

Abstract
We established a variant of MIAPaCa‐2 human pancreatic cancer cells that is resistant to 2′,2′‐difluorodeoxycytidine (gemcitabine, dFdCyd), MIAPaCa‐2/dFdCyd, and elucidated the biochemical characteristics and mechanism of dFdCyd‐resistance in these cells. We also evaluated 1‐(3‐C‐ethynyl‐β‐d‐ribo‐pentofuranosyl)cytosine (ECyd, TAS‐106, RNA polymerase inhibitor), a new anticancer ribonucleoside, for antitumor activity against the resistant cells in vitro and in vivo. MIAPaCa‐2/dFdCyd cells were 2541‐fold more resistant to dFdCyd than parental MIAPaCa‐2 cells, and the major mechanism of the dFdCyd‐resistance was found to be a decrease in the intracellular pool of dFdCyd and its active metabolites, which would result in a decrease in incorporation of dFdCyd triphosphate into DNA. This finding was confirmed by the discovery of decreased deoxycytidine kinase activity, increased cytidine deaminase and ribonucleotide reductase activity, and increased 5′‐nucleotidase mRNA expression in the MIAPaCa‐2/dFdCyd cells. The cytotoxicity of TAS‐106 as an antitumor nucleoside analog was similar in both parental and dFdCyd‐resistant cells, with IC50 values of 6.25 and 6.27 nM, respectively, and this finding was supported by similar intracellular uptake and metabolism of TAS‐106 in both cell lines. We also evaluated the in vivo antitumor activity of TAS‐106 against MIAPaCa‐2 and dFdCyd‐resistant MIAPaCa‐2/dFdCyd tumors implanted into nude mice. The tumor growth inhibition rate of weekly additions of TAS‐106 (7 mg/kg, iv) against parental and dFdCyd‐resistant tumors was 73% and 76%, respectively, while that of dFdCyd administered twice a week (240 mg/kg, iv) was 84% and 34%, respectively. These results suggest that TAS‐106 would contribute to the treatment of patients with advanced pancreatic carcinomas in whom dFdCyd‐based chemotherapy has failed. (Cancer Sci 2005; 96: 295 –302)
Pancreatic cancer is one of the most intractable cancers and has been associated with an increasing mortality rate in recent years. It remains the fifth leading cause of cancer mortality in the United States, with a 5‐year survival rate of 4%. (1) Thus there is an urgent need to develop effective drugs for the treatment of this cancer.
2′,2′‐Difluorodeoxycytidine (gemcitabine, dFdCyd) 2 , 3 is currently the standard chemotherapeutic drug for metastatic and advanced pancreatic cancer, but it only leads to a modest improvement in quality of life and survival. (4) Other cytotoxic drugs, including the anthracycline doxorubicin, have not shown any clinical benefit, and for these reasons many patients turn to alternative treatment modalities. Moreover, one of the problems with the use of dFdCyd, as in the case of other anticancer agents, is the induction of resistance. It was therefore considered worthwhile to evaluate the response of dFdCyd‐resistant tumors to new anticancer agents.
1‐(3‐C‐Ethynyl‐β‐d‐ribo‐pentofuranosyl)cytosine (ECyd, TAS‐106) is a new anticancer cytidine analog 5 , 6 , 7 , 8 found to possess a potent cytotoxic and antitumor activity in preclinical therapeutic models, and is undergoing a phase I clinical study in the United States. After incorporation into tumor cells TAS‐106 is converted rapidly to the monophosphate (ECMP) by uridine‐cytidine kinase (UCK, EC 2.7.1.48), (9) especially by uridine‐cytidine kinase 2 (UCK2, GenBank no. AF236637), and subsequently phosphorylated to the diphosphate (ECDP) and triphosphate (ECTP) forms. (10) ECTP is an active metabolite and inhibits RNA synthesis by non‐specifically blocking RNA polymerase. 11 , 12 , 13 Accordingly, the antitumor efficacy of the new antimetabolite TAS‐106 depends on intracellular accumulation of ECTP, which may be regulated by both cell membrane transport mechanisms and expression of UCK2. As it has been reported that UCK2 protein expression is higher in pancreatic tumor tissue than in other tumor tissues tested, (14) TAS‐106 holds great potential as a therapeutic agent for pancreatic cancer.
To investigate the efficacy of TAS‐106 against pancreatic cancers, we established a variant of MIAPaCa‐2 human pancreatic cancer cells resistant to dFdCyd (MIAPaCa‐2/dFdCyd). The present study was undertaken to identify factors involved in the development of resistance to dFdCyd by analyzing the intracellular metabolism of the drug in MIAPaCa‐2/dFdCyd cells, and to evaluate the efficacy of TAS‐106 against both parental (MIAPaCa‐2) and dFdCyd‐resistant pancreatic cancer cells both in vitro and in vivo.
Materials and Methods
Chemicals. TAS‐106 and dFdCyd were synthesized at Taiho Pharmaceutical Co. (Tokyo, Japan). [Cytosine‐5‐3H]TAS‐106 (4 Ci/mmol) was synthesized by Amersham International (Buckinghamshire, UK). [5–3H]dFdCyd (14 Ci/mmol), [5–3H(N)] cytidine (Cyd, 21 Ci/mmol), [5–3H(N)]2′‐deoxycytidine (dCyd, 23.5 Ci/mmol), and [methyl‐3H]dTTP (60 Ci/mmol) were purchased from Moravek Biochemicals (Brea, CA, USA). All other chemicals were commercial products of the highest quality available.
Cells and cell culture. The MIAPaCa‐2 human pancreatic carcinoma cell line was purchased from the American Type Culture Collection (Rockville, MD, USA) through Dainippon Pharmaceutical Co. (Osaka, Japan). A dFdCyd‐resistant variant of the MIAPaCa‐2 cell line, MIAPaCa‐2/dFdCyd cells, was developed in our laboratory by continuously exposing MIAPaCa‐2 cells to dFdCyd, with a starting concentration of 0.05 µM and stepwise increases up to 10 µM. The MIAPaCa‐2 cells and MIAPaCa‐2/dFdCyd cells were maintained in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum (ICN Biomedicals, Aurora, OH, USA) and 2.5% horse serum (Trace Scientific, Melbourne, Australia) at 37°C and 5% CO2. Under these conditions, acquired resistance to dFdCyd on MIAPaCa‐2/dFdCyd cells was maintained for at least 6 months.
Chemosensitivity test. The growth inhibitory effects of the drugs on human cancer cells were determined by colorimetric assay with 2‐(2‐methoxy‐4‐nitrophenyl)‐3‐(4‐nitrophenyl)‐5‐(2,4‐disulfophenyl)‐2H‐tetrazolium, monosodium salt (Cell Counting Kit‐8) reagent (Wako Pure Chemical Industries, Osaka, Japan). (15) Briefly, 180 µL aliquots of an exponentially growing cell suspension (1000 cells/180 µL/well) were incubated in 96‐well microplates for 24 h, and 20 µL volumes of various concentrations of the drugs were then added. After exposure to the drugs for 72 h, 20 µL of Cell Counting Kit‐8 reagent was added to each well, and the cell cultures were incubated at 37°C. After thorough mixing, the absorbance of each well was measured at 450 nm with a Model 3550 microplate reader (Bio‐Rad Laboratories, Hercules, CA, USA). The IC50 was defined as the drug concentration needed to produce a 50% reduction of growth relative to the control.
