

Abstract
We describe two unrelated children with de novo variants in the non-erythrocytic alpha-II-spectrin (SPTAN1) gene who have hypoplastic brain structures, intellectual disability, and both fine and gross motor impairments. Using agnostic exome sequencing, we identified a nonsense variant creating a premature stop codon in exon 21 of SPTAN1, and in a second patient we identified an intronic substitution in SPTAN1 prior to exon 50 creating a new donor acceptor site. Neither of these variants has been described previously. Although some of these patients’ features are consistent with the known SPTAN1 encephalopathy phenotype, these two children do not have epilepsy, in contrast to reports about nearly every other patient with heterozygous SPTAN1 variants and in all patients with a variant near the C-terminal coding region. Moreover, both children have abnormal thyroid function, which has not been previously reported in association with SPTAN1 variant. We present a detailed discussion of the clinical manifestations of these two unique SPTAN1 variants and provide evidence that both variants result in reduced mRNA expression despite different locations within the gene and clinical phenotypes. These findings expand the motor, cognitive, and behavioral spectrum of the SPTAN1-associated phenotype and invite speculation about underlying pathophysiologies.
Keywords: ataxia, cerebellar cognitive affective syndrome, cerebellar hypoplasia, early infantile epileptic encephalopathy, premature termination codon, splice acceptor site, SPTAN1
1 |. INTRODUCTION
De novo variants in non-erythrocytic alpha-II-spectrin (SPTAN1) cause infantile-onset epileptic encephalopathy type 5 (OMIM 613477) also referred to as SPTAN1 encephalopathy (Tohyama et al., 2015). SPTAN1 codes for the membrane structural protein alpha-II-spectrin, which forms heterotetramers with beta spectrin proteins (Bignone & Baines, 2003). These spectrin assemblies are important for the maintenance of the cytoskeleton (Weigand, Boeckel, Gellert, & Dimmeler, 2012). In neurons, spectrin complexes bind with syntaxins to play a putative role in presynaptic vesicular release (Nakano, Nogami, Sato, Terano, & Shirataki, 2001) and interact directly with Shank proteins that localize glutamate receptors in the post synaptic densities (Böckers et al., 2001). SPTAN1 expression is highest in the temporal and cerebellar cortices (He et al., 2016), explaining the decreased volumes of these structures in affected patients (Tohyama et al., 2015), and the consequent occurrence of early onset epilepsy and poor motor coordination.
Fewer than 20 patients have been described with heterozygous SPTAN1 variants, and almost all of them had a diagnosis of epilepsy, typically starting with infantile spasms (Campbell et al., 2012; Nicita et al., 2015; Tohyama et al., 2015). The phenotypic spectrum seen in this small pool of patients, specifically the range of severity and preponderance of epilepsy, suggests the possibility of nuanced genotype–phenotype correlations. The last two spectrin repeats in the C-terminus of SPTAN1 contain all of the known variants associated with epilepsy while variants elsewhere in the SPTAN1 gene are associated with more variable outcomes and an absence of epileptic encephalopathy (Tohyama et al., 2015). These final spectrin repeats are critically involved in the interaction of alpha-II-spectrin with beta-spectrin to form functional heterodimers (Zhang et al., 2010). Thus, C-terminal variants may alter binding and act in a dominant negative fashion, perhaps even causing aggregates within neurons (Saitsu et al., 2010). In contrast, variants outside the spectrin binding domains are considered less clinically devastating (Campbell et al., 2012; Tohyama et al., 2015).
We report two patients with novel SPTAN1 variants that expand our understanding of SPTAN1 encephalopathy in several ways. First, an observed variant in the last two spectrin repeat domains does not invariably predict an early life onset epileptic encephalopathy but is associated with a more pronounced cerebellar phenotype. Second, a variant situated within the SH3 domain is associated with a significant protean phenotype. Third, both these two variants lead to marked downregulation in SPTAN1 RNA production suggesting that any deleterious variant in SPTAN1 causes reduced mRNA expression.
2 |. MATERIALS AND METHODS
2.1 |. Patient enrollment
The patients were admitted to the National Institutes of Health (NIH) Clinical Center and enrolled in the NIH Undiagnosed Diseases Program (Gahl et al., 2012, 2016) and in protocol 76-HG-0238, “Diagnosis and Treatment of Patients with Inborn Errors of Metabolism or Other Genetic Disorders,” approved by the National Human Genome Research Institute (NHGRI) Institutional Review Board (IRB). Written informed consent was provided by the parents for their minor children, and the patients themselves provided verbal assent to study participation.
2.2 |. Genetic analyses
Genomic DNA was extracted from peripheral leukocytes of the two affected individuals, their unaffected parents, and unaffected siblings using the Gentra Puregene Blood Kit (Qiagen, Valencia, CA). The DNA of all family members was subjected to an integrated set of genomic analyses including high-density single nucleotide polymorphism (SNP) arrays and whole exome sequencing (WES). WES was performed on the nuclear family using the Illumina HiSeq2000 platform and the TrueSeq capture kit (Illumina, San Diego, CA). Sample library preparation, sequencing, and analysis were performed using the standard NIH Intramural Sequencing Center (NISC) pipeline (Markello et al., 2012). Sequence data were aligned to human reference genome (hg19) using Novoalign (Novocraft Technologies, Selangor, Malaysia). To test for copy number variants and to form segregation BED files for exome analysis, Omni Express 12 (hg18) SNP arrays were run on genomic DNA from all family members as described (Markello et al., 2012) and converted to hg19. Parentage was confirmed through SNP analysis. Variants listed in the Variant Call Files were filtered based on rarity, Mendelian segregation, and predicted deleteriousness. Allele frequencies were required to be <0.06 for the UDP patient cohort and <0.02 for the Exome Sequencing Project v.0.0.20 African Ancestry population, Exome Sequencing Project v.0.0.20 European Ancestry population, and dbSNP build 137. Homozygous and de novo variants were required to occur in fewer than two individuals in the UDP cohort. Variants needed to segregate with autosomal recessive, X-linked recessive, or de novo dominant modes of inheritance and be annotated as nonsynonymous, frame shift, premature stop, loss of start codon, loss of stop codon, near splice (within 20 base pairs of a canonical splice site), or splicing variants. Variants that passed filtration were ranked using Exomiser 2.0 (http://software.broadinstitute.org/software/igv/); the shared Human Phenotype Ontology terms of the patients were used as the phenotype input for Exomiser. Using the Integrative Genome Viewer (https://www.broadinstitute.org/igv/home), we assessed the quality of alignment and genotype call of variants. CLIA certification was performed using Sanger DNA sequencing (Gene Dx, Rockville, MD).
