

Abstract
The selective elimination of tumor cells by inducing apoptosis is one of the most important issues in cancer therapy. In this context, artificial expression of the p53 tumor‐suppressor gene has been an attractive approach and numerous studies have shown its efficacy in combination with other therapies such as radiation or chemotherapy. One of the critical issues for current cancer gene therapy is how to induce apoptosis in cancer cells without affecting normal cells. In the present study, we examined the potential of Noxa, a BH3‐only protein with proapoptotic activity that functions downstream of the p53‐mediated apoptotic pathway, to selectively induce apoptosis in tumor cells. We found that upon infection of a recombinant adenovirus contrived to express the Noxa gene, apoptosis was induced in vitro in several human breast cancer cell lines, but not in normal mammary epithelial cell lines. Furthermore, intratumoral injection of the Noxa‐expressing adenovirus resulted in marked shrinkage of the transplanted tumor derived from breast cancer cells without any notable adverse effect on the surrounding normal tissue. In contrast, the expression of Puma, another BH3‐only protein that also functions downstream of the p53 pathway, induced apoptosis in both cancer and normal cells. Thus, our results suggest a mechanism wherein Noxa, but not Puma, selectively induces apoptosis in human tumor cells. These data provide a new prospect for cancer therapy by the Noxa‐mediated selective elimination of malignant cells. (Cancer Sci 2009; 100: 759–769)
Cancer therapy consists of two main approaches, the surgical removal of tumor masses and induction of death in tumor cells. Conventionally, these approaches are combined for the effective and complete elimination of tumor cells, wherein the latter approach includes chemotherapy, radiation therapy, gene therapy, and immunotherapy.( 1 ) In the context of gene therapy, the p53 tumor‐suppressor gene has been extensively studied. This gene is functionally inactivated in approximately 50% of human tumors( 2 ) and its function in the induction of apoptosis in cancerous cells is considered as a major mechanism in tumor suppression.( 3 , 4 ) When p53 is activated by noxious stimuli such as DNA damage, hypoxia, and oncogenes, it mediates two types of cellular response, induction of cell cycle arrest followed by DNA repair and induction of apoptosis.( 5 ) It is thought that the fine balance of these two types of cellular response is a key event in the determination of cell fate in tumor suppression. Precisely how p53 mediates these cellular responses has been extensively studied; activation of its target genes is considered an essential mechanism. The induction of cell‐cycle arrest requires induction of the cyclin dependent kinase (CDK) inhibitor p21WAF1/Cip1,( 6 ) whereas the induction of apoptosis requires the induction of at least two genes for BH3‐only proteins of the Bcl‐2 family, namely, Noxa and Puma.( 7 , 8 , 9 , 10 )
Thus, reinstatement of p53 function is an attractive tumor‐specific therapeutic strategy, and p53 gene therapies for malignant tumors such as lung, breast, and head and neck cancer have been widely performed in animal experiments and clinical trials.( 11 , 12 , 13 , 14 , 15 ) In previous clinical trials, such tumors ceased their progression after intratumoral injection of an adenoviral vector, termed Ad‐p53, bearing the p53 gene, and they seemingly became more susceptible to other therapies such as radiation or chemotherapy.( 16 , 17 , 18 , 19 ) In many studies, however, tumors shrank only in the case of combining p53 expression with other therapies.( 16 , 17 , 18 , 19 ) As mentioned above, because activated p53 regulates both cell‐cycle arrest and apoptosis, its overexpression might have a certain drawback in the efficient regression of tumor.( 20 ) It is also known that tumor cells that express wild‐type p53 are resistant to Ad‐p53 treatment.( 20 , 21 ) Moreover, an attempt was made to utilize Puma, a downstream factor of p53 that functions in the limb of the apoptotic pathway, in order to devise a more effective cancer gene therapy.( 21 )
Noxa works in cooperation with Puma in the p53‐mediated apoptotic response.( 22 , 23 , 24 , 25 ) Interestingly, it was found that Noxa induces apoptosis in mouse embryonic fibroblasts (MEF) only when the cells are transformed by an oncogene.( 22 ) In view of the fact that Puma induces apoptosis in both transformed and untransformed cells, an interesting distinction can be made in which Noxa, but not Puma, can selectively induce apoptosis in a broader range of cancerous cells.( 26 ) Although the mechanism underlying selective induction of apoptosis in oncogene‐expressing cells is not yet clarified, it has been proposed that Noxa may require the cell state wherein cells are more sensitive to apoptotic induction by increased expression of other proapoptotic factors such as Bax.( 26 ) These observations therefore prompted us to investigate whether some, if not all, human cancer cells are even more susceptible than normal cells to undergoing apoptosis induced by Noxa. The rationale behind this investigation is that cancer cells are not fully “armed” during their development by gene mutations in terms of resistance of apoptotic responses such that this weakness may be exploited to induce effective cell death. Indeed, the rationale behind chemotherapy lies in a similar context; cancer cells, but not normal cells, are susceptible to cell death by mitotic catastrophe induced by chemotherapeutic agents.( 27 )
In the present study, we asked whether Noxa can also selectively induce apoptosis in human cancer cells, as this may become an interesting candidate for cancer gene therapy. We examined the effect of Noxa expression in several human cancer cell lines and normal cells in vitro, and studied tumor progression in mice after intratumoral expression of Noxa by an adenoviral vector or non‐viral vector, named polymeric micelle vector. Our results suggest the potential of Noxa in the selective induction of apoptosis in human cancer cells.
Materials and Methods
Cell lines and culture conditions. The human breast cancer cell lines HBC4, HBC5, MCF7, and HTB26 were kindly provided by Dr K. Yamazaki (Cancer Institute Hospital, Tokyo), and human normal fibroblasts (HF) were kindly provided by Dr K. Yoshimura (University of Tokyo Hospital). YMB‐1‐E and SK‐BR‐3 were provided by the Institute of Development, Aging and Cancer (Tohoku University). The human fibrosarcoma cell line HT1080 was purchased from Health Science Research Resources Bank (Osaka, Japan). Human mammary epithelial cells (HMEC) and the human mammary epithelial cell line MCF10A were purchased from American Type Culture Collection (Peoria, IL, USA). HBC4, MCF7, YMB‐1‐E, HF, and MEF were propagated in Dulbecco's Modified Eagle Medium with 10% fetal calf serum (FCS) and 50 µg/mL kanamycin. HBC5, HTB26, and SK‐BR‐3 were propagated in RPMI‐1640 with 10% FCS, 10 mM HEPES, and 50 µg/mL kanamycin. HT1080 was propagated in minimum essential medium with 10% FCS and 50 µg/mL kanamycin. HMEC and MCF10A were cultured in MEBM (Lonza, Valais, Switzerland) supplemented with MEGM SingleQuots (Lonza) in accordance with the manufacturer's protocol.
Recombinant adenovirus. The replication‐deficient adenoviral vectors for human Noxa and Puma tagged with the sequence encoding two consecutive hemagglutinin (HA) peptides (2 × HA), named Ad‐HA‐Noxa and Ad‐HA‐Puma, were constructed using the Adeno‐X Expressing System (BD Biosciences Clontech, Bedford, MA, USA) and purified in accordance with the manufacturer's protocol. The adenoviral vector expressing p53 (Ad‐p53) was kindly given by Dr T. Tokino (Sapporo Medical School). An empty adenoviral vector (Ad‐empty) was used as the control.
Detection of cell death. To quantify apoptotic death, cells were collected 16 h after adenoviral vector infection of each cell line. Subsequently, cells were stained with Annexin V‐Cy3 or Annexin‐fluorescein isothiocyanate (MBL, Nagoya, Japan) and analyzed by FACS caliber (Becton‐Dickinson, Cockeysville, MD, USA).