Evaluation of antitumor activity in vivo. Tumor fragments approximately 2 mm3 in size were transplanted subcutaneously into male BALB/cAJcl‐nu nude mice (CLEA Japan, Tokyo, Japan), and the mice were assigned randomly to a control group or to drug treatment groups, each consisting of six animals (day 0). Drugs were administered intravenously in a volume of 1 mL/100 g bodyweight starting the next day (day 1). In the group given the drug on days 1 and 8, the dose level of TAS‐106 was 7.0 mg/kg/day and in the groups given the drug on days 1, 4, 8 and 11, the dose level of dFdCyd was 240.0 mg/kg/day. Each drug was administered intermittently for 2 weeks according to the above schedules. To assess the antitumor effect of the drugs, tumor volume was calculated twice weekly using the formula LxW2/2, where L is the length (longest diameter) and W is the width (shortest diameter) of the tumor. Tumor volume was expressed as relative tumor volume (RTV) in the form of the ratio Vn/V0, where Vn is the volume on any given day and V0 is the volume on day 0. The dose of each drug used was the maximal tolerable dose, based on the results of a preliminary examination. In addition, 2 weeks after the initial injection (day 15), the tumor growth inhibition rate (IR, %) was calculated as the ratio of the mean RTV of the tumors treated to that of the control tumors (T/C) according to the formula (1‐T/C) × 100%.
Enzyme assay
Preparation of crude cell extracts. Cells were collected during logarithmic growth, and cell pellets were stored frozen at −135°C. Immediately before use, the pellets were thawed and homogenized with two volumes of 50 mM Tris‐HCl buffer (pH 8.0) containing 10 mM 2‐mercaptoethanol, 25 mM KCl, and 5 mM MgCl2. The homogenates were then centrifuged at 105 000 g for 1 h at 4°C, and the resultant supernatant (cytosol fractions) was used as an enzyme solution. The enzyme assays described below were carried out according to the method of Ikenaka et al. (16) with slight modification. The protein concentration in the cell extracts was measured by the Bradford method. (17)
Deoxycytidine kinase (DCK) activity. The reaction mixture (120 µL) consisted of 50 mM Tris‐HCl buffer (pH 8.0), 10 mM ATP, 5 mM MgCl2, 10 mM NaF, 50 µM [3H]dCyd (0.24 µCi/tube), 500 µM tetrahydrouridine (THU), and 48 µL of enzyme solution (cell cytosol). The reaction mixture was incubated at 37°C, and after stopping the reaction by the addition of 2 M perchloric acid (PCA) (24 µL), the incubation samples were centrifuged at 13 000 g . for 5 min at 4°C. The supernatant (96 µL) was then neutralized by the addition of 1 M KOH (24 µL), and after centrifugation at 13 000 g for 5 min at 4°C, the supernatant was analyzed by high‐performance liquid chromatography (HPLC). The extracts were applied to a DEAE‐2SW column (250 × 4.6 mm, Toso Co., Tokyo, Japan) and eluted with a 10 min linear gradient from 20 to 200 mM sodium phosphate buffer (pH 6.8) containing 10% acetonitrile, and then eluted for 20 min with 200 mM sodium phosphate buffer (pH 6.8) containing 10% acetonitrile at a rate of 1 mL/min.
Cytidine deaminase (CDA) activity. The assay method was similar to the DCK assay, except that the reaction mixture (120 µL) consisted of 60 mM potassium phosphate buffer (pH 7.6), 50 µM [3H]dCyd (0.24 µCi/tube) and 48 µL of enzyme solution.
Uridine/cytidine kinase (UCK) activity. The assay method was similar to the DCK assay, except that 50 µM [3H]Cyd (0.24 µCi/tube) was used instead of dCyd.
DNA polymerase activity. The reaction mixture (200 µL) consisted of 75 mM Tris‐HCl buffer (pH 7.1), 3 mM MgCl2, 1 mM dithiothreitol, 40 µM di‐sodium dihydrogen ethylenediamine tetraacetate, 75 µM dATP, 75 µM dGTP, 75 µM dCTP, 15 µM [methyl‐3H] dTTP (0.2 µCi/tube), 30 µg/tube salmon testes DNA, 100 µg/tube bovine serum albumin (BSA), and 20 µL of enzyme solution. After incubation at 37°C, the reaction was terminated by cooling the mixture in an ice‐bath, and to the entire mixture was added 0.5 M NaOH (50 µL), 10 mg/mL BSA (50 µL), and 50 µM sodium pyrophosphate containing 10% trichloro acetic acid (2 mL). After maintaining the mixture at 0°C for 10 min, it was centrifuged at 1200 g for 5 min, and the pellet was dissolved in 0.5 M NaOH (100 µL). The lysate was washed with 10% TCA (2 mL), and after centrifugation at 1200 g for 5 min, the resulting pellet was washed twice with 0.5 M NaOH (100 µL) and 10% TCA (2 mL). Next, the pellet was dissolved in 5% TCA (400 µL), and the solution was heated for 20 min at 90°C. The mixture was then centrifuged at 540 g for 5 min, and to the supernatant (200 µL) was added 10 mL of liquid scintillant AQUASOL‐2 (Amersham) to measure radioactivity with a Wallac 1414 WinSpectral liquid scintillation counter (Wallac Berthold Japan Co., Tokyo, Japan).
Ribonucleotide reductase (RNR) activity. RNR activity was determined using [(U)‐14C]‐CDP as the substrate according to the method described previously. (18)
Real‐time polymerase chain reaction (PCR). The mRNA levels of DCK (NM_000788), CDA (L27943), dCMP deaminase (DCD, NM_001921), equilibrative NBMPR‐sensitive nucleoside transporter (ENT1, AF079117), equilibrative NBMPR‐insensitive nucleoside transporter (ENT2, AF034102), ecto‐5′‐nucleotidase (NT5, NM_002526), 5′‐nucleotidase cytosolic III (NT5C3, NM_016489), DNA polymerase α (POLA, NM_016937), UCK2 (AF236637), and glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH, M33197) in the cancer cells were quantified by the real‐time PCR method with an ABI PRISM 7700 Sequence Detector System (PE Applied Biosystems, Foster City, CA, USA). Total RNA was extracted from approximately 1 × 107 cells with an RNeasy RNA Purification Kit (Qiagen, Hilden, Germany), and first‐strand cDNA were synthesized from 10 µg of total RNA with a Superscript Preamplification System for First Strand cDNA Synthesis (Gibco BRL, Gaithersburg, MD, USA). The PCR solution (25 µL) was composed of 10 µL of cDNA solution, 2.5 µL of primer/probe mixture (containing 5 pmol of each of the forward and reverse primers, 2.5 pmol of internal probe) and 12.5 µL of TaqMan Universal PCR Master Mix (Applied Biosystems, Foster City, CA, USA). The internal probes were labeled with a reporter dye, 6‐carboxyfluorescein (FAM), at the 5′‐end, and a quencher dye, 6‐carboxytetramethylrhodamine (TAMRA), at the 3′‐end. The primer and internal probe sequences are shown in Table 1. The NT5C3 and POLA primer and internal probe mixtures were purchased from Applied Biosystems. The PCR reaction conditions were 50°C for 2 min, 95°C for 10 min, and 45 cycles of 95°C for 15 s and 60°C for 1 min. The amounts of the PCR products were normalized to the GAPDH level.
