

Abstract
The aim of the present study was to investigate the safety and immunological responses of personalized peptide vaccination in combination with oral administration of a 5‐fluorouracil derivative (TS‐1) in advanced gastric or colorectal carcinoma patients. Eleven patients (four with gastric cancer and seven with colorectal cancer) who failed to improve by prior TS‐1‐based chemotherapies were enrolled in this study. Peptides to be administered to patients were determined based on the presence of peptide‐specific cytotoxic T lymphocyte (CTL) precursors in peripheral blood mononuclear cells and peptide‐specific IgG in the plasma of cancer patients. Patients were vaccinated with peptides (a maximum of four) biweekly in combination with or without three different doses of TS‐1 (20, 40 and 80 mg/m2/day). Although grade 3 toxicity, including anemia (one patient) and neutropenia (one patient) were observed, the combination therapy was generally well tolerated. An increase in peptide‐specific IgG after the sixth vaccination was observed in the vast majority of patients irrespective of the dose of TS‐1 used. In contrast, an increase in peptide‐specific interferon‐γ production by CTL was most evident in patients who were administered the highest dose of TS‐1. Furthermore, in the patients who received 80 mg/m2/day TS‐1, CTL‐mediated cytotoxicity against cancer cells was maintained at the prevaccination level. These results indicate that administration of the standard dose (80 mg/m2/day) of TS‐1 in combination with a personalized peptide vaccination does not necessarily impede immunological responses in cancer patients, and could actually maintain or augment them. (Cancer Sci 2007; 98: 1113–1119)
Recent advances in tumor immunology have resulted in the identification of a number of antigens and their peptides that are recognized by tumor‐reactive and human leukocyte antigen (HLA) class I‐restricted cytotoxic T lymphocytes (CTL).( 1 ) Cancer vaccines have emerged as a promising therapeutic approach,( 2 ) but their clinical responses have been limited.( 3 ) In order to develop a new treatment modality, we recently devised a new regime of peptide‐based vaccination that consists of measuring pre‐existing CTL precursors and IgG reactive to many kinds of vaccine candidates, followed by administration.( 4 , 5 , 6 , 7 , 8 ) In addition, we recently reported the benefits of the combination of this type of peptide vaccination with a low dose of estramustine phosphate in hormone refractory prostate cancer patients.( 9 ) This chemoimmunotherapy may break through the impasse in the clinical efficacy of cancer vaccines.
TS‐1 is an oral fluoropyrimidine derivative consisting of Tegafur (FT) and two modulators, 5‐chloro‐2,4‐dihydroxypyrimidine (CDHP) and potassium oxonate (Oxo). FT is a prodrug of 5‐fluorouracil (5‐FU) and CDHP is a reversible competitive inhibitor of an enzyme involved in the degradation of 5‐FU. Therefore, the FT‐derivative 5‐FU remains in tumor tissue a long time, resulting in an enhanced antitumor effect.( 10 , 11 ) Indeed, TS‐1 has been reported to be effective at prolonging the survival of patients with advanced gastric cancer.( 12 ) Furthermore, TS‐1 can be administered as an oral formulation that permits treatment on an outpatient basis, leading to improvement in the quality of life (QOL) of patients. In the present study, we investigated the safety and immunological responses of personalized peptide vaccination in combination with the oral administration of TS‐1 in advanced gastric or colorectal carcinoma patients.
Materials and Methods
Patients and eligibility criteria. The study protocol was approved by the Institutional Ethical Review Boards of Hokkaido University, Okayama University and Kurume University. Complete written informed consent was obtained from all patients at the time of enrolment. According to the protocol, patients were confirmed to be HLA‐A24 or HLA‐A2 positive and had a histologically confirmed lesion of gastric or colorectal carcinoma. In HLA‐A2+ patients, their HLA‐A2 genotypes were determined using sequence‐specific oligonucleotide DNA typing after polymerase chain reaction. All patients had failed to improve by prior chemotherapy with TS‐1. The other eligibility criteria included an age of 85 years or less, serum creatinine of less than 1.4 mg/dL, bilirubin of less than 1.5 mg/dL, a platelet count of 100 000 µL or more, hemoglobin of 8.0 g/dL or more, and total white blood cell count (WBC) of 3000 cells/µL or more. Hepatitis B surface antigen, hepatitis C antigen and human immunodeficiency virus (HIV) were required to be negative. All patients had been untreated for at least 4 weeks before the study, and had an Eastern Cooperative Oncology Group performance status of 0–2. Patients were assigned based on their factors, including performance status, age, site of metastasis, type of cancer and HLA‐A allele, at an independent data center. The treatment was carried out at Hokkaido University Hospital and Okayama University Hospital. All immunological analyses were carried out at the Department of Immunology, Kurume University School of Medicine.