Western blot analysis. Cells were harvested by scraping, washed with phosphate‐buffered saline (PBS), and resuspended in Nonidet (NP)‐40 lysis buffer (0.25% NP‐40, 142.5 mM KCl, 5 mM MgCl2, 1 mM ethylenediaminetetraacetic acid, and 10 mM Hepes at pH 7.4) containing protease inhibitors (1 mM phenylmethylsulfonyl fluoride [PMSF], 400 µM Na3VO4, 10 µg/mL aprotinin, and 10 µg/mL leupeptin). Extracts were centrifuged at 12 000 g for 10 min. Supernatants were collected and protein concentration was determined. Fifty‐microgram of protein sample was resolved by sodium dodecylsulfate–polyacrylamide gel electrophoresis (SDS‐PAGE) and transferred to polyvinylidene diflouride membranes by electroblotting. Membranes were probed with a primary mouse anti‐HA antibody (monoclonal, 12CA5; Roche Diagnostics, Rotkreuz, Switzerland), a rabbit anti‐Mcl‐1 antibody (S‐19; Santa Cruz Biotechnology, Santa Cruz, CA, USA), a rabbit anti‐Bcl‐xL antibody (S‐18; Santa Cruz), a rabbit anti‐Bax antibody (polyclonal, N‐20; Santa Cruz), a mouse anti‐Bax antibody (monoclonal; 6A7; Calbiochem, Darmstadt, Germany), a rabbit anti‐Bak antibody (06–536; Upstate Biotechnology, Lake Placid, NY, USA), a mouse anti‐β‐actin antibody (AC‐15; Sigma Chemical, St Louis, MO, USA), a rabbit anti‐β‐tubulin antibody (H‐235; Santa Cruz), a mouse anti‐Porin antibody (20B12; Santa Cruz), or a mouse anticytochrome c antibody (7H8.2C12; Pharmingen, SanDiego, CA, USA) followed by a peroxidase‐linked secondary antibody. Western Lightning Chemiluminescence Reagent Plus (PerkinElmer Life Sciences, Boston, MA, USA) was used to detect secondary probes.
Analysis of Bax–Bak oligomerization. Tumor cell lines and MCF10A were harvested in isotonic buffer (210 mM mannitol, 70 mM sucrose, 1 mM ethylene glycoltetraacetic acid (EGTA), and 10 mM Hepes, pH 7.5) supplemented with protease inhibitors (1 mM PMSF, 400 µM Na3VO4, 10 µg/mL aprotinin, and 10 µg/mL leupeptin). Then cells were homogenized through a 25‐G needle and the suspension was centrifuged at 600 g for 10 min at 4°C. This procedure was repeated four times, and supernatants from each step were collected and centrifuged at 8000 g for 10 min at 4°C. The resulting pellet was washed and resuspended in isotonic buffer to 0.5 mg/mL protein, and then 11 mM 1,6‐bismaleimidohexane (Pierce, Rockford, IL, USA) in dimethyl sulfoxide was added at a 1:11 dilution. After incubation at room temperature for 30 min, the cross‐linking reaction was quenched by adding dithiothreitol to a final concentration of 25 mM, followed by centrifugation to pellet mitochondria, which were subsequently analyzed by immunoblotting.
Analysis of activated Bax. Total cell lysates were prepared using chaps lysis buffer. Immunoprecipitation was carried out using anti‐Bax monoclonal antibody 6A7, followed by immunoblotting with the anti‐Bax polyclonal antibody N‐20.
Tumor cell xenograft transplantation and gene therapy. Mice were housed in specific pathogen‐free conditions at the Animal Facility of University of Tokyo and all experimental protocols were approved by the Animal Ethics Committee of the University of Tokyo. HBC4 (6 × 106 cells), HTB26 (6 × 106 cells), or HT1080 (4 × 106 cells) in 100 µL PBS were injected subcutaneously into both sides of the flanks of 6–8‐week‐old female nude mice (BALB/c‐nu/nµ). Two months later (HBC4 cells) or 2 weeks (HTB26 and HT1080 cells) later, tumors grew to be 6 mm in diameter, and 1 × 108 plaque‐forming units (pfu) Ad‐HA‐Noxa/HA‐Puma/p53/empty in 100 µL PBS were injected intratumorally five times every 3 days. The major axis, minor axis, and height of tumors were measured every 3 days. After 10 days from the last injection, the mice were killed, and then tumors were excised en bloc and frozen in OCT compound.
To examine the effect of Noxa on normal tissue, 1 × 108 pfu Ad‐HA‐Noxa/p53/empty were also injected subcutaneously near mammary glands of non‐tumor‐bearing 15‐week‐old female nude mice. Then, the specimens were taken from these tissues as described above.
Histopathological examinations. Microdissections were processed and fixed in 4% formaldehyde: 4 and 6 µm‐thick specimens were use for hematoxylin–eosin (HE) staining and TdT‐mediated dUTP‐biotin nick‐end labeling (TUNEL) staining and immunostaining, respectively. Apoptotic cell death in segments was measured by detecting nuclear morphological changes by HE staining and TUNEL with an In Situ Cell Death Detection Kit and fluorescein (Roche Diagnostics) followed by microscopy. To determine cells infected by Ad‐HA‐Noxa, HA‐Puma, or p53, specimens were immunostained with the anti‐HA‐antibody or antip53 antibody as the primary antibody and Alexa Fluor568 antimouse IgG as the secondary antibody (Invitrogen, Carlsbad, CA, USA).
Administration of polymeric micelle vectors. Polyethylene glycol polymer was kindly provided by Dr M. Ohba (University of Tokyo). After preincubation of the polymer and plasmid DNA (pDNA) (pEF‐HA‐Noxa/pEF‐HA‐Puma/pEF‐HA‐empty) overnight at room temperature, 200 µL of 2‐optical density (OD) pDNA‐micelle was injected intravenously into HBC4‐bearing nude mice. Tumor size was measured every 2 days. Histopathological examinations were carried out after 16 days of micelle‐type vector injections.
Preparation of recombinant TAT‐Noxa protein. The cDNA of human Noxa with a human immunodeficiency virus (HIV)‐1 TAT protein transduction domain (TAT‐PTD) sequence( 28 ) was cloned into pGEX4T3 (GE Healthcare Biosciences, Piscataway, NJ, USA) between the XhoI and NotI sites. It was expressed in TOP10F′ Chemically Competent Escherichia coli (Invitrogen) and purified using glutathione sepharose 4B resin (GE Healthcare Biosciences). Finally, product was desalted into PBS with a Sephadex PD‐10 column (GE Healthcare Biosciences).
Statistical analysis. Statistical analysis was carried out using Student's t‐test.
Results
Induction of apoptosis in human breast cancer cells by Noxa.
-
1
To investigate whether Noxa can induce apoptosis in human cancer cells, we studied the breast cancer cell lines SK‐BR‐3, MCF7, YMB‐1‐E, HBC4, HBC5, and MTB26, together with normal human mammary epithelial cells, termed HMEC, and MCF10A, a spontaneously immortalized cell line with normal characteristics of mammary epithelial cells.( 29 , 30 ) The status of the p53 gene is summarized in Table 1. At first, we established adenoviral vectors that express human Noxa or Puma, which were tagged with HA peptide. As shown in Figure 1(a), infection of HBC4 cells by these viruses resulted in expression of these molecules at similar levels. We also examined the efficiencies of adenovirus infection in these cell lines using a green fluorescent protein‐expressing adenovirus and found that all cells, including MCF10A cells but not YMB‐1‐E, were efficiently infected by this virus, suggesting that most of these cells are amenable to virus infection (Fig. 1b).
Table 1.
The status of p53 and expression of downstream genes.
| p53 states | cell type | Mcl‐1 | Bcl‐XL | Bax | Bak | p16INK4a | p21WAF1/Cip1 | Rb | c‐myc | Ras | HER2/neu | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SK‐BR‐3 (HTB‐30) | Deletion & mutation [aa175] | IDC† ER(‐)‡ PgR(‐)§ | ++ | + | + | +++ | + high | + low | + high | |||
| YMB‐1‐E (CRL1500, ZR‐75‐1) | intact | IDC ER(+) PgR(+) | ++ | ++ | + | + | + high | + high | + low | + high | ||
| MCF7 (HTB22) | intact | IDC ER(+) PgR(+) | ++ | + | + | + | − | + | wt | + high | + low | deletion/+ low |
| HBC4 | intact | ER(‐) | ++ | ++ | +++ | + | − | + high | + low | |||
| HBC5 | Mutation [C242F (ttc)] | +++ | +++ | + | ++ | |||||||
| HTB26 | Mutation [R284L (aaa)] | IDC ER(‐) PgR(‐) | +++ | +++ | ++ | ++ | − | + low | wt | + high | + low | + low |
| HMEC | intact | normal breast epitherial cell | + | ++ | + | + | + | wt | ND | ND | + low | |
| MCF10A | intact | spontaneously immortarized HMEC | + | +++ | + | + | + | wt | ND | ND | + low |
: invasive ductal carcinoma,
‡: ertrogen receptor,
§: progesterone receptor
Figure 1.
