

Abstract
Bufadienolides bufalin and cinobufagin are cardiotonic steroids isolated from the skin and parotid venom glands of the toad Bufo bufo gargarizans Cantor. They have been shown to induce a wide spectrum of cancer cell apoptosis. However, the detailed molecular mechanisms of inducing apoptosis in hepatocellular carcinoma (HCC) are still unclear. In the present study, the apoptosis‐inducing effect of bufalin and cinobufagin on HCC cell line HepG2 was investigated. We found bufalin and cinobufagin induced marked changes in apoptotic morphology and significantly increased the proportion of apoptotic cells. This apoptotic induction was associated with an increase in Fas, Bax and Bid expression, a decrease in Bcl‐2 expression, disruption of the mitochondrial membrane potential, release of cytochrome c, activation of caspase‐3, ‐8, ‐9 and ‐10, and the cleavage of poly(ADP‐ribose)polymerase (PARP), which indicated that bufalin and cinobufagin induced apoptosis through both Fas‐ and mitochondria‐mediated pathways. In addition, caspase activation during bufalin‐ and cinobufagin‐induced apoptosis was further confirmed by caspase‐3 inhibitor Z‐DEVD‐FMK, caspase‐8 inhibitor Z‐IETD‐FMK, caspase‐9 inhibitor Z‐LEHD‐FMK and caspase‐10 inhibitor Z‐AEVD‐FMK. The results showed that bufalin‐ and cinobufagin‐induced apoptosis was blocked by these inhibitors and particularly by caspase‐10 inhibitor. Taken together, bufalin and cinobufagin induce apoptosis of HepG2 cells via both Fas‐ and mitochondria‐mediated pathways, and a Fas‐mediated caspase‐10‐dependent pathway might play a crucial role. (Cancer Sci 2011; 102: 951–958)
Hepatocellular carcinoma (HCC) is one of the most common malignancies worldwide with 600 000 deaths per year, and its incidence is still on the rise.( 1 ) Surgical treatments, such as liver resection and transplantation, are the first‐line therapeutic strategies for HCC. However, the postoperative survival rate is only 30–40% at 5 years and recurrence is quite common in patients who have had a resection.( 2 ) In addition, because HCC is a relatively chemo‐resistant tumor and highly refractory to cytotoxic chemotherapy, systemic cytotoxic chemotherapy agents are minimally effective at improving the survival of patients with advanced HCC.( 3 , 4 ) Therefore, development of novel chemotherapeutic agents and more effective therapies for the treatment of HCC are urgently needed. Recently, traditional Chinese medicines and their active components have attracted a great deal of attention as candidates for HCC therapy.( 5 )
Chan Su is a traditional Chinese medicine obtained from the skin and parotid venom glands of the toad Bufo bufo gargarizans Cantor. It has been used to prepare many popular traditional Chinese medicines such as Liu‐Shen‐Wan and Niu‐Huang‐Xiao‐Yan‐Pian. These Chinese medicines have long been used in numerous areas such as China, Japan, South Korea and other parts of Asia.( 6 ) Bufadienolide type cardiotonic steroids bufalin and cinobufagin (Fig. 1A) are the major active components of Chan Su. They are known as Na+‐K+‐ATPase inhibitors that exhibit a variety of biological activities, such as cardiotonic, anesthetic, blood pressure stimulation and antineoplastic activities.( 7 ) Recently, many studies have focused on the anti‐cancer activities of bufalin and cinobufagin. They have been demonstrated to induce apoptosis in human leukemia, HCC and prostate cancer. Induction of apoptosis by bufalin and cinobufagin has been reported to associate with inhibition of Na+‐K+‐ATPase. Bufalin inhibits this enzyme and then induces apoptosis by activation of activator protein‐1 (AP‐1), NF‐kappaB, T lymphoma invasion and metastasis gene 1 (Tiam1), the c‐Jun N‐terminal protein kinase, Rac1, mitogen‐activated protein kinase,( 8 , 9 , 10 ) as well as by inhibition of Bcl‐2 and c‐myc( 11 ) in human leukemia cells. Bufalin and cinobufagin induce apoptosis of human prostate cancer cells in part with Fas stimulation, Δψm disruption, cytochrome c release and caspase activation.( 12 ) Bufalin has also been found to induce apoptosis by upregulating the expression of Bax in an orthotopic transplantation tumor model of HCC in nude mice.( 13 ) However, the detailed molecular mechanisms of apoptosis induced by bufalin and cinobufagin in HCC are still unclear.
Figure 1.