Screening of peptide‐specific CTL precursors. Thirty milliliters of peripheral blood was obtained before and after the sixth vaccination, and peripheral blood mononuclear cells (PBMC) were isolated by means of Ficoll–Conray density gradient centrifugation. Peptide‐specific CTL‐precursors in PBMC were detected using a previously reported culture method.( 13 ) Briefly, PBMC (1 × 105 cells/well) from cancer patients were incubated with 10 µM of a peptide in 200 µL of culture medium in U‐bottom‐type 96‐well microculture plates (Nunc, Roskilde, Denmark) in quadruplicate. The culture medium consisted of 45% RPMI‐1640 medium, 45% AIM‐V medium (Gibco BRL, Grand Island, NY, USA), 10% fetal calf serum, 100 U/mL interleukin‐2 (IL‐2) and 0.1 µM MEM non‐essential amino acid solution (Gibco BRL). Half of the medium was removed and replaced with new medium containing a corresponding peptide (20 µM) every 3 days. After incubation for 14 days, the cultured cells in one well were separated into four wells, with two wells being used for HIV peptide‐pulsed cells and the other two wells for the corresponding peptide‐pulsed cells. Then, their ability to produce interferon (IFN)‐γ in response to CIR‐A2402 (Dr M. Takiguchi, Kumamoto University, Japan) or T2 cells that were preloaded with either a corresponding peptide or HIV peptides (RYLRQQLLGI for HLA‐A24 and LLFGYPVYV for HLA‐A2) as a negative control, was examined. The level of IFN‐γ was determined by enzyme‐linked immunosorbent assay (limit of sensitivity: 10 pg/mL), and the background IFN‐γ production in response to the HIV peptide was subtracted. The result of the best well among four wells is shown.
Screening of peptide‐specific IgG. The levels of antipeptide IgG were measured using a Luminex system as reported previously.( 14 ) In brief, plasma was incubated with 25 µL peptide‐coupled color‐coded beads for 2 h at room temperature on a plate shaker. After incubation, the mixture was washed with a vacuum manifold apparatus and incubated with 100 µL biotinylated goat antihuman IgG (γ chain‐specific) for 1 h at room temperature. The plate was then washed, followed by the addition of 100 µL streptavidin–phycoerythrin (PE) into wells, and was incubated for 30 min at room temperature on a plate shaker. The bound beads were washed three times followed by the addition of 100 µL Tween–phosphate‐buffered saline into each well. Samples (50 µL) were analyzed using the Luminex system.
Vaccination and combination therapy. The peptides utilized in the present study were prepared by Multiple Peptide Systems (San Diego, CA, USA) under the conditions of Good Manufacturing Practice. They are listed in Table 1. SART3 93–101, SART3 109–118, CypB 91–99, Lck 486–494, Lck 488–497, MRP3 1293–1302, PAP 213–221, EZH2 291–299, PSCA 76–84 and PSA 248–257 peptides have been reported to induce HLA‐A24‐restricted and tumor‐specific CTL in PBMC of cancer patients( 15 , 16 , 17 , 18 , 19 , 20 , 21 ) and were used for HLA‐A24+ patients. SART3 302–310, Lck 246–254, WHS 103–111, HNR 140–148, MAP 294–302, UBE 43–51, EIF 51–59, EGFR 479–488, PSA 170–178 and HER2/neu 484–493 peptides have been reported to induce HLA‐A2‐restricted and tumor‐specific CTL in PBMC of cancer patients( 22 , 23 , 24 , 25 , 26 , 27 ) and were used for HLA‐A2+ patients. Both the CEA 390–399 and CEA 425–433 peptides and the CEA 605–613 peptide have the ability to induce HLA‐A24‐restricted or HLA‐A2‐restricted and tumor‐specific CTL in PBMC of cancer patients, respectively (K Itoh, unpublished observation). Montanide ISA‐51 adjuvant was manufactured by Seppic (Franklin Lake, NJ, USA). The peptides were supplied in vials containing 3 mg/mL sterile solution for injection. Three milligrams of peptide with sterile saline was added in a 1:1 volume to the Monotide ISA‐51, and then mixed in a Vortex mixer (Fisher, Alameda, CA, USA). The resulting emulsion was injected subcutaneously into the lateral thigh using a glass syringe. Up to four peptides were selected for each patient. Patients were vaccinated every 2 weeks, for a total of six injections. Patients also received oral administration of TS‐1 in one of several doses (20, 40 or 80 mg/m2/day) every day for 4 weeks. After cessation for 2 weeks, TS‐1 was administered again for an additional 4 weeks.
Table 1.
