, and enlarged lymph nodes. Over 2 years, this patient developed increasing hypoxia with radiographic progression to diffuse “crazy-paving” consolidation and centrilobular nodules despite treatments with anticytokine biologics, multiple JAK inhibitors, and emapalumab. Together, these illustrate the spectrum of dynamic changes in radiographic SJIA-LD findings.
Figure 5.
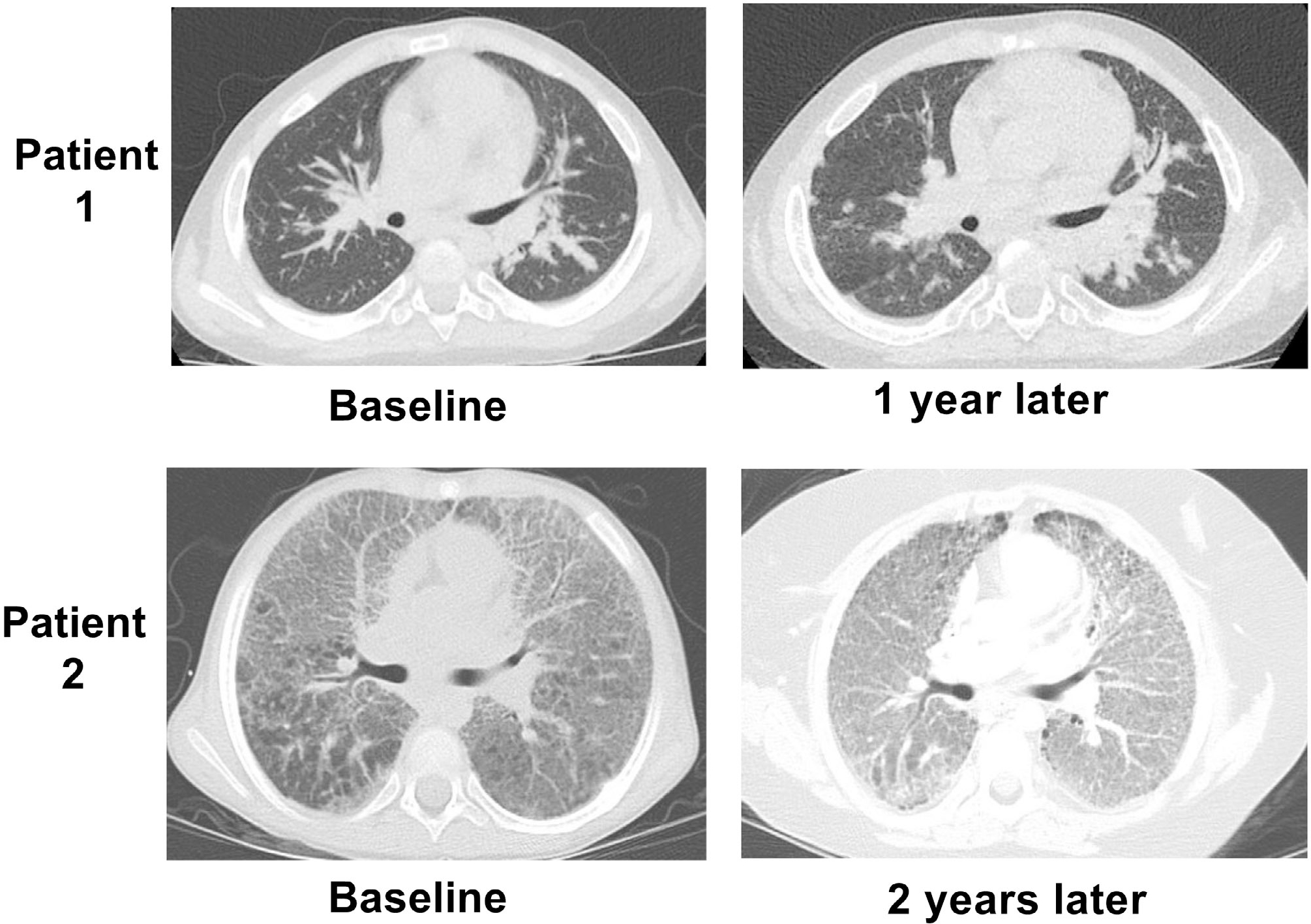
Variability in imaging progression in patients with SJIA-LD. For patient 1, baseline chest CT demonstrated extensive parenchymal pulmonary opacities with interstitial thickening. Repeat imaging showed an overall similar pattern with development of small cysts suggesting fibrosis despite stopping all anticytokine therapy. Patient 2 at baseline demonstrated extensive bilateral ground-glass opacities and interlobular septal thickening. Follow-up study 2 years later showed progression of findings, including more dependent consolidation despite treatment with multiple biologic and nonbiologic agents. CT, computed tomography; LD, lung disease; SJIA, systemic juvenile idiopathic arthritis.
