

Abstract
In 2005, a Japanese epidemiological study showed that increase in plasma glucose levels is a risk factor for gastric cancer. However, no animal model has hitherto shown any association between diabetes mellitus and neoplasia in the stomach. Diabetic (db/db) mice have obese and diabetic phenotypes, including hyperglycemia, because of disruption of the leptin receptor. In the present study, effects of hyperglycemia and/or hyperinsulinemia on the development of proliferative lesions were therefore examined in db/db mice given N‐methyl‐N‐nitrosourea (MNU). A total of 120 mice were assigned to four groups: Group A, 40 db/db mice with MNU; Group B, 40 + /db mice with MNU; Group C, 30 misty (wild‐type) mice with MNU; Group D, 10 db/db mice without MNU. MNU was given at 60 ppm in drinking water for 20 weeks. Subgroups of animals were sacrificed at weeks 21 and 30 and blood samples were collected to measure glucose, insulin, leptin, and adiponectin concentrations. The removed stomachs were fixed in formalin, and embedded in paraffin for histological examination and immunohistochemistry. At week 30 in Groups A, B, C and D, hyperplasia was observed in 100, 79, 57, and 0%, and dysplasia in 91, 43, 71, and 0%, respectively. Adenocarcinomas and pepsinogen‐altered pyloric glands (PAPG), putative preneoplastic lesions, were observed only in Group A, at an incidence of 45%. The serum levels of insulin and leptin were also elevated in Group A. Gastric carcinogenesis by MNU was enhanced in db/db mice, possibly in association with hyperinsulinemia and hyperleptinemia. (Cancer Sci 2009; 100: 1180–1185)
As in almost all countries in the world, type 2 diabetes mellitus (DM) has become a serious public health problem in Japan. Associations between DM and malignant neoplasms have been discussed for many years and several prospective cohort and case‐control studies have provided evidence of positive links.( 1 , 2 , 3 , 4 ) Recently, a Japanese prospective study showed high hazard ratios for cancers of the liver, pancreas, kidney, colon, ovaries and stomach.( 5 )
The Japanese population is characterized by a high morbidity from gastric cancer. In a prospective epidemiological study about gastric cancer and DM, described in 2005, Yamagata et al. surveyed 2466 cases, and concluded a modest increase in fasting plasma glucose levels to be a risk factor for gastric cancer among Helicobacter pylori‐seropositive subjects.( 6 ) Furthermore, insulin can stimulate insulin‐like growth factor 1 receptor (IGF‐1R),( 7 ) and leptin induces gastric cancer cell proliferation,( 8 , 9 ) while adiponectin inhibits the growth and peritoneal metastasis of gastric cancer.( 10 ) However, corroboratory experimental evidence is limited and no animal model has hitherto shown any association between DM and gastric cancer. In the present study we investigated whether hyperglycemia and/or hyperinsulinemia are risk factors for gastric cancer using genetically predisposed diabetic (db/db) mice treated with N‐methyl‐N‐nitrosourea (MNU), a carcinogen well established to induce adenocarcinomas in the stomachs of C3H mice, BALB/c mice, and Mongolian gerbils.( 11 , 12 , 13 ) db/db mice are of the C57BL strain( 14 ) and show symptoms similar to those of insulin‐resistant DM, such as severe obesity, hyperglycemia, hyperinsulinemia, and insulin resistance.( 15 ) db/db mice have a defective leptin receptor: Ob‐R.( 16 ) A point mutation in the Lepr gene causes abnormal splicing, leading to premature termination of the intracellular domain of the Ob‐Rb splice form and loss of its signal transducing function. Because of a deficiency in leptin‐mediated satiety signaling, hyperphagia occurs in homozygous db/db mice, resulting in the phenotypes stated above.( 17 ) db/db mice have two types of littermates: +/db, which is the heterozygote, and misty, whose chromosomes both have a normal Lepr gene. Heterozygotes (+/db) have normal body weight, blood glucose, and plasma insulin but increased metabolic efficiency.( 18 ) We therefore also investigated whether the occurrence of neoplastic lesions is associated with the serum levels of insulin, leptin and adiponectin. The results demonstrated the frequency of dysplasia and carcinoma to be significantly elevated with serum levels of insulin and leptin in db/db mice receiving MNU. Although leptin in DM patients is not as high as in db/db mice, hyperinsulinemia can be a risk factor for gastric cancer in DM patients.