Immune response during the therapy
| Patient number | Dose of TS‐1 (mg/m2/day) | Peptide | Anti‐peptide cellular response † | Anti‐peptide IgG ‡ | Best clinical response | Follow up (M) | Prognosis | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Pre | Post (sixth) | Increase after vaccine | Pre | Post (sixth) | Increase after vaccine | ||||||
| 1 | 80 | SART3 109–118 | 0 | 0 | – | 129 | 13 656 | + | PD | 5 | Dead |
| CypB 91–99 | 0 | 212 | + | 48 | 56 | – | |||||
| Lck 488–497 | 0 | 23 | – | 26 | 475 | + | |||||
| MRP3 1293–1302 | 0 | 153 | + | 29 | 40 | – | |||||
| 2 | 80 | SART3 302–310 | 0 | 275 | + | 113 | 2986 | + | SD | 17 | Alive |
| Lck 246–254 | 0 | 221 | + | 122 | 174 | – | |||||
| WHS 103–111 | 0 | 1253 | + | 212 | 250 | – | |||||
| HNR 140–148 | 0 | 0 | – | 114 | 115 | – | |||||
| 3 | 80 | Lck 486–494 | 0 | 222 | + | 696 | 1009 | + | PD | 11 | Dead |
| MRP3 1293–1302 | 0 | 0 | – | 144 | 301 | + | |||||
| PAP 213–221 | 0 | 0 | – | 317 | 4696 | + | |||||
| EZH2 291–299 | 0 | 0 | – | 400 | 10 329 | + | |||||
| 4 | 40 | SART3 109–118 | 15 | 188 | + | 356 | 9135 | + | PD | 30 | Alive |
| CEA 390–399 | 0 | 0 | – | 13 | 19 | – | |||||
| 5 | 40 | SART3 93–101 | 58 | 0 | – | 219 | 166 | – | PD | 5 | Dead |
| Lck 486–494 | 89 | 0 | – | 209 | 165 | – | |||||
| MRP3 1293–1302 | 0 | 0 | – | 303 | 247 | – | |||||
| PSCA 76–84 | 60 | 0 | – | 144 | 120 | – | |||||
| 6 | 40 | SART3 109–118 | 0 | 60 | + | 543 | 103 | – | PD | 7 | Dead |
| MRP3 1293–1302 | 0 | 0 | – | 224 | 142 | – | |||||
| PAP 213–221 | 0 | 0 | – | 819 | 266 | – | |||||
| CEA 425–433 | 9 | 0 | – | 195 | 67 | – | |||||
| 7 | 20 | MAP 294–302 | 0 | 0 | – | 104 | 1993 | + | SD | 8 | Dead |
| UBE 43–51 | 0 | 0 | – | 207 | 24 391 | + | |||||
| EIF 51–59 | 56 | 0 | – | 40 | 156 | + | |||||
| EGFR 479–488 | 0 | 0 | – | 112 | 276 | + | |||||
| 8 | 20 | UBE 43–51 | 0 | 0 | – | 580 | 20 163 | + | PD | 15 | Dead |
| PSA 170–178 | 729 | 0 | – | 499 | 990 | – | |||||
| HER2/neu 484–493 | 146 | 0 | – | 477 | 665 | – | |||||
| CEA 605–613 | 0 | 0 | – | 831 | 23 694 | + | |||||
| 9 | 0 | SART2 93–101 | 0 | 388 | + | 51 | 1649 | + | SD | 25 | Alive |
| SART3 109–118 | 0 | 0 | – | 594 | 481 | – | |||||
| Lck 488–497 | 0 | 0 | – | 100 | 361 | + | |||||
| MRP3 1293–1302 | 0 | 0 | – | 167 | 932 | + | |||||
| 10 | 0 | SART3 109–118 | 0 | 0 | – | 420 | 1838 | + | PD | 5 | Dead |
| MRP3 1293–1302 | 115 | 45 | – | 167 | 1399 | + | |||||
| CypB 91–99 | 314 | 0 | – | 119 | 779 | + | |||||
| PSA 248–257 | 160 | 0 | – | 124 | 149 | – | |||||
| 11 | 0 | Lck 422–430 | 0 | 172 | + | 120 | 115 | – | SD | 17 | Alive |
| MAP 294–302 | 0 | 0 | – | 48 | 248 | + | |||||
| UBE 43–51 | 348 | 0 | – | 91 | 113 | – | |||||
| EIF 51–59 | 227 | 0 | – | 26 | 2462 | + | |||||
Values indicate interferon (IFN)‐γ production of peptide‐stimulated peripheral blood mononuclear cells (PBMC) reactive to the corresponding peptide. An increase in IFN‐γ production after vaccination was judged positive when the level of IFN‐γ production was more than 50 pg/mL and showed an at least two‐fold increase after the sixth vaccination.
‡ Values indicate the fluorescence intensity of IgG reactive to the corresponding peptide. An increase in fluorescence intensity after vaccination was judged positive when the level increased more than two‐fold after the sixth vaccination. PD, progressive disease; PR, partial response; SD, stable disease.
Adverse events and clinical responses. Adverse events were monitored according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 3.0. The clinical response was evaluated based on clinical observations and radiological findings. All known sites of disease were evaluated on a monthly basis by computed tomography (CT)‐scan or magnetic resonance imaging (MRI) examination. The tumor size was estimated via direct measurement of the region of abnormal enhancement observed on CT‐scan or MRI examination. Patients were assigned a response category according to the Response Evaluation Criteria in Solid Tumors. Survival time was estimated from the date of the initial vaccination.
Cytotoxicity assay. Cytotoxic activity was measured using a standard 6‐h 51Cr‐release assay. In brief, cryopreserved PBMC before and after the sixth vaccinations were thawed simultaneously and stimulated repeatedly in vitro with the corresponding peptide in the presence of 100 U/mL IL‐2, as described above. After the 14th day of culture, peptide‐stimulated cells were harvested and cultured with 100 U/mL IL‐2 alone for an additional 10–14 days, and used for effector cells. The targets used for the 51Cr‐release assay were as follows: KATO‐III (HLA‐A24+ gastric cancer), KWS (HLA‐A24− gastric cancer), SW620 (HLA‐A24+ colon cancer), COLO201 (HLA‐A24− colon cancer), COLO320 (HLA‐A24+ colon cancer) and phytohemagglutinin (PHA)‐stimulated T‐cell blasts from PBMC of HLA‐A24+ healthy donors.
Flow cytometric analysis. PBMC were stained with fluorescein‐isothiocyanate‐conjugated anti‐CD4 monoclonal antibody (DAKO Cytomation, Glostrup, Denmark) and PE‐conjugated anti‐CD25 monoclonal antibody (DAKO Cytomation), and the percentage of positive CD4+ and CD25+ T cells was examined by flow cytometry.