DISCUSSION
Over the past decade, increasing numbers of children with SJIA have been recognized to have chronic LD. Although initial case series helped define the clinical spectrum of SJIA-LD, these retrospective reports had broad ranges of outcomes.13–15 In addition, only scattered case reports describe the clinical course and possible treatments of SJIA-LD after diagnosis.14,17–19,31,32 Here, we present an overview of our prospective cohort of children with SJIA-LD evaluated at our multidisciplinary clinic. Our key findings are first, that patients with SJIA-LD were young at SJIA diagnosis, with frequent occurrence of MAS, adverse reactions to biologics, and high serum IL-18 levels. Second, these patients have a highly varied clinical course, and although our overall mortality rate of 7% is lower than prior studies, 65% had overnight hypoxia and 37% at some point required respiratory support. Third, after LD detection, patients were frequently treated with alternative therapeutics, including JAK inhibitors and T cell targeting therapies. Fourth, we describe several dynamic measures of clinical disease status, including overnight oximetry, the PGALD, and a four-domain measure of change. Finally, although clinical measures and overnight oximetry changed over time in many patients, imaging findings were the most stable. Together, these findings provide key insights into the overall course of SJIA-LD, including modalities to follow disease progression and potential treatments.
This work contributes to the emerging understanding of both the clinical spectrum of SJIA-LD and the phenotype of patients with SJIA who develop LD. In line with prior work,14,15 children with SJIA-LD were young at diagnosis (median age 1.5 years), with high incidence of both MAS (85%) and adverse reactions to biologic medications (41%) and markedly elevated serum IL-18. Radiographically the most common features at LD diagnosis were septal thickening, lymphadenopathy, bronchial wall or peribronchovascular thickening, and ground-glass opacities; interestingly, there was a trend towards presence of ground-glass opacities and later progression to an oxygen requirement. Finally, 15% of patients had trisomy 21; although the connection between SJIA-LD and this disorder is unclear, it may relate to the emerging understanding of interferonopathy in Down syndrome.33,34 Indeed, along with elevated IL-18, some patients with SJIA-LD demonstrated elevated levels of the IFNγ-induced protein CXCL9. Notably, although CXCL9 is rarely elevated during SJIA flares, it is a specific marker of MAS.11,35 We also examined other proposed biomarkers for SJIA-LD that may be independent of the level of MAS activity.26 We found that levels of MMP7, a marker of lung injury, were significantly higher in patients with SJIA-LD compared with patients without LD but that the eosinophil chemotaxin CCL-11 was not. This could suggest that eosinophil activation is not a consistent feature of SJIA-LD, or differences in treatment approach in the present cohort compared with prior work, but this requires further study.16
Prior large series of patients with SJIA-LD have reported mortality rates ranging from 0% to 68%.13–15 Here, patients were prospectively enrolled at time of initial clinical assessment and followed for a median 2.9 years from LD diagnosis. Although, encouragingly, 93% of patients were alive at last follow-up, substantial numbers required supplemental respiratory support and/or had abnormal overnight oximetry readings. To better understand the clinical status of patients with SJIA-LD, we piloted two measures: the PGALD and a four-domain assessment of change over time. Together, these measures tell a mixed story, with many patients having low LD activity (PGALD score ≤2) but some with persistent high disease activity (PGALD ≥5) and several distinct patterns of disease evolution. Notably, radiographic features of disease were the least likely to improve over time, which could suggest that resolution of chest CT findings may be a relatively late finding and not as sensitive to short-term change or more fibrotic and chronic. Indeed, our work suggests that clinical measures such as the PGALD, or functional measures such as overnight oximetry, may be more amenable for determining responses to treatment.