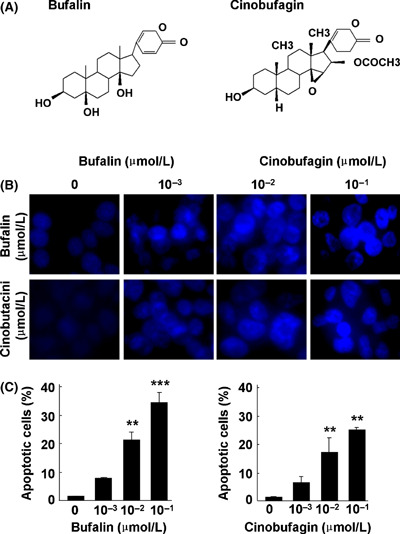
Bufalin and cinobufagin induce apoptosis in HepG2 cells. (A) Structure of bufalin and cinobufagin. (B) Representative microphotographs of Hoechst 33258 staining (original magnification, ×400). (C) Proportion of apoptotic cells. The data represent mean ± SD (n = 3). **P < 0.01 and ***P < 0.001 versus untreated controls.
Cinobufacini (Huachansu), a water‐soluble extract isolated from the skin of toad containing Chan Su, is a form of traditional Chinese medicine approved by the Chinese State Food and Drug Administration (SFDA) and widely used to treat patients with liver, lung, colon and pancreatic cancers at oncology clinics in China.( 14 , 15 ) Previous studies by the current authors indicated that cinobufacini induces apoptosis of HCC cell lines HepG2 and Bel‐7402 cells via a mitochondria‐mediated pathway.( 16 , 17 ) Furthermore, some active compounds like cinobufagin have been separated and identified from cinobufacini by the current authors.( 18 ) In the present study, possible pathways and related molecular mechanisms by which bufalin and cinobufagin induce apoptosis in the HCC cell line HepG2 are investigated.
Materials and Methods
Bufalin and cinobufagin. Bufalin and cinobufagin were purchased from Sigma–Aldrich (St Louis, MO, USA). For in vitro studies, bufalin and cinobufagin were dissolved in 1 mmol/L of dimethyl sulfoxide (DMSO) and aliquoted as a stock solution. To prepare working solutions, aliquots were further diluted in high glucose Dulbecco’s modified Eagle’s medium (DMEM; Gibco‐BRL, Gaithersburg, MD, USA) immediately before each experiment. The maximum DMSO concentration was 0.1% (v/v) in all experiments.
Cell culture. The HCC cell line HepG2 was purchased from the European Collection of Animal Cell Cultures (ECACC, Salisbury, UK). HepG2 cells were cultured in high glucose DMEM supplemented with 10% fetal bovine serum (FBS; Gibco‐BRL, Gaithersburg, MD, USA), 100 U/mL of penicillin and 100 μg/mL of streptomycin in a humidified atmosphere with 5% CO2 in air at 37°C.
Cell viability assay. A MTT (3‐[4, 5‐dimethylthiazol‐2‐yl]‐2, 5‐diphenyl‐tetrazolium bromide) assay was used to detect cell viability as previously described.( 19 ) Briefly, cells (6 × 103 per well) were plated in 96‐well plates and incubated with various concentrations of bufalin or cinobufagin for 24, 48 and 72 h. Cell viability was then detected using Cell Proliferation kit I (MTT; Roche Applied Science, Mannheim, Germany). The cell viability ratio was calculated by the following formula: cell viability (%) = average absorbance of treated group/average absorbance of control group × 100%, and the IC50 (concentration of drug that inhibits cell growth by 50%) value was calculated using SPSS.15.0 software (International Business Machines, New York, USA).
Detection of cell apoptosis. Hoechst 33258 staining was used to observe the morphology of apoptotic cells. After incubation with bufalin or cinobufagin (0, 10−3, 10−2 and 10−1 μmol/L) for 24 h, cells were stained with Hoechst 33258 solution (Dojindo Laboratories, Kumamoto, Japan). Morphological changes in cells were then observed and photographed using a fluorescence microscope at ×400 magnification. The proportion of apoptotic cells was determined by flow cytometry. Briefly, after incubation with bufalin or cinobufagin for 24 h, cells were trypsinized, fixed in 70% ice‐cold ethanol and then stored at −20°C for at least 24 h. Cells were then stained with PI/RNase Staining Buffer (BD Biosciences Pharmingen, San Diego, CA, USA) and analyzed using a FACScan flow cytometry system (Becton Dickinson, San Jose, CA, USA). To analyze the effect of caspase inhibitors on apoptosis, cells were pre‐incubated with 20 μmol/L caspase‐3 inhibitor Z‐DEVD‐FMK, caspase‐8 inhibitor Z‐IETD‐FMK, caspase‐9 inhibitor Z‐LEHD‐FMK and caspase‐10 inhibitor Z‐AEVD‐FMK (BD Biosciences Pharmingen, San Diego, CA, USA) for 2 h, respectively, and then exposed to 10−1 μmol/L of bufalin or cinobufagin for 24 h. Afterwards, cell apoptosis was detected using Hoechst 33258 staining and flow cytometry.