Materials and Methods
Animals. Five‐week‐old specific pathogen‐free male db/db mice (BKS.Cg‐m +/+ Leprdb/J; mice homozygous for the mutation of Lepr gene), + /db mice (heterozygotes) and misty mice (wild‐type with a normal Lepr gene) (Charles River Laboratories Japan, Inc., Yokohama, Japan) were housed in plastic cages on hardwood‐chip bedding in an air‐conditioned biohazard room with a 12:12 L : D cycle. They were given food (Oriental CRF‐1, Oriental Yeast Co., Ltd, Tokyo, Japan) and autoclaved distilled water ad libitum. Body weights and water intake were measured once a week. Animal experiments were performed according to the Laboratory Animal Guidelines of the University of Tokyo.
Chemical. MNU (Sigma Chemical Co., St. Louis, MO, US) was dissolved in distilled water at a concentration of 60 ppm three times per week and administered as drinking water in light‐shielded bottles ad libitum for 20 weeks.
Study design. A total of 120 mice were divided into four groups: Group A, 40 db/db mice treated with MNU for 20 weeks; Group B, 40 +/db mice similarly treated with MNU; Group C, 30 misty mice similarly treated with MNU; Group D, 10 db/db mice not treated with MNU. The animals were then sacrificed at 27 or 36 weeks of age (weeks 21 or 30) under anesthesia. Prior to sacrifice, mice were fasted for 24 h but had access to water. Two hours before necropsy, BrdU (200 mg/kg body wieght) in saline was injected intraperitoneally. At killing, blood samples were collected and glucose, insulin, leptin, and adiponectin concentrations were measured by suspension array at Genetic Lab Co., Ltd. (Sapporo, Japan). Necropsies were performed on all animals which died. The experimental design was approved by the animal care committee of The University of Tokyo, and the animals were cared for in accordance with institutional guidelines.
Histological examination. Excised stomachs were fixed in neutral‐buffered 10% formalin and cut into approximately seven strips in parallel to the greater curvature, which were then embedded in paraffin and cut into 5 µM sections. Other organs were carefully checked with the naked eye and if they contained tumors, were fixed in 10% buffered formalin for 24 h and embedded in paraffin.
Replicate sections were stained with H&E and periodic acid Schiff (PAS) for histologic examination.
Immunohistochemistry. To identify proliferating S‐phase nuclei and malignant cells, the following primary antibodies were used to stain paraffin‐embedded sections: biotinylated murine monoclonal anti‐BrdU IgG (Zymed Laboratories Inc, San Francisco, CA, US) and rabbit polyclonal IgG anti‐mouse p53 (1:500; Novocastra, Newcastle, UK), respectively. To detect parietal cells and mucous neck cells, murine monoclonal IgG anti‐H+/K+‐exchanging ATPase (H/K‐ATPase) (1:2000; a gift from Dr Adam Smolka, Medical University of South Carolina, Charleston, SC, US) and murine monoclonal IgM anti‐human trefoil factor 2 (TFF2) (1:100; a gift from Dr Nicholas Wright, Cancer Research UK, London, UK) were applied. To evaluate the expression of pepsinogen, anti‐rat Pg serum (1:10 000) was employed.( 19 )
Immunohistochemical staining was performed with the avidin‐biotin complex (ABC) method (Vectastain Elite ABC kit, Vector Laboratories, Burlingame, CA, US). Immune complexes were visualized by incubation with 0.01% H2O2 and 0.05% 3,3′‐diaminobenzidine tetrachloride. For the immunostaining of BrdU, a kit (Zymed Laboratories Inc.) was used.
For all immunostaining, nuclear counterstaining was conducted using Mayer's hematoxylin.
Histological evaluation. The glandular mucosae of the corpus and antrum were examined histologically. The pathological criteria used were as follows: hyperplasia was defined as an increase in the thickness of glands by two‐fold or more with no nuclear or structural abnormalities. Cystically dilated glands were defined by an increase in the width of glands by two‐fold or more or the presence of cysts in the mucosa. We diagnosed dysplasia including adenomas and adenocarcinomas according to the Japanese classification of gastric carcinoma.( 20 ) To show the degree of fundic gland loss with oxyntic atrophy, we calculated the proportion of fundic glands as follows: in one strip of tissue, we measured the length of the glandular stomach located between the forestomach and duodenum; then using HK‐ATPase staining, we measured the length of the fundic gland area with positive parietal cells; with seven strips for each mouse, we added the seven lengths of the glandular stomach and the seven lengths of the fundic gland area to calculate the rate for fundic glands as an index of fundic gland loss.