2.3 |. Skin fibroblasts
Skin fibroblasts were obtained from both patients to provide a source of RNA and protein. Control skin fibroblasts were procured from Coriell Institute for Medical Research (Camden, NJ). Fibroblasts were grown in high glucose DMEM (Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum and 1% Antibiotic-Antimycotic solution (Life Technologies, Carlsbad, CA). Cultured fibroblasts were incubated in a humidity-controlled environment at 37 °C with 95% O2 and 5% CO2, fed fresh media daily for 3 days, and were studied before passage 10.
2.4 |. Immunocytochemistry
Fibroblasts were fixed in 4% (w/v) paraformaldehyde for 10 min at room temperature, followed by three washes with phosphate buffered saline (PBS). After incubation in 0.1% (v/v) Triton X-100 in PBS (PBS-T) for 10 min at room temperature, the cells were incubated with blocking buffer (PBS-T containing 4% (v/v) horse serum and 1% (v/v) glycerol at room temperature for 20 min. Cells were stained with monoclonal anti-alpha fodrin (anti-SPTAN1 pre-SH3 segment) antibody (1:100, abcam, San Francisco, CA) and anti-actin (1:100, BD Biosciences), followed by appropriate secondary antibodies and 4′,6-diamidino-2-phenylindole (DAPI) nuclear staining. Images were acquired on an EVOS fluorescence microscope (AMG, Bothell, WA).
2.5 |. Digital droplet polymerase chain reaction (PCR)
Digital droplet PCR (ddPCR) analysis was performed on 50 ng of cDNA derived from patient and control fibroblasts’ RNA extracted via a standard Trizol protocol and cDNA generated using TaqMan Genotyping Mastermix (Life Technologies, Carlsbad, CA) and a customized assay for the two observed variants (shown in Supporting Information Table S1). The PCRs were subsequently amplified in a Biorad Mycycler PCR Thermal Cycler (BioRad, Hercules, CA) with cycling conditions summarized in Supporting Information Table S2. The amplified products (approximately 10 million mRNA-containing droplets per sample) were read on the Sense RainDrop Digital PCR System (RainDance Technologies, Billerica, MA) and analyzed using the Raindrop Analyst II software. Results for ddPCR are reported as mean number of counted droplets for each specific assay, where each counted droplet contained amplified mRNA.
2.6 |. Western blot assay
Cytosolic proteins were isolated from patient and control skin fibroblasts using citrate buffer with the addition of Complete Protease Inhibitor (Sigma-Aldrich, St. Louis, MO). Proteins were then resolved by SDS-PAGE and transferred to a nitrocellulose membrane. An overnight incubation was performed with the primary antibody, 1:10,000 anti-GAPDH (Abcam, Cambridge, MA) or 1:1,000 anti-SPTAN1 (BD Biosciences), followed by 1 hr incubation with an appropriate fluorescent secondary antibody. The quantity of protein from patient and control fibroblasts was identical (30 μg). Protein was visualized on an Odyssey CLx infrared imaging system (LI-COR Biosciences, Lincoln, NE). Quantification of the SPTAN1 band’s intensity was performed relative to the GAPDH intensity.
3 |. RESULTS
3.1 |. Patient reports
3.1.1 |. Patient 1
Patient 1 was a 10-year-old Caucasian male referred to the NIH Undiagnosed Diseases Program (UDP) with intellectual disability, aggressive behaviors, global developmental delay, dysmorphic features (notably microcephaly and bilateral ptosis) as well as multiple congenital anomalies including left hydronephrosis, right small kidney, undescended testes, bilateral fifth digit clinodactyly, and bilateral club feet. He has a healthy younger sister in a family of maternal German and Cherokee Native American ancestry and paternal German and English ancestry. The patient was born following a 37-week gestation complicated by first trimester spotting. A prenatal ultrasound in the fourth month of pregnancy revealed microcephaly, renal cysts, clubbed feet, and clinodactyly; follow-up studies noted persistent microcephaly. Apgar scores at birth were 8 at 1 min and 9 at 5 min. Birth weight was 3,061 g (23rd percentile), birth length 47 cm (6th percentile), and head circumference 31.5 cm (3rd percentile).
As a newborn, he had microcephaly (with a normal cranial ultrasound), low tone, clubbed feet, facial dysmorphism, undescended testes, and a grade 3/6 cardiac murmur with a small persistent ductus arteriosus. He had poor nipple attachment with slow feeding and emesis with choking episodes during feedings. After multiple changes in feeding and medication trials for presumed gastroesophageal reflux, a nasogastric feeding tube was placed at age 3 months and remained until 1 year of life. The child had a history of global developmental delay, including motor, language, and cognitive domains. He rolled over at age 7 months and sat without support at age 13 months. He cruised at 18 months, walked with a walker at 2.5 years, and walked independently at 3.5 years. Surgeries to correct his clubbed feet and bilateral heel tenotomies may have contributed to the delay in independent ambulation.
Regarding fine motor skills, pincer grasp was developed at 14 months but was delayed in use of a writing implement with age-appropriate dexterity. Language delays resulted in signing words at 18 months, with a vocabulary of three distinct signed words at 2 years. He began to use single spoken words at approximately 3 years; phrase speech at 8.5 years, and at age 10, he used a combination of spoken words and hand gestures (not American sign language) to communicate basic wants and needs. He continued to be delayed in daily living skills, and at the time of evaluation could not write his name, make simple drawings, or engage in dressing activities requiring zippers and buttons.