Table 1.
Sequences of polymerase chain reaction primers and sequence‐specific probes for target genes and glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH)
| Gene | Forward primer | Reverse primer | Internal probe | Corresponding cDNA sequence |
|---|---|---|---|---|
| DCK | ATCCAGCTTCCTTCTGTCATTCC | CAACGAAGTGAGAGGCACCAG | CTCTTTTGTCTTCCTCAGCAGGTTGGCTT | 1235–1314 |
| CDA | AGGCAAGTCATGAGAGAGTTTGG | GCATTCTCTGGCTGTCACTGAGT | TGGCCCGTGTACATGACCAAGCC | 424–572 |
| DCD | CAGCACTGTTGGTGTTCGGA | GAAGGTGATGCTTGTGTAGGTGAA | CTCTTCTGTGCCCTGGCTCCATGC | 1517–1586 |
| ENT1 | TCTTCATGGCTGCCTTTGC | GGCTTCACTTTCTTGGGCC | TCGCCAGCCTCTGCATGTGCTT | 1295–1373 |
| ENT2 | CAAGACCTCATGGAAAGGGTG | CCACTCTGAACCCTCTGGTCA | CCCCACCACCAGGTCTGCATTTGT | 1838–1961 |
| NT5 | TAATGGTATAAACACAGGATACCATCCT | CATTATCTACTACAGCTTGCTACCTGACT | TCTTGCAACACCCATGTGCCTTTGA | 2457–2541 |
| UCK2 | GTGATCATCCCTAGAGGTGCAGATA | GGCCCTCCATTCAGGATGT | TCTGGTGGCCATCAACCTCATCGTG | 634–716 |
| GAPDH | GAAGGTGAAGGTCGGAGTC | GAAGATGGTGATGGGATTTC | CAAGCTTCCCGTTCTCAGCC | 66–291 |
The probes were labeled with a reporter dye (FAM) situated at the 5′ end of the oligonucleotide and a quencher dye (TAMRA) located at the 3′ end.
Measurement of intracellular metabolism and ribonucleotide pools. After preincubating 2 × 106 cells with drug‐free medium for 24 h, cells were incubated with 1 µM [3H]‐dFdCyd and 1 µM [3H]‐TAS‐106 for 0, 2, 4 or 6 h, and with 1 µM [3H]‐Cyd and 1 µM [3H]‐dCyd for 0, 0.5, 1, 2 or 6 h. The acid‐soluble fraction containing cell nucleotides was extracted from the cells with 0.42 M PCA (150 µL). The mixture was mixed well, neutralized with 1 M K2HPO4 (150 µL), and then centrifuged at 20 400 g for 5 min at 4°C. The supernatant was passed through a 0.45‐µm filter and subjected to HPLC. Aliquots of the filtrate were applied to a Shodex Axpac WA‐624 weak anion‐exchange column (250 × 4.6 mm, Showa Denko K.K., Tokyo, Japan), and nucleotides were eluted with a linear gradient from 100% solution A (0.1 M sodium phosphate buffer, pH 6.8, containing 10% acetonitrile) to 80% solution B (0.5 M sodium phosphate buffer, pH 6.8, containing 20% acetonitrile) for the first 30 min followed by elution for 50 min from 80 to 100% solution B at a rate of 1 mL/min. The radioactivity associated with the respective nucleotides was measured with an on‐line radioactive flow detector (Radiomatic D515TR, Packard, St. Louis, MO, USA). The eluent was mixed automatically with scintillation fluid (Ultima FLOW AP, Packard) at a ratio of 1 : 3. The amount of each compound and its nucleotides in the extracts was calculated based on the specific activity. Ribonucleotides were measured with a UV detector (UV‐1575, JASCO) at the same time.
Incorporation of dFdCyd and TAS‐106 into DNA and RNA. The PCA‐insoluble fraction obtained by the above procedure was dissolved in formic acid (1 mL), and after mixing the entire volume of the sample with 10 mL of liquid scintillant ACS‐II (Amersham), radioactivity was measured with a Wallac 1414 WinSpectral liquid scintillation counter (Wallac Berthold Japan Co.).
Measurement of deoxyribonucleotide pools. After preincubating 8 × 106 cells with drug‐free medium for 24 h, cells were incubated with 1 µM dFdCyd and 1 µM TAS‐106 for 0, 2, 4 or 6 h. The cell pellets were then suspended with 0.42 M PCA (150 µL), the mixture was agitated well for 10 min at 4°C. The mixture was then neutralized by the addition of 1 M K2HPO4 (150 µL), centrifuged at 20 400 g for 5 min at 4°C, and passed through a 0.45‐µm filter. The PCA‐soluble fraction (100 µL) was mixed with 20 M 2′‐deoxyguanosine (25 µL) and 0.2 M NaIO4 (25 µL), and the mixture was incubated at 37°C for 5 min. At the end of the incubation period, it was mixed with 1 M rhamnose (4 µL) and 4 M methylamine (6 µL). The mixture was then incubated at 37°C for 5 min, and subjected to HPLC analysis. The mixture was applied to a Partisil 10 SAX anion‐exchange column (250 × 4.6 mm, Whatman, Clifton, NY, USA), eluted with 10% acetonitrile in a 0.35 M ammonium phosphate buffer, pH 3.0, at a rate of 2 mL/min, and the deoxyribonucleotides were measured with a UV detector (UV‐970, JASCO).
Statistical analysis. The significance of differences between the parental cells and dFdCyd‐resistant cells was assessed using Welch's test.
Results
Chemosensitivity of human pancreatic cancer cells. The IC50 values of dFdCyd and TAS‐106 are shown in Table 2. The IC50 value of dFdCyd and TAS‐106 was 0.00692 µM and 0.00625 µM, respectively, for MIAPaCa‐2 cells, and 17.6 µM and 0.00627 µM, respectively, for MIAPaCa‐2/dFdCyd cells. Based on these data, the relative resistance ratio to dFdCyd and TAS‐106 was calculated to be 2541 and 1.00, respectively. The MIAPaCa‐2/dFdCyd cell line showed no cross‐resistance to TAS‐106.
Table 2.