Suspension array. The system uses a Mouse Serum Adipokine LINCOplex KIT and LINCOplex Mouse Adipokine Immunoassay kit (Millipore Inc, MA, USA). This assay is based on the Luminex xMAP technology and uses microsphere bead sets that are uniquely labeled with a mixture of fluorescent dyes. The capture antibodies specific for each analyte are covalently coupled to individual bead sets. At the time of the assay, a mixture of beads was incubated with 10 µL of standards or mouse serum samples in a 96‐well filter‐bottom plate overnight at 4°C. The next day, the beads were washed; a biotinylated detection antibody cocktail was added and it was incubated for 30 min at room temperature, followed by the addition of streptavidin‐phycoerythrin and incubation for another 30 min. After a final wash, the resuspended beads were read on a Luminex 100 reader and the concentration of each analyte in samples was determined based on individual standard curves.( 21 )
Statistics. All statistical analyses were performed using the chi‐square test and the Tukey‐Kramer's Honestly Significant Difference test with JMP (R) 6.0 (SAS Institute. Inc. NC, USA) software. A P‐value < 0.05 was considered significant.
Results
Animals. Total mortality for each group is shown in Table 1. Approximately 70–80% of the mice in each group survived until they were killed, and consequently 90 mice were investigated. In the MNU‐treated groups, hemorrhaging from hemangioendothelial sarcomas in the spleen was one of the main causes of death. We also found digestive bleeding, hydronephrosis from kidney tumors, and duodenal tumors including gastrointestinal stromal tumors.
Table 1.
Mortality within 30 weeks and N‐methyl‐N‐nitrosourea (MNU) intake
| Number of mice | Death | Total MNU intake mg/mouse | 5‐week MNU intake* (mg/body weight (g)) | |
|---|---|---|---|---|
| A: db/db + MNU | 40 | 10 (25%) | 19.3 | 0.09 |
| B: +/db + MNU | 40 | 10 (25%) | 16.8 | 0.217 |
| C: misty + MNU | 30 | 9 (30%) | 13.8 | 0.182 |
| D: db/db | 10 | 1 (10%) | 0 | 0 |
MNU intake between week 16 and week 20 per mouse by weight.
The changes in body weight of each group are shown in Fig. 1. Average body weights (g) were 25.5 (SD 1.4), 21.5 (SD 1.1), 16.3 (SD 0.96), and 29.2 (SD 1.3) at week 1, 56.3 (SD 4.5), 22.5 (SD 2.7), 18.8 (SD 2.5), and 36.5 (SD 4.0) at week 21, and 61.7 (SD 5.6), 23.2 (SD 3.2), 18.9 (SD 3.1), and 34.9 (SD 3.3) at week 30 in Groups A–D, respectively. The weights were significantly different between each group except between B and C.
Figure 1.
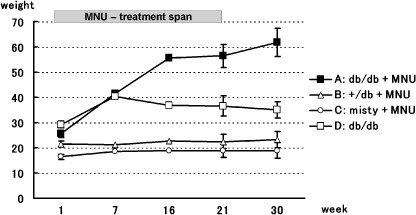
Changes in body weight in each group. The bars attached on each value show SD.
Total intake of MNU (mg) per mouse is shown in Table 1. As body weights of Group A increased until week 15 and stabilized between week 16 and week 20, we calculated MNU intake between week 16 and week 20 per mouse by weight. Although total intake of MNU per animal was slightly elevated in Group A, the amount of MNU per body weight was only half that in control mice.
Histological findings. Table 2 is a summary of histological findings. At week 21, cysts or dilated glands and hyperplasia were frequent in the groups treated with MNU (Fig. 2A,B). We observed two kinds of cells in hyperplastic areas: foveolar, recognized as an expansion of the PAS‐positive cell population located in the upper part of glands (Fig. 2G), and mucous cell, involving mostly TFF2‐positive cells located at the bases of glands (Fig. 2H). Parietal cells recognized as H/K‐ATPase‐positive were decreased or lacking in a hyperplastic area (Fig. 2F).
Table 2.
Histological findings
| Week | Group | Histological findings (number of mice) | ||||
|---|---|---|---|---|---|---|
| Survival | Cyst, dila* | Hyperplasia | Dysplasia | Adenocarcinoma | ||
| Week 21 | A: db/db + MNU | 19 | 15 (79%) | 10 (53%) | 7 (37%) | 0 |
| B: +/db + MNU | 16 | 13 (68%) | 8 (50%) | 1 (6%) ‡ | 0 | |
| C: misty + MNU | 12 | 8 (42%) | 5 (42%) | 1 (8%) ‡ | 0 | |
| D: db/db | 5 | 1 (5%) | 0 | 0 | 0 | |
| Week 30 | A: db/db + MNU | 11 | 11 | 11 | 10 (91%) | 5 (45%) |
| B: +/db + MNU | 14 | 11 (79%) | 11 (79%) | 6 (43%) § | 0 | |
| C: misty + MNU | 9 | 5 (56%) | 6 (67%) | 5 (56%) § | 0 | |
| D: db/db | 4 | 0 | 0 | 0 | 0 | |
Cyst or dilated glands.
