

Abstract
Cancer cells consume large amounts of glucose because of their specific metabolic pathway. However, cancer cells exist in tumor tissue where glucose is insufficient. To survive, cancer cells likely have the mechanism to elude their glucose addiction. Here we show that functional mitochondria are essential if cancer cells are to avoid glucose addiction. Cancer cells with dysfunctional mitochondria, such as mitochondrial DNA‐deficient ρ0 cells and electron transport chain blocker‐treated cells, were highly sensitive to glucose deprivation. Our data demonstrated that this sensitization was associated with failure of the unfolded protein response (UPR), an adaptive response mediated by the endoplasmic reticulum (ER). This study suggests a link between mitochondria and the ER during the UPR under glucose deprivation conditions and that mitochondria govern cell fate, not only through ATP production and apoptosis regulation, but also through modulating the UPR for cell survival.
(Cancer Sci 2010; 101: 1125–1132)
Glucose plays a major role in the metabolism of living cells, and has a central role as an energy source for cells. It is taken up by specific transporters and then it is finally converted to pyruvate by glycolysis, generating two ATP per glucose. In the absence of oxygen, pyruvate is reduced to lactate, which is exported from the cells. In the presence of oxygen, pyruvate is oxidized in the mitochondria respiratory chain through OxPhos, generating 36 additional ATP per glucose. Mitochondrial OxPhos is an efficient energy production system of normal cells under aerobic conditions.
Mitochondria are important organelles with an essential role in cell fate: ATP production during cell growth or cell division and apoptosis leading to cell death.( 1 , 2 ) Recent studies( 3 , 4 ) also suggest that mitochondria contribute to differentiation. Furthermore, they contribute to malignant transformation of normal cells. In fact the irreversible damage to respiration is associated with normal cells’ transformation to cancer cells.( 5 ) The first identified biochemical hallmark of cancer cells was a shift in glucose metabolism from OxPhos to glycolysis under aerobic conditions.( 5 , 6 )
Cancer cells tend to synthesize ATP mainly through glycolysis( 6 , 7 , 8 ) (Warburg effect( 5 , 9 )), even under aerobic conditions, although glycolysis is a less‐efficient system than OxPhos in generating ATP. Most primary and metastatic human cancers show significantly increased glucose uptake( 8 ) because of their enhanced glucose consumption. This characteristic is utilized for PET. Cancer cells are more dependent on glucose for energy production than normal cells. However, cancer cells in solid tumors are not supplied sufficient glucose because they are often distant from blood vessels.( 10 ) Although cancer cells can induce angiogenesis during tumor development, sufficient glucose is not supplied until new blood vessels are formed. These cells need to survive in tumor tissue with inadequate glucose levels, (11 ) and if they cannot then they will not survive under glucose deprivation conditions. Adaptive response mechanisms are required for cancer cells to survive in the tumor microenvironment.
The UPR is a survival response of cancer cells and is mediated by the ER.( 12 ) Newly synthesized proteins are folded in the ER; however, stress conditions cause unfolded protein accumulation in the ER, leading to ER stress. The UPR enables the cell to reduce unfolded proteins in the ER by attenuating translation and promoting protein folding, secretion, and ERAD.( 13 , 14 , 15 ) The UPR pathway is mediated by three master regulators (PERK, ATF6, and IRE1) located in the ER membrane.( 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 ) The activation of these molecules by unfolded proteins induces the activation of multiple transcription factors thereby resulting in the UPR. ATF4, cleaved ATF6, and XBP1 splicing variant are the transcription factors derived from PERK, ATF6, and IRE1 activation, respectively.( 11 , 12 , 14 , 15 , 16 , 17 ) PERK phosphorylates eIF2α to attenuate translation, and to up‐regulate ATF4 expression, leading to enhanced transcription of target genes such as CHOP. ATF6 is translocated to the Golgi apparatus and cleaved by proteases such as S1P and S2P, leading to enhanced transcription of ER chaperone genes. IRE1 splices XBP1 mRNA, and protein translated from mature XBP1 mRNA activates transcription of ERAD component genes.( 17 )
The UPR has been observed in tumors because of their particularly stressful microenvironment,( 11 , 19 , 20 , 21 ) which includes glucose deprivation. The UPR in cancer cells plays an important role in their survival( 11 , 19 , 20 , 21 ) and results in tumor malignancies and antitumor drug resistance.( 11 , 19 , 20 , 21 ) If cancer cells have no adaptive response, such as the UPR, they would not be able to escape death under glucose deprivation conditions, namely, glucose addiction. However, what drives the UPR under glucose deprivation conditions in the tumor microenvironment has not been elucidated. Here we show that functional mitochondria are essential to the UPR induction and cancer cell survival under glucose deprivation conditions, indicating that mitochondria contribute to cancer cells’ avoiding glucose addiction.
Materials and Methods
Cell culture. Human cancer cell lines were used as follows: fibrosarcoma HT‐1080, colorectal adenocarcinoma HT‐29, renal cell carcinoma OS‐RC‐2, pancreas epithelioid carcinoma PANC‐1, breast adenocarcinoma MCF7, hepatocellular carcinoma Hep G2, stomach cancer MKN74, glioblastoma A172, and glioblastoma multiforme T98G. Cells were cultured at 37°C in a humidified atmosphere containing 5% CO2. Cancer cells were cultured in RPMI‐1640 (Wako, Osaka, Japan) supplemented with 10% heat‐inactivated FBS, 2 mM L‐glutamine, and 100 μg/mL kanamycin (growth medium). Normal HMECs were cultured in D‐MEM/F‐12 (Invitrogen, Carlsbad, CA, USA) supplemented with 20 μg/mL BPE, 0.5 μg/mL hydrocortisone, 5 μg/mL insulin, 0.01 μg/mL epidermal growth factor, and 1× antibiotic‐antimycotic (Invitrogen). Establishment of ρ0 cells( 22 , 23 ) was performed in high glucose D‐MEM (Invitrogen) supplemented with 10% FBS, 100 μg/mL kanamycin, 50 μg/mL uridine, 1 mM sodium pyruvate, and 0.1 μg/mL (ρ0‐HT‐1080) or 1 μg/mL (ρ0‐HT‐29) ethidium bromide. Established ρ0 cells were cultured in growth medium supplemented with 50 μg/mL uridine and 1 mM sodium pyruvate.