IC50 values for MIAPaCa‐2 and its dFdCyd‐resistant cells, MIAPaCa‐2/dFdCyd
| Cell Line | Compound | IC50 value (µM) | Resistant ratio |
|---|---|---|---|
| MIAPaCa‐2 | dFdCyd | 0.00692 ± 0.00228 | – |
| TAS‐106 | 0.00625 ± 0.00110 | – | |
| MIAPaCa‐2/dFdCyd | dFdCyd | 17.58667 ± 1.63016 | 2541 |
| TAS‐106 | 0.00627 ± 0.00073 | 1.00 |
Growth inhibition was determined as described in Materials and Methods section. IC50 values are given as mean values (in µM) ± standard error of the mean (SEM) of three independent experiments. Resistant ratio was the ratio of IC50 values for resistant variants to that for the respective parental cell lines.
Antitumor activity in vivo. In addition to in vitro cytotoxicity, we evaluated the in vivo antitumor effect of dFdCyd and TAS‐106 against parental and dFdCyd‐resistant pancreatic tumors in nude mice (Fig. 1). The tumor growth IR of dFdCyd against MIAPaCa‐2 and MIAPaCa‐2/dFdCyd tumors was 84% and 34%, respectively, indicating the lack of antitumor activity of dFdCyd against pancreatic tumors after they have acquired resistance. On the other hand, TAS‐106 displayed potent antitumor activity against both MIAPaCa‐2 tumors and MIAPaCa‐2/dFdCyd tumors, with IR values 73% and 76%, respectively, suggesting the absence of cross‐resistance of dFdCyd‐resistant pancreatic tumors to TAS‐106, both under in vitro and in vivo conditions.
Figure 1.
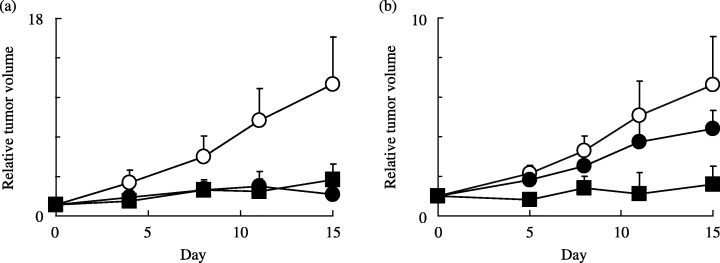
Antitumor activity of TAS‐106 and dFdCyd in nude mouse models with(a) MIAPaCa‐2 and (b) MIAPaCa‐2/dFdCyd tumors. Control (○), TAS‐106 (▪, 7 mg/kg once weekly, i.v.), and dFdCyd (•, 240 mg/kg twice weekly, i.v.) were administered for 2 weeks. Points are mean ± standard deviation for six mice.
Metabolic enzyme activities in parental and dFdCyd‐resistant pancreatic cancer cells. Table 3 shows the activity of enzymes involved in pyrimidine metabolism in the MIAPaCa‐2 cell line and its dFdCyd‐resistant subline. The DCK activity of the MIAPaCa‐2/dFdCyd cells was approximately three times lower (P < 0.01) than its activity level in the parental cells. On the other hand, the CDA and RNR activities were twofold higher (P < 0.01) than that in the parental cells. UCK activity and DNA polymerase activity were unchanged.
Table 3.
Metabolic enzyme activities of MIAPaCa‐2 and its dFdCyd‐resistant cells, MIAPaCa‐2/dFdCyd
| Enzyme | Enzyme activity (nmol/min/mg protein) | |
|---|---|---|
| MIAPaCa‐2 | MIAPaCa‐2/dFd | |
| dCyd kinase | 0.0436 ± 0.0017 | 0.0146 ± 0.0033* |
| Cyd deaminase | 1.20 ± 0.10 | 2.30 ± 0.22* |
| Ribonucleotide reductase | 0.0158 ± 0.00378 | 0.0370 ± 0.00566* |
| DNA polymerase | 0.0651 ± 0.0042 | 0.0679 ± 0.0080 |
| Cyd kinase | 5.46 ± 0.34 | 5.50 ± 0.23 |
Enzyme activities are mean values ± SEM of three individual experiments. *P < 0.01 vs value for MIAPaCa‐2.
Expression levels of mRNA of various genes in human pancreatic cancer cells. We measured the mRNA levels of DCK, CDA, DCD, ENT1, ENT2, NT5, NT5C3, POLA and UCK2 by real‐time PCR with primers specific for each transcript (Table 1). As shown in Table 4, expression of NT5 mRNA in dFdCyd‐resistant cells was amplified approximately threefold compared with its expression in parental cells, but the mRNA levels of DCK, CDA, DCD, ENT1, ENT2, NT5C3, POLA and UCK2 in the dFdCyd‐resistant cells were almost the same as in the parental cells.
Table 4.
Gene expression of MIAPaCa‐2 and its dFdCyd‐resistant cells, MIAPaCa‐2/dFdCyd
| Gene | Gene expression | |
|---|---|---|
| MIAPaCa‐2 | MIAPaCa‐2/dFdCyd | |
| DCK | 0.31 ± 0.03 | 0.31 ± 0.02 |
| CDA | 0.93 ± 0.09 | 1.07 ± 0.24 |
| DCD | 1.15 ± 0.06 | 1.25 ± 0.09 |
| ENT1 | 1.04 ± 0.04 | 1.27 ± 0.06 |
| ENT2 | 0.20 ± 0.03 | 0.17 ± 0.01 |
| NT5 | 1.00 ± 0.04 | 2.52 ± 0.18 |
| NT5C3 | 0.60 ± 0.06 | 0.60 ± 0.10 |
| POLA | 0.88 ± 0.18 | 1.11 ± 0.03 |
| UCK2 | 1.02 ± 0.05 | 1.36 ± 0.37 |
A relative target gene expression value was obtained by division of the target gene value by the value for glyceraldehyde‐3‐phosphate dehydrogenase as an internal reference gene. Gene expression levels are mean values ± SEM of at least three experiments.
Intracellular metabolism and ribonucleotide pools. Total cellular uptake of TAS‐106, dFdCyd, deoxycytidine, cytidine and their phosphorylated forms in parental and dFdCyd‐resistant pancreatic cancer cells after exposure to the drugs are shown in Fig. 2. The intracellular concentrations of the nucleoside and nucleotide forms of each compound are also shown in Fig. 3, and the incorporation of TAS‐106 and dFdCyd into the acid‐insoluble fraction of the cells is shown in Fig. 4. The metabolites of dFdCyd accumulated linearly in the parental cells for the first 6 h, and dFdCyd triphosphate (dFdCTP) was the predominant metabolite in the cells. Incorporation of dFdCTP into the acid‐insoluble fraction plateaued at the level of 16 pmol/106 cells, but the total amount of metabolites, including predominant dFdCTP, in the dFdCyd‐resistant cells was approximately 0.1%, the amount formed in the parent cells. The intracellular metabolites derived from TAS‐106 increased linearly for the first 6 h in both the parent and dFdCyd‐resistant cells reaching steady‐state levels of about 250 pmol/106 cells. ECTP was the major metabolite, and was followed by ECDP, with these two metabolites accounting for the majority of the metabolites. Incorporation of ECTP into the acid‐insoluble fraction also increased linearly for the first 6 h of exposure in both the parent and the dFdCyd‐resistant cells, and reached 30 pmol/106 cells or more after incubation for 6 h. Intracellular accumulation of dCyd and Cyd peaked at 2 h and decreased thereafter; however, their incorporation into the acid‐insoluble fraction continued to increase linearly even after 2 h in both the parent and dFdCyd‐resistant cells (data not shown).