Mitochondrial membrane potential (Δψm) measurement. Δψm was measured with a MitoCapture Mitochondrial Apoptosis Detection kit (BioVision, Mountain View, CA, USA) as previously described.( 17 ) Briefly, after the cells were treated with bufalin or cinobufagin for 24 h, 200 μL of pre‐warmed incubation buffer containing 0.2 μL MitoCapture was added to each well and cells were incubated at 37°C in a 5% CO2 incubator for 15 min. Afterwards, cells were observed under fluorescence microscopy at a magnification of ×400.
Caspase activity assay. Following the protocols of the Caspase Colorimetric Assay kits (BioVision), the activity of caspase‐3, ‐8, ‐9 and ‐10 was detected by cleavage of chromogenic caspase substrates as previously described.( 17 ) Briefly, after treatment with bufalin or cinobufagin for 24 h, cells were harvested and lysed in the supplied lysis buffer. The lysed cells were centrifuged at 10 000 g for 1 min, and 100 μg proteins were incubated with 4 mmol/L caspase substrates at 37°C for 2 h. Then, pNA light emission was quantified using a microplate reader at 405 nm. To analyze the effects of caspase inhibitors on caspase activity, the cells were pre‐incubated with 20 μmol/L caspase‐3, ‐9, ‐8 and ‐10 inhibitors for 2 h, respectively, and then treated with 0.1 μmol/L bufalin or cinobufagin for 24 h. After that, the activity of caspase‐3, ‐8, ‐9 and ‐10 was detected by cleavage of chromogenic caspase substrates.
Western blot analysis. After cells were treated with specified concentrations of various agents, total cell lysates and cytosolic fractions were prepared as previously described.( 17 ) Thirty micrograms of total cellular proteins were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) and transferred onto polyvinylidene fluoride (PVDF) transfer membranes by western blotting.( 20 ) The results were quantified using Image J 1.43 (C) (National Institutes of Health, Bethesda, MD, USA). The following antibodies were used: Bax, Bcl‐2, Bid, Fas, cytochrome c, caspase‐9 and caspase‐3 (Santa Cruz Biotechnology, Santa Cruz, CA, USA); caspase‐8 (BioVision); caspase‐10 (Medical & Biological Laboratories, Nagoya, Japan); full‐length 116 kDa poly(ADP‐ribose)polymerase (PARP) and 89 kDa cleavage fragments of PARP (Cleaved PARP; Cell Signaling Technology, Danvers, MA, USA).
Statistical analysis. All experiments were performed in triplicate and the results are expressed as mean ± SD. Statistical analysis was performed with one‐way analysis of variance (ANOVA) and the Student’s t‐test using SPSS.15.0 software. P < 0.05 was indicative of a significant difference.
Results
Bufalin and cinobufagin inhibit cell viability and induce apoptosis in HepG2 cells. After cells were treated with various concentrations of bufalin or cinobufagin for 24, 48 and 72 h, cell viability decreased significantly in a dose‐ and time‐dependent manner (Table 1). At concentrations higher than 10−3 μmol/L, bufalin or cinobufagin treatment significantly inhibited cell proliferation. The IC50 values of bufalin and cinobufagin were in the range of 0.12–0.81 and 0.17–1.03 μmol/L, respectively. Thus, a range of concentrations (≤10−1 μmol/L) was applied for all subsequent experiments. Hoechst 33258 staining and flow cytometric analysis were used to detect apoptosis. After cells were treated with different concentrations of bufalin or cinobufagin for 24 h, marked morphological changes in chromatin morphology such as crenation, condensation and fragmentation were observed, and this was especially true with a concentration of 10−1 μmol/L (Fig. 1B). Flow cytometric analysis showed that the apoptotic cell population increased significantly in a dose‐dependent manner with bufalin or cinobufagin treatment (Fig. 1C). When the cells were treated with 10−1 μmol/L bufalin or cinobufagin for 24 h, the apoptotic cell population was 34.31% and 28.36% of cells, respectively. These results suggest that both bufalin and cinobufagin exhibited a significant apoptosis‐inducing effect on HepG2 cells and the apoptotic effect of bufalin might be greater than that of cinobufagin.