Stress conditions. Glucose‐free RPMI‐1640 (Invitrogen) supplemented with 10% FBS (GS medium) or 10 mM 2DG (2DG) were used for glucose deprivation. Five μg/mL TM or 30 nM TG was used as a chemical ER stress inducer. AnaeroPack (Mitsubishi Gas Chemical Co., Tokyo, Japan) was used to induce hypoxia.
RNA preparation. Total RNA was extracted from cultured cells using a QIAshredder (Qiagen, Valencia, CA, USA) and RNeasy Mini Kit (Qiagen). Quality of the RNA was checked with a 2100 Bioanalyser (Agilent Technologies, Santa Clara, CA, USA).
Real‐time RT‐PCR. Purified RNA was examined by real‐time RT‐PCR using an ABI Prism 700 (Applied Biosystems, Foster City, CA, USA), according to the manufacturer’s instructions. For internal control, we used B2M.
Immunoblotting. Whole cell lysates were subjected to SDS‐PAGE and electroblotted onto a nitrocellulose membrane. ND6 protein, COX II, GRP78, β‐actin, Hsp90, and PARP p85 fragment were detected using a mouse monoclonal antibody against OxPhos Complex I 20‐ kDa subunit (Invitrogen), OxPhos Complex IV subunit II (Invitrogen), KDEL (Stressgen, Ann Arbor, MI, USA), β‐actin (Sigma‐Aldrich, St. Louis, MO, USA), Hsp90 (BD Biosciences, San Jose, CA, USA), and a rabbit polyclonal antibody against p85 fragment of PARP (Promega, Madison, WI, USA), respectively. ATF4 was detected with a rabbit polyclonal antibody against CREB‐2 (C‐20) (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Proteins were visualized with horseradish peroxidase‐conjugated second antibody (Amersham Biosciences, Buckinghamshire, UK) under chemiluminescence detection. Exogenous ATF6 was detected by anti‐FLAG M2 mouse monoclonal antibody‐peroxidase conjugate (Sigma‐Aldrich) and visualized.
Viability assay. Overgrowth‐induced cell death and stress sensitivity were examined by calcein‐AM assay. Cells were plated into 96‐well plates in the appropriate medium. After 72 h, fluorescence derived from viable cells was detected spectrofluorometrically. For the study of ETCBs, 3 × 103 cells in growth medium were plated into 96‐well plates. After 24 h, the medium was replaced with medium containing Rot or AA. Cells were then incubated for 72 h, and calcein‐AM assay was performed. Cell viability was estimated as the rate of no ETCBs‐treated cells (% live cells). For the study of HMECs, 7 × 103 cells in appropriate medium were plated in 96‐well plates. After 72 h, the medium was replaced with medium containing TM or 2DG. Cells were then incubated for 72 h, and a alamarBlue (Biosource International, Camarillo, CA, USA) assay was performed.
ATP assay. Intracellular ATP content was measured by using an Intracellular ATP Kit (BioThema, Handen, Sweden). Cells (3 × 104) in growth medium were plated in 96‐well plates. After 24 h, the medium was replaced with GS medium. Cells were then incubated for 8 or 18 h, and ATP assay was performed according to the instruction manual. Ratio = ATP content in GS medium/ATP content in growth medium.
GRP78 reporter assay. Cells were cultured overnight in six‐well plates (4 × 105 cells/well). Then, growth medium was changed to antibiotic‐free RPMI‐1640 supplemented with 5% FBS. Transient transfection was performed using FuGENE 6 Transfection Reagent (Roche Applied Science, Mannheim, Germany). In brief, the cells were incubated for 6 h with a complex mixture containing 500 ng of the firefly luciferase‐containing reporter plasmids pGRP78pro160‐Luc( 24 ) and 1 ng of phRL‐CMV (Promega), as an internal control reporter vector. pGL3‐Control Vector (Promega) was used in place of pGRP78pro160‐Luc as a negative control. The medium was then replaced with fresh growth medium, and the cells were incubated for another 2 h. After the transfection, cells were exposed to stress conditions for 18 h with or without ETCBs. Relative firefly‐to‐Renilla luciferase activity was determined using a Dual‐Glo Luciferase Assay System (Promega Corporation).
Microarray analysis. HT‐1080 or HT‐29 cells were exposed to stress with or without inhibitor (3 μM VST, 10 mM Met, 300 μM Bu, 100 μM Phen, 100 nM Rot, 10 ng/mL of AA) for 18 h. (GS‐treated ρ0‐HT‐29 and corresponding ρ+‐HT‐29 (HT‐29/GS#3) were exposed to GS for 14 h because ρ0‐HT‐29 was severely damaged by 18‐h exposure.) Purified RNA was examined by microarray using the Affymetrix GeneChip Human Genome U133 Plus 2.0 Array (Affymetrix, Santa Clara, CA, USA). Probes with low (less than 100) signal intensity in all arrays were excluded. The UPR‐related genes were selected as previously described.( 25 ) Clustering analysis was performed by complete linkage analysis using Cluster 3.0 software.
Transient transfection of ATF6. The plasmid of 7× FLAG‐tagged ATF6( 26 ) was transfected into HT‐1080 cells using FuGENE 6, according to the instruction manual.
Detection of XBP1 mRNA splicing. XBP1 cDNA was amplified with the primers reported previously ( 24 ) using the SuperScript III One‐Step RT‐PCR System with Platinum Taq DNA Polymerase (Invitrogen). PCR products were analyzed by PAGE.