At week 21, P < 0.01 compared with Group A.
At week 30, P < 0.01 compared with Group A.
Figure 2.
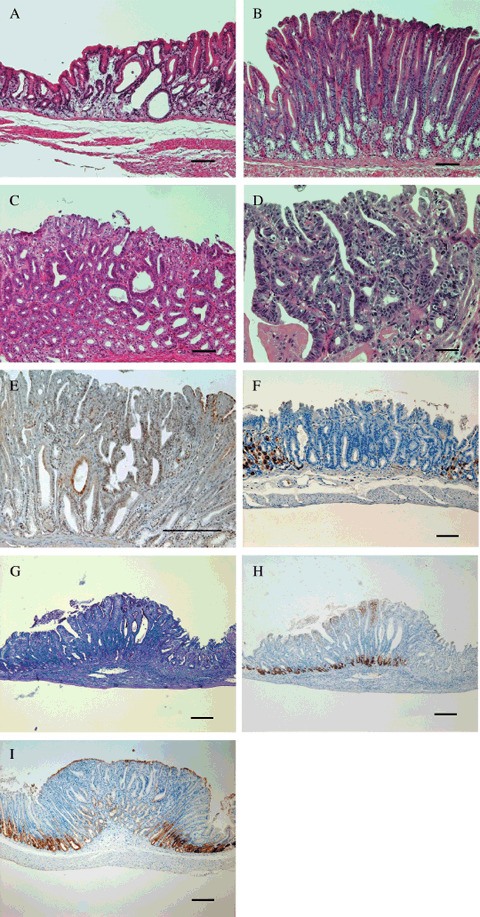
Cysts (a) or dilated glands (A) and hyperplasia (B) were frequent in the groups treated with N‐methyl‐N‐nitrosourea (MNU), dysplasia (C) being significantly increased in diabetic (db/db) mice treated with MNU. At week 30, adenocarcinomas were observed in 45% of db/db mice treated with MNU (D,E). H&E (d), p53 (E). Parietal cells recognized as H+/K+‐exchanging ATPase (H/K‐ATPase)‐positive were decreased or lacking in a hyperplastic area (F). Foveolar hyperplasia was recognized by a proliferation of periodic acid‐Schiff‐positive cells (G) and mucous cell hyperplasia, by mostly trefoil factor 2 (TFF2)‐positive cells (H). A tumor in which the expression of pepsinogen was decreased or lacking (I). Bar = 100 µM in (A–C, E–I), 20 µM in (D).
Hyperplasia was more widespread in Group A than the other groups. In mice with hyperplasia, the average number of hyperplasias per body was 3.2 (SD 1), 2.4 (SD 1.7), 2 (SD 1.4) at week 21, 4.6 (SD 1.5), 1.7 (SD 1), 2.8 (SD 1.4) at week 30 in Groups A–C, respectively. At week 30, average number of hyperplasias per body in Group A was significantly higher than those in Groups B and C (P < 0.05) (supplementary Table 1).
Moreover, the frequency of dysplasia was significantly higher in Group A (Fig. 2C, Table 2). At week 30, we could see the same trends for dilated glands, hyperplasia and dysplasia as at week 21. However, at week 30, we also observed adenocarcinoma in 45% of db/db mice treated with MNU (Fig. 2D,E). One lesion showed invasion into the lamina propria of the mucosa. The remaining four lesions were in situ. The expression of pepsinogen was decreased or disappeared in all of these lesions (Fig. 2I). The tumors were mostly found in the areas where hyperplasia was observed, as BrdU staining clearly revealed (Fig. 3C,D). Most instances of dysplasia were also found in hyperplastic areas. In the normal‐appearing fundic gland area, the average number of BrdU‐positive nuclei per gland was 1.9 (SD 0.93), 1.2 (SD 0.69), 0.95 (SD 0.34), 1.7 (SD 0.01) in Groups A–D, respectively. However, there was a lot of individual variation in the number of BrdU‐positive nuclei even in the same group. When compared with Group A, there was no significant difference between the groups, although we found the BrdU labeling of the misty control were weaker than in db/db mice (Fig. 3A,B).
Figure 3.
