

Abstract
Helicobacter pylori (H. pylori) infection causes chronic gastritis and is also related to gastric carcinoma. The present study focused on severity of H. pylori‐induced gastritis as a determinant of carcinogenesis. Seven‐week‐old male Mongolian gerbils were inoculated with H. pylori at experimental weeks 0, 12, or 18, then given N‐methyl‐N‐nitorosourea (MNU) from weeks 20–40. At week 70, stomachs were then excised for histological examination 70, 58, or 52 weeks after H. pylori inoculation, respectively (Groups A, B, and C for long‐, middle‐, and short‐term). The respective incidences of glandular stomach adenocarcinomas were 65.0% (13/20), 20.0% (2/10), and 23.0% (3/13) (P < 0.05). Higher scores of infiltration of inflammatory cells, hyperplasia, intestinal metaplasia and mucosal bromodeoxyuridine (BrdU) labeling index in antrum and corpus mucosa, were seen in group A than B or C (P < 0.05) and serum anti‐H. pylori IgG titer and gastrin levels were also significantly higher, along with mRNA levels for mucosal interleukin‐1β (IL‐1β), tumor necrosis factor‐α (TNF‐α), cyclooxygenase‐2 (COX‐2), and inducible nitric oxide synthase (iNOS). The results demonstrated the term and severity of H. pylori infection to play important roles in gastric carcinogenesis, with essential involvement of chronic inflammation, especially increased rates of cell proliferation, in H. pylori‐associated carcinogenesis. (Cancer Sci 2007; 98: 478–483)
A large body of evidence points to a close relationship between Helicobacter pylori (H. pylori) infection and the development of chronic gastritis, peptic ulcers and gastric carcinomas.( 1 , 2 ) Recently the concept that inflammation is a critical component of tumor progression has received a great deal of attention. Many cancers arise from sites of infection, chronic irritation and inflammation,( 3 ) and H. pylori can induce cytokine release from epithelial cells which leads to recruitment of inflammatory cells. Reactive oxygen species generated by neutrophils causes lipid peroxidation, as well as protein and DNA oxidation. In addition, persistent inflammation has an impact on cellular turnover.( 4 , 5 )
It is clear that not all H. pylori infections are linked to gastric cancer, since approximately 50% of the world population is infected, and only a very small minority of infected subjects suffer from an associated neoplasm. The reasons for this phenomenon are unknown although different mechanisms have been proposed.( 6 , 7 ) In humans, studies have shown that H. pylori gastritis is associated with increased risk, and this decreases following cure of bacterial infection.( 8 ) The gastritis model in Mongolian gerbils has advantages in that the human pathogen H. pylori is used and the consequent chronic gastritis, intestinal metaplasia, and gastric cancer closely mimic those in humans.( 9 , 10 ) Previous studies showed that H. pylori infection induces chronic active gastritis, expression of various cytokines and regenerative epithelial cell responses in gerbils.( 11 , 12 ) However, the level of gastric mucosal injury has received only limited attention in studies of H. pylori‐induced gastritis. For this study, we therefore designed an experiment with different timing and periods of infection in animals of the same ages. Our hypothesis was that early inoculation and long‐term colonization of H. pylori would result in more severe chronic active gastritis, and therefore a greater yield of gastric carcinomas.
Materials and Methods
Animal and H. pylori inoculation. Seven‐week‐old male specific‐pathogen‐free Mongolian gerbils (Meriones unguiculatus; MGS/Sea) were purchased from Seac Yoshitomi, Ltd. (Fukuoka, Japan) and maintained in an air‐conditioned biohazard room with free access to a commercial rodent diet (Oriental CRF‐1; Oriental Yeast Co., Tokyo, Japan) and water ad libitum. H. pylori ATCC 43504 [American Type Culture Collection (ATCC), Manassas, VA, USA] were grown in Brucella broth supplemented with 7% fetal calf serum at 37°C under microaerophilic conditions for 48 h. Animals were inoculated with 0.8 mL of broth culture containing 1 × 108 colony‐forming units (cfu) of H. pylori by gastric intubation three times at 48‐h intervals. The animals were fasted for 24 h before the first inoculation. All experiments and procedures carried out on the animals were approved by the Animal Care Committee of Aichi Cancer Center Research Institute.
Chemical. N‐methyl‐N‐nitrosourea (MNU) (Sigma Chemical Co., St Louis, MO) was dissolved in distilled water at a concentration of 10 p.p.m. (solution was freshly prepared three times per week) for administration in light‐shielded bottles as drinking water ad libitum.
Experimental protocol. The animals were divided into six H. pylori ‐inoculated and two control groups (Fig. 1). H. pylori were inoculated for groups A and E at 0 weeks, groups B and F at 12 weeks, and groups C and G at 18 weeks, representative of early long‐term, middle, and short‐term infection, respectively. Since the early long‐term Group A was predicated that it might be of poorer survival rate at the end of the experiment, more animals were added into Group A than Groups B and C. Groups D and H received Brucella broth without H. pylori. At 20 weeks, Groups A, B, C, and D were given MNU. Before the MNU administration, subgroups of animals in groups A, B, and C (a1, b1, and c1) were sacrificed at 20, 12, and 2 weeks post‐infection, respectively. For these gerbils, 5′‐bromo‐2′‐deoxyuridine (BrdU) at a dose of 100 mg/kg, was injected intraperitoneally, 60 min before the sacrifice. All animals were subjected to deep ether anesthesia after 24 h fasting, laparotomized, and exsanguinated from the inferior vena cava, after which their stomachs were excised.