Results
Glucose addiction of r0 cells. To investigate the effects of glucose metabolism alteration in cancer cells, we established mitochondrial DNA (mtDNA)‐deficient (ρ0) cells derived from two human cancer cell lines: fibrosarcoma HT‐1080 and colorectal adenocarcinoma HT‐29. The mtDNA codes for several genes essential to the respiratory chain. Therefore, ρ0 cells do not possess the functional respiratory chain. These cells are dependent on glycolysis for their energy production,( 27 , 28 ) as are cancer cells in tumor tissue. ND6 protein and COX2, which are encoded in circular mtDNA, were not detected in these cells at either the mRNA (Fig. 1a) or protein level (Fig. 1b). Established ρ0 cells did not maintain their mtDNA.
Figure 1.
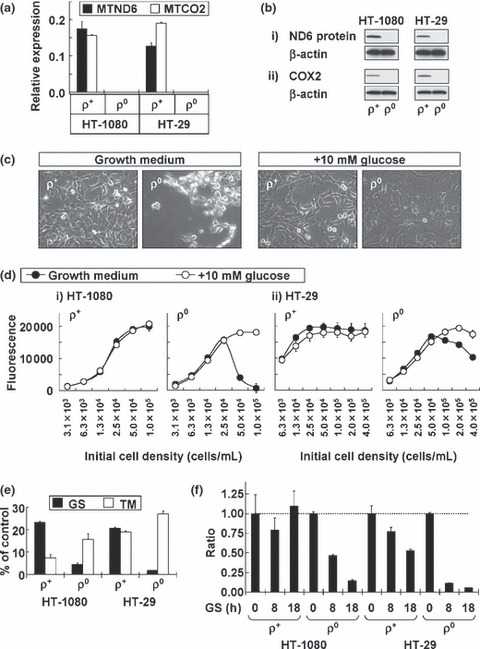
Characterization of ρ0 cells. (a) Mito‐chondrial DNA‐coded transcripts (MTND6 and MTCO2) in HT‐1080 and HT‐29 cells examined by real‐time RT‐PCR analysis. Data were normalized by b2‐microglobulin (B2M) mRNA expression. Results shown are the means of three independent experiments; bars, ±SD. (b) Mitochondrial DNA‐coded proteins (ND6 protein and cyclooxygenase‐2 [COX2]) in HT‐1080 and HT‐29 cells examined by immunoblotting. The amount of β‐actin is used as a loading control. (c) Phase contrast images of HT‐1080 cells. (d) Overgrowth‐induced cell death in ρ0 cells and its inhibition by additional glucose. Results shown are the means of three independent experiments; bars, ± SD. (e) Stress sensitivity. 2.5 ×104 cells/mL of HT‐1080 cells or 5.0 × 104 cells/mL of HT‐29 cells were plated into 96‐well plates in glucose starvation (GS) medium or tunicamycin (TM)‐containing medium. After 72 h, calcein‐AM assay was performed. Cell viability was indicated as the rate of the data in growth medium (% of control). (f) Intracellular adenosine triphosphate (ATP) content alteration under glucose deprivation conditions. ATP content was indicated as the rate of the data in growth medium (ratio).
Under normal culture conditions, cancer cell growth arrest is induced when cell density is increased too much. Cancer cells maintain their numbers and are not immediately killed by their dense population alone. Figure 1(c) illustrates the morphology of HT‐1080 cells. Cells were seeded (1 × 105 cells/mL) in a culture dish and observed under the microscope after 3 days. Morphological changes were not observed in parental (ρ+) cells. However, ρ0 cells were fatally damaged by overgrowth. The results of our viability assay suggested that growth arrest was induced in ρ+ cells but not ρ0 cells (Fig. 1d). This overgrowth‐induced cell death in ρ0 cells could be suppressed by additional glucose (Fig. 1c,d), indicating that this cell death was caused by glucose deprivation. These data suggest that ρ0 cells show glucose addiction.
To confirm this, we cultured ρ0 cells in GS medium instead of normal medium for 3 days. Most of the ρ0 cells died (Fig. 1e), indicating that ρ0 cells are highly sensitive to glucose deprivation stress. On the other hand, proliferation of ρ+ cells in GS medium was arrested, and their cell number decreased to about 20% of the cells in the normal culture medium (Fig. 1e). Initial cell density in this experiment was determined on the data in Figure 1(d) to avoid the effect of overgrowth‐induced cell death in normal culture medium. Therefore, death in ρ0 cells was induced by GS medium and not by glucose deprivation with overgrowth. Further, we examined the TM sensitivity of the ρ0 cells. TM is a glycosylation inhibitor leading to unfolded protein accumulation in the ER( 17 ) and it is often used as a chemical ER stress inducer. In contrast to glucose deprivation, ρ0 cells were no more sensitive than ρ+ cells (Fig. 1e). Intracellular ATP in ρ0 cells decreased more rapidly than in ρ+ cells under glucose deprivation conditions (Fig. 1f). Taken together, these results suggest that functional mitochondria are essential to cancer cell survival specifically under glucose deprivation conditions. Glucose deprivation‐sensitive ρ0 cells would lose their responsiveness of growth arrest as we observed in ρ+ cells.
Increased sensitivity to glucose deprivation. The data from ρ0 cells suggests that the functional respiratory chain is required for cancer cell survival under glucose deprivation conditions. We further examined the effects of Rot (Complex I inhibitor) and AA (Complex III inhibitor) on glucose deprivation sensitivity of various human cancer cell lines. If cancer cells acquire a resistant phenotype to glucose deprivation through the respiratory chain, these cells could be sensitized by the ETCBs that inhibit the respiratory chain. We used not only HT‐1080 and HT‐29 but also human cancer cell lines derived from the kidney, pancreas, breast, liver, stomach, and brain. The increased ETCB sensitivities were observed in all cell lines tested (Fig. 2a).