Figure 2.
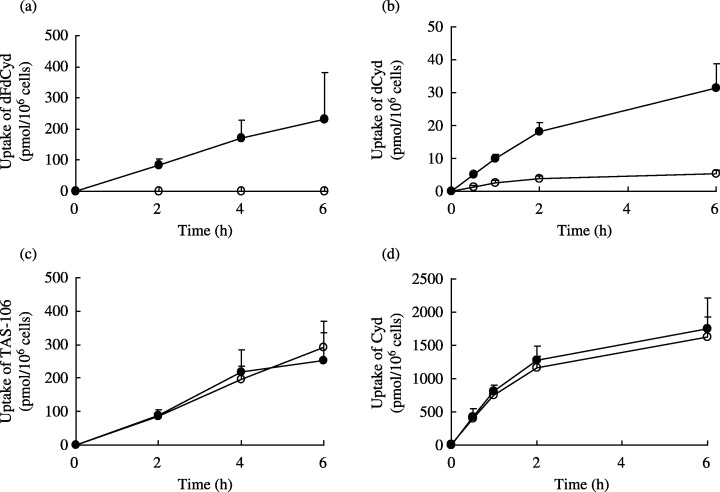
Total cellular uptake of (a) dFdCyd, (b) dCyd, (c) TAS‐106 and (d) Cyd in MIAPaCa‐2 (•) and MIAPaCa‐2/dFdCyd (○) cells. Uptake of each compound was showed as the sum of intracellular nucleoside, nucleotides and the acid‐insoluble fraction. Values are mean ± standard deviation for three experiments.
Figure 3.
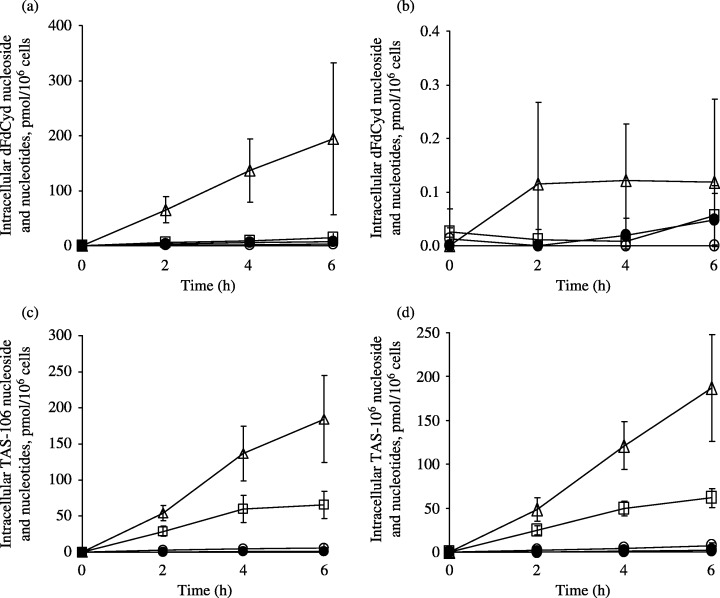
Level of intracellular nucleosides and nucleotides in (a,c) MIAPaCa‐2 and (b,d) MIAPaCa‐2/dFdCyd cells. After incubation with (a,b) dFdCyd and (c,d) TAS‐106, intracellular concentrations of the drug (•), its monophosphate (○), diphosphate (□) and triphosphate (▵) form was measured by high‐performance liquid chromatography as described in Materials and Methods. Values are mean ± standard deviation for three experiments.
Figure 4.
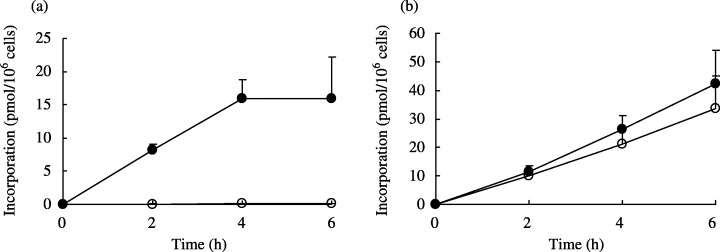
Incorporation of (a) dFdCyd and (b) TAS‐106 into the acid‐insoluble fraction of MIAPaCa‐2 (•) and MIAPaCa‐2/dFdCyd (○) cells. Values are mean standard deviation for three experiments.
Effect of dFdCyd and TAS‐106 on natural intracellular ribonucleotide and deoxyribonucleotide pools. The intracellular ribonucleoside triphosphate (NTP) pools and deoxyribonucleoside triphosphate (dNTP) pools following exposure to dFdCyd and TAS‐106 are shown in Figs 5 and 6, respectively. In the parental cells, the NTP pools doubled in size within 0.5 h after exposure to dFdCyd and TAS‐106, whereas the size of the NTP pools in the dFdCyd‐resistant cells was almost unchanged. The NTP pools in dFdCyd‐resistant cells were two times higher than those in the parental cells at 0 h. Measurement of the dNTP pools in the MIAPaCa‐2 cells following exposure to dFdCyd showed a time‐dependent increase in the size of the dTTP pool, whereas dATP and dGTP had disappeared completely and the dCTP pool size had decreased slightly. These changes were less marked in the dFdCyd‐resistant cells. By contrast, no significant changes in the size of the dNTP pools following exposure of TAS‐106 was observed in either the parental or the dFdCyd‐resistant cells.
Figure 5.
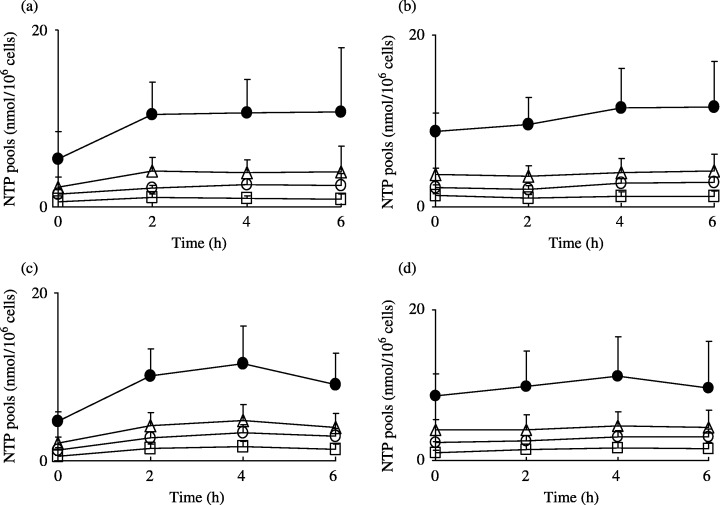
Changes in NTP pools in (a,c) MIAPaCa‐2 and (b,d) MIAPaCa‐2/dFdCyd cells after incubation with (a,b) dFdCyd and (c,d) TAS‐106. •, ATP; □, CTP; ○, GTP; and ▵, UTP. Values are mean ± standard deviation for three experiments.