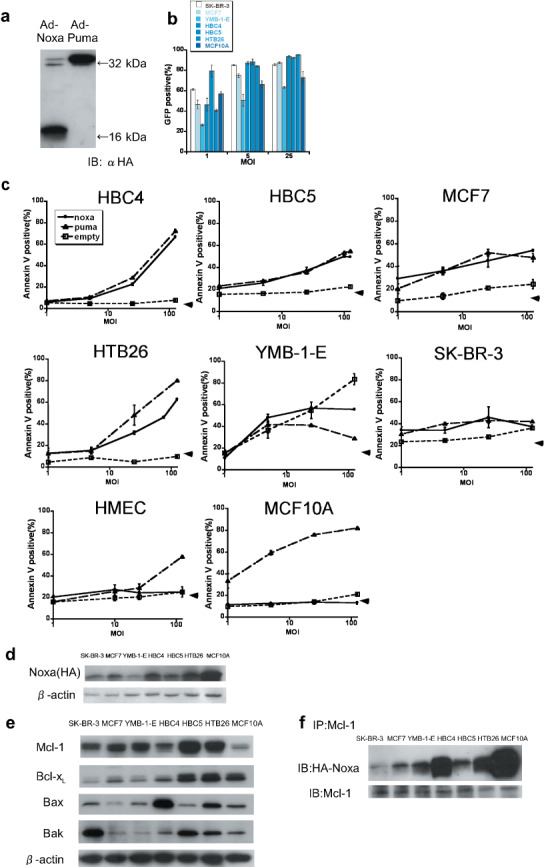
Forced expression of Noxa and Puma and apoptosis induction in human breast cancer cell lines and mammary epithelial cells. (a) HBC4 cells were infected with 10 multiplicity of infection (MOI) of Ad‐HA‐Noxa and Ad‐HA‐Puma. Twelve hours later, whole‐cell lysates were collected and western blotting was carried out with anti‐HA‐antibody as the primary antibody. (b) The efficiency of adenoviral forced expression. SK‐BR‐3, MCF7, YMB‐1‐E, HBC4, HBC5, HTB26, and MCF10A cells were infected with 1, 5, and 25 MOI of green fluorescent protein (GFP)‐expressing adenovirus. The percentages of GFP‐expressing cells were measured 16 h after infection. (c) The apoptotic cell death of Ad‐HA‐Noxa‐, Ad‐HA‐Puma‐, and Ad‐empty‐infected cancer cell lines HMEC and MCF10A. Sixteen hours after infection with 1, 5, 25, and 125 MOI of adenovirus, the percentages of Annexin V‐positive cells were examined. (d) Expression levels of Noxa protein. Twelve hours after infection with 5 MOI of Ad‐HA‐Noxa, whole‐cell lysates of cells were western blotted with anti‐HA antibody. (e) Expression of pro‐survival and pro‐apoptotic Bcl‐2 family proteins. β‐Actin was used as an internal standard. (f) The association between Mcl‐1 and Noxa. Whole extracts of cells were immunoprecipitated using anti‐Mcl‐1 antibody, and then immunoblotted with anti‐HA and anti‐Mcl‐1 antibodies.
We then infected these cancer cells with Ad‐HA‐Noxa and Ad‐HA‐Puma. Sixteen hours after the virus infection, comparable rates of apoptosis were observed between Noxa‐infected and Puma‐infected cells: 54.26 ± 0.83 and 47.92 ± 3.53% of MCF7, 66.94 ± 0.97 and 72.37 ± 1.65% of HBC4, 49.52 ± 0.24 and 54.84 ± 0.52% of HBC5, and 62.68 ± 0.81 and 80.25 ± 0.25% of HTB26 apoptotic cells by Ad‐HA‐Noxa and Ad‐HA‐Puma (at 125 multiplicity of infection), respectively (Fig. 1c). In all cases, the virus‐induced apoptosis occurred in a dose‐dependent manner (Fig. 1c). SK‐BR‐3 and YMB‐1‐E cells infected with the control adenoviral vector died of unknown causes; therefore, these cells were not studied further. Although untransformed HMEC and MCF10A cells died following Ad‐HA‐Puma infection, both were resistant to Ad‐HA‐Noxa infection. This is similar to our previous observation that mouse NIH3T3 cells are susceptible to apoptosis induced by Puma but not to that induced by Noxa, yet Noxa can induce apoptosis when these cells express the adenovirus‐derived E1A oncoprotein.( 26 ) We also examined apoptosis induction by Ad‐HA‐Noxa and Ad‐HA‐Puma in the fibrosarcoma cell line HT1080 and normal HF. Interestingly, it was found that Puma induced apoptosis in both cell types, whereas Noxa did so only in HT1080 cells (Supporting Fig. S1a,b).
To examine whether the difference in apoptotic responses to Noxa correlate with the expression levels of Noxa in these cells, we compared the expression levels among these cell lines infected with Ad‐HA‐Noxa. As shown in Figure 1(c), the Noxa expression level was high in MCF10A cells, which showed resistance to Noxa for apoptosis. On the other hand, the expression level of Noxa in Noxa‐resistant HF was almost the same as that in Noxa‐sensitive HT1080 cells (Supporting Fig. S1c). These results suggest that the levels of Noxa expression do not closely correlate with apoptosis induction.
Thus, these observations in toto point to the interesting possibility that Noxa can selectively induce apoptosis in cancer cells without affecting normal cells.
Expression of prosurvival and proapoptotic Bcl‐2 family members in Noxa‐sensitive cancer cells. Many cancer cells express prosurvival members of the Bcl‐2 family, thereby rendering cells resistant to apoptosis.( 7 , 8 , 9 , 10 ) We therefore examined by immunoblotting analysis whether the differential sensitivity to apoptosis observed in the above cell lines correlated with the expression levels of prosurvival Bcl‐2 family proteins. It was found that although the expression level of prosurvival Bcl‐xL is high in the Noxa‐sensitive cell lines, it is also so in the untransformed, Noxa‐resistant MCF10A cells (Table 1; Fig. 1e). We next measured the expression level of Mcl‐1, which is a critical target of Noxa in MCF10A cells, and found that it is even lower than that observed in other cells (Fig. 1e). Moreover, Noxa's association with Mcl‐1 was not impaired in MCF10A cells (Fig. 1f).
When HT1080 cells and HF were examined, the expression of Mcl‐1 and Bcl‐xL were found to be even lower in Noxa‐resistant HF than in Noxa‐sensitive HT1080 cells (Supporting Fig. S1d). Furthermore, an association between Mcl‐1 and Noxa was also observed in the Noxa‐resistant HF (Supporting Fig. S1e). We also examined the expression of pro‐apoptotic proteins Bax and Bak. The expression levels of these proteins did not correlate with cellular sensitivity to Ad‐HA‐Noxa (Fig. 1e; Supporting Fig. S1d).
These results suggest that sensitivity to Noxa is independent of the expression levels of the Bcl‐2 family proteins and the association of these proteins with Noxa.
Induction of apoptosis in the breast cancer xenografts. We carried out subcutaneous xenografting of HBC4 cells to bilateral flanks of nude mice, followed by intratumoral injection of Ad‐HA‐Noxa, Ad‐HA‐Puma, or Ad‐p53 to one side, and the control virus (Ad‐empty) to the other side (Fig. 2a). Changes in relative tumor volume were monitored (n = 4 for each condition). As shown in Figure 2(b,c), injection of Noxa‐expressing and Puma‐expressing viruses resulted in a marked decrease in tumor volume. Interestingly, the expression of p53 had a less pronounced effect than Noxa and Puma expression, inducing only a growth delay of the grafted tumor (Fig. 2d). Moreover, a previous report showed that tumor cells carrying wild‐type p53 are relatively resistant to p53 therapy.( 21 ) These facts suggest that Noxa and Puma are more effective than p53 in abrogating tumor growth. Actually, tumors injected with Ad‐p53 grew similarly to those with the control virus for the first 2 weeks, but then began to cease their progression (Fig. 2d); this growth delay remained for 10 days of follow up after the last injection (Supporting Fig. S2a–f).