Figure 2.
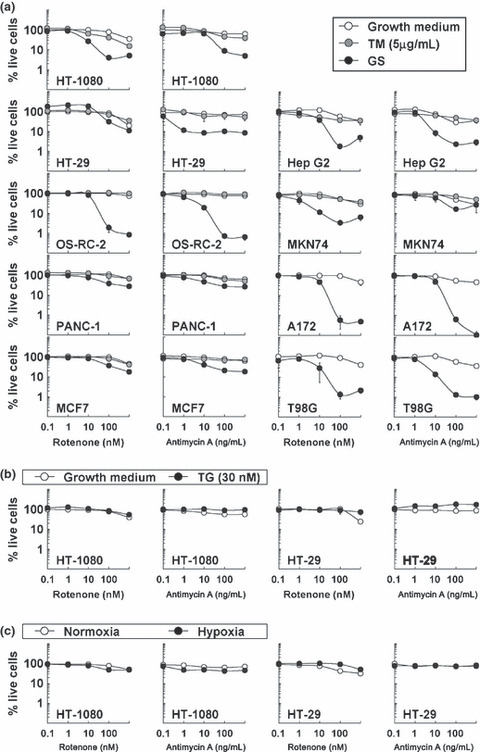
Increased sensitivity to glucose deprivation by electron transport chain blockers (ETCBs) in human cancer cell lines. The effect of ETCBs on the growth of human cancer cell lines under stress conditions: (a) tunicamycin (TM) or glucose starvation (GS); (b) thapsigargin (TG); (c) Hypoxia). (a–c) Results shown are the means of three independent experiments; bars, ±SD.
We also examined the cytotoxicity of TM (Fig. 2a) and TG (Fig. 2b) with or without ETCBs. ETCBs induced no sensitization to these chemical stress inducers (Fig. 2a,b). We could not detect the effect of ETCBs on TM treatment in brain tumor cell lines (A172 and T98G) because these cell lines were highly sensitive to TM alone. Cancer cells in tumor tissue were not supplied sufficient oxygen or glucose. Therefore, we also examined the effect of ETCBs on hypoxia sensitivity in HT‐1080 and HT‐29 cells. ETCBs did not enhance cytotoxicity under hypoxic conditions (Fig. 2c).
Mitochondria dependency of glucose deprivation‐induced UPR. Ahmad et al. reported( 29 ) that ETCBs enhanced glucose deprivation‐induced cytotoxicity with increasing oxidative stress in some human cancer cells, suggesting that intracellular oxidative stress could be a cause of the glucose deprivation‐induced cytotoxicity. The results using ρ0 cells suggested another possibility, that mitochondrial dysfunction leads to an adaptation disorder during glucose deprivation (Fig. 1). Glucose deprivation sensitivity may be regulated not only by intracellular oxidative stress but also by the potential of cells to respond to stress conditions. To investigate whether ρ0 cells could respond to stress, we first examined GRP78 up‐regulation by immunoblotting since this is generally observed under stress conditions.( 30 ) 2DG and TM were used as chemical ER stress inducers in this assay. 2DG is a glucose analogue that mimics glucose deprivation in competition with normal glucose.( 17 , 31 ) In ρ+ cells, all the stresses increased GRP78 expression (Fig. 3a). However, the amount of GRP78 protein was not increased in ρ0 cells under glucose deprivation conditions, such as 2DG treatment or in GS medium (Fig. 3a). On the other hand, TM treatment caused GRP78 up‐regulation in ρ0 cells (Fig. 3a).
Figure 3.
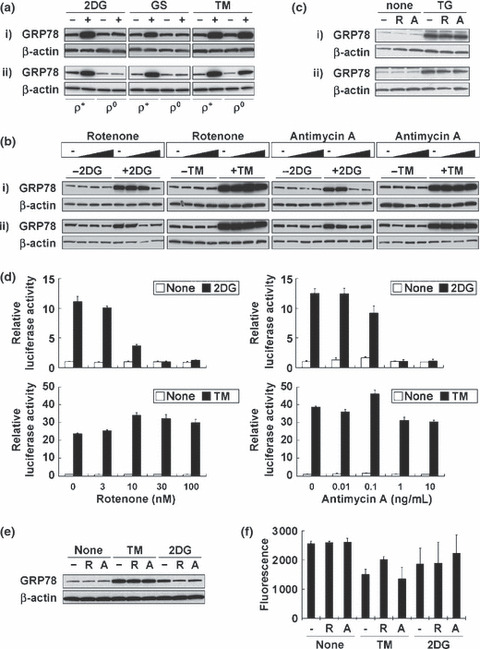
Respiratory chain is required for GRP78 up‐regulation induced by glucose deprivation. (a) GRP78 up‐regulation under stress conditions for 18 h. (b) The effect of electron transport chain blockers (ETCBs) on GRP78 up‐regulation under stress conditions for 18 h. Rotenone: 0, 10, 100, 1000 nM. Antimycin A: 0, 1, 10, 100 ng/mL. (c) The effect of ETCBs on thapsigargin (TG)‐induced GRP78 up‐regulation (18 h). –, no inhibitor; R, 100 nM rotenone; A, 10 ng/mL antimycin A. (a–c) The amount of β‐actin is used as a loading control. (i) HT‐1080; (ii) HT‐29. (d) The effect of ETCBs on GRP78 up‐regulation under stress conditions in HT‐1080 cells using GRP78 reporter assay. Results shown are the means of three independent experiments; bars, ±SD. (e) The effect of ETCBs on GRP78 up‐regulation under stress conditions for 18 h in HMECs. (f) The effect of ETCBs on the growth of HMECs under stress conditions. Results shown are the means of three independent experiments; bars, ±SD. (e,f) –, no inhibitor; R, 100 nM rotenone; A, 10 ng/mL antimycin A.