Figure 6.
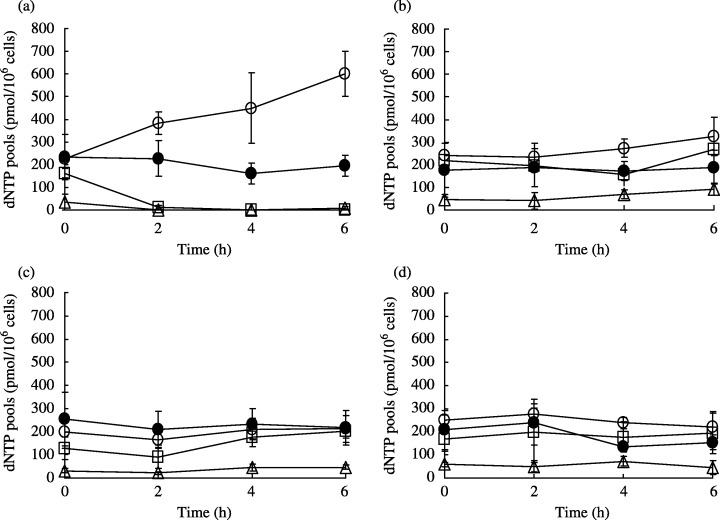
Changes in dNTP pools in (a,c) MIAPaCa‐2 and (b,d) MIAPaCa‐2/dFdCyd cells after incubation with (a,b) dFdCyd and (c,d) TAS‐106. □, dATP; •, dCTP; ▵, dGTP; and ○, dTTP. Values are mean ± standard deviation for three experiments.
Discussion
Pancreatic cancer continues to represent a significant oncological challenge. Because of the high mortality due to pancreatic cancer and few effective therapeutic agents, we assessed TAS‐106 for efficacy against human pancreatic cancer cell line MIAPaCa‐2 and the dFdCyd‐resistant subline MIAPaCa‐2/dFdCyd. The results showed that TAS‐106 would be an effective drug against pancreatic cancer (Table 2). Moreover, the present study shows TAS‐106 to be useful in the treatment of pancreatic cancer, as revealed by the experiments performed in nude mice subcutaneously implanted with the human pancreatic carcinoma cell line MIAPaCa‐2/dFdCyd resistant to dFdCyd (Fig. 1). The tumor growth inhibitory effect of TAS‐106 was not affected by a cross‐resistance for gemcitabine. We therefore analyzed why dFdCyd‐resistant cells did not show cross‐resistance to TAS‐106.
The major factor responsible for the resistance of MIAPaCa‐2/dFdCyd cells to dFdCyd seems to be decreased uptake of dFdCyd (Fig. 2) and an associated decrease in the incorporation of the dFdCyd‐triphosphate (Fig. 3) of the drug into nuclear fractions of cancer cells (Fig. 4). It has been reported that dFdCyd is incorporated into RNA as well as DNA, and that, whereas its incorporation into DNA is both time and concentration dependent, its incorporation into RNA is only concentration dependent. (19) In view of previous findings, total radioactivity in the acid‐insoluble fraction measured in the present study may be concluded to reflect the total amount of dFdCyd incorporated into DNA and RNA. The decrease in the intracellular pool of dFdCyd seems to represent a decrease in the expression of nucleoside transporter or alternation of its function, which is thought to serve as the route of uptake of many antitumor nucleosides by cancer cells. It has been reported that the nucleoside transporter‐mediated uptake of dFdCyd varies from cell to cell. 20 , 21 , 22 Although the transporter participating in the incorporation of dFdCyd into MIAPaCa‐2/dFdCyd cells is unknown, the magnitude of the decrease in the amount of deoxycytidine metabolites in MIAPaCa‐2/dFdCyd cells was about half of that observed in the parent cell line (data not shown), while in the case of dFdCyd the decrease of its intracellular metabolites was much more pronounced. This indicates that among various subtypes of nucleoside transporters reported to date, different types of transporters might be involved in controlling intracellular concentration of dCyd and dFdCyd. Although, the expression levels of ENT1 and ENT2 participating in active cellular transport (22) were similar in parental and dFdCyd‐resistant cells (Table 4), the function of ENT1 and ENT2 could be changed in the process of acquiring resistance for dFdCyd. However, overexpression of NT5, which localizes to the cell membrane, occurred in the dFdCyd‐resistant cells (Table 4). This means that after dephosphorylation of an active metabolite (phosphorylated dFdCyd species) by NT5, the dFdCyd released could be effluxed through nucleoside transporters. However, it is still unclear whether NT5 overexpression participates in an increased efflux of gemcitabine, and this issue will be examined in detail in the future.
Decrease in DCK activity and increase in CDA and RNR activity in MIAPaCa‐2/dFdCyd cells indicates that cells were regulated to avoid damage by dFdCyd incorporated into the cell. dFdCyd is a relatively good substrate of CDA, while TAS‐106 hardly undergoes the deamination process. The change in CDA activity appeared to have occurred at the protein translation stage because there was no change in the level of gene expression. In MIAPaCa‐2/dFdCyd cells the factor that caused the decreased DCK activity was identified as a point mutation, resulting in the change in amino acid composition, and in enzymatic activity as well (data not shown).
When the changes in the intracellular NTP pools in parental MIAPaCa‐2 cells following exposure to TAS‐106 and dFdCyd were analyzed (Fig. 5), the amount of ATP (a phosphate donor) was found to have increased markedly. Moreover the CTP, UTP and GTP pools increased as a result of exposure to dFdCyd. The increase in the NTP pools seems to favor dFdCyd phosphorylation, and the CTP, UTP and ATP pools do play an important role in the accumulation of dFdCyd triphosphate in solid cancers. (23) Moreover, UTP has been reported to be the optimal phosphate donor for phosphorylation of the deoxynucleoside by deoxycytidine kinase. (24) On the other hand, when MIAPaCa‐2/dFdCyd cells were exposed to dFdCyd, there was scarcely any change in the size of the NTP pools during the first 6 h of exposure. This is probably caused by a decrease in the intracellular pools of dFdCyd and its metabolites associated with the development of resistance. When MIAPaCa‐2/dFdCyd cells were exposed to TAS‐106, Cyd (data not shown) and dCyd (data not shown), no change in the size of the NTP pools was seen. In the dFdCyd‐resistant cells, the NTP pool size before exposure of dFdCyd was approximately twice that in the parent cells, and the same as the size of the pool observed in the parent cells after exposure to each of the nucleosides. It therefore seems likely that the MIAPaCa‐2/dFdCyd cells did not require any new phosphate donor following exposure to each of the nucleosides. When the changes in the size of the dNTP pools were analyzed (Fig. 6), dATP and dGTP were found to have completely disappeared from the MIAPaCa‐2 cells following exposure to dFdCyd. This phenomenon has been reported in solid cancers exposed to dFdCyd. 25 , 26 It has also been reported that the size of dNTP pools is maintained by de novo enzyme (RNR) and salvage pathway enzymes (nucleoside/nucleotide kinase), and that purine deoxyribonucleotide pools are dependent on RNR, whereas pyrimidine dNTP pools are dependent on the salvage pathway enzymes. (25) It therefore seems possible that exposure to dFdCyd inhibited RNR, which led to a loss of dATP and dGTP. Exposure of cells to 2‐chlorodeoxyadenosine (a DNA synthesis inhibitor) has been reported to reduce the amounts of dATP and dGTP and increase the amount of dTTP, suggesting that the resulting imbalance among the different dNTP triggered inhibition of DNA synthesis. (27) Therefore, the increase in amount of dTTP observed in the present study seems to act as a factor that triggers the inhibition of DNA synthesis following exposure to dFdCyd. The absence of changes in size of the dNTP pools following exposure of dFdCyd to MIAPaCa‐2/dFdCyd cells is probably explained by the absence of any effect of dFdCyd on the de novo and salvage pathway enzymes owing to the reduced intracellular pools of dFdCyd and its metabolites. On the other hand, TAS‐106 had little effect on the size of the intracellular dNTP pools, probably due to a lack of inhibitory activity on the enzymatic pathways that alter the size of the dNTP pools.