After LD diagnosis, patients were treated with a wide variety of immunomodulatory therapies. Although slightly fewer patients were treated with anti–IL-1/6 biologics after diagnosis, and some permanently discontinued such therapy, most were treated with combinations including both biologics and adjunctive therapies. These adjunctive therapies including medications that target T cell activation (abatacept, azathioprine, calcineurin inhibitors, and mycophenolate) as well as those targeting other cytokines, including interferons (JAK inhibitors and emapalumab). Other common therapies were directed at improving pulmonary function, including prophylactic antimicrobials, bronchodilators, inhaled corticosteroids, and airway clearance. Azithromycin was also commonly used; this macrolide antibiotic has antiinflammatory effects and has been shown in randomized trials to reduce pulmonary exacerbations in children with cystic fibrosis (CF), non-CF bronchiectasis, and HIV-associated LD.29,36,37 Given the diversity of treatment regimens in our cohort, it is impossible to judge the effectiveness of any one therapy. Notably, however, comparing patients who permanently discontinued anti–IL-1/6 biologics with those who continued such therapy, there were no differences in mortality rate or PGALD score. Additionally, at least one patient developed an oxygen requirement after stopping biologics, and others who attempted to discontinue therapy were reported to have disease flares and restarted biologics. However, this critical question requires more study and may be impacted by LD chronicity.
This study has several important limitations. First, this is a single-center cohort, and although patients were referred from numerous centers in the US, inclusion may be biased by such referral patterns. However, a bias towards referral of more severe/complicated patients may cause an overestimation of disease severity, making these higher survival rates more noteworthy. Although we assessed for overt drug reactions leading to medication discontinuation, we were unable to quantify the incidence of less significant drug reactions or of eosinophilia or more subtle features that could represent delayed drug reactions. We also lack a similar longitudinal comparison with patients with SJIA without LD, which is urgently needed. Our proposed SJIA-LD definitions, PGALD, and four-domain measure of change have not been validated, and thus, their clinical and research utility is uncertain. We have not measured free IL-18, and, although there are limited data about this in SJIA, it is the biologically active form of the cytokine. Finally, as we did not have access to electronic CT images, we were unable to perform any quantitative assessment of changes over time.
Our findings underscore the most pressing question related to SJIA-LD: To what degree do anti–IL-1/6 biologics contribute to the development of SJIA-LD, possibly through a delayed-type hypersensitivity or DRESS reaction? Recent work has reported that some patients with SJIA treated with these biologics have unusual clinical features, including eosinophilia, atypical rashes, and cytopenias that would fit the definition of DRESS, before development of LD.16 This phenotype was strongly linked to presence of an HLA haplotype including HLA-DRB1*15, which was also identified in 83% of our cohort (including 100% of those with biopsy-proven LD). The DRESS hypothesis is provocative and would explain the increasing recognition of SJIA-LD in the past decade and noted association with adverse drug reactions; however, it fails to fully explain the relative absence of these features in other diseases treated with such biologics, the development of LD in some patients never exposed to biologics,38 and observation of both improvement in some patients who remain on these therapies and lack of improvement in some others upon discontinuation (Figure 5). Notably, most of the adverse reactions noted in the present cohort were overt, including severe local reactions, anakinra-induced liver injury, and tocilizumab infusion reactions. An alternative hypothesis was recently proposed, termed the cytokine plasticity hypothesis,39 in which biologic treatment alters T cell phenotypes towards those promoting allergic features, and by creating an altered cytokine milieu that alters phenotypes of lung macrophages to cause PAP.14 Finally, it should be considered whether children with the disorder we term “SJIA-LD” in fact represent a unique phenotype of the disorders classified as SJIA. This would explain the distinct features of these children at diagnosis (early age, frequently minimal to absent arthritis, high IL-18, incidence of Down syndrome), the presence of a specific genetic marker, and idiosyncratic responses to anticytokine biologics. Regardless of the underlying causes of SJIA-LD, anticytokine biologics as currently used do not prevent this complication, and as such, careful investigations of other treatment strategies are urgently needed. Our work here describes important clinical, radiographic, and biomarker assessment of SJIA-LD that will be critical measures of future interventional studies.