We next examined the effect of ETCBs on GRP78 up‐regulation. Both Rot and AA remarkably diminished 2DG‐induced up‐regulation in a dose‐dependent manner, whereas they only slightly diminished TM‐ or TG‐induced up‐regulation (Fig. 3b,c). Then, we performed the reporter assay to detect GRP78 promoter activity in HT‐1080 cells. GRP78 promoter activation was detected when cells were treated with 2DG or TM (Fig. 3d). Both ETCBs inhibited 2DG activation in a dose‐dependent manner, but not TM‐induced activation (Fig. 3d). We further examined the effects of ETCBs on GRP78 up‐regulation using HMECs. Both Rot and AA diminished 2DG‐induced up‐regulation, whereas they did not induce any changes in TM‐induced up‐regulation (Fig. 3e). However, increased cellular sensitivity to 2DG by ETCBs was not observed in HMECs (Fig. 3f).
We used two experimental systems for the dysfunctional respiratory chain: genetic deficiency in ρ0 cells and chemical inhibition by ETCBs. In both cases, the cells could not induce GRP78 up‐regulation (Fig. 3) and did not survive (1, 2) under glucose deprivation conditions. GRP78 up‐regulation occurs in various cell lines as a result of the UPR.( 30 ) Therefore, these results strongly suggest that the UPR leading to cell survival also requires the functional respiratory chain.
We further examined gene expression profiles using microarray and clustering analysis (Fig. 4a). HT‐1080 and HT‐29 cells were exposed to stress (GS, 2DG, or TM) with or without a UPR inhibitor. The UPR inhibitor is thought to be the selective antitumor drug.( 11 , 32 ) VST is a UPR inhibitor( 24 , 25 ) derived from Streptomyces versipellis. Cytotoxic activity of VST is potentiated under glucose deprivation conditions( 24 , 25 ) in vitro, and its antitumor activity is also confirmed in an in vivo xenograft model. Biguanides (Met (33), Bu, and Phen) were also shown to be UPR inhibitors.( 25 ) Met is an oral antihyperglycemic drug widely used to treat diabetes.( 33 ) After stress treatment with or without inhibitors, purified RNA was examined. Twenty‐six arrays were analyzed by complete linkage analysis. We previously reported( 25 ) the glucose deprivation signature that captured the UPR upon glucose deprivation. This signature consists of 246 probes that detect genes which expressions alter under glucose deprivation conditions. These genes overlap with the genes induced by TM. We analyzed these UPR‐related genes using this signature (Fig. 4a). Stress‐treated samples in the presence of the UPR inhibitor (biguanides or VST) (Group A in Table 1) were in Cluster 1, indicating that Cluster 1 is the UPR‐negative group. Stress‐treated samples in the absence of the UPR inhibitor (Group B in Table 1) were in Cluster 2, indicating that Cluster 2 is the UPR‐positive group. The ρ0 cells or ρ+ cells in the presence of Rot or AA (Group C in Table 1) were assigned to Cluster 1 (Fig. 4a). On the other hand, TM‐treated cells (Groups B and D in Table 1) were assigned to Cluster 2 regardless of their respiratory chain status (Fig. 4a). These results indicate that a functional respiratory chain is specifically required for the UPR under glucose deprivation conditions.
Figure 4.
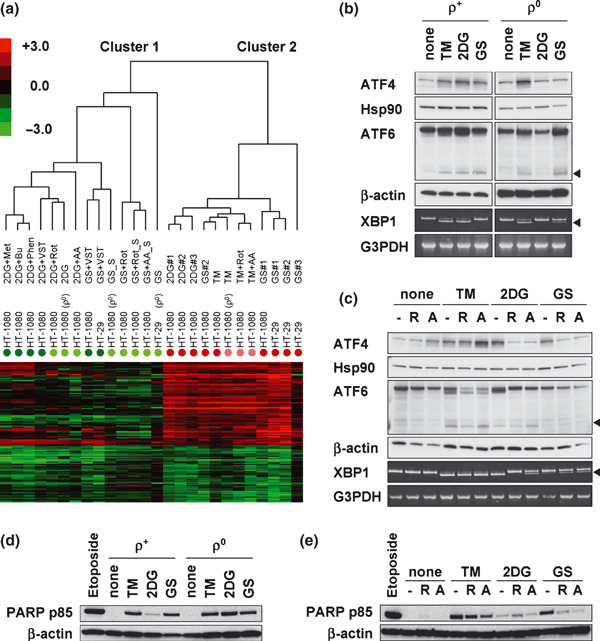
Respiratory chain is required for the unfolded protein response (UPR) induced by glucose deprivation. (a) Clustering analysis using the glucose deprivation signature. Colored, closed circles indicate each group shown in Table 1. Colored bar indicates signal log ratio. Capital S (_S) indicates the supernatant of sample including floating cells. (b,c) Activation of the UPR‐related transcription factors (ATF4, ATF6, and XBP1) under stress conditions for 6 h in HT‐1080 cells with or without electron transport chain blockers (ETCBs). Arrowheads indicate activated ATF6 or splicing variant of XBP1 mRNA. The amounts of HSP90, β‐actin, and G3PDH are used as controls, respectively. (d) The expression of poly(ADP‐ribose) polymerase (PARP) p85 fragment in stress‐treated (48 h) ρ+‐ or ρ0‐HT‐1080 cells. (e) The effect of ETCBs on stress‐treated (48 h) PARP p85 fragment expression in ρ+‐HT‐1080 cells. (c,e) –, no inhibitor; R, 100 nM rotenone; A, 10 ng/mL antimycin A. (d,e) Whole cell lysate of 10 μg/mL etoposide‐treated (24 h) cells was used as control of apoptosis induction.
Table 1.