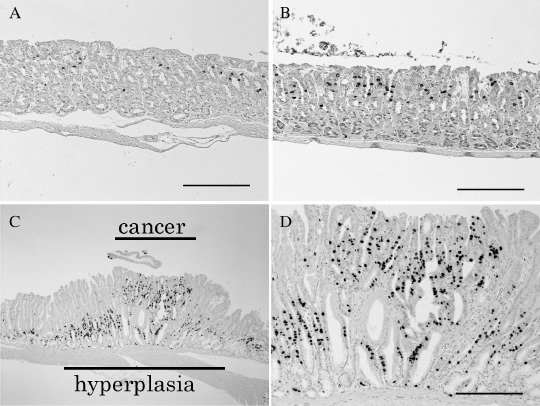
BrdU staining in the normal fundic gland of Group C (A), and Group A (B). In hyperplasia cases, BrdU staining showed an expansion of the proliferating S‐phase cell compartment (×40; C, ×100; D). Bar = 100 µM.
The fundic gland area was reduced in Group A because of ‘antralization’ where TFF2‐positive mucous metaplasia (spasmolytic polypeptide expressing metaplasia: SPEM) and oxyntic atrophy were observed. To show this phenomenon clearly, we calculated the index of fundic gland loss as mentioned above. The results are shown in Table 3. In Group A, we observed more oxyntic atrophy than in controls.
Table 3.
Index of fundic gland loss
| Week | Group | Number of mice | Mean † | SD |
|---|---|---|---|---|
| week 21 | A: db/db + MNU | 19 | 0.661 | 0.097 |
| B: +/db + MNU | 16 | 0.780 ‡ | 0.064 | |
| C: misty + MNU | 12 | 0.738 | 0.097 | |
| D: db/db | 5 | 0.811 ‡ | 0.026 | |
| Week 30 | A: db/db + MNU | 11 | 0.638 | 0.088 |
| B: +/db + MNU | 14 | 0.787 § | 0.053 | |
| C: misty + MNU | 9 | 0.748 § | 0.059 | |
| D: db/db | 4 | 0.817 § | 0.026 |
Mean of Index of fundic gland loss.
n = strip number (1–7).
An = the length of the fundic gland area.
Bn = the length of the glandular stomach, 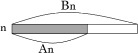 Index of fundic gland loss = (A1 + A2 + A3 + A4 + A5 + A6 + A7)/(B1 + B2 + B3 + B4 + B5 + B6 + B7).
Index of fundic gland loss = (A1 + A2 + A3 + A4 + A5 + A6 + A7)/(B1 + B2 + B3 + B4 + B5 + B6 + B7).
At week 21, P < 0.05 compared with Group A.
At week 30, P < 0.05 compared with Group A.
Blood tests. The results of blood testing are shown in Table 4. Levels of insulin were significantly higher in Group A than in Groups B, C, or D. The average concentration of leptin in Group D appeared to be similar to that in Group A, but omitting one Group D mouse whose leptin concentration was extremely high, the average became 8.7 (SD 3.4) from 56.1 (SD 125.3) and was significantly lower than that of Group A. Adiponectin levels were low in Groups A, B and D. Glucose levels were high in Groups A and D, and those in Group D were significantly higher than those in Group A.
Table 4.
The results of blood testing
| Insulin (pg/mL) average (SD) | Leptin (ng/mL) average (SD) | Adiponectin (µg/mL) average (SD) | Glucose (mg/dL) average (SD) | |
|---|---|---|---|---|
| A: db/db + MNU | 6318 (4303) | 65.5 (84.7) | 7.6 (2.5)** | 309 (144) |
| B: +/db + MNU | 375 (690)* | 0.716 (0.718)* | 12 (3.2)** | 76 (22)* |
| C: misty + MNU | 195 (184)* | 0.188 (0.185)* | 19 (14) | 120 (25)* |
| D: db/db | 941 (938)* | 56.1 (125.3) | 8.5 (4.8)** | 567 (108)* |
P < 0.05 compared with Group A.
P < 0.05 compared with Group C.
Discussion
In db/db mice treated with MNU, oxyntic atrophy and hyperplasia were observed earlier and were more widespread than in controls (Table 2). Dysplasia was observed significantly more frequently in this group. Moreover, adenocarcinoma were found only in this group, mostly located in areas where hyperplasia was observed. The expression of pepsinogen was decreased or lacking in all of these lesions (Fig. 2I), pyloric mucosa with focal pepsinogen enzyme 1 (Pg 1) decrease being generally considered preneoplastic and called pepsinogen‐altered pyloric glands (PAPGs).( 22 , 23 ) All of these data indicated enhanced stomach neopasia in db/db mice exposed to MNU.
In the present study, we found cancerous lesions in db/db mice at week 30, almost the same time point as that in experiments using C3H (30–50 weeks) or BALB/C (20–40 weeks) mice.( 11 , 12 ) In Mongolian gerbils with H. pylori, tumor development usually takes about 50 weeks.( 13 ) Although the amount of MNU per animal was here found to be slightly increased as compared to controls, the ratio to body weight was much lower in the db/db mice. Yamachika et al. earlier examined the influence of the dose of MNU on tumor development in the BALB/c strain and concluded that the induction of stomach cancers depends not on the total quantity but rather the body concentration of the carcinogen.( 24 ) Therefore, we think that controlling the concentration of MNU and period of exposure is more important. The degree of DNA damage caused by MNU should correlate with the frequency with which MNU is in contact with the DNA.