Figure 1.

Experimental design. Animals, 7‐week‐old male Mongolian gerbils, ▾, H. pylori (i.g.), ▪, N‐methyl‐N‐nitorosourea in drinking water at the concentration of 10 p.p.m.
Histopathological evaluation. Multiple 4 µm‐thick histologic sections were obtained from routinely processed 4% paraformaldehyde‐fixed and paraffin‐embedded tissues. Sections were stained with hemotoxylin and eosin (H&E) or with Alcian blue (pH 2.5)‐periodic acid Schiff (AB‐PAS) for detection of mucin‐containing cells. The glandular mucosa of the antrum and corpus was examined histologically for inflammatory and epithelial changes. Active chronic gastritis was characterized by infiltration of neutrophils, mononuclear cells, hyperplasia, and intestinal metaplasia. The degree of change was graded on a scale from 0 to 3, [0 (normal), 1 (mild), 2 (moderate), and 3 (marked)], based on the Updated Sydney System.( 13 ) Epithelial cell proliferation was assessed by BrdU labeling, visualized using a mouse monoclonal anti‐BrdU antibody (1:50, Dako, Glostrup, Denmark) as described previously.( 14 ) The numbers of BrdU‐labeled cells in the gastric mucus of antrum and corpus were counted under a microscope with a ×40 objective lens, and indices were determined as the mean percentages of positive cells among totals of more than 1000 cells. Mucosal thickness was assessed using NIH Image version J1.272 (National Institutes of Health, USA).
Analysis of cytokines by real time quantitative PCR. Total RNA was extracted from the glandular stomach mucosa at the border between the antrum and corpus using an RNA extraction kit (Isogen, Nippon Gene, Tokyo, Japan). After DNase treatment, first strand cDNAs were synthesized using the Thermoscript RT‐PCR System (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. Relative quantitative PCR of interleukin‐1β (IL‐1β), tumor necrosis factor‐α (TNF‐α), cyclooxygenase‐2 (COX‐2), and inducible nitric oxide synthase (iNOS),( 15 ) was performed with the LightCycler system (Roche Diagnostics, Mannheim, Germany), using the gerbil‐specific glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) gene as an internal control.( 16 ) PCR was performed basically as described earlier using a QuantiTect SYBR Green PCR (QIAGEN) kit with optimal Mg2+ concentration at 2.5 mM.( 17 ) The 5′‐ and 3′‐primer sequences are listed in Table 1. Specificity of the PCR reaction was confirmed using the melting program provided with the LightCycler software. To further confirm that there was no obvious primer dimer formation or amplification of any extra bands, the samples were electrophoresed in 3% agarose gels and visualized with ethidium bromide after the LightCycler reaction. Relative quantification was performed as previously established using an internal control without the necessity for external standards.( 17 ) The expression levels of cytokine mRNAs were expressed relative to 1.0 in the control group (group H).
Table 1.
PCR primers used for real‐time quantitative RT‐PCR analysis
| Gene | Primer | Product size (bp) |
|---|---|---|
| GAPDH | F: 5′‐AACGGCACAGTCAAGGCTGAGAACG‐3′ R: 5′‐CAACATACTCGGCACCGGCATCG‐3′ | 118 |
| IL‐1β | F: 5′‐TGACTTCACCTTGGAATCCGTCTCT‐3′ R: 5′‐GGCAACAAGGGAGCTCCATCAC‐3′ | 91 |
| TNF‐α | F: 5′‐GCTGCCCCCACCTCGTGCTC‐3′ R: 5′‐CTTGATGGCAGACAGGAGGCTGACC‐3′ | 89 |
| COX‐2 | F: 5′‐GCCGTCGAGTTGAAAGCCCTCTACA‐3′ R: 5′‐CCCCGAAGATGGCGTCTGGAC‐3′ | 97 |
| iNOS | F: 5′‐GCATGACCTTGGTGTTTGGGTGCC‐3′ R: 5′‐GCAGCCTGTGTGAACCTGGTGAAGC‐3′ | 110 |
GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; IL‐1β, interleukin‐1β; TNF‐α, tumor necrosis factor‐α; COX‐2, cyclooxygenase‐2; iNOS, inducible nitric oxide synthase; F, forward; R, reverse.
Serology. Serum samples were used to measure the titers of anti‐H. pylori IgG antibodies by enzyme‐linked immunosorbent assay, and serum gastrin levels were measured at 70 weeks using radioimmunoassay. The antibody titers were expressed using an arbitrary index, and values greater than the cut‐off of 1.5 were considered to be positive for H. pylori infection.( 18 )
Statistical analyses. Results for the gastritis scores, BrdU labeling indices, mRNA expression levels, and antibody and gastrin levels are given as means ± SE. The Fisher's exact test and the Bonferroni multiple‐comparison test were performed to establish the significance of differences with the cut‐off at P < 0.05.