Upon physical examination, the patient was found to be dysmorphic and exhibited perseverative behaviors. His weight was 27.4 kg (10th percentile), height 128.1 cm (2nd percentile), and head circumference 49 cm (−3.22SD 0th percentile). He had brachycephaly and microcephaly. His mid-face was flat with a broad nasal tip and prominent nasal bridge. He had prominent epicanthal folds and mild ptosis bilaterally status post bilateral frontalis sling procedures (Figure 1). Ophthalmic examination revealed microphthalmia, microcornea, mild optic nerve hypoplasia, and mild myopia bilaterally. He had short fingers with clinodactyly of the fifth digit and flexion of the proximal and distal joints. He had mild right foot inversion and hindfoot varus, and pes planus. His left foot appeared to have less inversion and hindfoot varus. His right foot had an overlapping second toe and mild 2–3 syndactyly. His left foot had third toe clinodactyly and 2–3 syndactyly. Neurological examination noted static encephalopathy that limited examination. He used two-three-word phrases and needed redirection to remain on task. Mild trunk hypotonia was present. Strength was preserved. His gait was “shuffling” without being ataxic; upper extremity testing revealed no dysmetria. Reflexes were symmetric, proportionate, and elicited without spread.
FIGURE 1.

Photographs of Patient 1 showing dysmorphic features including flat mid-face, a broad nasal tip, prominent nasal bridge, epicanthal folds, and mild ptosis bilaterally
The only notable abnormal laboratory findings were a microcytic anemia in conjunction with low serum iron and a low prealbumin level, all attributable to malnutrition with a specific deficiency in iron-enriched foods.
Renal sonography noted mild-to-moderate left hydronephrosis with a small right kidney but this and numerous previous studies did not confirm evidence of the renal cysts seen in prenatal imaging. There is no family history of renal disorders.
A thyroid ultrasound revealed a borderline enlarged thyroid gland; thyroid hormone studies showed dysregulated TSH secretion (less than 50% increase in nocturnal TSH secretion). Peripheral T3 and T4 values were normal. The consultant endocrinologist recommended against supplementation because the patient had no symptoms of hypothyroidism and advised annual monitoring of thyroid function.
The brain magnetic resonance imaging (MRI) study confirmed microcephaly with small optic nerves and an otherwise normal-appearing brain (Figure 2). The cerebellum and brainstem had normal appearance and proportional volume to the overall small brain. Myelination appeared normal. A magnetic resonance spectroscopy (MRS) study revealed no abnormalities in the basal ganglia, cortex, and brain stem. Electromyelogram (EMG) and Nerve Conduction Studies (NCS) were normal. There was historical concern that infrequent staring spells were potentially seizures, but appropriate response to stimulus during the behavior suggested a non-epileptic etiology. Several prior electroencephalogram (EEG) studies had revealed no epileptic discharges, and the EEG obtained during the NIH admission showed diffuse slowing without epileptiform abnormalities.
FIGURE 2.
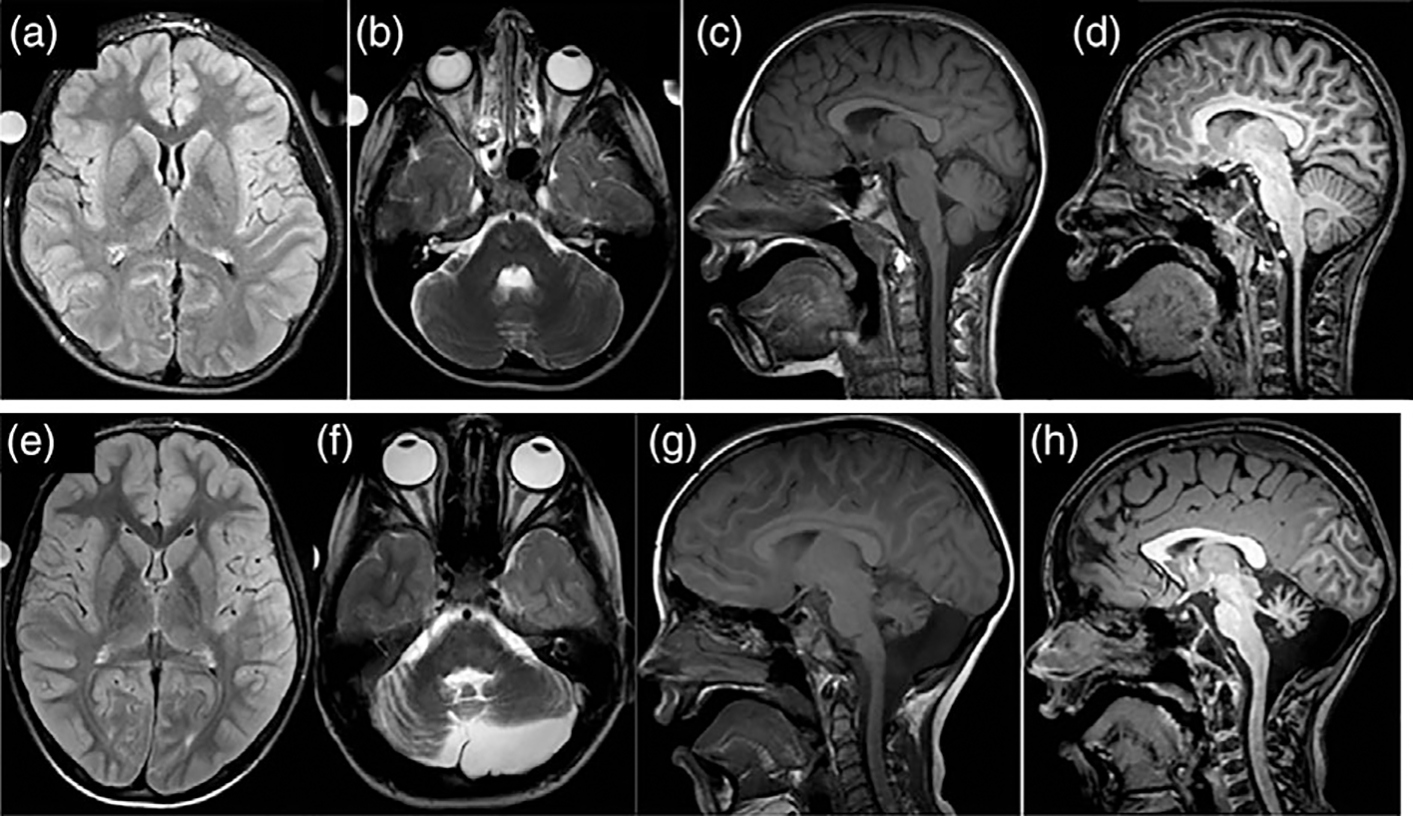
(a–d) MRI images from Patient 1 demonstrating microcephaly, small optic nerves bilaterally and otherwise normal neuroanatomy. (e–f ) MRI images from Patient 2 showing small volume cerebellum with left hemisphere more affected than right hemisphere
Neuropsychological testing occurred at age 12 years, 6 months with mostly short phrases as a primary mode of communication, augmented by gestures that included pointing, nodding of his head, and use of descriptive gestures. At evaluation, speech was marked by enunciation difficulties, including omitting the beginning sounds and final consonants in words, and he stammered when trying to use two-to three-word phrases. During the evaluation, he was mostly cooperative, socially interactive, and engaged. The Vineland Adaptive Behavior Skills, 2nd ed. (Vineland-II; Sparrow, Cicchetti, & Balla, 2005), is a caregiver report of adaptive behavior in everyday life. His adaptive behavior was significantly impaired in all areas, with gross motor skills being the most adversely affected domain (Table 1). Visual reception and fine motor skills were assessed using the Mullen Scales of Early Learning (MSEL; Mullen, 1995) and were scored at 50 months and 36 months, respectively. Using the MSEL, both his nonverbal and verbal Developmental Quotient (DQ)s, derived from age equivalents divided by chronological age and multiplied by 100, averaged around 30 (with the mean of the average range set at a value of 100). Age equivalent skills for receptive vocabulary tested with gesturing on the Peabody Picture Vocabulary Test (PPVT) indicated comprehension for single words at 5.6 years. This profile was indicative of a diagnosis of Intellectual Disability in the severe range.