Classification of samples used in microarray analysis
| Color (in Fig. 4a) | Stress | UPR inhibitor | Respiratory chain | |
|---|---|---|---|---|
| Group A | Green | GS, 2DG, TM | + | Functional |
| Group B | Red | GS, 2DG, TM | − | Functional |
| Group C | Light green | GS, 2DG | − | Dysfunctional |
| Group D | Pink | TM | − | Dysfunctional |
2DG, 2‐deoxyglucose; GS, glucose starvation; TM, tunicamycin; UPR, unfolded protein response.
Figure 4(b,c) showed the activation of the UPR‐related transcription factors (ATF4, ATF6, and XBP1) in HT‐1080 cells. Endogenous ATF4 was detected by immunoblotting. The plasmid of FLAG‐tagged ATF6( 26 ) was transfected into HT‐1080 cells, and then exogenous ATF6 was detected by immunoblotting. XBP1 mRNA splicing was examined using RT‐PCR followed by native PAGE. ATF4 activation was observed in ρ+ cells regardless of the stressor (Fig. 4b). However, this activation did not occur in 2DG‐treated ρ0 cells or ρ0 cells in GS medium (Fig. 4b), and it was inhibited by ETCBs under these stress conditions (Fig. 4c). TM‐induced ATF4 activation was also observed in both ρ0 cells (Fig. 4b) and ETCB‐treated ρ+ cells (Fig. 4c). A positive correlation with GRP78 up‐regulation or the UPR induction was not observed in ATF6 activation or XBP1 splicing (Fig. 4b,c). To explore the relationships between the UPR and apoptosis which is one of the downstream elements of the UPR, we examined PARP cleavage (Fig. 4d,e). PARP (116 kDa) is known to be cleaved to 85 kDa fragment (p85) as a result of apoptosis induction. A positive correlation with the UPR was also not observed in PARP cleavage.
Discussion
We demonstrated that cancer cells with dysfunctional respiratory chains could not survive (Fig. 1e and Fig. 2a) and could not induce the UPR (Fig. 4a) in GS medium or by 2DG treatment. As shown in Figure 4(a), glucose deprivation‐induced UPR did not occur in ρ0 cells or ρ+ cells in the presence of Rot or AA (Group C in Table 1), because these samples were assigned to Cluster 1, UPR‐negative group (Fig. 4a). On the other hand, TM‐induced UPR also occurred in cells with a dysfunctional respiratory chain (Group D in Table 1). These results indicate that a functional respiratory chain is specifically required for the UPR under glucose deprivation conditions. ATF4 activation, as well as the UPR, also did not occur in cells with mitochondrial dysfunction under glucose deprivation conditions (Fig. 4b,c). These results suggested that mitochondria regulated ATF4 activation under glucose deprivation conditions.
Respiratory chain dysfunction did not enhance the sensitivities to chemical stress inducers (TM or TG) or hypoxia (Figs 1e and 2). Furthermore, respiratory chain dysfunction did not inhibit GRP78 up‐regulation induced by chemical stressors (Fig. 3a–c). These results indicate that functional mitochondria are essential to UPR induction, leading to cancer cell survival specifically under glucose deprivation conditions (Fig. 5a). On the other hand, ETCBs did not enhance 2DG sensitivity in HMECs (Fig. 3f) despite their inhibitory effects on GRP78 up‐regulation (Fig. 3e). These data suggest a link between the UPR and survival of cancer cells, but not normal cells, under glucose deprivation conditions.
Figure 5.
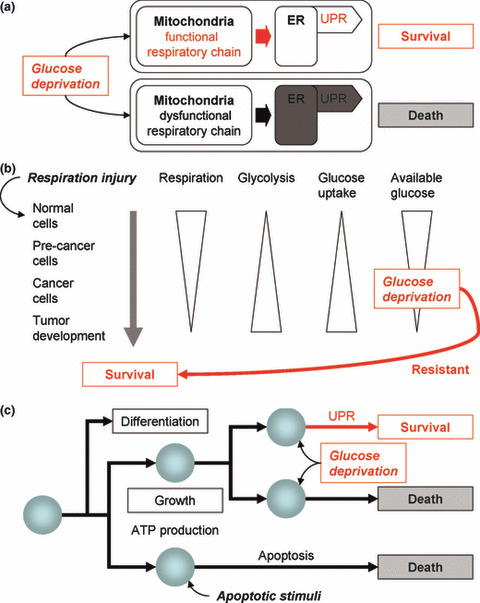
Cancer cell survival under glucose deprivation conditions. (a) Mitochondria are required for the unfolded protein response (UPR) leading to cancer cell survival under glucose deprivation conditions. (b) The shift in glucose metabolism during cancer cell generation and its survival in tumors. Cancer cells are exposed to glucose deprivation caused by abundant glucose uptake or in the tumor microenvironment. The glucose deprivation‐resistant phenotype is essential for their survival. (c) Cell behavior in multicellular organisms and mitochondrial functions. Mitochondria are not only involved growth, differentiation, and death, but also in the survival of cancer cells.
The increased ETCB sensitivities were observed in all human cancer cell lines tested (Fig. 2a), suggesting that this is general characteristic of cancer cells regardless of their origin. The balance between the utilization of respiratory and glycolytic pathways is modulated by p53,( 34 ) one of the most frequently mutated genes in cancers. Therefore, we used wild‐type p53 A172 and mutant‐type p53 T98G brain tumor cell lines. Both cell lines were similarly sensitized to glucose deprivation by ETCBs (Fig. 2a), suggesting that glucose deprivation resistance by mitochondria did not depend on p53 status. The targets of these ETCBs in the respiratory chain complex are different; therefore, these data suggest that cancer cell survival under glucose deprivation conditions requires the whole respiratory chain system. The UPR inhibitor Met was reported to inhibit respiratory chain complex I,( 35 ) raising the possibility that the UPR inhibitory effect of Met was caused by respiratory chain dysfunction as well as ETCBs. Complex I could be the regulatory factor of mitochondria‐mediated UPR.