The body weights of Group D were here found to be lower than for Group A. In the normal case with db/db mice food intake results in an uncontrolled rise in blood sugar and chronic hyperglycemia leads to high concentrations of interleukin‐1‐beta within the islets, which in turn worsens beta cell function( 25 ). Around week 30 of age, severe depletion of the insulin‐producing beta‐cells of the pancreatic islets occurs in db/db mice,( 26 ) leading to severe weight loss. This does not appear to be the case when the animals are exposed to MNU. In this study, serum glucose levels in Group D were significantly higher than those in Group A (Table 4). That means DM in Group D was more severe than in Group A. MNU causes loss of appetite, so DM was likely to progress more slowly. Depletion of the insulin‐producing beta‐cells was likely to be suppressed and the mice continued gaining weight in Group A. We also believe that higher serum insulin levels were observed in Group A because the insulin‐producing beta‐cells were saved.
The biological mechanism by which DM influences carcinogenesis remains to be clarified, but the associated metabolic and hormonal abnormalities clearly play important roles. In this study, serum levels of insulin and leptin were elevated only in db/db mice treated with MNU. When inflammation due to MNU occurs, db/db mice seem to produce these hormones significantly more than controls do. The tendency for proliferation may thus be due to these hormones. Impairment of the immune system can also affect inflammation, and delayed food transit can extend the duration of exposure of carcinogens (MNU in this study).
In guinea pigs, the proliferation of gastric epithelial cells can be enhanced by insulin( 27 ) and at high concentrations, insulin can bind to and activate IGF‐1R.( 7 ) This receptor is present in multiple cell types and tissues( 28 ) and overexpression can result in the ability of cells to form tumors when injected into mice.( 29 ) Mitogenic effects of exogenous IGF‐1 and IGF‐2 on human gastric cancer cell lines LIM‐1839, St16, St42, and MKN45 have also been reported.( 30 , 31 ) Many patients with insulin‐dependent diabetes mellitus have decreased serum IGF‐I concentrations.( 32 ) We obsereved IGF‐1R by immunostaining in all epithelial cells of the stomach of db/db mice (data not shown). Our finding that the serum insulin level was significantly high in db/db treated with MNU supports the idea that hyperinsulinemia leads to activation of IGF‐1R. The results of BrdU staining showed there was no significant difference between groups, although there seems to be a tendency towards increased proliferating cells in the normal‐appearing fundic gland area in groups with a high level of insulin. Higher insulin levels can cause an increase in proliferating cells. However, its effect appears to be much stronger in the pyloric gland area or antralized area than the fundic gland area. In the former area, most hyperplasia, dysplasia or neoplasia occurred. Some suppressing factors may still have good control in the fundic gland area.
Both normal gastric epithelium and carcinoma cells express significant levels of leptin,( 33 , 34 , 35 ) a hormone known to induce gastric cancer cells to proliferate.( 8 , 9 ) The leptin receptor Ob‐R has at least five splicing variants (Ob‐Ra, Ob‐Rb, Ob‐Rc, Ob‐Rd and Ob‐Re). Though db/db mice have a defective Ob‐Rb,( 16 ) the other splicing variants, Ob‐Ra, Ob‐Rc, Ob‐Rd, and Ob‐Re, are preserved.( 16 ) In this study, the concentration of leptin was elevated in serum. So there is a possibility that leptin can perform signal transduction through any of these receptors and may promote cell proliferation. However, since the functions of these receptors have not been elucidated, it is difficult to definitely conclude that leptin acted as a promoter in this study. Moreover, even if leptin had some effects on cell proliferation in this study, it is difficult to extrapolate the mechanism in DM patients, in which serum leptin is not as high as in db/db mice in most cases.
Recently, the level of adiponectin has been shown to be reduced in the plasma of patients with gastric cancer,( 36 ) raising the possibility that adiponectin is functionally involved in neoplastic progression. In our study, serum adiponectin levels did not show any association with cancer, but the situation was complicated by the significant difference in the weights of mice. The level of adiponectin has been reported to be inversely related to body mass index (BMI).( 37 )
In conclusion, we here observed that the MNU‐induced generation of neoplastic lesions was enhanced in db/db mice. Though the db/db mouse is not a complete model for DM, this is the first animal model to show an association between DM and gastric cancer. Since serum levels of insulin were significantly high in this group, this cell proliferation factor may have been responsible for the observed enhancement.