TABLE 1.
Patient neuropsychological characteristics
| Patient 1 | Patient 2 | |
|---|---|---|
| Chronological age (year) | 12.5 | 7.2 |
| DQ | 30 | 25.71 |
| NVDQ | 28.67 | 25 |
| VDQ | 31.33 | 24.42 |
| Nonverbal mental age (month) | 43 | 21.5 |
| Verbal mental age (month) | 47 | 21 |
| Visual reception age equivalent (month) a | 50 | 27 |
| Fine motor age equivalent (month) a | 36 | 16 |
| Communication SS b | 54 | 67 |
| Daily living skills SS b | 58 | 66 |
| Socialization SS b | 55 | 68 |
| Adaptive behavior composite SS b | 59 | 66 |
| Adaptive gross motor age equivalent (month) b | 33 | 21 |
| Adaptive fine motor age equivalent (month) b | 29 | 32 |
| Receptive vocabulary (month)c | 67 | 44 |
Abbreviations: SS = standard score (mean = 100, SD = 15); DQ, developmental quotient; NVDQ, nonverbal developmental quotient; VDQ, verbal developmental quotient.
Measured using the Mullen Scales of Early Learning.
Measured using the Vineland Adaptive Behavior Scales, 2nd ed.
Measured using the Peabody Picture Vocabulary Test, 4th ed.
3.1.2 |. Patient 2
Patient 2 was a 7-year 2-month old Caucasian male who presented to the NIH UDP with ataxia, global developmental delay, small cerebellum, hypotonia, and hypothyroidism. He had two healthy siblings in a non-consanguineous family of maternal Germanic and paternal Irish ancestry. Following an uncomplicated term pregnancy, at birth he had Apgar scores of 4 at 1 min (with deductions for being diffusely pale, limp, and having bradycardia,) 7 at 5 min, and 9 at 10 min. Birth weight was 3,317 g (40th percentile), length 49.5 cm (25th percentile), and head circumference 32.5 cm (1st percentile).
He had postnatal feeding difficulties including excessive oral secretions and vomiting that improved with an elemental formula. At age 6 months, the patient could not hold his head erect, and when placed prone he could not support his weight using his arms. He also had a paucity of vocalizations and did not imitate sounds despite passing a newborn hearing examination. Early Intervention services at age 6 months detected low tone, language delay, and poor swallowing coordination; therapy yielded developmental progress. He spoke his first specific word at 3 years (“mama”) and used gestures to indicate being finished or indicate he wanted more at mealtimes. He began to use single words reliably at 36 months; he currently uses up to 20 single recognizable words. He sat without support at 11 months, crawled at 16 months, and walked unaided at 30 months. His gait was notably ataxic, and his family reported persistent instability with frequent falls especially on inclined surfaces; thus, he was prescribed a wheeled walker for use when outside the home. His ambulation remained stably ataxic. He has never had seizures.
At age 15 months, the thyroid stimulating hormone (TSH) level was elevated, with a normal T4 level; thyroid hormone supplementation normalized the serum TSH.
On evaluation, he was not dysmorphic, his head was normocephalic, and he resembled his biological parents. His height was 112 cm (1st percentile), weight 22.2 kg (26th percentile), and head circumference 52 cm (35th percentile). His general physical examination was remarkable for dental crowding and a fractured incisor. He was hyperkinetic with poor attention, meeting diagnostic criteria for Attention-Deficit Hyperactivity Disorder not otherwise specified. Oculomotor exam showed saccadic intrusions into pursuit, hypermetric saccades, and failure of suppression of the vestibular ocular reflex cancellation test. Eyes were quiet in primary position, there was no gaze-evoked nystagmus, and he had preserved oculokinetic nystagmus. His speech was severely dyspraxic and dysarthric. He attempted to use hand gestures or signs to augment his single word utterances with limited success. He had a hyperactive gag reflex, and sialorrhea was noted. Tone was decreased throughout, strength largely preserved except for poor trunk control attributed to mild core muscle weakness. Finger to nose testing produced end-point dysmetria and oscillation at the elbows. Heel to shin testing showed slowing and jerking in the plane. He had truncal titubation with standing, gait was wide based and mildly unstable with lurching, extra steps to turn, and frequent loss of balance without external support. Deep tendon reflexes were normal and the plantar responses both flexor. Sensation was normal.
Laboratory studies, including cerebrospinal fluid evaluations of neurotransmitters, were normal. Brain MRI obtained at 23 months demonstrated a small pons and cerebellum with left cerebellar hemisphere smaller than the right, mildly progressive on repeat MRI at 29 months of age (Figure 2). There was also suggestion of a stable left posterior fossa arachnoid cyst. The cerebral hemispheres were morphologically normal, and myelination patterns were normal. At age 7 and 11, follow-up brain MRI scans revealed stable global cerebellar and pontine volume loss, and a MRS study revealed elevated creatine in the pons and decreased N-acetyl-aspartate in the superior cerebellar vermis. An EEG revealed “a poorly organized background rhythm that was diffusely slow” and “no definitive epileptiform discharges.” EMG and NCS were normal.