Results
Histopathology. The survival rates in all groups were >85%, with no differences among groups. All gastric mucosal specimens from control gerbils demonstrated a normal histomorphology. Histological findings for gastric mucosal specimens from H. pylori‐infected gerbils are summarized in Table 2. At 20 weeks, the early/long‐term H. pylori‐infected gerbils (group a1) showed greater lymphoplasmocytic infiltration and submucosal lymphoid follicle formation than the middle and short‐term H. pylori‐infected groups in the antral mucosa (Table 2; Fig. 2a,b). At 70 weeks, there was a change over time in topography of the gastritis from predominantly antral gastritis to pangastritis in H. pylori‐infected gerbils. Lesions of gastric mucosa were more marked in the long‐term infection groups than in the middle and short‐term infected groups (Table 2; Fig. 2c,d). Heterotopic proliferative glands (HPGs) were observed in all H. pylori‐infected gerbils at 77 weeks,( 19 ) limited to the antrum and the junctional region between the antrum and the body (Fig. 2c).
Table 2.
Histopathological response in gerbils
| Group | No. | Antrum | Corpus | Anti‐Hp IgG titer (A.I) | Serum gastin levels (pg/mL) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mucosal thinckness (mm) | Neutrophils | Mono‐nuclear cells | Hyperplasia | Intestinal metaplasia | BrdU labeling index (%) | Mucosal thinckness (mm) | Neutrophils | Mono‐nuclear cells | Hyperplasia | Intestinal metaplasia | BrdU labeling index (%) | ||||
| A | 20 | 0.82 ± 0.25* | 2.1 ± 0.6* | 3.0 ± 0.2* | 2.9 ± 0.3* | 2.3 ± 0.7* | 24.6 ± 5.9* | 1.04 ± 0.26* | 1.3 ± 0.3* | 2.4 ± 0.3* | 2.4 ± 0.2* | 1.1 ± 0.4* | 14.1 ± 3.3* | 288.1 ± 44.7* | 1235.1 ± 373.1* |
| a1 | 6 | 0.35 ± 0.09 † | 2.9 ± 0.2 ‡ | 3.0 ± 0.0 ‡ | 1.3 ± 0.3 † | 0.0 ± 0.0 | 20.2 ± 6.3 ‡ | 0.64 ± 0.07 † | 1.0 ± 0.1 † | 1.0 ± 0.3 ‡ | 1.1 ± 0.2 ‡ | 0.0 ± 0.0 | 4.5 ± 1.5 | 14.3 ± 3.5 ‡ | ND |
| B | 10 | 0.48 ± 0.20 | 1.7 ± 0.3 | 2.3 ± 0.2 | 1.8 ± 0.6 | 0.7 ± 0.4 | 13.4 ± 4.2 | 0.77 ± 0.29 | 1.2 ± 0.3 | 1.6 ± 0.6 | 1.6 ± 0.5 | 0.3 ± 0.3 | 9.6 ± 2.1 | 175.8 ± 23.1 | 323.6 ± 114.4 |
| b1 | 7 | 0.30 ± 0.11 | 2.4 ± 0.2 | 2.4 ± 0.4 | 1.4 ± 0.2 | 0.0 ± 0.0 | 10.4 ± 3.8 | 0.54 ± 0.06 | 1.0 ± 0.0 | 1.1 ± 0.2 | 1.1 ± 0.3 | 0.0 ± 0.0 | 4.4 ± 1.0 | 8.8 ± 1.4 | ND |
| C | 13 | 0.60 ± 0.17 | 1.4 ± 0.3 | 2.1 ± 0.3 | 1.8 ± 0.3 | 0.6 ± 0.2 | 8.3 ± 1.8 | 0.77 ± 0.21 | 1.1 ± 0.0 | 1.5 ± 0.4 | 1.4 ± 0.2 | 0.5 ± 0.4 | 9.3 ± 2.3 | 137.1 ± 19.6 | 395.0 ± 68.3 |
| c1 | 7 | 0.19 ± 0.02 | 1.0 ± 0.1 | 1.0 ± 0.1 | 0.7 ± 0.3 | 0.0 ± 0.0 | 7.2 ± 1.8 | 0.45 ± 0.08 | 0.5 ± 0.0 | 0.5 ± 0.0 | 0.5 ± 0.0 | 0.0 ± 0.0 | 3.5 ± 1.3 | 0.8 ± 0.3 | ND |
| D | 16 | 0.20 ± 0.04 | 0.0 ± 0.0 | 0.4 ± 0.1 | 0.0 ± 0.0 | 0.0 ± 0.1 | 4.4 ± 1.9 | 0.42 ± 0.04 | 0.0 ± 0.0 | 0.3 ± 0.1 | 0.0 ± 0.0 | 0.0 ± 0.0 | 3.0 ± 0.8 | 1.4 ± 0.3 | 206.2 ± 18.6 |
| E | 5 | 0.62 ± 0.13 § | 1.9 ± 0.2 ¶ | 3.0 ± 0.0 ¶ | 2.8 ± 0.3 ¶ | 2.7 ± 0.3 § | 20.4 ± 4.8 § | 1.02 ± 0.18 ¶ | 1.5 ± 0.4 ¶ | 2.4 ± 0.2 ¶ | 2.0 ± 0.4 ¶ | 1.1 ± 0.4 ¶ | 13.7 ± 1.6 § | 498.4 ± 74.0 § | 807.6 ± 117.0 § |
| F | 5 | 0.41 ± 0.09 | 1.6 ± 0.2 | 2.5 ± 0.4 | 2.3 ± 0.3 | 1.0 ± 0.0 | 10.2 ± 2.4 | 0.86 ± 0.05 | 1.2 ± 0.3 | 2.0 ± 0.0 | 1.7 ± 0.6 | 0.7 ± 0.3 | 5.9 ± 2.1 | 115.2 ± 36.5 | 396.3 ± 61.6 |
| G | 5 | 0.34 ± 0.08 | 1.3 ± 0.3 | 2.0 ± 0.0 | 1.9 ± 0.4 | 0.6 ± 0.2 | 4.5 ± 1.6 | 0.71 ± 0.22 | 1.0 ± 0.1 | 1.4 ± 0.2 | 1.0 ± 0.0 | 0.5 ± 0.1 | 3.9 ± 1.6 | 102.0 ± 37.5 | 375.2 ± 89.1 |
| H | 6 | 0.19 ± 0.03 | 0.0 ± 0.0 | 0.3 ± 0.1 | 0.0 ± 0.0 | 0.0 ± 0.0 | 2.1 ± 0.4 | 0.44 ± 0.06 | 0.0 ± 0.0 | 0.3 ± 0.1 | 0.0 ± 0.0 | 0.0 ± 0.0 | 2.7 ± 0.9 | 1.3 ± 0.4 | 244.7 ± 8.8 |
P < 0.05 to group B and C;
P < 0.05 to group b1;
P < 0.05 to group b1 and c1;
P < 0.05 to group F and G;
P < 0.05 to group G; ND, not determined; Hp, H.pylori; BrdU, bromodeoxyuridine
Figure 2.