A glucose deprivation‐resistant phenotype is essential for cancer cell to generate and survive within a tumor. Figure 5(b) illustrates the process of cancer cell generation. They originate from normal cells by “respiration injury,” as O. Warburg described.( 5 ) However, respiration injury alone is not sufficient since ρ0 cells showed glucose addiction (Fig. 1c–e). Respiration‐injured (pre‐cancer) cells gradually acquire a glycolysis‐dominant phenotype to produce sufficient ATP.( 5 ) The shift in glucose metabolism of pre‐cancer cells leads to abundant glucose uptake resulting in glucose deprivation. Furthermore, cancer cells survive in tumor tissue with inadequate glucose.( 11 ) This means that from their generation to tumor formation, cancer cells frequently must survive under glucose deprivation conditions. In ρ0 cells, cellular ATP content remarkably decreased compared with parental cells (Fig. 1f). However, all the UPR‐related changes did not stop in ρ0 cells under glucose deprivation conditions as shown in Figure 4. Therefore, it was suggested that ATP depletion is not the only reason for glucose addiction of ρ0 cells. Taken together, mitochondria help cancer cells avoid glucose addiction, thereby developing a glucose deprivation‐resistant phenotype.
Stress sensitivities to glucose deprivation by mitochondrial dysfunction (Fig. 1e and Fig. 2a) were positively correlated with the UPR induction (Fig. 4a); that is, the cells in UPR‐negative group were sensitive. However, the appearance of p85 fragment of PARP (Fig. 4d,e) did not correlate with the UPR induction (Fig. 4a) nor stress sensitivities (Figs 1e and 2a), indicating that stress sensitivities were not only caused by apoptosis, one of the downstream elements of the UPR. These results suggested that cancer cells under glucose deprivation conditions survive through activating the multiple branches of the UPR mediated by mitochondria. In addition to ATP production and apoptosis execution, the UPR regulation is positioned as novel function of mitochondria (Fig. 5c). The respiratory chain may be a futile system for glycolysis‐dominant cancer cells to produce ATP; however, it is essential to regulate the UPR.
Mitochondria‐mediated UPR and/or cell survival could have a role in cancer treatment strategies that focus on the tumor microenvironment. First, under stress conditions, UPR inhibition can induce selective cytotoxicity in the tumor microenvironment. Second, it can eradicate cancer‐initiating (stem) cells. Conventional chemotherapies can be effective against most cancer cells; however, they are less effective against cancer‐initiating cells because of the cells’ resistant phenotype. These dormant cells can survive in stressful microenvironments, such as glucose deprivation, and contribute to tumor malignancy. Therefore, recovering the glucose addiction phenotype may lead to the eradication of these sleeping cancer cells, and cancer recurrence can be prevented. Furthermore, respiratory chain‐related molecules are also possible antitumor molecular targets, because mitochondrial alteration is a major distinction between normal and cancer cells.( 5 , 6 , 7 , 8 , 9 , 28 ) It was recently reported( 36 ) that mtDNA mutations leading to respiratory chain alteration could contribute to tumor progression by enhancing the metastatic potential of cancer cells. Cancer cells could acquire their malignant properties through mitochondrial alteration induced by genetic alterations. We anticipate this study to provide insights into the precise mechanisms of the cellular stress response against glucose deprivation and to develop novel strategies for the treatment of several UPR‐related diseases,( 11 , 14 , 17 , 18 , 19 , 37 , 38 ) including cancer,( 11 , 19 ) diabetes,( 37 ) and neurodegenerative disorders.( 38 )
Acknowledgments
We thank Dr Ryohei Katayama (JFCR, Japan) for help with the HMEC experiments, and Dr Yasuo Harada (Otsuka Pharmaceutical, Japan) for help with the ATP assay. This work was supported in part by a Grant‐in‐Aid for Cancer Research (21‐3‐1) from the Ministry of Health, Labour and Welfare of Japan (A.T.), a Grant‐in‐Aid for Scientific Research on Priority Areas for Cancer (T.T. and A.T.), a grant from the New Energy and Industrial Technology Development Organization (NEDO) of Japan.
Disclosure Statement
The authors have no conflict of interest.
Abbreviations
- 2DG
2‐deoxyglucose
- AA
antimycin A
- ATP
adenosine triphosphate
- B2M
β2‐microglobulin
- Bu
buformin
- COX2
cytochrome c oxidase subunit II
- ER
endoplasmic reticulum
- ERAD
ER‐associated degradation
- ETCB
electron transport chain blocker
- FBS
fetal bovine serum
- GS
glucose starvation
- HMEC
human mammary epithelial cell
- Met
metformin
- mtDNA
mitochondrial DNA
- OxPhos
oxidative phosphorylation
- PAGE
polyacrylamide gel electrophoresis
- PARP
poly(ADP‐ribose) polymerase
- PET
positron emission tomography
- Phen
phenformin
- Rot
rotenone
- S1P
site 1 protease
- S2P
site 2 protease
- TG
thapsigargin
- TM
tunicamycin
- UPR
unfolded protein response
- VST
versipelostatin
References
- 1. Haga N, Fujita N, Tsuruo T. Mitochondrial aggregation precedes cytochrome c release from mitochondria during apoptosis. Oncogene 2003; 22: 5579–85. [DOI] [PubMed] [Google Scholar]
- 2. Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med 2000; 6: 513–9. [DOI] [PubMed] [Google Scholar]