Figure 2.
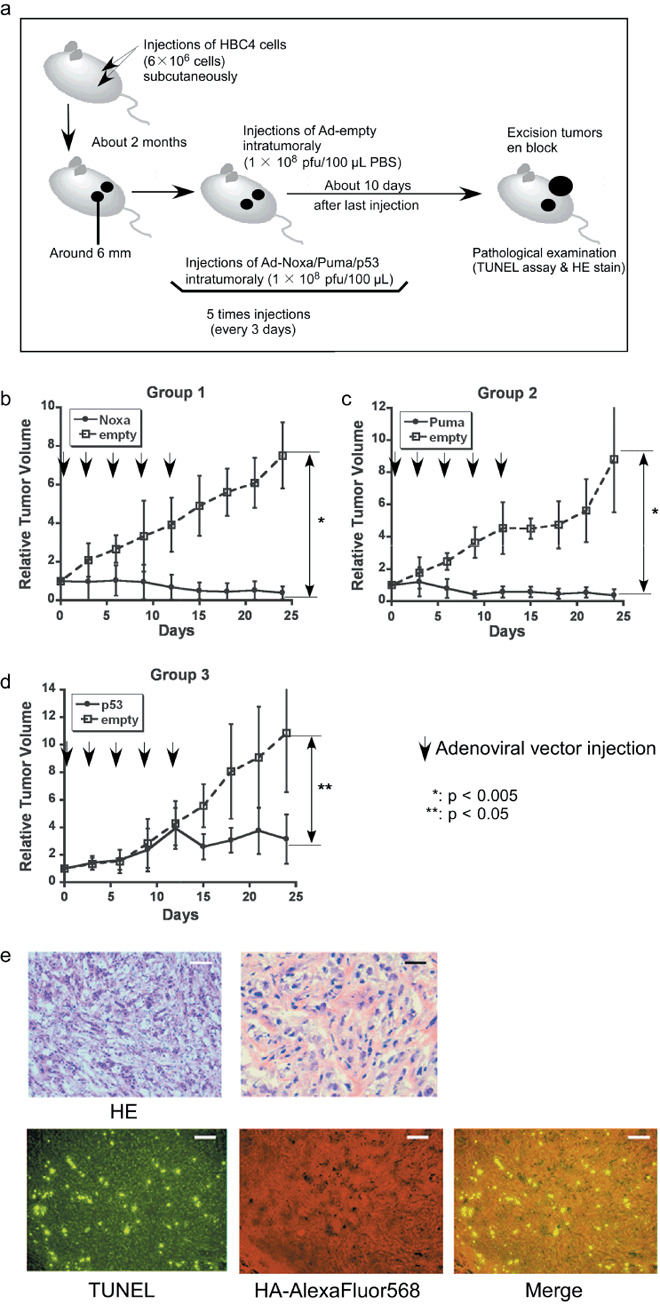
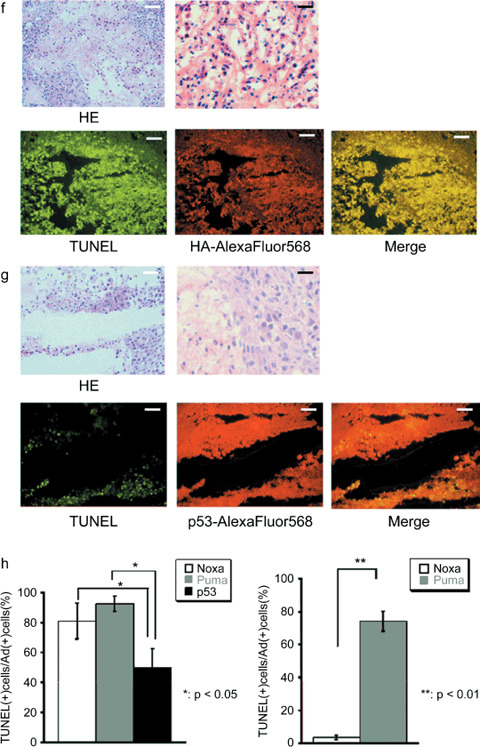
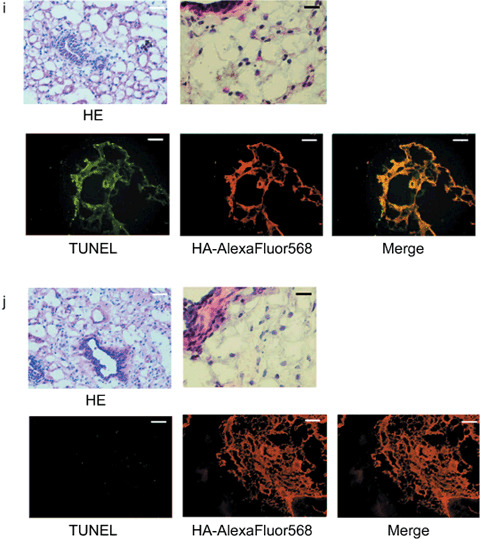
Antitumor effect of adenoviral proapoptotic gene expression in vivo. (a) Assay procedures of adenoviral gene expression in a xenograft tumor model. Suppression of tumor growth by treatment with (b) Ad‐HA‐Noxa, (c) Ad‐HA‐Puma, and (d) Ad‐empty. At the indicated points, the major axis, minor axis, and height of tumors were measured. Then, tumor volume was estimated approximately as ellipsoid and normalized by the volume of the tumor just before adenoviral gene therapy. The experiments were carried out in quadruplicate and the results are presented as the mean ± SD. (e–g,i,j) Histopathological examinations of tumors and their peripheral tissues. These (e–g) tumors and (i,j) peripheral tissues were sliced and stained with hematoxylin–eosin (HE), TdT‐mediated dUTP‐biotin nick‐end labeling (TUNEL), and immunohistochemistry using anti‐HA‐antibody after adenoviral gene therapy with (e,j) Ad‐HA‐Noxa, (f,i) Ad‐HA‐Puma, and (g) Ad‐empty. White bars = 200 µm, black bars = 50 µm. (h) Apoptotic cell death in adenovirus‐infected tumors (left graph) and their peripheral tissues (right graph). The rates of TUNEL‐positive cells were measured in 10 independent focuses. The results are presented as the mean + SD.
Histopathological examinations of virus‐injected tumor regions were carried out. As shown in Figure 2(e), many cells had nuclei showing condensed chromatin and the cytoplasm apparently cohered in an Ad‐HA‐Noxa‐injected tumor specimens. These observations are consistent with an apoptotic response of these cells. Similar observations were made in an Ad‐HA‐Puma‐injected tumor specimen (Fig. 2f). On the other hand, there were several apoptotic cells in an Ad‐p53‐injected tumor specimen, but many nonapoptotic cells were also detectable (Fig. 2g). Immunofluorescence staining analysis revealed that 80.96 ± 11.96% of Ad‐HA‐Noxa‐infected cells, 92.57 ± 5.08% of Ad‐HA‐Puma‐infected cells, and 49.64 ± 12.82% of Ad‐p53‐infected cells were TUNEL positive (Fig. 2h). Taken together, the results of HE staining and TUNEL analyses correlate with the growth‐suppressive effects of these viruses on the tumors described above, indicating that Puma and Noxa are more effective than p53 in inducing apoptosis of tumor cells in vivo.
It is noteworthy that TUNEL‐positive cells were also seen in portions of the tumor injected with Ad‐empty, but nearly no TUNEL‐positive cells were detected around the tumor (Supporting Fig. S3a). There were some TUNEL‐positive cells in the center of the tumor injected with 100 µL PBS only (Supporting Fig. S3b). These observations suggest that cell death is partially attributable to a non‐specific event such as the pressure created by the injection or injected reagent, rather than due to the immunoreactive response to the adenoviral vector.