Results
Patient characteristics. Between January 2004 and July 2005, 11 patients positive for HLA‐A24 or HLA‐A2 with advanced gastric or colorectal cancer entered into the clinical trial. Their median ages were 61.5 (35–71) and 65.0 (51–75) years, respectively. Only one patient (patient 1) was female. Seven patients and four patients were positive for HLA‐A24 and HLA‐A2, respectively. Their Eastern Cooperative Oncology Group performance status was 0 for seven patients, 1 for three patients and 2 for one patient. All patients other than patient 11 suffered from metastases, and received surgical operation and chemotherapy consisting of at least TS‐1. 2, 3 summarize the clinical characteristics of all 11 patients.
Table 2.
Summary of patients
| Characteristic | Gastric cancer | Colon cancer |
|---|---|---|
| Number of patients | 4 | 7 |
| Age (years) | ||
| Median | 61.5 | 65 |
| Range | 35–71 | 51–75 |
| Sex (male/female) | 3/1 | 7/0 |
| Performance status † | ||
| 0 | 2 | 5 |
| 1 | 1 | 2 |
| 2 | 1 | 0 |
| Stage | IV: 4 | I: 1 |
| IIIb: 3 | ||
| IV: 3 | ||
Performance status by Eastern Cooperative Oncology Group (ECOG) score.
Table 3.
Patient characteristics
| Patient number | Age (year) | Sex | HLA‐A allele | Cancer | PS | Metastasis | Previous treatment | |
|---|---|---|---|---|---|---|---|---|
| Surgery | Chemotherapy † | |||||||
| 1 | 35 | F | A24 | Gastric | 1 | Peritoneum | None | TS‐1, TXT |
| 2 | 55 | M | A*0206 | Colon | 0 | LN | Done | CPT‐11, TS‐1 |
| 3 | 75 | M | A24 | Colon | 0 | Lung | Done | UFT, CPT‐11, DFUR |
| 4 | 74 | M | A24 | Colon | 0 | Lung, LN | Done | TS‐1, 5FU, LV, CPT‐11 |
| 5 | 65 | M | A24 | Colon | 0 | Peritoneum, Liver, LN | Done | 5FU, I‐LV, TS‐1 |
| 6 | 65 | M | A24 | Colon | 1 | Liver, Lung | Done | 5FU, UFT, CPT‐11, TS‐1 |
| 7 | 71 | M | A*0201/0207 | Gastric | 0 | Liver, LN | None | 5FU, CDDP, TS‐1 |
| 8 | 51 | M | A*0201/0206 | Colon | 0 | Liver, Lung, LN | Done | CPT‐11, 5FU, TS‐1 |
| 9 | 55 | M | A24 | Gastric | 0 | LN | Done | TS‐1 |
| 10 | 70 | M | A24 | Colon | 1 | Liver, LN | Done | 5FU, UFT, CPT‐11, TS‐1 |
| 11 | 68 | M | A*0201/0207 | Gastric | 2 | None | Done | TS‐1 |
Chemotherapy: CDDP, cisplatin; CPT‐11, irinotecan hydrochloride; DFUR, doxifluridine; 5‐FU, fluorouracil; I‐LV, levofolinate calcium; LV, leucocorin; TS‐1, 5‐fluorouracil derivative; TXT, docetaxel; UFT, tegafur‐uracil.
Vaccination and toxicity. All 11 patients were evaluated for all common toxicities, and the overall toxicities are shown in Table 4. As grade 3 adverse events, one case had anemia and another case had neutropenia. One case had fatigue as a grade 2 adverse effect. As for grade 1 adverse events, four, two, one and one cases had dermatologic reaction, fatigue, diarrhea and anemia, respectively, using the National Cancer Institute common toxicity criteria.
Table 4.
Adverse events
| Toxicity | G1 | G2 | G3 | G4 | Total |
|---|---|---|---|---|---|
| Dermatologic reaction | 4 | 0 | 0 | 0 | 4 |
| Fatigue | 2 | 1 | 0 | 0 | 3 |
| Diarrhea | 1 | 0 | 0 | 0 | 1 |
| Anemia (hemoglobin < 8 mg/dL) | 1 | 0 | 1 | 0 | 2 |
| Neutropenia | 0 | 0 | 1 | 0 | 1 |
Toxicities based on the National Cancer Institute common toxicity scale (version 2). Some patients had more than one toxic reaction. G, grade.