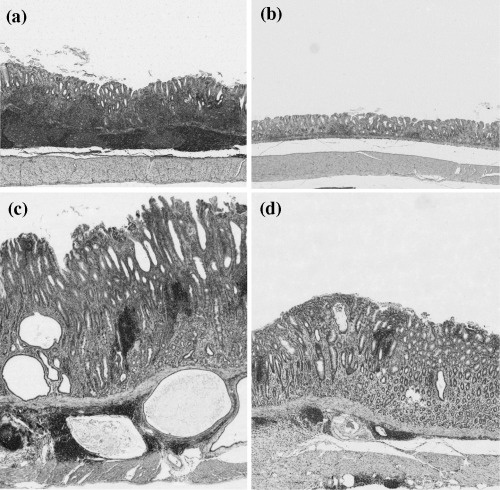
Histopathological findings of pyloric mucosa after inoculation of H. pylori. (a) Marked lymphoid follicle formation in a group a1 gerbil (H&E; × 25); (b) Mild gastritis in a group c1 gerbil (H&E; × 25); (c) Marked gastritis with heterotopic proliferative glands (HPGs) at 70 weeks post‐infection (group A, H&E; × 25); (D) Moderate gastritis in a group C gerbil (H&E; × 25).
Incidence of adenocarcinomas. Data for adenocarcinomas are summarized in Table 3. Both well differentiated (Fig. 3a) and signet ring‐cell carcinomas (Fig. 3b) were found, mainly located in the antrum or at the border between the antrum and the corpus. Whereas 13 of 20 (65%) in the long‐term H. pylori + MNU group (group A) had adenocarcinomas in the glandular stomach, this was the case for only two of 11 (20.0%) in the middle‐term H. pylori + MNU group (group B), and three of 13 (23.1%) in the late short‐term H. pylori + MNU group (group C). The difference was statistically significant. In the MNU‐alone group (group D), H. pylori‐alone groups (groups E, F, and G), and controls (group H), no tumors developed in the glandular stomach.
Table 3.
Incidence of gastric carcinoma
| Groups | Age colonized | Duration of colonization | Age sacrificed | No. of animals | Carcinoma | ||
|---|---|---|---|---|---|---|---|
| Dif. | Undif. | Incidence (%) | |||||
| A Hp 20 w + MNU | 7 w | 70 w | 77 w | 20 | 12 | 1 | 13 (65.0)* |
| a1 before MNU treatment | 7 w | 20 w | 27 w | 6 | 0 | 0 | 0 |
| B Hp 8 w + MNU | 19 w | 58 w | 77 w | 10 | 2 | 0 | 2 (20.0) |
| b1 before MNU treatment | 19 w | 8 w | 27 w | 7 | 0 | 0 | 0 |
| C Hp 2 w + MNU | 25 w | 52 w | 77 w | 13 | 2 | 1 | 3 (23.1) |
| c1 before MNU treatment | 25 w | 2 w | 27 w | 7 | 0 | 0 | 0 |
| D MNU | (‐) | (‐) | 77 w | 16 | 0 | 0 | 0 |
| E Hp 70 w | 7 w | 70 w | 77 w | 5 | 0 | 0 | 0 |
| F Hp 58 w | 19 w | 58 w | 77 w | 5 | 0 | 0 | 0 |
| G Hp 52 w | 25 w | 52 w | 77 w | 5 | 0 | 0 | 0 |
| H Control | (‐) | (‐) | 77 w | 6 | 0 | 0 | 0 |
P < 0.05 between groups A, B and C, P < 0.05 to group B and group C, P < 0.001 to group D, by Fisher"s exact test. Hp, H. pylori infection (i.g.); Dif, differentiated adenocarcinoma; Undif, undifferentiated adenocarcinoma. MNU, N‐methyl‐N‐nitorosourea.
Figure 3.
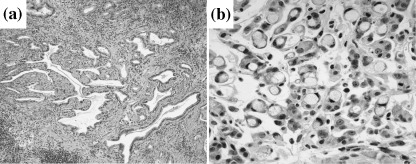
Histology of adenocarcinomas. (a) Well differentiated adenocarcinoma in glandular stomach at 70 weeks post‐H. pylori infection in a group A gerbil (H&E; × 80). (b) Signet ring‐cell carcinoma at 70 weeks post‐infection in a group A gerbil (H&E; × 400).