Table 1.
Inhibitory effect on HepG2 cell proliferation by bufalin and cinobufagin
| Reagents (μmol/L) | Cell viability (%) | ||
|---|---|---|---|
| 24 h | 48 h | 72 h | |
| Bufalin | |||
| 0 | 100.00 | 100.00 | 100.00 |
| 10−4 | 91.99 ± 0.68* | 85.61 ± 2.07*** | 82.43 ± 0.85*** |
| 10−3 | 84.11 ± 4.24*** | 79.32 ± 3.66*** | 74.22 ± 1.34*** |
| 10−2 | 76.02 ± 4.32*** | 72.74 ± 2.82*** | 67.44 ± 1.79*** |
| 10−1 | 63.14 ± 2.75*** | 61.72 ± 1.71*** | 48.89 ± 1.02*** |
| 1 | 41.61 ± 2.31*** | 37.30 ± 3.62*** | 26.35 ± 0.88*** |
| 10 | 33.51 ± 2.05*** | 29.70 ± 2.55*** | 19.56 ± 0.98*** |
| 100 | 25.16 ± 0.74*** | 25.21 ± 0.60*** | 14.69 ± 0.95*** |
| Cinobufagin | |||
| 0 | 100.00 | 100.00 | 100.00 |
| 10−4 | 92.91 ± 3.29* | 91.61 ± 0.42*** | 90.86 ± 1.89** |
| 10−3 | 88.54 ± 5.26** | 81.95 ± 2.37*** | 71.21 ± 2.33*** |
| 10−2 | 78.77 ± 5.98*** | 73.02 ± 0.17*** | 63.71 ± 3.01*** |
| 10−1 | 67.68 ± 4.33*** | 62.21 ± 2.45*** | 57.83 ± 5.00*** |
| 1 | 49.36 ± 1.64*** | 42.20 ± 2.20*** | 40.20 ± 2.30*** |
| 10 | 35.14 ± 4.64*** | 25.63 ± 1.21*** | 26.52 ± 2.43*** |
| 100 | 23.50 ± 2.72*** | 22.30 ± 0.75*** | 15.07 ± 1.66*** |
Data are shown as mean ± SD (n = 3). *P < 0.05, **P < 0.01 and ***P < 0.001 versus untreated controls.
Bufalin and cinobufagin regulate Bax and Bcl‐2 expressions, disrupt Δψm and induce cytochrome c release in HepG2 cells. The Bcl‐2 family proteins Bax and Bcl‐2 play important roles in initiating mitochondrial death cascade.( 21 ) To investigate the underlying mechanism of apoptosis induced by bufalin and cinobufagin, the expression of pro‐apoptotic protein Bax and anti‐apoptotic protein Bcl‐2 was measured by western blot analysis. The protein expression of Bax was upregulated and Bcl‐2 was downregulated with an increase in the Bax/Bcl‐2 ratio (Fig. 2A–C). Following the changes of Bcl‐2 family members, Δψm decreases and cytochrome c is released from the mitochondria.( 22 ) As shown in Figure 2A,B, the protein expression of cytosolic cytochrome c increased in a dose‐dependent manner. As shown in Figure 2D,E, a Δψm loss was observed as fluorescence gradually shifted from red to green with an increase in the green/red fluorescence intensity ratio. Western blot analysis was used to detect the release of cytochrome c. These suggest that mitochondrial dysfunction was involved in the apoptosis induced by bufalin and cinobufagin in HepG2 cells.
Figure 2.