To further validate the above data, similar experiments were also carried out with other cancer cell lines. We performed xenografting of HTB26, a breast cancer cell line carrying an inactivated p53 gene, followed by intratumoral injection of adenoviral vectors (Supporting Fig. S4a–c). It was found that HTB26‐derived tumors grew faster than those derived from HBC4 cells in nude mice. Both Ad‐HA‐Noxa and Ad‐HA‐Puma injections resulted in a significant tumor growth inhibition compared with Ad‐empty injection, although they could not markedly reduce tumor volume as observed in HBC4 tumor. The Ad‐p53 injection also showed a less‐effective tumor growth inhibition than in HBC4 tumor‐bearing mice. We infer that if the signaling pathway downstream of p53 is not impaired in p53‐defective HTB26 cells, p53 overexpression can induce apoptosis in these cells (see Supporting Fig. S4c).
We also examined HT1080 cells, which are fast‐growing fibrosarcoma cells, and found that intratumoral injection of Ad‐HA‐Noxa or Ad‐HA‐Puma also delayed tumor growth (Supporting Fig. S5a,b). These results therefore underscore the potential of Ad‐HA‐Noxa and Ad‐HA‐Puma in the inhibition of tumors in vivo by inducing apoptosis.
Selective effect of Noxa in vivo. To examine the effect of Ad‐HA‐Puma or Ad‐HA‐Noxa on non‐cancerous tissues, these viruses were injected subcutaneously around mammary glands. The Ad‐HA‐Puma‐injected tissue showed a lot of cells with condensed chromatin inside their nuclei and cohering cytoplasm, as revealed by HE staining, and many TUNEL‐positive cells were observed (74.17 ± 6.16% of Ad‐HA‐Puma‐infected cells) (Fig. 2h,i). On the other hand, apoptotic cells were rarely found in Ad‐HA‐Noxa‐injected tissues (3.46 ± 1.34% of Ad‐HA‐Noxa‐infected cells) (Fig. 2h,j). These results are congruent with our in vitro data showing that Noxa induces apoptosis in cancerous cells but not in normal cells (Fig. 1c; Supporting Fig. S1a,b). Thus, both Ad‐HA‐Noxa and Ad‐HA‐Puma could reduce tumor volume by induction of apoptosis in vivo, but Ad‐HA‐Puma also induces apoptosis in normal subcutaneous tissue.
Complexity of the mechanism of tumor‐specific induction of apoptosis by Noxa. Is there any common mechanism operating in cancer cells wherein Noxa selectively induces apoptosis? Previous reports suggest that NIH3T3 cells become sensitive to Noxa‐induced apoptosis when active‐form Bax oligomerization is triggered by the expression of E1A oncoprotein.( 26 ) We therefore studied Bax and Bak oligomerizations among these breast cancer cell lines and MCF10A cells. As shown in Figure 3(a), some Noxa‐sensitive cancer cell lines, HBC4 and HBC5, were found to constitutively express oligomerized Bax and its level increased upon Noxa expression. However, somewhat unexpectedly, oligomerized Bax and Bak levels also increased in Noxa‐resistant MCF10A cells after Noxa overexpression, indicating that the increase in oligomerized Bax and Bak levels does not solely account for the cellular sensitivity to Noxa.
Figure 3.
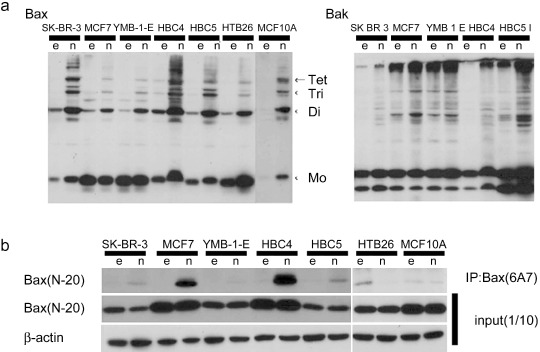
Differences in the activation of Bax and Bak depend on cell lines. Tumor cells and MCF10A were infected with 5 multiplicity of infection of Ad‐empty (e) or Ad‐HA‐Noxa (n) for 12 h, and then whole‐cell lysates were collected. (a) Polymer formation of Bax or Bak protein. The mitochondrial fraction was collected from cell lysates, followed by cross‐linking between Bax molecules and 1,6‐bismaleimidohexane. Then, fractions were immunoblotted with anti‐Bax (left panel) or anti‐Bak (right panel) antibodies. (b) Immunoblot of activated Bax. Whole‐cell lysates were analyzed by immunoprecipitation (IP) with anti‐Bax (6A7) antibody, followed by immunoblotting with anti‐Bax (N‐20) antibody (upper panel). To verify that these cells expressed Bax protein, whole‐cell lysates were also immunoblotted using anti‐Bax (N‐20) and anti‐β‐actin antibodies (middle and lower panels).
The active form of Bax was also seen in the Noxa‐sensitive cell lines HBC4, HBC5, and HTB26, even before Noxa overexpression, and its level increased after Ad‐HA‐Noxa infection (Fig. 3b). However, other Noxa‐sensitive cell lines such as MCF7 did not show Bax oligomerization without Noxa overexpression and this form of Bax was not observed in Noxa‐sensitive HTB26 cells even after Noxa overexpression. Furthermore, Bax oligomerization was induced by Ad‐HA‐Noxa in Noxa‐resistant MCF10A cells. HT1080 cells and HF expressed oligomerized Bax and its level was increased further by Noxa overexpression (Supporting Fig. S5c,d). However, the level of oligomerized Bax was higher in Noxa‐resistant HF than in Noxa‐sensitive HT1080 cells. We further studied cytochrome c release in these cells, as this is a key event of the apoptotic signaling pathway, but we could not detect any difference in the capability of cytochrome c release between normal and cancer cells (data not shown). Neither did we find any significant difference with the downstream caspase activation pathway among these normal and cancer cells (data not shown). These results therefore indicate that the functional mechanism of Noxa in the selective induction of apoptosis in cancer cells is complex and that Bax and Bak oligomerizations may explain only partly, if that, the mechanism.
Expression of Noxa by non‐viral vectors. In the interest of future prospects of Noxa‐mediated cancer therapy, we also developed two non‐viral vectors: a non‐viral nanometer‐size polymeric vector and a cell‐penetrating peptide vector.
A polymeric vector, named polymeric micelle, was used for Noxa and Puma expression (termed micelle‐Noxa and micelle‐Puma, respectively) and we examined the effect of this gene delivery system on HBC4‐xenografted nude mice. As shown in Figure 4(a), a bolus intravenous injection of the polymeric micelle vector including HA‐Noxa or HA‐Puma cDNA resulted in a marked reduction in tumor volume for 10 days, although the tumors began to regrow afterwards (Fig. 4a).
Figure 4.