Supporting information
Table S1. The number of hyperplasias per body
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Acknowledgments
We thank Dr Nicholas Wright and Dr Adam Smolka for antibodies.
References
- 1. Rousseau M, Parent M, Pollak M, Siemiatycki J. Diabetes mellitus and cancer risk in a population‐based case‐control study among men from Montreal. Can Int J Cancer 2006; 118: 2105–9. [DOI] [PubMed] [Google Scholar]
- 2. Batty G, Shipley M, Marmot M, Smith G. Diabetes status and post‐load plasma glucose concentration in relation to site‐specific cancer mortality: findings from the original Whitehall study. Cancer Causes Control 2004; 15: 873–81. [DOI] [PubMed] [Google Scholar]
- 3. Coughlin S, Calle E, Teras L, Petrelli J, Thun M. Diabetes mellitus as a predictor of cancer mortality in a large cohort of US adults. Am J Epidemiol 2004; 159: 1160–7. [DOI] [PubMed] [Google Scholar]
- 4. Gapstur SM, Gann PH, Lowe W, Liu K, Colangelo L, Dyer A. Abnormal glucose metabolism and pancreatic cancer mortality. JAMA 2000; 283: 2552–8. [DOI] [PubMed] [Google Scholar]
- 5. Inoue M, Iwasaki M, Otani T, Sasazuki S, Noda M, Tsugane S. Diabetes mellitus and the risk of cancer: results from a large‐scale population‐based cohort study in Japan. Arch Intern Med 2006; 166: 1871–7. [DOI] [PubMed] [Google Scholar]
- 6. Yamagata H, Kiyohara Y, Nakamura S et al . Impact of fasting plasma glucose levels on gastric cancer incidence in a general Japanese population: the Hisayama study. Diabetes Care 2005; 28: 789–94. [DOI] [PubMed] [Google Scholar]
- 7. Le Roith D. Seminars in medicine of the Beth Israel Deaconess Medical Center. Insulin‐like growth factors. N Engl J Med 1997; 336: 633–40. [DOI] [PubMed] [Google Scholar]
- 8. Schneider R, Bornstein S, Chrousos G, Boxberger S, Ehninger G, Breidert M. Leptin mediates a proliferative response in human gastric mucosa cells with functional receptor. Horm Metab Res 2001; 33: 1–6. [DOI] [PubMed] [Google Scholar]
- 9. Pai R, Lin C, Tran T, Tarnawski A. Leptin activates STAT and ERK2 pathways and induces gastric cancer cell proliferation. Biochem Biophys Res Commun 2005; 331: 984–92. [DOI] [PubMed] [Google Scholar]
- 10. Ishikawa M, Kitayama J, Yamauchi T et al . Adiponectin inhibits the growth and peritoneal metastasis of gastric cancer through its specific membrane receptors AdipoR1 and AdipoR2. Cancer Sci 2007; 98: 1120–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tatematsu M, Ogawa K, Hoshiya T et al . Induction of adenocarcinomas in the glandular stomach of BALB/c mice treated with N‐methyl‐N‐nitrosourea. Jpn J Cancer Res 1992; 83: 915–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tatematsu M, Yamamoto M, Iwata H et al . Induction of glandular stomach cancers in C3H mice treated with N‐methyl‐N‐nitrosourea in the drinking water. Jpn J Cancer Res 1993; 84: 1258–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tatematsu M, Yamamoto M, Shimizu N et al . Induction of glandular stomach cancers in Helicobacter pylori‐sensitive Mongolian gerbils treated with N‐methyl‐N‐nitrosourea and N‐methyl‐N′‐nitro‐N‐nitrosoguanidine in drinking water. Jpn J Cancer Res 1998; 89: 97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Coleman D. Obese and diabetes: two mutant genes causing diabetes‐obesity syndromes in mice. Diabetologia 1978; 14: 141–8. [DOI] [PubMed] [Google Scholar]
- 15. Garris D. Hypercytolipidemia‐induced cellular lipoapoptosis: cytostructural and endometabolic basis of progressive organo‐involution following expression of diabetes (db/db) and obese (ob/ob) mutation syndromes. Prog Histochem Cytochem 2006; 40: 181–231. [DOI] [PubMed] [Google Scholar]
- 16. Lee G, Proenca R, Montez J et al . Abnormal splicing of the leptin receptor in diabetic mice. Nature 1996; 379: 632–5. [DOI] [PubMed] [Google Scholar]
- 17. Chen H, Charlat O, Tartaglia L et al . Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell 1996; 84: 491–5. [DOI] [PubMed] [Google Scholar]
- 18. Coleman D. Obesity genes: beneficial effects in heterozygous mice. Science 1979; 203: 663–5. [DOI] [PubMed] [Google Scholar]