- 3. Griguer CE, Oliva CR, Gobin E et al. CD133 is a marker of bioenergetic stress in human glioma. PLoS ONE 2008; 3: e3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen C, Liu Y, Liu R et al. TSC‐mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J Exp Med 2008; 205: 2397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Warburg O. On the origin of cancer cells. Science 1956; 123: 309–14. [DOI] [PubMed] [Google Scholar]
- 6. Shaw RJ. Glucose metabolism and cancer. Curr Opin Cell Biol 2006; 18: 598–608. [DOI] [PubMed] [Google Scholar]
- 7. Kim JW, Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res 2006; 66: 8927–30. [DOI] [PubMed] [Google Scholar]
- 8. Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer 2004; 4: 891–9. [DOI] [PubMed] [Google Scholar]
- 9. Warburg O. On respiratory impairment in cancer cells. Science 1956; 124: 269–70. [PubMed] [Google Scholar]
- 10. Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer 2006; 6: 583–92. [DOI] [PubMed] [Google Scholar]
- 11. Ma Y, Hendershot LM. The role of the unfolded protein response in tumour development: friend or foe? Nat Rev Cancer 2004; 4: 966–77. [DOI] [PubMed] [Google Scholar]
- 12. Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem 2005; 74: 739–89. [DOI] [PubMed] [Google Scholar]
- 13. Mori K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell 2000; 101: 451–4. [DOI] [PubMed] [Google Scholar]
- 14. Zhao L, Ackerman SL. Endoplasmic reticulum stress in health and disease. Curr Opin Cell Biol 2006; 18: 444–52. [DOI] [PubMed] [Google Scholar]
- 15. Bernales S, Papa FR, Walter P. Intracellular signaling by the unfolded protein response. Annu Rev Cell Dev Biol 2006; 22: 487–508. [DOI] [PubMed] [Google Scholar]
- 16. Xu C, Bailly‐Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest 2005; 115: 2656–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yoshida H. ER stress and diseases. Febs J 2007; 274: 630–58. [DOI] [PubMed] [Google Scholar]
- 18. Marciniak SJ, Ron D. Endoplasmic reticulum stress signaling in disease. Physiol Rev 2006; 86: 1133–49. [DOI] [PubMed] [Google Scholar]
- 19. Feldman DE, Chauhan V, Koong AC. The unfolded protein response: a novel component of the hypoxic stress response in tumors. Mol Cancer Res 2005; 3: 597–605. [DOI] [PubMed] [Google Scholar]
- 20. Tsuruo T, Naito M, Tomida A et al. Molecular targeting therapy of cancer: drug resistance, apoptosis and survival signal. Cancer Sci 2003; 94: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tomida A, Suzuki H, Kim HD, Tsuruo T. Glucose‐regulated stresses cause decreased expression of cyclin D1 and hypophosphorylation of retinoblastoma protein in human cancer cells. Oncogene 1996; 13: 2699–705. [PubMed] [Google Scholar]
- 22. King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science 1989; 246: 500–3. [DOI] [PubMed] [Google Scholar]
- 23. Gamen S, Anel A, Montoya J, Marzo I, Pineiro A, Naval J. mtDNA‐depleted U937 cells are sensitive to TNF and Fas‐mediated cytotoxicity. FEBS Lett 1995; 376: 15–8. [DOI] [PubMed] [Google Scholar]
- 24. Park HR, Tomida A, Sato S et al. Effect on tumor cells of blocking survival response to glucose deprivation. J Natl Cancer Inst 2004; 96: 1300–10. [DOI] [PubMed] [Google Scholar]
- 25. Saito S, Furuno A, Sakurai J et al. Chemical Genomics Identifies the Unfolded Protein Response as a Target for Selective Cancer Cell Killing during Glucose Deprivation. Cancer Res 2009; 69: 4225–34. [DOI] [PubMed] [Google Scholar]
- 26. Tsukumo Y, Tomida A, Kitahara O et al. Nucleobindin 1 controls the unfolded protein response by inhibiting ATF6 activation. J Biol Chem 2007; 282: 29264–72. [DOI] [PubMed] [Google Scholar]
- 27. Chandel NS, Schumacker PT. Cells depleted of mitochondrial DNA (rho0) yield insight into physiological mechanisms. FEBS Lett 1999; 454: 173–6. [DOI] [PubMed] [Google Scholar]
- 28. Pelicano H, Xu RH, Du M et al. Mitochondrial respiration defects in cancer cells cause activation of Akt survival pathway through a redox‐mediated mechanism. J Cell Biol 2006; 175: 913–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ahmad IM, Aykin‐Burns N, Sim JE et al. Mitochondrial O2*‐ and H2O2 mediate glucose deprivation‐induced stress in human cancer cells. J Biol Chem 2005; 280: 4254–63. [DOI] [PubMed] [Google Scholar]
- 30. Lee AS. GRP78 Induction in Cancer: Therapeutic and Prognostic Implications. Cancer Res 2007; 67: 3496–9. [DOI] [PubMed] [Google Scholar]
- 31. Xu RH, Pelicano H, Zhou Y et al. Inhibition of glycolysis in cancer cells: a novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res 2005; 65: 613–21. [PubMed] [Google Scholar]
- 32. Mann MJ, Hendershot LM. UPR activation alters chemosensitivity of tumor cells. Cancer Biol Ther 2006; 5: 736–40. [DOI] [PubMed] [Google Scholar]
- 33. Bailey CJ, Turner RC. Metformin. N Engl J Med 1996; 334: 574–9. [DOI] [PubMed] [Google Scholar]
- 34. Matoba S, Kang JG, Patino WD et al. p53 regulates mitochondrial respiration. Science 2006; 312: 1650–3. [DOI] [PubMed] [Google Scholar]
- 35. El‐Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem 2000; 275: 223–8. [DOI] [PubMed] [Google Scholar]
- 36. Ishikawa K, Takenaga K, Akimoto M et al. ROS‐generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008; 320: 661–4. [DOI] [PubMed] [Google Scholar]
- 37. Ozcan U, Cao Q, Yilmaz E et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004; 306: 457–61. [DOI] [PubMed] [Google Scholar]
- 38. Rao RV, Bredesen DE. Misfolded proteins, endoplasmic reticulum stress and neurodegeneration. Curr Opin Cell Biol 2004; 16: 653–62. [DOI] [PMC free article] [PubMed] [Google Scholar]