On neuropsychological assessment, Patient 2’s primary method of communication was gesturing, which included pointing and specific signs; he less frequently used word approximations and some single intelligible words. He was unable to write or draw. He was generally happy and social, with a strong desire to share interests. His adaptive behavior on the Vineland test (Sparrow et al., 2005) was significantly impaired in all areas, most prominently in gross motor skills (Table 1). Visual reception and fine motor skills, assessed through the MSEL (Mullen, 1995), were 27 months and 16 months, respectively, with calculated verbal and nonverbal DQs around 25 months, with an average score of 100. The PPVT, which requires very few motor demands (pointing only), was used to evaluate his receptive vocabulary for single words and revealed his most advanced achievement with comprehension approximately equivalent to that of a four-year-old. Patient 2’s cognitive profile indicated a diagnosis of Intellectual Disability in the severe range, in addition to Attention Deficit Hyperactivity Disorder.
3.2 |. Genetic studies
Both patients underwent Comprehensive Genomic Hybridization (CGH) arrays, mitochondrial DNA sequencing analysis, and whole exome sequencing. The CGH array and mitochondrial DNA sequencing tests did not identify pathological deletions or duplications for either patient. Though Patient 2 had a 667 kilobase duplication of chromosome 2p22.3, this duplication was also present in the patient’s healthy biological father. A comprehensive mitochondrial DNA analysis of Patient 2 showed 79% heteroplasmy for an uncommon variant in ATP6 (m.9025G>A, p.G167F) which was interpreted as a polymorphism. This variant does not have a known associated phenotype. Fragile X testing and POLG sequencing were normal for both patients.
WES revealed de novo heterozygous SPTAN1 variants in both patients that met criteria for being potentially disease causing. Sanger sequencing by a CLIA certified laboratory confirmed the next generation sequencing findings of de novo heterozygous variants in SPTAN1 for each of these two patients. Patient 1 had a single nucleotide change (NM_001130438.2; [2950C>T]) in exon 21 of SPTAN1 that leads to a premature stop codon (NP_001123910.1: [R984X]) within the SH3 domain of the alpha-II-spectrin protein. Patient 2 had an intronic missense variant (NM_001130438.2: [6690–17G>A]) that creates a premature splice acceptor site before exon 50 and is predicted to result in the translation of 15 formerly intronic nucleotide bases into five amino acids added into the final protein (NP_001123910.1: [Arg2229_Thr2230insSerAlaLeuHisArg]). This specific intronic nucleotide is highly conserved through many species (Table 2). SCA8 testing of the ATXN8OS/ATXN8 locus for Patient 2 revealed a finding of 978 CTG/CAG repeats, with expansions larger than 250 repeats considered pathogenic (Ayhan, Ikeda, Dalton, Day, & Ranum, 2014).
TABLE 2.
Conservation of G nucleotide at chromosome 9: Position 128,627,908 across multiple species
| Patient 2 | CTTAG-A---- |
|---|---|
| Human (reference) | CTTGG-A---- |
| Chimp | CTTGG-A---- |
| Gorilla | CTTGG-A---- |
| Mouse | CTTGG-A---- |
| Rat | CTTGG-A---- |
| Rabbit | CTTGG-A---- |
| Pig | CTTGG-A---- |
| Cow | CTTGG-A---- |
| Sheep | CTTGG-A---- |
| Dog | CTTGG-A---- |
| Pigeon | TTTGG-A---- |
| Chicken | TTTGG-A---- |
| Turkey | TTTGG-A---- |
| Lizard | TTTGG-G---- |
| Zebrafish | CTTGGGA---- |
3.3 |. Cellular studies
Fibroblasts from these patients with SPTAN1 variants were morphologically similar to those from control patients and did not have visible differences in spectrin or actin staining patterns (Figure 3). Evaluation of fibroblast SPTAN1 expression using ddPCR for both patients revealed a significant reduction in all wild type sequence RNAs for both patients relative to control fibroblast expression levels (Figure 3). Probes specific for each patient’s variant (located at exon 21 and exon 50, respectively) revealed small but consistently measurable quantities of variant sequences that were not seen in control fibroblasts. A Western blot comparison of the quantity of alpha-II-spectrin protein in fibroblasts showed that fibroblasts from Patient 1 had a significantly reduced amount of protein relative to all control patients (Figure 3).
FIGURE 3.
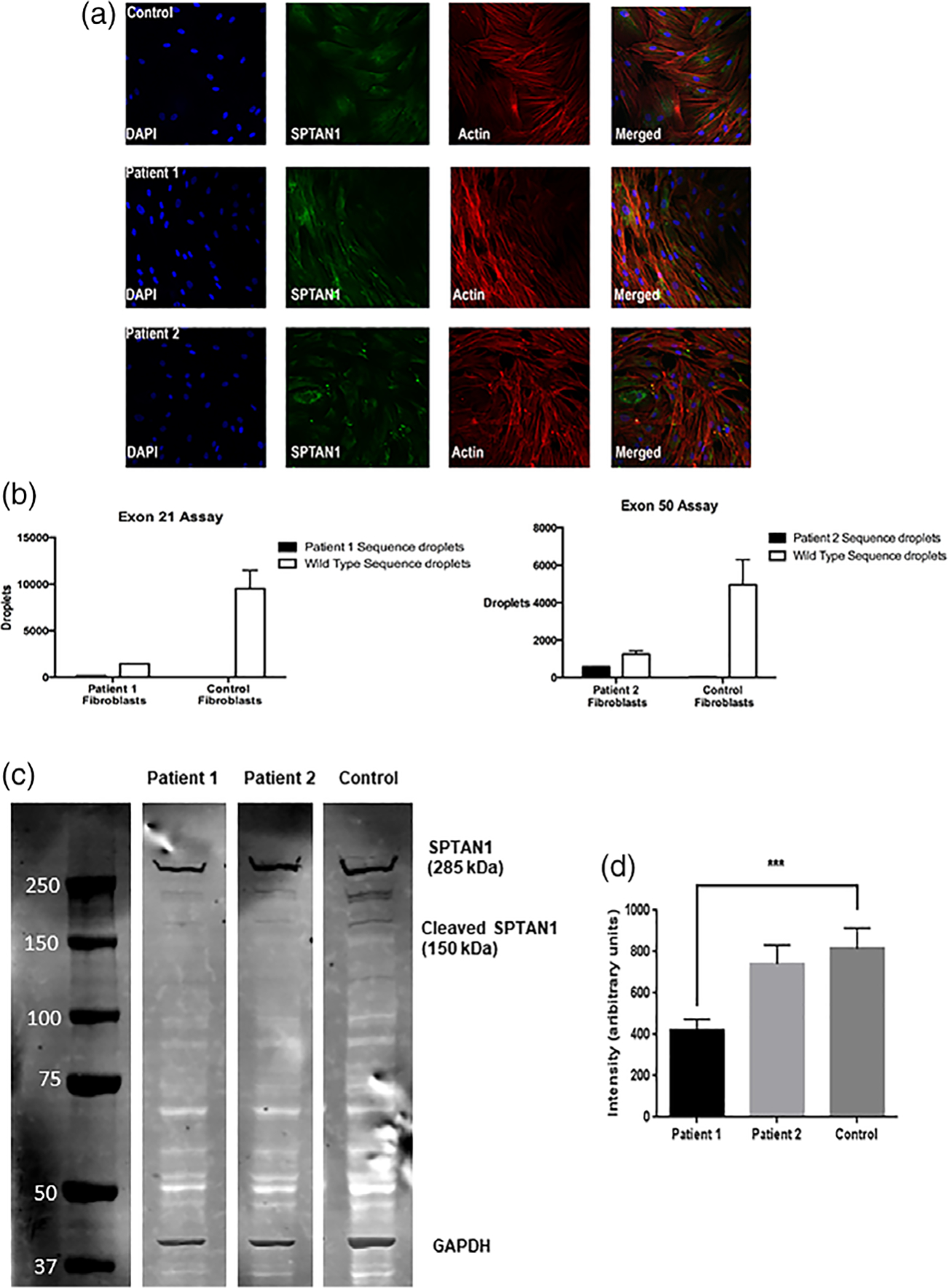