BrdU labeling indices for epithelial cells. At 20 weeks, antrum BrdU labeling indices in group a1 were significantly greater than in groups b1 and c1 (P < 0.01), with no significant increase in the corpus. At 70 weeks, both antrum and corpus BrdU labeling indices in the long‐term infection groups (A and E) were significantly elevated as compared to middle and short infection groups (B and F, and C and F) (P < 0.05) (Table 1).
Expression of cytokine mRNAs in gastric mucosa of gerbils. IL‐1β, TNF‐α, COX‐2 and iNOS mRNA levels were very low in uninfected gerbils at 77 weeks of age. Infection with the H. pylori resulted in significant increase in transcripts at 20 and 70 weeks in long‐term infection gerbils compared with middle and short‐term infection gerbils (Fig. 4). The mRNA levels at 70 week post‐infection were greater than at 20 weeks post‐infection. In H. pylori‐infected gerbils, IL‐1β, TNF‐α, COX‐2, and iNOS mRNA levels correlated significantly with chronic active inflammation scores in the gastric mucosa.
Figure 4.
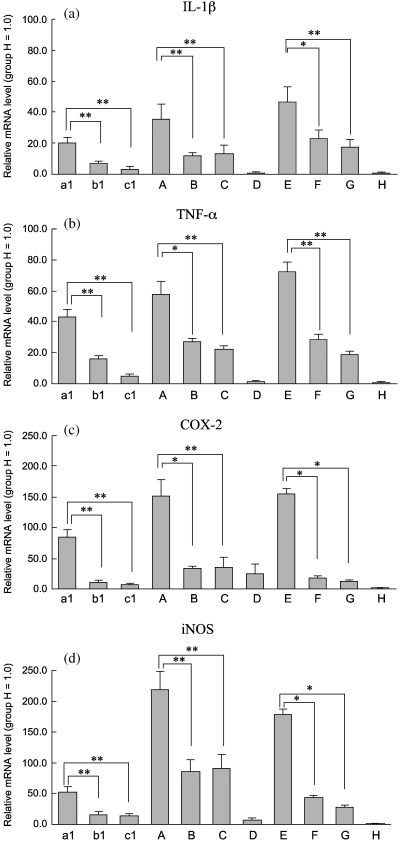
Relative expression levels of IL‐1β, TNF‐α, COX‐2 and iNOS mRNAs in glandular stomachs of gerbils. (a) IL‐1β; (b) TNFα; (c) COX‐2; (d) iNOS. Values were set at 1.0 in group H and expressed as mean ± SE relative values. Note increase in relative IL‐1β, TNFα, COX‐2 and iNOS mRNA expression levels in group A as compared to groups B and C; in group a1 as compared to groups b1 and c1; in group E as compared to groups F and G. *P < 0.05 and **P < 0.01, by the Bonferroni multiple‐comparison test.
Anti‐H. pylori IgG titers and gastrin levels. The serum anti‐H. pylori IgG titers and gastrin levels were significantly higher in the long‐term infection group than in the middle and short‐term groups (Table 2).
Discussion
At 20 and 70 weeks post‐infection, histologic examination revealed more severe neutrophil and mononuclear cells infiltration and submucosal lymphoid follicle formation in the antrum mucosa in early/long‐term groups than in the middle and short‐term H. pylori‐infected groups, and this correlated with the yield of adenocarcinomas. The results thus provide support for the conclusion that infiltration of polymorphonuclear neutrophils and mucosa‐associated lymphoid tissue, as well as damage to the epithelial cells, characterizes the first phase of gastric carcinogenesis in this model.( 20 ) Neutrophil infiltration is an almost invariable finding in H. pylori‐associated gastritis and is topographically related to H. pylori colonization. It is now evident that inflammatory cells have powerful effects on gastric mucosal injury, neutrophils being recruited to the gastric mucosa and generate reactive oxygen and nitrogen species and proteases.( 21 ) However, neutrophils are not able to kill the H. pylori that live in the gastric mucus, and compounds produced by activated neutrophils themselves are potentially harmful for normal tissue. Active neutrophil infiltration in gastric H. pylori infection may contribute to the increased levels of mutation also observed in Big Blue transgenic mice.( 22 )
The type of host immune response against H. pylori is considered crucial for the outcome of the infection.( 23 ) One possible mechanism involves up‐regulation of inflammatory responses through increase of synthesis and release of pro‐inflammatory mediators, such as iNOS and COX‐2, via effects on nuclear factor κB (NF‐κB).( 24 , 25 ) A predominant H. pylori‐specific Th1 response, characterized by high TNF‐α, IL‐1β and interferon‐γ (IFN‐γ) production is associated with gastritis and peptic ulcer.( 26 ) We found mRNA levels of mucosal IL‐1β and TNF‐α mRNA levels were significantly higher in gerbils with long‐term H. pylori‐infection than in those with middle and the short‐term infection. Yamaoka et al. reported that IL‐1β and IFN‐γ play important roles in the acute phase of pyloric inflammation in H. pylori‐infected gerbils.( 12 ) H. pylori stimulates NF‐κB activation and chemokine interleukin‐8 (IL‐8) expression in gastric epithelial cells. Epithelial IL‐8 can be induced not only by direct stimulation of H. pylori, but also following exposure to the endogenous proinflammatory mediators IL‐1 and TNF‐α in the gastric mucosa.( 27 , 28 )
COX‐2 and iNOS may play important roles in stomach tumor growth and progression. COX‐2 is frequently undetectable in normal tissues but is induced by cytokines, growth factors, reactive oxygen species and tumor promoters,( 29 , 30 ) and catalyzes the committed step in the conversion of arachidonic acid to protumorigenic eicosanoids, such as prostaglandin E2, which are involved in the maintenance of tumor integrity.( 30 , 31 ) Several reports have documented H. pylori up‐regulation of COX‐2 mRNA expression and release of prostaglandin E2 from a gastric cancer cell line, as well as in the gastric mucosa of gerbil models and in humans.( 32 , 33 , 34 ) In the present study, the fact that long‐term H. pylori infection significantly up‐regulated COX‐2 expression is of clear interest.