Supplementary Material
SIGNIFICANCE & INNOVATIONS.
Systemic juvenile idiopathic arthritis–associated lung disease (SJIA-LD) is an increasingly recognized disease complication, but little is known about disease course, morbidity, mortality, and treatments after diagnosis.
This study represents a needed prospective cohort with defined clinical outcomes and demonstrates lower mortality than prior reports but frequent hypoxia with need for respiratory support and changes in treatment approaches after diagnosis.
This report describes needed measures for tracking SJIA-LD over time, including overnight oximetry measurements, the Physician Global Assessment of Lung Disease, and a four-domain measure of clinical change.
ACKNOWLEDGMENTS
We thank the patients and families for participating in this research project.
Supported by an ARC Award by the Cincinnati Children’s Research Foundation, the Systemic Juvenile Idiopathic Arthritis Foundation, and the Jellen Family Foundation. Dr. Schulert was supported by National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health (grant K08-AR-072075). Processing and storage of biosamples was supported by the Cincinnati Pediatric Rheumatology Tissue Repository (PRTR) (grant P30-AR-047363).
Footnotes
Additional supplementary information cited in this article can be found online in the Supporting Information section (http://onlinelibrary.wiley.com/doi/10.1002/acr.25234).
Author disclosures are available at https://onlinelibrary.wiley.com/doi/10.1002/acr.25234.
REFERENCES
- 1.Mellins ED, Macaubas C, Grom AA. Pathogenesis of systemic juvenile idiopathic arthritis: some answers, more questions [review]. Nat Rev Rheumatol 2011;7:416–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee JJY, Schneider R. Systemic juvenile idiopathic arthritis [review]. Pediatr Clin North Am 2018;65:691–709. [DOI] [PubMed] [Google Scholar]
- 3.Woo P, Southwood TR, Prieur AM, et al. Randomized, placebo-controlled, crossover trial of low-dose oral methotrexate in children with extended oligoarticular or systemic arthritis. Arthritis Rheum 2000;43:1849–1857. [DOI] [PubMed] [Google Scholar]
- 4.De Benedetti F, Brunner HI, Ruperto N, et al. ; PRINTO; PRCSG. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N Engl J Med 2012;367:2385–2395. [DOI] [PubMed] [Google Scholar]
- 5.Ruperto N, Brunner HI, Quartier P, et al. ; PRINTO; PRCSG. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N Engl J Med 2012;367:2396–2406. [DOI] [PubMed] [Google Scholar]
- 6.Quartier P, Allantaz F, Cimaz R, et al. A multicentre, randomised, double-blind, placebo-controlled trial with the interleukin-1 receptor antagonist anakinra in patients with systemic-onset juvenile idiopathic arthritis (ANAJIS trial). Ann Rheum Dis 2011;70:747–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nigrovic PA, Mannion M, Prince FH, et al. Anakinra as first-line disease-modifying therapy in systemic juvenile idiopathic arthritis: report of forty-six patients from an international multicenter series. Arthritis Rheum 2011;63:545–555. [DOI] [PubMed] [Google Scholar]
- 8.Beukelman T, Tomlinson G, Nigrovic P, et al. First-line options for systemic juvenile idiopathic arthritis treatment: an observational study of Childhood Arthritis and Rheumatology Research Alliance Consensus Treatment Plans. Pediatr Rheumatol Online J 2022;20:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schulert GS, Grom AA. Pathogenesis of macrophage activation syndrome and potential for cytokine- directed therapies [review]. Annu Rev Med 2015;66:145–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Put K, Avau A, Brisse E, et al. Cytokines in systemic juvenile idiopathic arthritis and haemophagocytic lymphohistiocytosis: tipping the balance between interleukin-18 and interferon-γ. Rheumatology (Oxford) 2015;54:1507–1517. [DOI] [PubMed] [Google Scholar]