- 19. Furihata C, Saito D, Fujiki H, Kanai Y, Matsushima T, Sugimura T. Purification and characterization of pepsinogens and a unique pepsin from rat stomach. Eur J Biochem 1980; 105: 43–50. [DOI] [PubMed] [Google Scholar]
- 20. Japanese Gastric Cancer A. Japanese classification of gastric carcinoma – 2nd english edition. Gastric Cancer 1998; 1: 10–24. [DOI] [PubMed] [Google Scholar]
- 21. Hildesheim A, Ryan R, Rinehart E et al . Simultaneous measurement of several cytokines using small Volumes of biospecimens. Cancer Epidemiol Biomarkers Prev 2002; 11: 1477–84. [PubMed] [Google Scholar]
- 22. Tsukamoto T, Mizoshita T, Tatematsu M. Animal models of stomach carcinogenesis. Toxicol Pathol 2007; 35: 636–48. [DOI] [PubMed] [Google Scholar]
- 23. Yamamoto M, Furihata C, Ogiu T et al . Independent variation in susceptibilities of six different mouse strains to induction of pepsinogen‐altered pyloric glands and gastric tumor intestinalization by N‐methyl‐N‐nitrosourea. Cancer Lett 2002; 179: 121–32. [DOI] [PubMed] [Google Scholar]
- 24. Yamachika T, Nakanishi H, Inada K et al . N‐methyl‐N‐nitrosourea concentration‐dependent, rather than total intake‐dependent, induction of adenocarcinomas in the glandular stomach of BALB/c mice. Jpn J Cancer Res 1998; 89: 385–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Maedler K, Sergeev P, Ris F et al . Glucose‐induced beta cell production of IL‐1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest 2002; 110: 851–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Berglund O, Frankel B, Hellman B. Development of the insulin secretory defect in genetically diabetic (db/db) mouse. Acta Endocrinol (Copenh) 1978; 87: 543–51. [DOI] [PubMed] [Google Scholar]
- 27. Ogihara S, Yamada M, Saito T, Shono M, Rokutan K. Insulin potentiates mitogenic effect of epidermal growth factor on cultured guinea pig gastric mucous cells. Am J Physiol 1996; 271: G104–12. [DOI] [PubMed] [Google Scholar]
- 28. De Meyts P, Whittaker J. Structural biology of insulin and IGF1 receptors: implications for drug design. Nat Rev Drug Discov 2002; 1: 769–83. [DOI] [PubMed] [Google Scholar]
- 29. Baserga R, Peruzzi F, Reiss K. The IGF‐1 receptor in cancer biology. Int J Cancer 2003; 107: 873–7. [DOI] [PubMed] [Google Scholar]
- 30. Thompson M, Cox A, Whitehead R, Jonas H. Autocrine regulation of human tumor cell proliferation by insulin‐like growth factor II: an in‐vitro model. Endocrinology 1990; 126: 3033–42. [DOI] [PubMed] [Google Scholar]
- 31. Durrant L, Watson S, Hall A, Morris D. Co‐stimulation of gastrointestinal tumour cell growth by gastrin, transforming growth factor alpha and insulin like growth factor‐I. Br J Cancer 1991; 63: 67–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hall K, Johansson B, Póvoa G, Thalme B. Serum levels of insulin‐like growth factor (IGF) I, II and IGF binding protein in diabetic adolescents treated with continuous subcutaneous insulin infusion. J Intern Med 1989; 225: 273–8. [DOI] [PubMed] [Google Scholar]
- 33. Mix H, Widjaja A, Jandl O et al . Expression of leptin and leptin receptor isoforms in the human stomach. Gut 2000; 47: 481–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bado A, Levasseur S, Attoub S et al . The stomach is a source of leptin. Nature 1998; 394: 790–3. [DOI] [PubMed] [Google Scholar]
- 35. Brzozowski T, Konturek P, Konturek S et al . Leptin in gastroprotection induced by cholecystokinin or by a meal. Role of vagal and sensory nerves and nitric oxide. Eur J Pharmacol 1999; 374: 263–76. [DOI] [PubMed] [Google Scholar]
- 36. Ishikawa M, Kitayama J, Kazama S, Hiramatsu T, Hatano K, Nagawa H. Plasma adiponectin and gastric cancer. Clin Cancer Res 2005; 11: 466–72. [PubMed] [Google Scholar]
- 37. Yang W, Lee W, Funahashi T et al . Plasma adiponectin levels in overweight and obese Asians. Obes Res 2002; 10: 1104–10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. The number of hyperplasias per body
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item