Personalized peptide vaccination and immune responses. We previously identified a panel of peptide candidates applicable for vaccination of epithelial cancer patients, and devised a new regime of peptide‐based vaccination that we termed the personalized peptide vaccination: measurement of CTL precursors and IgG reactive to many kinds of vaccine candidates, followed by administration.( 8 ) When no peptide‐specific CTL precursors were detected, the peptides vaccinated were determined based on the results of peptide‐specific IgG. We applied this regime in the present study. A maximum of four peptides were selected for injection to each patient and the results of the selection are listed in Table 1. All patients other than patient 4 were vaccinated with four different kinds of peptide. In HLA‐A24+ patients, the most frequently used peptide was MRP3 1293–1302 (5/7), followed by SART3 109–118 (4/7). In HLA‐A2+ cancer patients, the most frequently used peptide was UBE 43–51 (3/4), followed by MAP 294–302 (2/4). HLA‐A2 alleles consist of several subtypes. Among the four HLA‐A2 patients, three of them were positive for HLA‐A*0201 molecules, and patient 2 was positive only for HLA‐A*0206 molecules. This patient received vaccination of the SART3 302–310, Lck 246–254, WHS 103–111 and HNR 140–148 peptides, and we have already reported that all of these peptides can be presented by HLA‐A*0206 molecules.( 22 , 23 , 24 )
We next examined whether or not personalized peptide vaccination in combination with TS‐1 could augment peptide‐specific CTL or IgG in patients after the sixth vaccination (Table 1). The vaccination increased peptide‐specific CTL response to two, three and one of the four vaccinated peptides in patients 1, 2 and 3, respectively, who had no CTL activity in prevaccination PBMC and received the vaccination combined with a maximum dose (80 mg/m2/day) of TS‐1. Two of three patients who received peptide vaccination alone or vaccination with a medium dose (40 mg/m2/day) of TS‐1 showed increased peptide‐specific CTL responses to one of the four vaccinated peptides, whereas no augmentation was observed in the two patients who received peptide vaccination with a low dose (20 mg/m2/day) of TS‐1. However, the level of peptide‐specific IgG increased in all of the patients except for two patients (patients 5 and 6) who received peptide vaccination with a medium dose (40 mg/m2/day) of TS‐1. The levels of IgG reactive to each of the 22 of 42 (52%) vaccinated peptides increased in the postvaccination plasma, and it seemed to be independent of the dose of TS‐1 administered. These results indicate that oral administration of TS‐1 does not necessarily impede immunological responses in cancer patients under the personalized peptide vaccination, but rather could maintain or augment CTL activity when a dose of 80 mg/m2/day of TS‐1 is administered.
We further examined the tumor‐reactive cytolytic activity of PBMC by means of 51Cr‐release assay. Pre‐ and post (sixth)‐vaccination PBMC were stimulated in vitro with the corresponding peptide, and examined for their cytotoxicity against gastric or colon cancer cells (Fig. 1). In only 4 of 11 patients (patients 1, 2, 3 and 10), both the pre‐ and postvaccination PBMC proliferated under the culture conditions used to test for their cytotoxicity. In patient 1, the PBMC stimulated with CypB 91–99 peptide showed a higher level of cytotoxicity against the HLA‐A24+ gastric cancer cell line KATO‐III than against the HLA‐A24− gastric cancer cell line KWS. Although the level of cytotoxicity against KATO‐III was almost equal between pre‐ and postvaccination PBMC, that against KWS decreased after the therapy, suggesting that the combination therapy increased cytotoxicity against HLA‐A24+ gastric cancer cells. No difference in cytotoxicity against HLA‐A24+ PHA‐stimulated T‐cell blasts was observed. A similar result was observed in the MRP3 1293–1302 peptide‐stimulated PBMC of this patient. In patient 2, HLA‐A2‐restricted CTL activity against tumor cells was not detectable in either prevaccination or postvaccination PBMC (data not shown). In patient 3, both pre‐ and postvaccination PBMC stimulated with Lck 486–494 peptide showed a higher cytotoxicity against the HLA‐A24+ colon cancer cell line SW620 than against either the HLA‐A24− colon cancer cell line COLO201 or HLA‐A24+ PHA‐stimulated T‐cell blasts. Such HLA‐A24‐restricted cytotoxicity was induced in the postvaccination PBMC stimulated with PAP 213–221 peptide (Fig. 1) as well as in those stimulated with MRP3 1293–1302 peptide (data not shown). We also examined the tumor‐reactive cytolytic activity of a patient who received peptide vaccination without TS‐1. In patient 10, both pre‐ and postvaccination PBMC stimulated with CypB 91–99 peptide showed a higher cytotoxicity against the HLA‐A24+ colon cancer cell lines SW620 and COLO320 than against either the HLA‐A24− colon cancer cell line COLO201, or HLA‐A24+ PHA‐stimulated T‐cell blasts. Although the cases were very limited due to the availability of samples, these results indicate that the TS‐1 administration, even at a maximum dose, in combination with the personalized peptide vaccine does not necessarily impede HLA‐A24‐restricted CTL activity against tumor cells.
Figure 1.
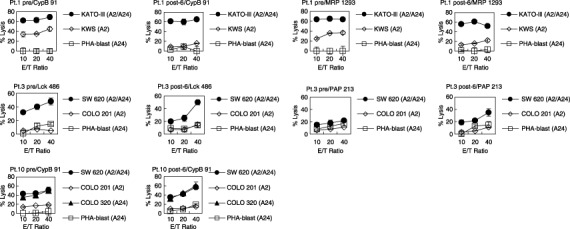
Cytotoxicity against cancer cells before and after the personalized peptide vaccination with TS‐1. Pre‐ and post (sixth)‐vaccination peripheral blood mononuclear cells (PBMC) from three patients (patients 1, 3 and 10) were stimulated repeatedly with the corresponding peptide in the presence of interleukin (IL)‐2 for 14 days, followed by the culture with IL‐2 alone for an additional 10–14 days. Thereafter, these cultured cells were used as effector cells in the cytotoxicity assay against the indicated targets. The 6‐h 51Cr‐release assay was carried out in triplicate, and the values are shown as the mean SD.