- 11.Bracaglia C, de Graaf K, Marafon DP, et al. Elevated circulating levels of interferon-γ and interferon-γ-induced chemokines characterise patients with macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. Ann Rheum Dis 2017;76:166–172. [DOI] [PubMed] [Google Scholar]
- 12.De Benedetti F, Grom AA, Brogan PA, et al. Efficacy and safety of emapalumab in macrophage activation syndrome. Ann Rheum Dis 2023;82:857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kimura Y, Weiss JE, Haroldson KL, et al. ; Childhood Arthritis Rheumatology Research Alliance Carra Net Investigators. Pulmonary hypertension and other potentially fatal pulmonary complications in systemic juvenile idiopathic arthritis. Arthritis Care Res (Hoboken) 2013;65:745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schulert GS, Yasin S, Carey B, et al. Systemic juvenile idiopathic arthritis–associated lung disease: characterization and risk factors. Arthritis Rheumatol 2019;71:1943–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saper VE, Chen G, Deutsch GH, et al. ; Childhood Arthritis and Rheumatology Research Alliance Registry Investigators. Emergent high fatality lung disease in systemic juvenile arthritis. Ann Rheum Dis 2019;78:1722–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saper VE, Ombrello MJ, Tremoulet AH, et al. ; Drug Hypersensitivity Consortium; INCHARGE Consortium. Severe delayed hypersensitivity reactions to IL-1 and IL-6 inhibitors link to common HLA-DRB1*15 alleles. Ann Rheum Dis 2022;81:406–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bader-Meunier B, Hadchouel A, Berteloot L, et al. Effectiveness and safety of ruxolitinib for the treatment of refractory systemic idiopathic juvenile arthritis like associated with interstitial lung disease: a case report. Ann Rheum Dis 2022;81:e20. [DOI] [PubMed] [Google Scholar]
- 18.Sato S, Hosokawa T, Kawashima H. Successful treatment of plasma exchange for refractory systemic juvenile idiopathic arthritis complicated with macrophage activation syndrome and severe lung disease. Ann Rheum Dis 2022;81:e61. [DOI] [PubMed] [Google Scholar]
- 19.Horne A, von Bahr Greenwood T, Chiang SC, et al. Efficacy of moderately dosed etoposide in macrophage activation syndrome - hemophagocytic lymphohistiocytosis (MAS-HLH). J Rheumatol 2021;48: 1596–1602. [DOI] [PubMed] [Google Scholar]
- 20.Petty RE, Southwood TR, Manners P, et al. ; International Leage of Associations for Rheumatology. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol 2004;31:390–392. [PubMed] [Google Scholar]
- 21.DeWitt EM, Kimura Y, Beukelman T, et al. ; Juvenile Idiopathic Arthritis Disease-specific Research Committee of Childhood Arthritis Rheumatology and Research Alliance. Consensus treatment plans for new-onset systemic juvenile idiopathic arthritis. Arthritis Care Res (Hoboken) 2012;64:1001–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ravelli A, Minoia F, Davì S, et al. ; Paediatric Rheumatology International Trials Organisation; Childhood Arthritis and Rheumatology Research Alliance; Pediatric Rheumatology Collaborative Study Group; Histiocyte Society. 2016 Classification criteria for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a European League Against Rheumatism/American College of Rheumatology/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Arthritis Rheumatol 2016;68:566–576. [DOI] [PubMed] [Google Scholar]
- 23.Erkens R, Esteban Y, Towe C, et al. Pathogenesis and treatment of refractory disease courses in systemic juvenile idiopathic arthritis: refractory arthritis, recurrent macrophage activation syndrome and chronic lung disease [review]. Rheum Dis Clin North Am 2021;47:585–606. [DOI] [PubMed] [Google Scholar]