In a study on dFdCyd‐resistant ovarian cancer cells, van Haperen et al. claimed that the main mechanism of the resistance of their cells to dFdCyd was caused by DCK deficiency. (28) As no DCK deficiency was detected (data not shown) in the dFdCyd‐resistant MIAPaCa‐2/dFdCyd cells in our study, the mechanism of resistance of the dFdCyd‐resistant cells established in the present work was different than that reported previously. Moreover, Goan et al. investigated dFdCyd‐resistant nasopharyngeal carcinoma KB cells and claimed that the main mechanism of the resistance acquired by their cell line to dFdCyd was caused by the overexpression of RNR, and the expansion of dATP and dCTP pools. (29) In the present study, we also observed the increase of RNR activity; however, changes in the size of the dNTP pools were not observed in the MIAPaCa‐2/dFdCyd cell line. Achiwa et al. suggested that H23/GEM‐R, a dFdCyd‐resistant lung carcinoma cell, became resistant due to a significant decrease of dCK gene expression. (30) However, no change of dCK expression in the MIAPaCa‐2/dFdCyd cell line was found. Therefore, the mechanism of resistance of MIAPaCa‐2/dFdCyd to dFdCyd was also different than for those already reported. 28 , 29 , 30
The mechanism of development of resistance to dFdCyd in clinical cases involves various factors. The general mechanisms of resistance to nucleoside analogs have been described in cell lines and in clinical specimens. (31) In addition to these reports on multiple factors responsible for the development of resistance, the sensitivity of clinical tumor specimens to cytosine arabinoside, which is a deoxycytidine derivative like dFdCyd, has been reported to correlate with the level of expression of the nucleoside transporter. (32) This finding strongly suggests that the nucleoside transporter level also may play a major role in the mechanism of development of dFdCyd resistance clinically, although the exact mechanism still remains unknown.
In conclusion, TAS‐106 exerted strong antitumor effects against several pancreatic cancer cell lines, including a subline resistant to dFdCyd, a compound currently used as a first‐line drug in the treatment of pancreatic cancer patients. Thus, TAS‐106 is a promising drug for the treatment of pancreatic cancer patients.
References
- 1. Greenlee RT, Hill‐Harmon MB, Murray T, Thun M. Cancer statistics. CA Cancer J Clin 2001; 51: 15–36. [DOI] [PubMed] [Google Scholar]
- 2. Hertel LW, Boder GB, Kroin JS, Rinzel SM, Poore GA, Todd GC, Grindey GB. Evaluation of the antitumour activity of gemcitabine (2′,2′‐difluoro‐2′‐deoxycytidine). Cancer Res 1990; 50: 4417–22. [PubMed] [Google Scholar]
- 3. Braakhuis BJM, Van Dongen GAMS, Vermorken JB, Snow GB. Preclinical in vivo activity of 2′,2′‐difluoro‐2′‐deoxycytidine (gemcitabine) against human head and neck cancer. Cancer Res 1991; 51: 211–14. [PubMed] [Google Scholar]
- 4. Heinemann V. Gemcitabine: progress in the treatment of pancreatic cancer. Oncology 2001; 60: 8–18. [DOI] [PubMed] [Google Scholar]
- 5. Matsuda A, Sasaki T. Antitumor activity of sugar‐modified cytosine nucleosides. Cancer Sci 2004; 95: 105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hattori H, Tanaka M, Fukushima M, Sasaki T, Matsuda A. Nucleosides and nucleotides. 158. 1‐(3‐C‐ethynyl‐β‐d‐ribo‐pentofuranosyl) cytosine, 1‐(3‐C‐ethynyl‐β‐d‐ribo‐pentofuranosyl) uracil, and their nucleobase analogues as new potential multifunctional antitumor nucleosides with a broad spectrum of activity. J Med Chem 1996;. 39: 5005–11. [DOI] [PubMed] [Google Scholar]
- 7. Tabata S, Tanaka M, Matsuda A, Fukushima M, Sasaki T. Antitumor effect of a novel multifunctional antitumor nucleoside, 3′‐ethynylcytidine, on human cancers. Oncol Rep 1996; 3: 1029–34. [DOI] [PubMed] [Google Scholar]
- 8. Shimamoto Y, Fujioka A, Kazuno H, Murakami Y, Ohshimo H, Kato T, Matsuda A, Sasaki T, Fukushima M. Antitumor activity and pharmacokinetics of TAS‐106, 1‐(3‐C‐ethynyl‐β‐d‐ribo‐pentofuranosyl) cytosine. Jpn J Cancer Res 2001; 92: 343–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Koizumi K, Shimamoto Y, Azuma A, Wataya Y, Matsuda A, Sasaki T, Fukushima M. Cloning and expression of uridine/cytidine kinase cDNA from human fibrosarcoma cells. Int J Mol Med 2001; 8: 273–8. [PubMed] [Google Scholar]
- 10. Shimamoto Y, Kazuno H, Murakami Y, Azuma A, Koizumi K, Matsuda A, Sasaki T, Fukushima M. Cellular and biochemical mechanisms of the resistance of human cancer cells to a new anticancer ribo‐nucleoside, TAS‐106. Jpn J Cancer Res 2002; 93: 445–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tabata S, Tanaka M, Endo Y, Obata T, Matsuda A, Sasaki T. Anti‐tumor mechanisms of 3′‐ethynyluridine and 3′‐ethynylcytidine as RNA synthesis inhibitors: development and characterization of 3′‐ethynyluridine‐resistant cells. Cancer Lett 1997; 116: 225–31. [DOI] [PubMed] [Google Scholar]
- 12. Takatori S, Kanda H, Takenaka K, Wataya Y, Matsuda A, Fukushima M, Shimamoto Y, Tanaka M, Sasaki T. Antitumor mechanisms and metabolism of the novel antitumor nucleoside analogues, 1‐(3‐C‐ethynyl‐β‐d‐ribo‐pentofuranosyl) cytosine and 1‐(3‐C‐Ethynyl‐β‐d‐ribo‐pentofuranosyl) uracil. Cancer Chemother Pharmacol 1999; 44: 97–104. [DOI] [PubMed] [Google Scholar]