CD4+ and CD25+ T cells in the treated patients. We next tested the possibility that the TS‐1 administration influenced regulatory CD4+ T cells in the treated patients, because there is increasing evidence that these cells play a negative role in antitumor immune responses in vivo. ( 28 , 29 ) Then, we examined the positive percentage of CD4+ and CD25+ T cells in the PBMC of patients who received the peptide vaccination with TS‐1 (Fig. 2). As a result, although the positive percentage of CD4+ and CD25+ T cells in the PBMC of patient 2 did not decrease after the sixth peptide vaccination with 80 mg/m2/day TS‐1, that of patient 5 evidently decreased after the fifth peptide vaccination with 40 mg/m2/day TS‐1. Such a decrease was also observed in patient 11 who received the peptide vaccination without TS‐1.
Figure 2.
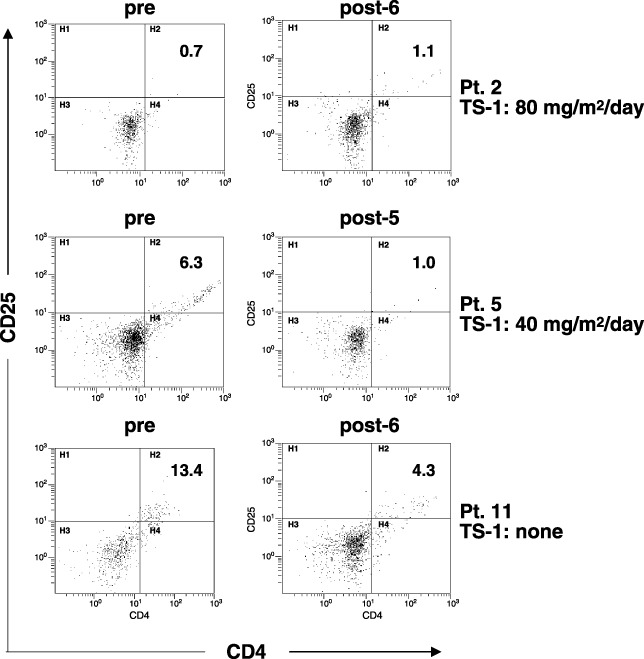
Flow cytometric analysis of peripheral blood mononuclear cells (PBMC) of the vaccinated patients. PBMC of three patients pre‐ and postvaccination with or without TS‐1 were stained with fluorescein‐isothiocyanate‐conjugated anti‐CD4 and phycoerythrin (PE)‐conjugated anti‐CD25 monoclonal antibodies, and were analyzed by flow cytometry. Patients 2 and 5 received peptide vaccination with 80 and 40 mg/m2/day of TS‐1, respectively, and patient 11 received peptide vaccination without TS‐1. The number represents the positive percentage of CD4+ and CD25+ cells.
Clinical responses. Because all patients had failed to respond to prior TS‐1‐based chemotherapy, evaluation of clinical responses was not a major objective of this study. However, description of the clinical responses could be important for the next stage of the clinical trials. Although no partial response was observed in any of patients, patients 2, 7, 9, and 11 showed stable disease for 17, 8, 25 and 17 months, respectively, but the other seven patients had rapid tumor progression (Table 1). At the present time, only four patients are alive, with two of these patients (patients 2 and 11) having survived more than 1 year and one patient (patient 9) having survived more than 2 years. The other seven patients have died, and all deaths were attributable to cancer progression. Patient 4 had progressive disease, but survived more than 30 months.
Discussion
Although recent advances in tumor immunology have enabled us to carry out anticancer vaccination against various types of cancers, the clinical responses of the treatments have been very limited. To overcome this problem, several devices have been developed, and one promising approach is vaccination in combination with chemotherapy. In our clinical trials against hormone‐refractory prostate cancer, peptide‐based vaccination in combination with estramustine phosphate, a mixture of anticancer and antihormone drugs, showed a superior antitumor effect compared to peptide vaccination alone.( 9 ) This type of chemoimmunotherapy may break through the impasse in the clinical efficacy of anticancer vaccines.( 8 ) This idea is based on the notion that cancer is an extremely complex and heterogeneous disease that exhibits a high level of robustness against a range of both host‐defense systems and therapeutic efforts.( 30 , 31 ) In fact, we previously observed that the expressions of HLA class I molecules, multidrug‐resistance 1 protein and androgen receptor in prostate cancer tissues at a pretreatment state were very heterogeneous in each sample (S Homma, Y Komohara, M Harada, H Saya, S Todo, M Noguchi, K Itoh, unpublished data). These three factors are necessary for the antitumor effects induced by peptide‐specific CTL, anticancer drugs and antihormone drugs, respectively. Combination therapy targeting separate molecules is thus theoretically reasonable.
Another finding supporting the efficacy of combination therapy is the revelation of immunologically suppressive cells, including regulatory CD4+ T cells and immature myeloid suppressor cells. There is increasing evidence that these cells play a negative role in antitumor immune response in vivo. ( 28 , 29 , 32 , 33 ) There is a possibility that administration of TS‐1 induces apoptosis of cancer cells, resulting in not only augmentation of the cross‐presentation by antigen‐presenting cells of tumor‐associated antigen peptides to CD8+ CTL but also a decrease in regulatory activity of suppressive cells. Although the cases were very limited, we examined the positive percentage of CD4+ and CD25+ T cells in the pre‐ and postvaccination PBMC of patients. As a result, a decrease in the positive percentage of CD4+ and CD25+ T cells in the PBMC was observed in one out of two patients who received the TS‐1 treatment. However, a similar decrease was also observed in a patient who received the peptide vaccination without the TS‐1 treatment. The number of cases was too small to conclude the effect of the TS‐1 treatment on regulatory CD4+ T cells.