(a) Immunocytochemistry staining of fibroblasts from Patients 1 and 2 and a control patient. There were no gross morphologic distinctions between patients and control in SPTAN1 amount or distribution. DAPI stains nuclei; actin stains actin filaments. (b) ddPCR using probes specific for the variant sequences of Patients 1 and 2 demonstrated reduced expression of healthy RNA sequences and detectible expression of both patients’ variant sequences. (c) Western blot assay showing detection of alpha-II spectrin protein in both patients’ and control fibroblasts. SPTAN1 can be seen at ~285 kDa and a fainter band corresponding to the major splice isoform of SPTAN1 is present at 150 kDa. An additional band was expected at ~140 kDa for the truncated SPTAN1 protein for Patient 1 but was not seen. (d) The intensity of the 285 kDa band for Patient 1 was decreased by over 25% relative to the 285 kDa band of a control patient in an average across three Western blots (p < .001)
4 |. DISCUSSION
We report two previously unpublished patients with de novo monoallelic variants in SPTAN1 resulting in multiple neurological symptoms without seizures. These two patients have features not previously reported in patients with pathogenic variants in SPTAN1 and suggest an expansion of the clinical phenotype.
Per the public domain GTEx database, the highest levels of expression for SPTAN1 occur within the cerebellar hemispheres, the cerebellum, and the cerebral cortex (Lonsdale et al., 2013). It is therefore not surprising that the motor and cognitive functions assigned to these structures are impaired in patients with pathogenic changes of the SPTAN1 gene. Gross and fine motor functions were disproportionately delayed for both patients, although the older child (Patient 2) made greater progress in language development relative to a stagnant motor performance. Both patients showed relatively stronger receptive language (comprehension) compared to expressive language; this discrepancy is likely related to significant oral motor impairments and cerebellum-specific linguistic difficulties as discussed below.
Patient 1 has a premature stop codon within the SH3 domain of SPTAN1, and his primary symptoms are intellectual disability, global developmental delay, microcephaly, and dysmorphic features. The expected consequence of this variant would be haploinsufficiency via a reduction in RNA and protein. The ddPCR assessment of this patient’s fibroblast RNA shows low expression of both the variant and normal allele. Expression of the normal allele RNA is significantly reduced compared with that of healthy control fibroblasts and appears well below a predicted 50% reduction. The alpha-II-spectrin protein content of fibroblasts from Patient 1 is reduced, confirming that haploinsufficiency is present. We do not expect the shortened RNA from the variant allele to be translated and did not see bands at the molecular weight (~150 kD) predicted for the truncated alpha-II-spectrin protein in any Western blot assay performed on Patient 1’s fibroblasts.
The phenotype of Patient 1 is a mixture of what has been observed in other patients with SPTAN1 variants. Patient 1 has microcephaly, developmental delay, and ocular abnormalities, features in common with most of the patients who have any pathogenic variants in SPTAN1, but he lacks the infantile-onset epilepsy, significant volume loss in the cerebellum, and hypomyelination that are typically present in patients who have deletions within SPTAN1 (Syrbe et al., 2017). We posit that the phenotype of Patient 1 represents the consequences of isolated SPTAN1 haploinsufficiency and hypothesize that epilepsy, hypomyelination, and cerebellar/brainstem abnormalities require the presence of a SPTAN1 variant near the C-terminus and translation of a protein with a disruption in or near the spectrin dimerization site.
Patient 2 has findings typical (small cerebellum, motor delays) and atypical (cognitive behavioral abnormalities, lack of epilepsy) for the SPTAN1 encephalopathy described previously in patients with variants near the C-terminus. The variant in Patient 2 is predicted to add five amino acids to the alpha-II-spectrin protein with an undetermined impact on protein structure. We hypothesize that this small intronic change alters cerebellar anatomy but fails to recapitulate the epilepsy of SPTAN1 epileptic encephalopathy. The uniqueness of this variant and the patient’s phenotype provides a potential window into the significant cerebellar contribution to SPTAN1 encephalopathy. His poor impulse control, deficient attention, and linguistic processing deficits are consistent with the Cerebellar Cognitive Affective Syndrome (Schmahmann & Sherman, 1998; Schmahmann, Weilburg, & Sherman, 2007) making this the first reported patient with SPTAN1 encephalopathy to have this additional clinical diagnosis.