A number of previous findings also suggest that iNOS contributes to H pylori‐associated gastric carcinogenesis in mice and man. Increased iNOS activity has been observed in patients with chronic gastritis and gastric adenocarcinoma patients,( 35 ) nitric oxide (NO) endogenously produced by this family of enzymes causing mutations and DNA deamination.( 36 , 37 ) Our present observations suggest that COX‐2 and iNOS are both induced in H. pylori‐positive gastritis and thus may modulate the inflammation and alterations in epithelial cell growth that occur with this disease. The increase in proliferative activity in the mucosa is another feature of interest. At 20 and 70 weeks post‐infection, BrdU‐labeled cells represented almost 20–25% of the antrum epithelial cells in long‐term infected gerbils (groups a1 and A). Early inoculation and long‐term H. pylori infection thus appeared to increase epithelial cell apoptosis and proliferation, and this would be expected to lead to increased susceptibility to carcinogens.
Immunohistochemical study showed increased expression of iNOS and COX‐2 genes in H. pylori gastritis in humans: iNOS protein was detected in epithelium, endothelium, and lamina propria inflammatory cells, and COX‐2 protein localized to mononuclear and fibroblast cells in the lamina propria.( 24 ) Thus, iNOS as well as other inflammatory cytokines may be expressed not only in inflammatory cells but also in epithelial cells in the H. pylori‐infected Mongolian gerbils.
Group A showed lower anti‐H. pylori IgG titer than that of the MNU untreated group (group E), although not statistically significant (P < 0.08). This tendency was not consistent between H. pylori + MNU and H. pylori alone groups (Groups B vs. F; Groups C vs. G) in anti‐H. pylori IgG titers and gastrin levels (Table 2). Thus, the effects of MNU on mucosal atrophy could not be considered significant.
Of note, gerbils infected with H. pylori developed an antral‐predominant gastritis, which progressed to corpus gastritis at 70 weeks. This pattern is typical of what is seen in humans living in regions of high gastric cancer incidence, and explains why H. pylori‐related gastric cancer in gerbils develops most often in the antrum.( 38 ) In the present study, we examined the inflammation status at two experimental timing points: 20 and 70 weeks. Among the 20‐week groups (Groups a1, b1, and c1), Group a1 showed the highest inflammatory and proliferative responses, which might affect MNU‐induced tumorigenesis. Thus, it could be concluded that the severity of H. pylori‐induced inflammation may play important roles in gastric carcinogenesis.
Long‐term H. pylori infection was here accompanied by significantly increased anti‐H. pylori serum IgG antibody titer, highlighting the importance of the immune response of the host in the development of H. pylori‐related gastric lesions. The results also suggest that the anti‐H. pylori serum IgG antibody titer may be used as a marker of the severity of the H. pylori‐infected chronic active gastritis.( 18 ) A previous study demonstrated that acid secretion is decreased in gerbils infected with H. pylori.( 39 ) In the present case, long‐term H. pylori infection induced chronic pangastritis and may have suppressed parietal cell function, causing compensatory gastrin release corresponding with a lower gastric acid secretion. Furthermore, increased gastrin levels could contribute to gastric barrier dysfunction in H. pylori infection.( 40 ) Wang et al. found that chronic hypergastrinemia can synergize with H. pylori infection and contribute to eventual parietal cell loss and progression to gastric cancer in insulin‐gastrin (INS‐GAS) transgenic mice.( 41 )
In summary, our study shows that the severity of chronic gastritis induced by pathogen H. pylori is linked with glandular gastric carcinogenesis. The gastric mucosal injury demonstrated in the long‐standing H. pylori‐infected gerbil clearly can lead to increased susceptibility to carcinogenic substances and also contribute to immune responses, perpetuation of mucosal inflammation and cancer development. This phenomenon is not only dependent on the timing and age at H. pylori infection, but also the period of infection.( 42 ) The pathological changes present at the early stage of H. pylori infection seems to persist and become aggravated during the life span of the animals, thus contributing to the multifactorial processes underlying gastric neoplasia.
Acknowledgments
This work was supported in part by a Grant‐in‐Aid for Cancer Research from the Ministry of Health, Labor and Welfare, Japan, and a Grant‐in‐Aid from the Ministry of Education, Culture, Science, Sports and Technology of Japan. X. C. was a recipient of a postdoctoral fellowship from the Japan Society for the Promotion of Science during the performance of this research.