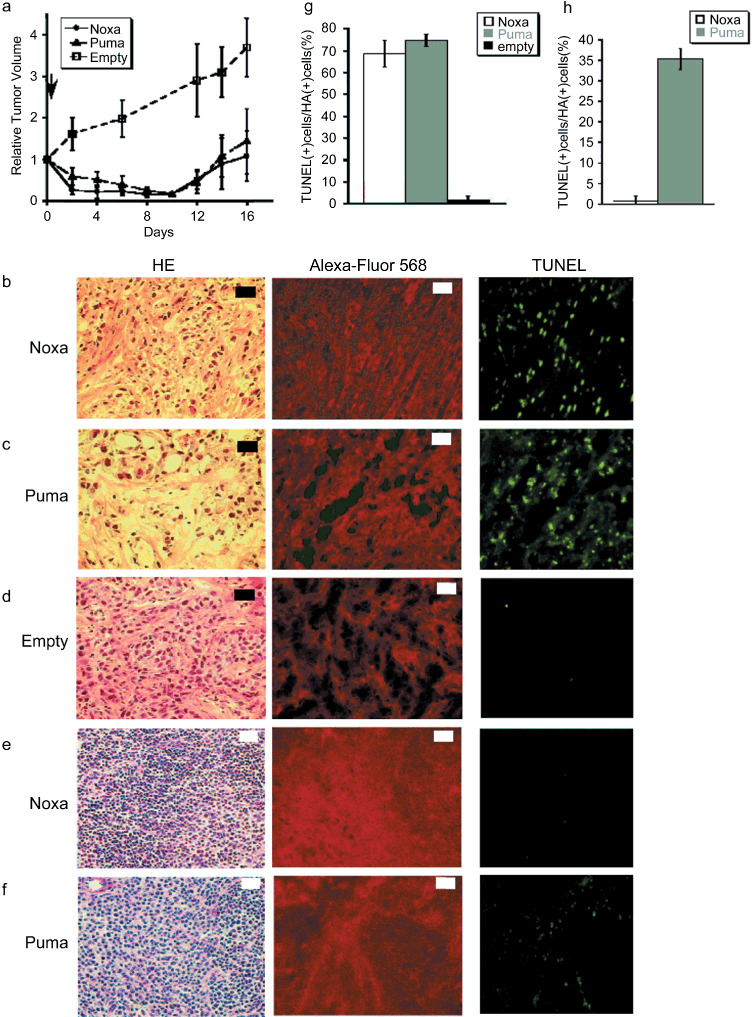
Forced expression of Noxa with viral‐free vectors. (a) The polymeric micelle vector including HA‐Noxa, HA‐Puma cDNA, or empty was injected intravenously into tumor‐transplanted mice and then the tumor size was measured. The experiments were carried out in quadruplicate and the results are presented as the mean ± SD. (b–f) Histopathological examinations of micelle vector‐injected tumor and the surrounding tissue. Micelle vectors contained cDNA of (b,e) HA‐Noxa, (c,f) HA‐Puma, and (d) empty. The sliced tumors and tissues were stained with hematoxylin–eosin (HE), anti‐HA antibody, or TdT‐mediated dUTP‐biotin nick‐end labeling (TUNEL). White bars = 200 µm, black bars = 50 µm. Induction of apoptotic cell death in micelle vectors used to treat (g) tumors and (h) surrounding tissue. The rates of TUNEL‐positive and HA‐positive cells were measured in 10 independent fields. The results are presented as the mean + SD.
As expected, histopathological examinations revealed that many cells had condensed chromatin and apparently cohering cytoplasm in the tumors that received micelle‐Noxa or micelle‐Puma (Fig. 4b–d). In fact, 68.60 ± 6.08% and 74.65 ± 2.83% of tumor cells were TUNEL‐positive for micelle‐Noxa and micelle‐Puma, respectively, whereas only 1.71 ± 1.91% of cells were TUNEL‐positive for the control vector (Fig. 4b–d,g). We also examined the spleen, liver, and kidney of mice even though all of the mice showed no mortality. There seemed to be no change in the liver and kidney, as shown by HE and TUNEL staining (data not shown), but the spleen of micelle‐HA‐Puma‐injected mice showed some TUNEL‐positive cells (35.28 ± 2.48% of HA‐positive cells; Fig. 4e,h), although the spleen of micelle‐HA‐Noxa‐injected mice rarely showed damaged cells (0.73 ± 1.26% of HA‐positive cells; Fig. 4f,h).
Finally, we tried to deliver Noxa into cells as a recombinant protein. Because most types of macromolecules cannot pass the plasma membrane of cells, we made a recombinant Noxa protein conjugated with a HIV‐1 TAT protein transduction domain, termed TAT‐PTD( 28 ) (Fig. 5a). Their purity was confirmed as a single band by SDS‐PAGE stained with Coomassie Brilliant Blue (Supporting Fig. S6). To examine the biological activity of this recombinant Noxa protein (TAT‐Noxa), we added 2 µM TAT‐Noxa or the same volume of PBS into the cell culture supernatant. Sixteen hours after, cells were harvested and the population of Annexin V‐positive cells was measured. In both HBC4 and HTB‐26 tumor cells, the treatment with TAT‐Noxa significantly increased the population of Annexin V‐positive cells compared with the PBS‐treated control cells: 38.83 ± 9.68% and 19.07 ± 5.45% of HBC4, 45.45 ± 23.21% and 14.56 ± 2.59% of HTB26 by TAT‐Noxa and PBS, respectively (Fig. 5b). On the other hand, MCF10A did not show any sensitivity for TAT‐Noxa. Equal extents of Annexin V‐positive cells were detected in TAT‐Noxa‐ or PBS‐treated HTB26 cells (Fig. 5b). These results are also consistent with the above results using the Noxa gene‐expressing vectors in that Noxa overexpression selectively induces apoptosis in cancer cells.
Figure 5.
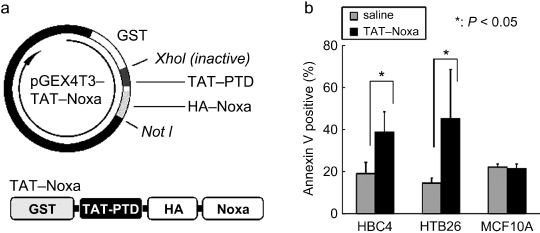
(a) Schematic structures of the TAT‐Noxa expression vector and recombinant protein. GST, glutathione S‐transferase tag for protein purification. (b) Induction of cancer cell‐selective apoptosis by treatment with TAT‐Noxa. HBC4, HTB‐26, and MCF10A cells were incubated with 2 µM TAT‐Noxa. Control cells received phosphate‐buffered saline alone. Sixteen hours later, cells were collected, stained with Annexin–fluorescein isothiocyanate and analyzed. Values shown are mean + SD from triplicate (HBC4 and MCF10A) or quadruplicate (HTB‐26) experiments.
Discussion
The present study stems from our previous finding that Noxa, a p53‐target gene, can induce apoptosis in mouse NIH3T3 cells only when the E1A oncoprotein is expressed. Puma, another target gene of p53, induces apoptosis in these cells irrespective of E1A expression. In the present study, an attempt was made to verify our hypothesis using several cancer cell lines and untransformed cells, both in vitro and in vivo. Gene therapies of some solid malignant tumors with p53 overexpression were studied in mice and in human clinical trials in many countries, resulting in only partial cures or growth inhibition instead of complete cures.( 11 , 12 , 13 , 14 , 15 ) It is worth nothing that tumors treated with Ad‐p53 often showed increased expression of p21WAF1/Cip1 mRNA,( 31 ) thereby suggesting the possibility that the overexpresssion of p53 alone may sometimes result in cell‐cycle arrest, rather than apoptosis.( 16 , 17 , 18 , 19 ) Furthermore, it is known that Ad‐p53 therapy is not effective in tumors in which p53 is not mutated.( 11 , 12 , 13 , 14 , 15 ) Our present findings suggest that the expression of Noxa by virus or by delivery vector may offer another means of cancer therapy with the advantage of selectivity to tumor cells.
Our in vivo studies demonstrated that Noxa induced apoptosis only in tumor cells, not normal mammary epithelial cells or surrounding subcutaneous cells, and that it could reduce tumor volume, without the need of any other additional therapies. This could be more effective than p53 gene therapy strategies. However, it remains to be rigorously examined whether the cancer cell‐specific induction of apoptosis by Noxa, herein tested using five cancer cell lines (HBC4, HBC5, MCF7, HTB26, HT1080) and three normal cell lines (HMEC, MCF10A, HF) can apply to other cells, particularly for fresh cancer cells. Although Puma is also potent in the induction of apoptosis, our results suggest that it might damage even normal tissues, unless the gene is specifically targeted to the tumors.( 21 )
One of the key future issues of the Noxa‐mediated induction of apoptosis is the mechanism underlying it selectivity for transformed cells. As shown in a previous study, expression of the oncogene E1A induces Bax oligomerizaion in NIH3T3 cells, and Noxa enhances this event.( 26 ) However, as shown in our present study, this does not account for the apoptosis of cancer cells studied here. We infer that the mechanism that underlies susceptibility of cancer cells to apoptosis upon overexpression of Noxa is complex and involves multiple cellular events. Although further study is obviously required, we hypothesize the following possibilities. Generally, tumor cells have mutations accumulated in multiple genes in order to gain the hallmarks of cancer.( 32 ) Such weaknesses are not selected against during tumor development in vivo, but can be the target of therapeutic intervention.( 33 ) An enhanced sensitivity to proapoptotic factors such as Noxa, provided that it is overexpressed in a cell, may be one such weakness in certain, if not all, tumors. This proposition obviously requires further verification.