- 24.Yasin S, Fall N, Brown RA, et al. IL-18 as a biomarker linking systemic juvenile idiopathic arthritis and macrophage activation syndrome. Rheumatology 2020;59:361–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee BS, Rust NA, McMichael AJ, et al. HLA-DR2 subtypes form an additional supertypic family of DR beta alleles. Proc Natl Acad Sci U S A 1987;84:4591–4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen G, Deutsch GH, Schulert GS, et al. Identification of distinct inflammatory programs and biomarkers in systemic juvenile idiopathic arthritis and related lung disease by serum proteome analysis. Arthritis Rheumatol 2022;74:1271–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huaux F, Gharaee-Kermani M, Liu T, et al. Role of Eotaxin-1 (CCL11) and CC chemokine receptor 3 (CCR3) in bleomycin-induced lung injury and fibrosis. Am J Pathol 2005;167:1485–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vos R, Verleden SE, Ruttens D, et al. Azithromycin and the treatment of lymphocytic airway inflammation after lung transplantation. Am J Transplant 2014;14:2736–2748. [DOI] [PubMed] [Google Scholar]
- 29.Valery PC, Morris PS, Byrnes CA, et al. Long-term azithromycin for Indigenous children with non-cystic-fibrosis bronchiectasis or chronic suppurative lung disease (Bronchiectasis Intervention Study): a multicentre, double-blind, randomised controlled trial. Lancet Respir Med 2013;1:610–620. [DOI] [PubMed] [Google Scholar]
- 30.Harrington JT. The uses of disease activity scoring and the physician global assessment of disease activity for managing rheumatoid arthritis in rheumatology practice. J Rheumatol 2009;36:925–929. [DOI] [PubMed] [Google Scholar]
- 31.Rood JE, Rezk A, Pogoriler J, et al. Improvement of refractory systemic juvenile idiopathic arthritis-associated lung disease with single-agent blockade of IL-1β and IL-18. J Clin Immunol 2023;43:101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yasin S, Solomon K, Canna SW, et al. IL-18 as therapeutic target in a patient with resistant systemic juvenile idiopathic arthritis and recurrent macrophage activation syndrome. Rheumatology (Oxford) 2020;59:442–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malle L, Bogunovic D. Down syndrome and type I interferon: not so simple [review]. Curr Opin Immunol 2021;72:196–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chung H, Green PH, Wang TC, et al. Interferon-driven immune dysregulation in Down syndrome: a review of the evidence [review]. J Inflamm Res 2021;14:5187–5200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Matteis A, Colucci M, Rossi MN, et al. Expansion of CD4dimCD8+ T cells characterizes macrophage activation syndrome and other secondary HLH. Blood 2022;140:262–273. [DOI] [PubMed] [Google Scholar]
- 36.Ferrand RA, McHugh G, Rehman AM, et al. ; BREATHE Trial Group. Effect of once-weekly azithromycin vs placebo in children with HIV-associated chronic lung disease: the BREATHE randomized clinical trial. JAMA Netw Open 2020;3:e2028484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stick SM, Foti A, Ware RS, et al. ; COMBAT CF Study Group. The effect of azithromycin on structural lung disease in infants with cystic fibrosis (COMBAT CF): a phase 3, randomised, double-blind, placebo-controlled clinical trial. Lancet Respir Med 2022;10:776–784. [DOI] [PubMed] [Google Scholar]
- 38.Schultz R, Mattila J, Gappa M, et al. Development of progressive pulmonary interstitial and intra-alveolar cholesterol granulomas (PICG) associated with therapy-resistant chronic systemic juvenile arthritis (CJA). Pediatr Pulmonol 2001;32:397–402. [DOI] [PubMed] [Google Scholar]
- 39.Binstadt BA, Nigrovic PA. The conundrum of lung disease and drug hypersensitivity-like reactions in systemic juvenile idiopathic arthritis [review]. Arthritis Rheumatol 2022;74:1122–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.