References
- 1. Kuipers EJ, Uyterlinde AM, Pena AS et al. Long‐term sequelae of Helicobacter pylori gastritis. Lancet 1995; 345: 1525–8. [DOI] [PubMed] [Google Scholar]
- 2. Parsonnet J, Friedman GD, Vandersteen DP et al. Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med 1991; 325: 1127–31. [DOI] [PubMed] [Google Scholar]
- 3. Coussens LM, Werb Z. Inflammation and cancer. Nature 2002; 420: 860–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yoshida N, Granger DN, Evans DJ Jr. et al. Mechanisms involved in Helicobacter pylori‐induced inflammation. Gastroenterology 1993; 105: 1431–40. [DOI] [PubMed] [Google Scholar]
- 5. Yoshikawa T, Naito Y. The role of neutrophils and inflammation in gastric mucosal injury. Free Radic Res 2000; 33: 785–94. [DOI] [PubMed] [Google Scholar]
- 6. Huang JQ, Sridhar S, Chen Y, Hunt RH. Meta‐analysis of the relationship between Helicobacter pylori seropositivity and gastric cancer. Gastroenterology 1998; 114: 1169–79. [DOI] [PubMed] [Google Scholar]
- 7. Uemura N, Okamoto S, Yamamoto S et al. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med 2001; 345: 784–9. [DOI] [PubMed] [Google Scholar]
- 8. Rad R, Brenner L, Bauer S et al. CD25(+) /Foxp3(+) T cells regulate gastric inflammation and Helicobacter pylori colonization in vivo. Gastroenterology 2006; 131: 525–37. [DOI] [PubMed] [Google Scholar]
- 9. Hirayama F, Takagi S, Yokoyama Y, Iwao E, Ikeda Y. Establishment of gastric Helicobacter pylori infection in Mongolian gerbils. J Gastroenterol 1996; 31 (Suppl 9): 24–8. [PubMed] [Google Scholar]
- 10. Tatematsu M, Yamamoto M, Shimizu N et al. Induction of glandular stomach cancers in Helicobacter pylori‐sensitive Mongolian gerbils treated with N‐methyl‐N‐nitrosourea and N‐methyl‐N′‐nitro‐N‐nitrosoguanidine in drinking water. Jpn J Cancer Res 1998; 89: 97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Crabtree JE, Court M, Aboshkiwa MA, Jeremy AH, Dixon MF, Robinson PA. Gastric mucosal cytokine and epithelial cell responses to Helicobacter pylori infection in Mongolian gerbils. J Pathol 2004; 202: 197–207. [DOI] [PubMed] [Google Scholar]
- 12. Yamaoka Y, Yamauchi K, Ota H et al. Natural history of gastric mucosal cytokine expression in Helicobacter pylori gastritis in Mongolian gerbils. Infect Immun 2005; 73: 2205–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dixon MF, Genta RM, Yardley JH, Correa P. Classification and grading of gastritis. The updated sydney system. International workshop on the histopathology of gastritis, Houston 1994. Am J Surg Pathol 1996; 20: 1161–81. [DOI] [PubMed] [Google Scholar]
- 14. Ikeno T, Ota H, Sugiyama A et al. Helicobacter pylori‐induced chronic active gastritis, intestinal metaplasia, and gastric ulcer in Mongolian gerbils. Am J Pathol 1999; 154: 951–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Matsubara S, Shibata H, Takahashi M et al. Cloning of Mongolian gerbil cDNAs encoding inflammatory proteins, and their expression in glandular stomach during H. pylori infection. Cancer Sci 2004; 95: 798–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Otsuka T, Tsukamoto T, Tanaka H et al. Suppressive effects of fruit‐juice concentrate of Prunus mume Sieb. et Zucc. (Japanese apricot, Ume) on Helicobacter pylori‐induced glandular stomach lesions in Mongolian gerbils. Asian Pac J Cancer Prev 2005; 6: 337–41. [PubMed] [Google Scholar]
- 17. Tsukamoto T, Fukami H, Yamanaka S et al. Hexosaminidase‐altered aberrant crypts, carrying decreased hexosaminidase alpha and beta subunit mRNAs, in colon of 1,2‐dimethylhydrazine‐treated rats. Jpn J Cancer Res 2001; 92: 109–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kumagai T, Yan J, Graham DY et al. Serum immunoglobulin G immune response to Helicobacter pylori antigens in Mongolian gerbils. J Clin Microbiol 2001; 39: 1283–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nozaki K, Shimizu N, Tsukamoto T et al. Reversibility of heterotopic proliferative glands in glandular stomach of Helicobacter pylori‐infected Mongolian gerbils on eradication. Jpn J Cancer Res 2002; 93: 374–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peek RM Jr, Crabtree JE. Helicobacter infection and gastric neoplasia. J Pathol 2006; 208: 233–48. [DOI] [PubMed] [Google Scholar]
- 21. Siomek A, Rytarowska A, Szaflarska‐Poplawska A et al. Helicobacter pylori infection is associated with oxidatively damaged DNA in human leukocytes and decreased level of urinary 8‐oxo‐7,8‐dihydroguanine. Carcinogenesis 2006; 27: 405–8. [DOI] [PubMed] [Google Scholar]