In addition to the SPTAN1 variant, Patient 2 has two genetic findings that could be associated with motor symptoms, a polymorphism in ATP6 and a potentially pathogenic expansion at the spinocerebellar ataxia type 8 (SCA8) locus, ATXN8OS/ATXN8.
It is unlikely that the ATP6 variant is causing any of the symptoms noted in Patient 2. Reports of patients with pathogenic ATP6 variants describe a mitochondrial disease phenotype akin to Leber’s hereditary optic neuropathy such as lactic acidosis, striatal necrosis, and progressive blindness or deafness. Patient 2 has normal serum and cerebrospinal fluid lactate levels, normal striatal appearance, normal hearing, and normal vision making a mitochondrial disease doubtful as a unifying diagnosis.
Although pathogenic ATXN8OS/ATXN8 expansions typically present as an adult-onset neurodegenerative ataxia (i.e., SCA8), one report suggests that ATXN8OS/ATXN8 expansions can potentiate childhood-onset ataxia symptoms due to other genetic causes (Ohnari, Aoki, Uozumi, & Tsuji, 2008). Hence, it is possible that this ATXN8OS/ATXN8 expansion interacts with the SPTAN1 encephalopathy in Patient 2 and is potentiating this patient’s pronounced early onset ataxia. The patient’s confirmed biological parents are healthy adults and presumably do not have a repeat expansion in the pathogenic range.
We do not believe the ATXN8OS/ATXN8 variant alone accounts for the phenotype of Patient 2. Expansions in the SCA8 locus cause an adult-onset progressive degenerative cerebellar ataxia not an infantile onset static encephalopathy. Furthermore, attempts using the CRISPR/Cas 9 technique to introduce Patient 2’s unique SPTAN1 variant into a mouse model were successful. Unlike other mice heterozygous for SPTAN1 variants that survived to maturity and appeared phenotypically normal (Stankewich et al., 2011), mice with copies of Patient 2’s SPTAN1 variant were born in the expected ratio (1:2:1) and appeared anatomically normal but were tremulous and died before weaning, regardless of whether they were heterozygous or homozygous for the variant (data not shown). Thus, we believe Patient 2’s SPTAN1 variant is pathogenic, and the patient’s cerebellar symptoms might be augmented by the expansion in ATXN8OS/ATXN8.
The unique nature of Patient 2’s variant provides support to the hypothesis that C-terminal variants always cause cerebellar volume reduction whereas the pathophysiology of SPTAN1 epilepsy is a complex outcome of the variant location and resultant change in the protein. Data obtained in mouse cortical neurons suggest that some variants in the C-terminal region of SPTAN1, which is essential to forming a heterodimer with beta-spectrin, yield protein aggregates (Saitsu et al., 2010). A recent publication supports this hypothesis of a phenotypic spectrum attributable to aggregation. Syrbe et al. (2017) reported that aggregate formation occurs only when variants are situated within the heterodimer formation region, and that these variants are associated with the most severe phenotypes that include epilepsy. This report provided phenotypes of several patients with deletions and duplications near the locus of Patient 2’s missense variant and all had small cerebellar volumes.
We propose introducing the known C-terminal SPTAN1 variants into cultured human cortical neurons to determine if aggregation is a consequence of SPTAN1 variants in the C-terminus. If other pathogenic C-terminal variants cause cortical aggregates but the variant of Patient 2 does not, that would support the hypothesis that aggregate formation is a pathogenic contributor to epileptogenesis in SPTAN1 encephalopathy, while any C-terminal variant could be sufficient to cause cerebellar dysgenesis.
Fibroblasts from Patient 2 similarly exhibit a deficiency in wild type SPTAN1 RNA as measured by ddPCR, although levels of alpha-II-spectrin protein appeared equivalent to those of normal control cells. As the antibody we used should recognize both the variant and wild type spectrin proteins, it is possible there is a significant reduction in wild type protein quantity that cannot be visualized using this antibody. Attempts to create an antibody capable of recognizing just Patient 2’s variant spectrin were unsuccessful; quantification of the relative content of the heathy and variant alpha-II-spectrins within Patient 2’s cells will require further investigation. Fibroblasts may not be the optimal target for investigations because they produce far less alpha-II-spectrin than neurons (Lonsdale et al., 2013), and thus cultured neurons might represent a more robust and mechanistically appropriate source for future protein assessments.
Both Patients 1 and 2 had evidence of altered thyroid function, a finding not previously reported in patients with SPTAN1 variants. The thyroid is enriched with respect to non-erythrocytic spectrin (Ishimura et al., 1987). Thyroid alpha-II-spectrin appears to be critical for the exocytosis of thyroid hormone in response to thyroid-stimulating hormone (Gabrion et al., 1990). Thus, alpha-II spectrin deficiency could result in impaired release of thyroid hormone, and patients with SPTAN1 variants should have routine hypothyroidism screening. Further investigation of thyroid hormone secretion and thyroid alpha-II-spectrin protein content in patients with SPTAN1 encephalopathy would be of great utility especially considering how crucial thyroid hormone is to neurodevelopment.
In conclusion, we present two patients with novel SPTAN1 variants who exhibit potentially important differences from other published cases and expand the SPTAN1 encephalopathy phenotype. The two patients have clinical features that overlap with prior descriptions of patients with SPTAN1 variants but may reveal isolated aspects of the SPTAN1 encephalopathy syndrome, specifically, haploinsufficiency and C-terminal disruption without aggregation. The absence of seizures in these two patients provides confirmation that pathogenic SPTAN1 variants do not always cause epilepsy. Both patients have abnormal thyroid hormone secretion, a finding not appreciated in any prior published patients. One of the patients meets criteria for Cerebellar Cognitive Affective Syndrome, and this diagnosis should be considered in other patients with SPTAN1 variants. These patients’ presentations expand the known spectrum of clinical findings present in SPTAN1 encephalopathy and provide evidence for two independent spectrin-related pathologies, hypoplasia and dysfunction, in the cerebellum and thyroid gland, two organs highly dependent on robust levels of normal alpha-II-spectrin.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the patients and their families for participating in this research. The authors thank Andrea De Biase for extensive technical assistance with the RainDance ddPCR assays. The authors thank Gretchen Golas for the expert clinical care she provided to both patients. These studies were funded by the intramural program of the National Human Genome Research Institute of the National Institutes of Health and by the Undiagnosed Diseases Network.