In addition, because the induction of apoptosis by Noxa is mild but sufficient to elicit a response, we considered the possibility of systematically administrating Noxa in the treatment of solid tumors. Because systemic adenoviral vector administration sometimes causes immunological reaction in host patients,( 34 ) we also examined the polymeric micelle vector and a recombinant Noxa protein, TAT‐Noxa, which is fused with the protein transduction domain of HIV‐1 TAT protein.
The former vector is theoretically based on the enhanced permeability and retention effect, that is, the leakiness of tumor vessels to macromolecular agents.( 35 , 36 , 37 , 38 ) We found that a bolus intravenous injection of the micelle‐type vector including HA‐Noxa or HA‐Puma cDNA resulted in a reduction in tumor volume for 10 days, after which time the tumor began to regrow. This may be due to the feature of the xenograft of the human mammary cell line; that is, the xenograft was relatively fibrous inside the tumor. The induction rate of the polymeric micelle vectors is considered to depend on the amount of vascularity and thin fibrosis inside the tumor. We also found that micelle‐Puma injection induced apoptosis in some cells in the spleen, although there seemed to be no apoptotic cells in micelle‐Noxa‐injected mice. Because the delivery efficiency of a nanoparticle generally depends on its size, we must await the advancement of a smaller polymeric micelle vector to deliver target genes for apoptosis induction to fibrosis tumor cells. In addition, we showed that recombinant TAT‐Noxa protein induces apoptotic cell death in cancer cells. Therefore, the tumor specificity of Noxa‐induced apoptosis was also confirmed by these experiments. In recent years, various biological protein agents have become important therapeutic options in the systemic treatment of breast cancer;( 39 ) hence, we envisage that TAT‐Noxa may also be useful for cancer therapy.
In conclusion, our present study offers an interesting possibility in which to exploit Noxa as a viable target of gene therapy strategy for some malignant solid cancer cells without any other chemotherapeutic stimulus or radiation. The Noxa‐mediated cancer therapy may also be applicable to the elimination of residual cancerous cells after surgical removal of tumors. However, these possibilities must be examined further in other cancer cells, particularly with respect to its efficacy in such cells developing in vivo.
Supporting information
Fig. S1. The induction of apoptotic cell in human fibrosarcoma and untransformed fibroblast by forced expression of Noxa and Puma. (a) HT1080 human fibrosarcoma cells and (b) human fibroblasts (HF) were infected with Ad‐HA‐Noxa, Ad‐HA‐Puma, and Ad‐empty using the indicated concentrations. Sixteen hours after, the rate of apoptotic cell death was measured with Annexin V staining. (c) Forced expression of Noxa in HT1080 and HF by 5 multiplicity of infection of Ad‐HA‐Noxa. (d) Expression of Bcl‐2 family proteins. (e) Association between Noxa and Mcl‐1.
Fig. S2. Suppression of tumor growth by adenoviral forced expression of proapoptotic genes. These mice were injected five times every 3 days with (a,b) Ad‐empty and Ad‐HA‐Noxa, (c,d) Ad‐HA‐Puma or (e,f) Ad‐p53 to generate tumors. (a,c,e) Tumor xenograft mice at days 0 and 24. (b,d,f) Change in relative tumor size. Relative tumor size was estimated as described above. Adenoviral therapy was carried out on the indicated days.
Fig. S3. Histopathological examinations of generated tumors and peripheral tissue. Tumors and peripheral tissues were injected five times every 3 days with (a) Ad‐empty or (b) phosphate‐buffered saline. Eleven days after the last injection, they were frozen, sliced, and stained with hematoxylin–eosin and TdT‐mediated dUTP‐biotin nick‐end labeling. White bars = 200 µm, black bars = 50 µm.
Fig. S4. Xenograft of HTB26 tumor cells in nude mice. The xenografting, subsequent adenoviral gene therapy, and estimation of tumor volumes were carried out as described above. These mice were treated with (a) Ad‐empty and Ad‐HA‐Noxa, (b) Ad‐HA‐Puma, and (c) Ad‐p53.
Fig. S5. (a,b) Xenograft of HT1080 fibrosarcoma cell and intratumoral adenoviral gene therapy in nude mice. The tumors were injected with (a) Ad‐empty, Ad‐HA‐Noxa, or (b) Ad‐HA‐Puma three times every 3 days. The changes in tumor size are shown. (c) Oligomerization of Bax and Bak in HT1080 and human fibrosarcoma cells transfected with Ad‐empty or Ad‐HA‐Noxa. (d) Expression of activated Bax.
Fig. S6. Coomassie Brilliant Blue staining of TAT‐Noxa. Purified product was separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, followed by Coomassie Brilliant Blue staining.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Acknowledgments
We thank Mr Y. Morishita for his great support for pathological experiments; Drs M. Ohba and M.R. Kano for the polymeric micelle vector and their kind advice; Drs A. Takaoka, T. Tamura and H. Yanai for their support; Dr. K. Yoshimura for human fibroblasts and his support; Drs T. Yamashita and T. Tokino for adenovirus expressing p53; Mr M. Shishido and Ms R. Takeda for technical assistance; and Dr D. Savitsky for kind advice and critical reading of the manuscript. This work was supported by Grant‐in‐Aid for Scientific Research on Priority Areas ‘Integrative Research Toward the Conquest of Cancer’, from MEXT, the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
References
- 1. Lucas R, Keisari Y. Innovative cancer treatments that augment radiotherapy or chemo‐therapy by the use of immunotherapy or gene therapy. Recent Patents Anticancer Drug Discov 2006; 1: 201–8. [DOI] [PubMed] [Google Scholar]
- 2. Hollstein M, Sidransky D, Vogelstein B et al . p53 mutations in human cancers. Science 1991; 253: 49–53. [DOI] [PubMed] [Google Scholar]
- 3. Martin CP, Brown‐Swigart L, Evan GI. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell 2006; 127: 1323–34. [DOI] [PubMed] [Google Scholar]
- 4. Levesque AA, Eastman A. p53‐based cancer therapies: is defective p53 the Achilles heel of the tumor? Carcinogenesis 2007; 28: 13–20. [DOI] [PubMed] [Google Scholar]
- 5. Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat Rev Cancer 2002; 2: 594–604. [DOI] [PubMed] [Google Scholar]
- 6. Gartel AL, Tyner AL. The role of the cyclin‐dependent kinase inhibitor p21 in apoptosis. Mol Cancer Ther 2002; 1: 639–49. [PubMed] [Google Scholar]
- 7. Shibue T, Taniguchi T. BH3‐only proteins. Integrated control of apoptosis. Int J Cancer 2006; 119: 2036–43. [DOI] [PubMed] [Google Scholar]
- 8. Adams JM. Ways of dying: multiple pathways to apoptosis. Genes Dev 2003; 17: 2481–95. [DOI] [PubMed] [Google Scholar]
- 9. Daniel NN, Korsmeyer SJ. Cell death: critical control points. Cell 2004; 116: 205–19. [DOI] [PubMed] [Google Scholar]
- 10. Labi V, Erlacher M, Kiessling S, Villunger A. BH3‐only proteins in cell death initiation, malignant disease and anticancer therapy. Cell Death Diff 2006; 13: 1325–38. [DOI] [PubMed] [Google Scholar]
- 11. Fujiwara T, Tanaka N, Kanazawa S et al . Multicenter phase I study of repeated intratumoral delivery of adenoviral p53 in patients with advanced non‐small cell‐lung cancer. J Clin Oncol 2006; 24: 1689–99. [DOI] [PubMed] [Google Scholar]
- 12. Seth P, Katayose D, Li Z et al . A recombinant adenovirus expression wild type p53 induces apoptosis in drug‐resistant human breast cancer cells: a gene therapy approach for drug‐resistant cancers. Cancer Gene Ther 1997; 4: 383–90. [PubMed] [Google Scholar]