- 13. Matsuda A, Fukushima M, Wataya Y, Sasaki T. A new antitumor nucleoside, 1‐(3‐C‐ethynyl‐β‐d‐ribo‐pentofuranosyl) cytosine (ECyd), is a potent inhibitor of RNA synthesis. Nucleosides Nucleotides 1999; 18: 811–14. [DOI] [PubMed] [Google Scholar]
- 14. Shimamoto Y, Koizumi K, Okabe H, Kazuno H, Murakami Y, Nakagawa F, Matsuda A, Sasaki T, Fukushima M. Sensitivity of human cancer cells to the new anticancer ribo‐nucleoside TAS‐106 is correlated with expression of uridine‐cytidine kinase 2. Jpn J Cancer Res 2002; 93: 825–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ishiyama M, Miyazono Y, Sasamoto K, Ohkura Y. A highly water‐soluble disulfonated tetrazolium salt as a chromogenic indicator for NADH as well as cell viability. Talanta 1997; 44: 1299–305. [DOI] [PubMed] [Google Scholar]
- 16. Ikenaka K, Fukushima M, Nakamura H, Okamoto M, Shirasaka T, Fujii S. Metabolism of pyrimidine nucleosides in various tissues and tumor cells from rodents. Gann 1981; 72: 590–7. [PubMed] [Google Scholar]
- 17. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein‐dye binding. Anal Biochem 1976; 72: 248–54. [DOI] [PubMed] [Google Scholar]
- 18. Fukushima M, Fujioka A, Uchida J, Nakagawa F, Takechi T. Thymidylate synthase (TS) and ribonucleotide reductase (RNR) may be involved in acquired resistance to 5‐fluorouracil (5‐FU) in human cancer xenografts in vivo . Eur J Cancer 2001; 37: 1681–7. [DOI] [PubMed] [Google Scholar]
- 19. Ruiz van Haperen VWT, Veerman G, Vermorken JB, Peters GJ. 2′,2′‐Difluorodeoxycytidine (gemcitabine) incorporation into RNA and DNA of tumor cell lines. Biochem Pharmacol 1993; 46: 762–6. [DOI] [PubMed] [Google Scholar]
- 20. Burke T, Lee S, Ferguson PJ, Hammond JR. Interaction of 2′,2′‐Difluorodeoxycytidine (gemcitabine) and formycin B with the Na+‐dependent nucleoside transporters of ehrlich ascites tumour cells. J Pharmacol Exp Ther 1998; 286: 1333–40. [PubMed] [Google Scholar]
- 21. Hammond JR, Lee S, Ferguson PJ. [3H]Gemcitabine uptake by nucleoside transporters in a human head and neck squamous carcinoma cell line. J Pharmacol Exp Ther 1999; 288: 1185–91. [PubMed] [Google Scholar]
- 22. Mackey JR, Mani RS, Selner M, Mowles D, Young JD, Belt JA, Crawford CR, Cass CE. Functional nucleoside transporters are required for gemcitabine influx and manifestaction of toxicity in cancer cell lines. Cancer Res 1998; 58: 4349–57. [PubMed] [Google Scholar]
- 23. Ruiz van Haperen VWT, Veerman G, Vermorken JB, Pinedo HM, Peters GJ. Regulation of phosphorylation of deoxycytidine and 2′,2′‐difluorodeoxycytidine (gemcitabine): Effects of cytidine 5′‐triphosphate and uridine 5′‐triphosphate in relation to chemosensitivity for 2′,2′‐difluorodeoxycytidine. Biochem Pharmacol 1996; 51: 911–18. [DOI] [PubMed] [Google Scholar]
- 24. Hughes TL, Hahn TM, Reynolds KK, Shewach DS. Kinetic analysis of human deoxycytidine kinase with the true phosphate donor uridine triphosphate. Biochemistry 1997; 36: 7540–7. [DOI] [PubMed] [Google Scholar]
- 25. Shewach DS, Hahn TM, Chang E, Hertel LW, Lawrence TS. Metabolism of 2′,2′‐difluoro‐2′‐deoxycytidine and radiation sensitization of human colon carcinoma cells. Cancer Res 1994; 54: 3218–23. [PubMed] [Google Scholar]
- 26. Heinemann V, Xu YZ, Chubb S, Sen A, Hertel LW, Grindey GB, Plunkett W. Inhibition of ribonucleotide reduction in CCRF‐CEM cells by 2′,2′‐difluorodeoxycytidine. Mol Pharmacol 1990; 38: 567–72. [PubMed] [Google Scholar]
- 27. Hirota Y, Yoshioka A, Tanaka S, Watanabe K, Otani T, Minowada J, Matsuda A, Ueda T, Wataya Y. Imbalance of deoxyribonucleoside triphosphates, DNA double‐strand breaks, and cell death caused by 2‐chlorodeoxyadenosine in mouse FM3A cells. Cancer Res 1989; 49: 915–19. [PubMed] [Google Scholar]
- 28. Ruiz van Haperen VWT, Veerman G, Eriksson S, Boven E, Stegman APA, Hermsen M, Vermorken JB, Pinedo HM, Peters GJ. Development and molecular characterization of a 2′,2′‐difluorodeoxycytidine‐resistant variant of the human ovarian carcinoma cell line A2780. Cancer Res 1994; 54: 4138–43. [PubMed] [Google Scholar]
- 29. Goan YG, Zhou B, Hu E, Mi S, Yen Y. Overexpression of ribonucleotide reductase as a mechanism of resistance to 2,2‐difluorodeoxycytidine in the human KB cancer cell line. Cancer Res 1999;. 59 : 4204–7. [PubMed] [Google Scholar]
- 30. Achiwa H, Oguri T, Sato S, Maeda H, Niimi T, Ueda R. Determinants of sensitivity and resistance to gemcitabine: The roles of human equilibrative nucleoside transporter 1 and deoxycytidine kinase in non‐small cell lung cancer. Cancer Sci 2004; 95: 753–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Galmarini CM, Mackey JR, Dumontet C. Nucleoside analogue: mechanisms of drug resistance and reversal strategies. Leukemia 2001; 15: 875–90. [DOI] [PubMed] [Google Scholar]
- 32. Wright AM, Paterson AR, Sowa B, Akabutu JJ, Grundy PE, Gati WP. Cytotoxicity of 2‐chlorodeoxyadenosine and arabinosylcytosine in leukaemic lymphoblasts from paediatric patients: significance of cellular nucleoside transporter content. Br J Haematol 2002; 116: 528–37. [DOI] [PubMed] [Google Scholar]