- 22. Jenks PJ, Jeremy AH, Robinson PA, Walker MM, Crabtree JE. Long‐term infection with Helicobacter felis and inactivation of the tumour suppressor gene p53 cumulatively enhance the gastric mutation frequency in Big Blue transgenic mice. J Pathol 2003; 201: 596–602. [DOI] [PubMed] [Google Scholar]
- 23. Roth KA, Kapadia SB, Martin SM, Lorenz RG. Cellular immune responses are essential for the development of Helicobacter felis‐associated gastric pathology. J Immunol 1999; 163: 1490–7. [PubMed] [Google Scholar]
- 24. Fu S, Ramanujam KS, Wong A et al. Increased expression and cellular localization of inducible nitric oxide synthase and cyclooxygenase 2 in Helicobacter pylori gastritis. Gastroenterology 1999; 116: 1319–29. [DOI] [PubMed] [Google Scholar]
- 25. Nozaki K, Tanaka H, Ikehara Y et al. Helicobacter pylori‐dependent NF‐kappa B activation in newly established Mongolian gerbil gastric cancer cell lines. Cancer Sci 2005; 96: 170–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. D’Elios MM, Manghetti M, De Carli M et al. T helper 1 effector cells specific for Helicobacter pylori in the gastric antrum of patients with peptic ulcer disease. J Immunol 1997; 158: 962–7. [PubMed] [Google Scholar]
- 27. Keates S, Hitti YS, Upton M, Kelly CP. Helicobacter pylori infection activates NF‐kappa B in gastric epithelial cells. Gastroenterology 1997; 113: 1099–109. [DOI] [PubMed] [Google Scholar]
- 28. Yoshino N, Ishihara S, Rumi MA et al. Interleukin‐8 regulates expression of Reg protein in Helicobacter pylori‐infected gastric mucosa. Am J Gastroenterol 2005; 100: 2157–66. [DOI] [PubMed] [Google Scholar]
- 29. Liu F, Pan K, Zhang X et al. Genetic variants in cyclooxygenase‐2: expression and risk of gastric cancer and its precursors in a Chinese population. Gastroenterology 2006; 130: 1975–84. [DOI] [PubMed] [Google Scholar]
- 30. Prescott SM, Fitzpatrick FA. Cyclooxygenase‐2 and carcinogenesis. Biochim Biophys Acta 2000; 1470: M69–78. [DOI] [PubMed] [Google Scholar]
- 31. Hansen‐Petrik MB, McEntee MF, Jull B, Shi H, Zemel MB, Whelan J. Prostaglandin E(2) protects intestinal tumors from nonsteroidal anti‐inflammatory drug‐induced regression in Apc (Min/+) mice. Cancer Res 2002; 62: 403–8. [PubMed] [Google Scholar]
- 32. Ristimaki A, Honkanen N, Jankala H, Sipponen P, Harkonen M. Expression of cyclooxygenase‐2 in human gastric carcinoma. Cancer Res 1997; 57: 1276–80. [PubMed] [Google Scholar]
- 33. Romano M, Ricci V, Memoli A et al. Helicobacter pylori up‐regulates cyclooxygenase‐2 mRNA expression and prostaglandin E2 synthesis in MKN 28 gastric mucosal cells in vitro. J Biol Chem 1998; 273: 28560–3. [DOI] [PubMed] [Google Scholar]
- 34. Sakai T, Fukui H, Franceschi F et al. Cyclooxygenase expression during Helicobacter pylori infection in Mongolian gerbils. Dig Dis Sci 2003; 48: 2139–46. [DOI] [PubMed] [Google Scholar]
- 35. Rajnakova A, Moochhala S, Goh PM, Ngoi S. Expression of nitric oxide synthase, cyclooxygenase, and p53 in different stages of human gastric cancer. Cancer Lett 2001; 172: 177–85. [DOI] [PubMed] [Google Scholar]
- 36. Arroyo PL, Hatch‐Pigott V, Mower HF, Cooney RV. Mutagenicity of nitric oxide and its inhibition by antioxidants. Mutat Res 1992; 281: 193–202. [DOI] [PubMed] [Google Scholar]
- 37. Wink DA, Kasprzak KS, Maragos CM et al. DNA deaminating ability and genotoxicity of nitric oxide and its progenitors. Science 1991; 254: 1001–3. [DOI] [PubMed] [Google Scholar]
- 38. Sepulveda A, Peterson LE, Shelton J, Gutierrez O, Graham DY. Histological patterns of gastritis in H. pylori‐infected individuals with a family history of gastric cancer. Am J Gastroenterol 2002; 97: 1365–70. [DOI] [PubMed] [Google Scholar]
- 39. Takashima M, Furuta T, Hanai H, Sugimura H, Kaneko E. Effects of Helicobacter pylori infection on gastric acid secretion and serum gastrin levels in Mongolian gerbils. Gut 2001; 48: 765–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fukui H, Franceschi F, Penland RL et al. Effects of Helicobacter pylori infection on the link between regenerating gene expression and serum gastrin levels in Mongolian gerbils. Laboratory Invest 2003; 83: 1777–86. [DOI] [PubMed] [Google Scholar]
- 41. Wang TC, Dangler CA, Chen D et al. Synergistic interaction between hypergastrinemia and Helicobacter infection in a mouse model of gastric cancer. Gastroenterology 2000; 118: 36–47. [DOI] [PubMed] [Google Scholar]
- 42. Cao X, Tsukamoto T, Nozaki K et al. Earlier Helicobacter pylori infection increases the risk for the N‐methyl‐N‐nitrosourea‐induced stomach carcinogenesis in Mongolian gerbils. Jpn J Cancer Res 2002; 93: 1293–8. [DOI] [PMC free article] [PubMed] [Google Scholar]