- 13. Shaw P, Bovey R, Tardy S et al . Induction of apoptosis by wild‐type p53 in human colon tumor‐derived cell line. Proc Natl Acad Sci USA 1992; 89: 4495–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Moon C, Oh Y, Roth JA. Current status of gene therapy for lung cancer and head and neck cancer. Clin Cancer Res 2003; 9: 5055–67. [PubMed] [Google Scholar]
- 15. Clayman GL, El‐Naggar AK, Lippman SM et al . Adenovirus‐mediated p53 gene transfer in patients with advanced recurrent head and neck squamous cell carcinoma. J Clin Oncol 1998; 16: 2221–32. [DOI] [PubMed] [Google Scholar]
- 16. Blagoskolonny MV, El‐Deiry WS. Acute overexpression of WT p53 facilitates anticancer drug‐induced death of cancer and normal cells. Int J Cancer 1998; 75: 933–40. [DOI] [PubMed] [Google Scholar]
- 17. Yoo GH, Piechocki MP, Oliver J et al . Enhancement of Ad‐p53 therapy with docetaxel in head and neck cancer. Laryngoscope 2004; 114: 1871–9. [DOI] [PubMed] [Google Scholar]
- 18. Ganjavi H, Gee M, Narendran A et al . Adenovirus‐mediated p53 gene therapy in pediatric soft‐tissue sarcoma cell lines: sensitization of cisplatin and doxorubicin. Cancer Gene Ther 2005; 12: 397–406. [DOI] [PubMed] [Google Scholar]
- 19. Cristofanilli M, Krishnamurthy S, Guerra L et al . A nonreplicating adenoviral vector that contains the wild‐type p53 transgene combined with chemotherapy for primary breast cancer: safety, efficacy, and biologic activity of a novel gene‐therapy approach. Cancer 2006; 107: 935–44. [DOI] [PubMed] [Google Scholar]
- 20. Gomez‐Manzano C, Fueyo J, Kyritsis AP et al . Characterization of p53 and p21 functional interactions in glioma cells en route to apoptosis. J Natl Cancer Inst 1997; 89: 1036–44. [DOI] [PubMed] [Google Scholar]
- 21. Ito H, Kanazawa T, Miyoshi T et al . Therapeutic efficacy of PUMA for malignant glioma cells regardless of the p53 status. Hum Gene Ther 2005; 16: 685–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Oda E, Ohki R, Murasawa H et al . Noxa, a BH3‐only member of the bcl‐2 family and candidate mediator of p53‐induced apoptosis. Science 2000; 288: 1053–8. [DOI] [PubMed] [Google Scholar]
- 23. Shibue T, Takeda K, Oda E et al . Integral role of Noxa in p53‐mediated apoptotic response. Genes Dev 2003; 17: 2233–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 2001; 7: 683–94. [DOI] [PubMed] [Google Scholar]
- 25. Yu J, Zhang L, Hwang PM et al . PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell 2001; 7: 673–82. [DOI] [PubMed] [Google Scholar]
- 26. Shibue T, Suzuki S, Okamoto H et al . Differential contribution of Puma and Noxa in dual regulation of p53‐mediated apoptotic pathways. EMBO J 2006; 25: 4952–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mansilla S, Bataller M, Portugal J. Mitotic catastrophe as a consequence of chemotherapy. Anticancer Agents Med Chem 2006; 6: 589–602. [DOI] [PubMed] [Google Scholar]
- 28. Nagahara H et al . Transduction of full‐length TAT fusion proteins into mammalian cells: TAT‐p27Kip1 induces cell migration. Nat Med 1998; 4: 1449–52. [DOI] [PubMed] [Google Scholar]
- 29. Soule HD et al . Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF‐10. Cancer Res 1990; 50: 6075–86. [PubMed] [Google Scholar]
- 30. Tait L et al . Ultrastructural and immunocytochemical characterization of an immortalized human breast epithelial cell line, MCF‐10. Cancer Res 1990; 50: 6087–94. [PubMed] [Google Scholar]
- 31. Ohtani S, Kagawa S, Tango Y et al . Quantitative analysis of p53‐targeted gene expression and visualization of p53 transcriptional administration of adenoviral p53 in vivo . Mol Cancer Ther 2004; 3: 93–100. [PubMed] [Google Scholar]
- 32. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100: 57–70. [DOI] [PubMed] [Google Scholar]
- 33. Mizuguchi H, Hayakawa T. Targeted adenovirus vectors. Hum Gene Ther 2004; 15: 1034–44. [DOI] [PubMed] [Google Scholar]
- 34. Raper ES, Chirmule F, Lee FS et al . Fatal systemic inflammatory response syndrome in an ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol Genet Metab 2003; 80: 148–58. [DOI] [PubMed] [Google Scholar]
- 35. Fukushima S, Miyata K, Nishiyama N et al . PEGylated polyplex micelles from triblock catiomers with spatially ordered layering of condensed pDNA and buffering units for enhanced intracellular gene delivery. J Am Chem Soc 2005; 127: 2810–11. [DOI] [PubMed] [Google Scholar]
- 36. Nishiyama N, Kataoka K. Current state, achievements, and future prospects of polymeric micelles as nanocarriers for drug and gene delivery. Pharmaco Ther 2006; 112: 630–48. [DOI] [PubMed] [Google Scholar]
- 37. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res 1986; 46: 6387–92. [PubMed] [Google Scholar]
- 38. Maeda H, Matsumura Y. Tumoritropic and lymphotropic principles of macromolecular drugs. Crit Rev Ther Drug Carrier Syst 1989; 6: 193–210. [PubMed] [Google Scholar]
- 39. Chu D, Lu J. Novel therapies in breast cancer: what is new from ASCO 2008. J Hematol Oncol 2008; 1: 16–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. The induction of apoptotic cell in human fibrosarcoma and untransformed fibroblast by forced expression of Noxa and Puma. (a) HT1080 human fibrosarcoma cells and (b) human fibroblasts (HF) were infected with Ad‐HA‐Noxa, Ad‐HA‐Puma, and Ad‐empty using the indicated concentrations. Sixteen hours after, the rate of apoptotic cell death was measured with Annexin V staining. (c) Forced expression of Noxa in HT1080 and HF by 5 multiplicity of infection of Ad‐HA‐Noxa. (d) Expression of Bcl‐2 family proteins. (e) Association between Noxa and Mcl‐1.
Fig. S2. Suppression of tumor growth by adenoviral forced expression of proapoptotic genes. These mice were injected five times every 3 days with (a,b) Ad‐empty and Ad‐HA‐Noxa, (c,d) Ad‐HA‐Puma or (e,f) Ad‐p53 to generate tumors. (a,c,e) Tumor xenograft mice at days 0 and 24. (b,d,f) Change in relative tumor size. Relative tumor size was estimated as described above. Adenoviral therapy was carried out on the indicated days.
Fig. S3. Histopathological examinations of generated tumors and peripheral tissue. Tumors and peripheral tissues were injected five times every 3 days with (a) Ad‐empty or (b) phosphate‐buffered saline. Eleven days after the last injection, they were frozen, sliced, and stained with hematoxylin–eosin and TdT‐mediated dUTP‐biotin nick‐end labeling. White bars = 200 µm, black bars = 50 µm.
Fig. S4. Xenograft of HTB26 tumor cells in nude mice. The xenografting, subsequent adenoviral gene therapy, and estimation of tumor volumes were carried out as described above. These mice were treated with (a) Ad‐empty and Ad‐HA‐Noxa, (b) Ad‐HA‐Puma, and (c) Ad‐p53.
Fig. S5. (a,b) Xenograft of HT1080 fibrosarcoma cell and intratumoral adenoviral gene therapy in nude mice. The tumors were injected with (a) Ad‐empty, Ad‐HA‐Noxa, or (b) Ad‐HA‐Puma three times every 3 days. The changes in tumor size are shown. (c) Oligomerization of Bax and Bak in HT1080 and human fibrosarcoma cells transfected with Ad‐empty or Ad‐HA‐Noxa. (d) Expression of activated Bax.
Fig. S6. Coomassie Brilliant Blue staining of TAT‐Noxa. Purified product was separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, followed by Coomassie Brilliant Blue staining.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item