The primary purpose of the present study was to investigate the safety and immunological responses of personalized peptide vaccinations in combination with oral administration of TS‐1 in advanced gastric or colon carcinoma patients. TS‐1 is a fluoropyrimidine derivative and can be administered as an oral formulation, allowing treatment on an outpatient basis. Our findings indicate that oral administration in combination with peptide vaccine does not necessarily impede the vaccine‐induced peptide‐specific immune response even when administered at a dose of 80 mg/m2/day (a clinically standard dose), and both cellular and humoral immune responses were augmented in all three cases receiving this dose. These observations encourage us to carry out additional trials with a larger number of patients.
We used both the IFN‐γ production assay in response to peptide‐loaded cells and the 51Cr‐release assay in response to cancer cells. The results from the two assays did not always correlate with each other. We have no clear explanation for this discrepancy, but an important point is that the duration of in vitro culture was different, being 14 days for the former assay and 24–28 days for the latter. For the latter assay, the peptide‐stimulated PBMC were cultured an additional 10–14 days to obtain enough effector cells to do the 51Cr‐release cytotoxicity assay. Another possibility is that prior chemotherapy including TS‐1 may have influenced the ability of the peptide‐stimulated T cells to survive in vitro during the additional culture. As a result, the T‐cell repertoire in the cultured cells might be changed. However, further studies are needed to clarify this issue.
In contrast to cellular responses, the level of peptide‐specific IgG increased in all but two of the patients (patients 5 and 6) who received peptide vaccination with a medium dose (40 mg/m2/day) of TS‐1. These results suggest that TS‐1 did not suppress the humoral immunity induced by the personalized peptide vaccination, although the biological roles of increased peptide‐specific IgG have not been determined.
In conclusion, TS‐1 administration in combination with a personalized peptide vaccination did not necessarily impede the immunological responses in cancer patients, and a dose of 80 mg/m2/day of TS‐1 actually maintained or augmented CTL activity. These results should encourage further clinical trials of the personalized peptide vaccine in combination with oral administration of TS‐1 for the treatment of patients with gastric and colorectal cancer.
Acknowledgments
This study was supported in part by Grants‐in‐Aid (KAKENHI) from the Ministry of Education, Science, Sport, Culture and Technology of Japan (no. 17016074 to K. I., and no. 18591449 to M. H.), from the Research Center of Innovative Cancer Therapy of the 21st Century COE Program for Medical Science (to K. I.), and from the Ministry of Health, Labor and Welfare, Japan (no. 15–17 to M. H.).
References
- 1. Novellino L, Castelli C, Parmiani G. A listing of human tumor antigens recognized by T cells: March 2004 update. Cancer Immuno Immunother 2005; 54: 187–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rosenberg SA. A new era for cancer immunotherapy based on the genes that encode cancer antigens. Immunity 1999; 10: 281–7. [DOI] [PubMed] [Google Scholar]
- 3. Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med 2004; 10: 909–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sato Y, Shomura H, Maeda Y et al . Immunological evaluation of peptide vaccination for patients with gastric cancer based on pre‐existing cellular response to peptide. Cancer Sci 2003; 94: 802–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mine T, Gouhara R, Hida N et al . Immunological evaluation of CTL precursor‐oriented vaccines for advanced lung cancer patients. Cancer Sci 2003; 94: 548–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tsuda N, Mochizuki K, Harada M et al . Vaccination with predesignated or evidence‐based peptides for patients with recurrent gynecologic cancers. J Immunother 2004; 27: 60–72. [DOI] [PubMed] [Google Scholar]
- 7. Mine T, Sato Y, Noguchi M et al . Humoral responses to peptides correlated with overall survival in advanced cancer patients vaccinated with peptides based on pre‐existing, peptide‐specific cellular responses. Clin Cancer Res 2004; 10: 929–37. [DOI] [PubMed] [Google Scholar]
- 8. Yamada A, Itoh K. Personalized peptide vaccines: a new therapeutic modality for cancer. Cancer Sci 2006; 97: 970–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Noguchi M, Itoh K, Yao A et al . Immunological evaluation of individualized peptide vaccination with a low dose of estramustine for HLA‐A24+ HRPC patients. Prostate 2005; 63: 1–12. [DOI] [PubMed] [Google Scholar]
- 10. Shirasaka T, Nakano K, Takechi T et al . Antitumor activity of 1 M tegafur‐0.4 M 5‐chloro‐2,4‐dihydroxypridine–1 M potassium oxonate (S‐1) against human colon carcinoma orthotopically implanted into nude rats. Cancer Res 1996; 56: 2602–6. [PubMed] [Google Scholar]