Footnotes
CONFLICTS OF INTEREST
The authors report no relevant conflicts of interest.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of the article.
REFERENCES
- Ayhan F, Ikeda Y, Dalton JC, Day JW, Ranum LPW. 2014. Spinocerebellar Ataxia Type 8. GeneReviews. Retrieved from https://www.ncbi.nlm.nih.gov/books/NBK1268/
- Bignone PA, & Baines AJ (2003). Spectrin alpha II and beta II isoforms interact with high affinity at the tetramerization site. Biochemical Journal, 374(Pt. 3), 613–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böckers TM, Mameza MG, Kreutz MR, Bockmann J, Weise C, Buck F, … Kreienkamp HJ (2001). Synaptic scaffolding proteins in rat brain. Ankyrin repeats of the multidomain shank protein family interact with the cytoskeletal protein alpha-fodrin. Journal of Biological Chemistry, 276(43), 40104–40112. [DOI] [PubMed] [Google Scholar]
- Campbell IM, Yatsenko SA, Hixson P, Reimschisel T, Thomas M, Wilson W, … Scaglia F (2012). Novel 9q34.11 gene deletions encompassing combinations of four Mendelian disease genes: STXBP1, SPTAN1, ENG, and TOR1A. Genetics in Medicine, 14(10), 868–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrion JB, Barrière H, Nguyen Than Dao B, Chambard M, Mauchamp J, Regnouf F, & Pradel LA (1990). Absence of fodrin (spectrin-like protein) under the pseudopod membrane in stimulated thyroid cells. European Journal of Cell Biology, 52(2), 282–290. [PubMed] [Google Scholar]
- Gahl WA, Markello TC, Toro C, Fuentes Fajardo K, Sincan M, Gill F, … Adams D (2012). The NIH undiagnosed diseases program: Insights into rare diseases. Genetics in Medicine, 14(1), 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahl WA, Mulvihill JJ, Toro C, Markello TC, Wise AL, Ramoni RB, … UDN. (2016). The NIH Undiagnosed Diseases Program And Network: Applications to modern medicine. Molecular Genetics and Metabolism, 117(4), 393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Zhou R, Wu Z, Carrasco MA, Kurshan PT, Farley JE, … Zhuang X (2016). Prevalent presence of periodic actin-spectrin-based membrane skeleton in a broad range of neuronal cell types and animal species. Proceedings of the National Academy of Sciences of the USA, 113(21), 6029–6034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimura K, Senda T, Kitajima K, Fujita H, Fujio Y, & Sobue K (1987). Immunocytochemical localization of calspectin (a non-erythroid spectrin-like protein) in thyroid glands of normal and TSH-treated rats. Histochemistry, 86(6), 537–539. [DOI] [PubMed] [Google Scholar]
- Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, … for the GTEx Consortium. (2013). The Genotype-Tissue Expression (GTEx) project. Nature Genetics, 45(6), 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markello TC, Han T, Carlson-Donohoe H, Ahaghotu C, Harper U, Jones M, … Boerkoel CF (2012). Recombination mapping using Boolean logic and high-density SNP genotyping for exome sequence filtering. Molecular Genetics and Metabolism, 105(3), 382–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen EM (1995). Mullen scales of early learning. Circle Pines, MN: American Guidance Service. [Google Scholar]
- Nakano M, Nogami S, Sato S, Terano A, & Shirataki H (2001). Interaction of syntaxin with alpha-fodrin, a major component of the submembranous cytoskeleton. Biochemical and Biophysical Research Communications, 288(2), 468–475. [DOI] [PubMed] [Google Scholar]
- Nicita F, Ulgiati F, Bernardini L, Garone G, Papetti L, Novelli A, & Spalice A (2015). Early myoclonic encephalopathy in 9q33-q34 deletion encompassing STXBP1 and SPTAN1. Annals of Human Genetics, 79(3), 209–217. [DOI] [PubMed] [Google Scholar]
- Ohnari K, Aoki M, Uozumi T, & Tsuji S (2008). Severe symptoms of 16q-ADCA coexisting with SCA8 repeat expansion. Journal of Neurological Sciences, 273(1–2), 15–18. [DOI] [PubMed] [Google Scholar]
- Saitsu H, Tohyama J, Kumada T, Egawa K, Hamada K, Okada I, … Matsumoto N (2010). Dominant-negative mutations in alpha-II spectrin cause West syndrome with severe cerebral hypomyelination, spastic quadriplegia, and developmental delay. American Journal of Human Genetics, 86, 881–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmahmann JD, & Sherman JC (1998). The cerebellar cognitive affective syndrome. Brain, 121, 561–579. [DOI] [PubMed] [Google Scholar]
- Schmahmann JD, Weilburg JB, & Sherman JC (2007). The neuropsychiatry of the cerebellum – Insights from the clinic. The Cerebellum, 6, 254–267. [DOI] [PubMed] [Google Scholar]
- Sparrow SS, Cicchetti VD, & Balla AD (2005). Vineland adaptive behavior scales (2nd ed.). Circle Pines, MN: American Guidance Service. [Google Scholar]
- Stankewich MC, Cianci CD, Stabach PR, Ji L, Nath A, & Morrow JS (2011). Cell organization, growth, and neural and cardiac development require αII-spectrin. Journal of Cell Science, 124(23), 3956–3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syrbe S, Harms FL, Parrini E, Montomoli M, Mütze U, Helbig KL, … Guerini R (2017). Delineating SPTAN1 associated phenotypes: From isolated epilepsy to encephalopathy with progressive brain atrophy. Brain, 140(9), 2322–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohyama J, Nakashima M, Nabatame S, Gaik-Siew C, Miyata R, Rener-Primec Z, … Saitsu H (2015). SPTAN1 encephalopathy: Distinct phenotypes and genotypes. Journal of Human Genetics, 60(4), 167–173. [DOI] [PubMed] [Google Scholar]
- Weigand JE, Boeckel JN, Gellert P, & Dimmeler S (2012). Hypoxia-induced alternative splicing in endothelial cells. PLoS One, 7(8), e42697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Resneck WG, Lee PC, Randall WR, Bloch RJ, & Ursitti JA (2010). Characterization and expression of a heart-selective alternatively spliced variant of alpha II-spectrin, cardi+, during development in the rat. Journal of Molecular and Cellular Cardiology, 48(6), 1050–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.