- 11. Shirasaka T, Shimamoto Y, Ohshimo H et al . Development of a novel form of an oral 5‐fluorouracil derivative (S‐1) directed to the potentiation of the tumor selective cytotoxicity of 5‐fluorouracil by two biochemical modulators. Anticancer Drugs 1995; 7: 548–57. [DOI] [PubMed] [Google Scholar]
- 12. Osigi H, Takada N, Takemura M et al . Oral fluoropyrimidine anticancer drugs TS‐1 for gastric cancer patients with peritoneal dissemination. Oncol Rep 2002; 9: 811–15. [PubMed] [Google Scholar]
- 13. Hida N, Maeda Y, Katagiri K et al . A new culture protocol to detect peptide‐specific cytotixic T lymphocyte precursors in the circulation. Cancer Immunol Immunother 2002; 51: 219–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Komatsu N, Shichijo S, Nakagawa M, Itoh K. New multiplexed flow cytometric assay to measure anti‐peptide antibody: a novel tool for monitoring immune responses to peptides used for immunization. Scand J Clin Lab Invest 2004; 64: 1–11. [DOI] [PubMed] [Google Scholar]
- 15. Wang Y, Harada M, Yano H et al . Prostatic acid phosphatase as a target molecule in specific immunotherapy for patients with nonprostate adencarcinoma. J Immnother 2005; 8: 5341. [DOI] [PubMed] [Google Scholar]
- 16. Matsueda S, Yao A, Ishihara Y et al . A prostate stem cell antigen‐derived peptide immunogenic in HLA‐A24+ prostate cancer patients. Prostate 2004; 60: 205–13. [DOI] [PubMed] [Google Scholar]
- 17. Gomi S, Nakao M, Niiya F et al . A cyclophilin B gene encodes antigenic epitopes recognized by HLA‐A24‐restricted tumor‐specific CTLs. J Mmunol 1999; 163: 4994–5004. [PubMed] [Google Scholar]
- 18. Ogata R, Matsueda S, Yao A, Noguchi M, Itoh K, Harada M. Identification of polycomb group protein enhancer of zeste homolog 2 (EZH2)‐derived peptides immunogenic in HLA‐A24+ prostate cancer patients. Prostate 2004; 60: 273–81. [DOI] [PubMed] [Google Scholar]
- 19. Yamada A, Kawano K, Koga M, Matsmoto T, Itoh K. Multidrug resistance‐associated protein 3 is a tumor rejection antigen recognized by HLA‐A2402‐restricted cytotoxic T lymphocytes. Cancer Res 2001; 61: 6459–66. [PubMed] [Google Scholar]
- 20. Yang D, Nakao M, Shichijo S et al . Identification of a gene coding for a protein possessing shared tumor epitopes capable of inducing HLA‐A24‐restricted cytotoxic T lymphocytes in cancer patients. Cancer Res 1999; 59: 4056–63. [PubMed] [Google Scholar]
- 21. Harashima N, Tanaka K, Sasatomi T et al . Recognition of the Lck tyrosine kinase as a tumor antigen by cytotoxic T lymphocytes of cancer patients with distant metastases. Eur J Immunol 2001; 31: 323–32. [DOI] [PubMed] [Google Scholar]
- 22. Ito M, Shichijo S, Tsuda N et al . Molecular basis of T cell‐mediated recognition of pancreatic cancer cells. Cancer Res 2001; 61: 2038–46. [PubMed] [Google Scholar]
- 23. Imai N, Harashima N, Ito M et al . Identification of Lck‐derived peptides capable of inducing HLA‐A2‐restricted and tumor‐specific CTLs in cancer patients with distant metastases. Int J Cancer 2001; 94: 237–42. [DOI] [PubMed] [Google Scholar]
- 24. Ito M, Shichijo S, Miyagi Y et al . Identification of SART3‐derived peptides capable of inducing HLA‐A2‐restricted and tumor‐specific CTLs in cancer patients with different HLA‐A2 subtypes. Int J Cancer 2000; 88: 633–9. [DOI] [PubMed] [Google Scholar]
- 25. Ishihara Y, Harada M, Azuma K et al . HER2/neu‐derived peptides recognized by both cellular and humoral immune systems in HLA‐A2+ cancer patients. Int J Oncol 2004; 24: 967–75. [PubMed] [Google Scholar]
- 26. Harada M, Matsueda S, Yao A, Ogata R, Noguchi M, Itoh K. Prostate‐related antigen‐derived new peptides having the capacity of inducing prostate cancer‐reactive CTLs in HLA‐A2+ prostate cancer patients. Oncol Rep 2004; 12: 601–7. [PubMed] [Google Scholar]
- 27. Shomura H, Shichijo S, Matsueda S et al . Identification of epidermal growth factor receptor‐derived peptides immunogenic for HLA‐A2+ cancer patients. Br J Cancer 2004; 90: 1563–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sakaguchi S. Naturally arising CD4+ regulatory T cells for immunologic self‐tolerance and negative control of immune responses. Annu Rev Immunol 2004; 22: 531–62. [DOI] [PubMed] [Google Scholar]
- 29. Chattopadhyay S, Chakraborty NG, Mukherji B. Regulatory T cells and tumor immunity. Cancer Immunol Immunother 2005; 54: 1153–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kitano H. Cancer robustness: tumor tactics. Nature 2003; 426: 125. [DOI] [PubMed] [Google Scholar]
- 31. Kitano H. Cancer as a robust system: implications for anticancer therapy. Nat Rev 2004; 4: 227–35. [DOI] [PubMed] [Google Scholar]
- 32. Li Q, Pan PY, Gu P, Xu D, Cehn SH. Role of immature myeloid Gr‐1+ cells in the development of antitumor immunity. Cancer Res 2004; 64: 1130–9. [DOI] [PubMed] [Google Scholar]
- 33. Kusmartsev S, Nagaraj S, Gabrilovich DI. Tumor‐associated CD8+ T cell tolerance induced by bone marrow‐derived immature myeloid cells. J Immunol 2005; 175: 4583–92. [DOI] [PMC free article] [PubMed] [Google Scholar]