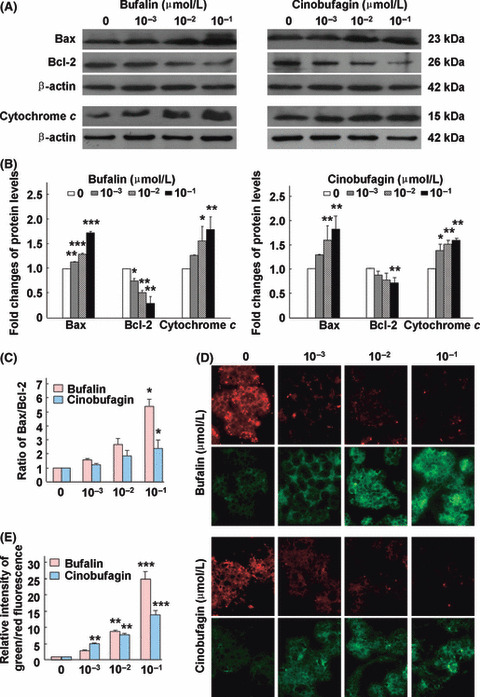
Bufalin and cinobufagin regulate Bax and Bcl‐2 protein expression, disrupt mitochondrial membrane potential (Δψm) and induce cytochrome c release in HepG2 cells. (A) Protein expression of Bax, Bcl‐2 and cytochrome c. (B) Fold changes in Bax, Bcl‐2 and cytochrome c levels. (C) Ratio of Bax/Bcl‐2. (D) Disruption of Δψm. (E) The relative ratio of green/red fluorescence intensity. Data are shown as mean ± SD (n = 3). *P < 0.05, **P < 0.01 and ***P < 0.001 versus untreated controls.
Bufalin and cinobufagin induce caspase activation, Fas upregulation and Bid and PARP cleavage in HepG2 cells. In the mitochondria‐mediated apoptotic pathway, the disruption of Δψm and release of cytochrome c are followed by activation of a caspase cascade including caspase‐9 and ‐3 and cleavage of PARP.( 23 ) As shown in Figure 3, caspase‐9 and ‐3 were activated and PARP (a substrate of caspase‐3) was cleaved from the full‐length 116 kDa form to its cleaved 89 kDa form. To investigate the Fas‐mediated apoptotic pathway induced by bufalin and cinobufagin in HepG2 cells, the activation of caspase‐8 and ‐10 and the protein expression of Fas were detected. As shown in Figure 4, Fas was upregulated and caspase‐8 and ‐10 were activated with bufalin or cinobufagin treatment. A BH‐3 domain‐containing protein Bid, a specific proximal substrate of caspase‐8 and ‐10, can be cleaved by caspase‐8 or ‐10 into a truncated bid and translocates from the cytosol to mitochondria triggering the release of caspase‐activating factors.( 24 ) Thus, caspase‐8 and ‐10 can directly or through cleaving Bid activate downstream caspases including caspase‐3.( 25 , 26 ) As shown in Figure 4B, bufalin and cinobufagin treatment induced a significant decrease in the Bid protein levels in the cytosol.
Figure 3.
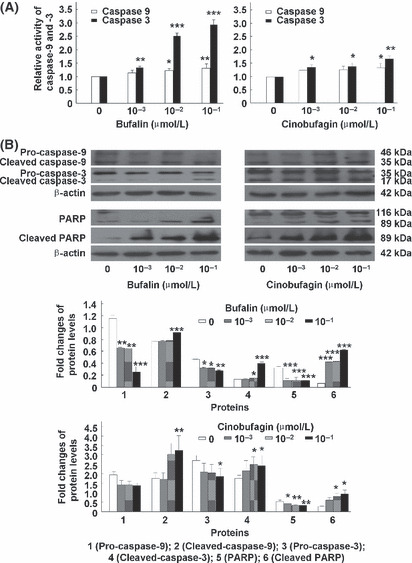
Bufalin and cinobufagin induce caspase‐9 and ‐3 activation and PARP cleavage in HepG2 cells. (A) Relative activity of caspase‐9 and ‐3. (B) Protein expression of caspase‐9, caspase‐3, poly(ADP‐ribose)polymerase (PARP) and cleaved PARP. Data are shown as mean ± SD (n = 3). *P < 0.05, **P < 0.01 and ***P < 0.001 versus untreated controls.
Figure 4.
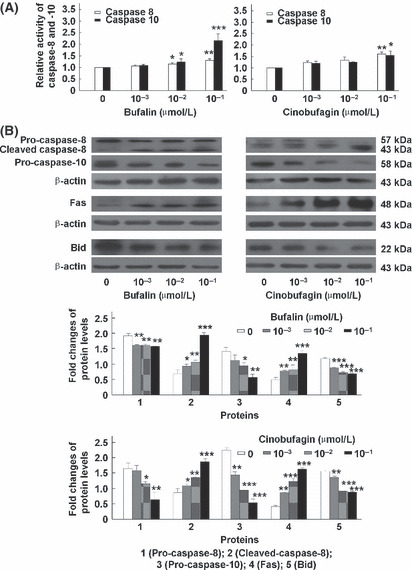
Bufalin and cinobufagin induce caspase‐8 and ‐10 activation, Fas upregulation and Bid cleavage in HepG2 cells. (A) Relative activity of caspase‐8 and ‐10. (B) Protein expression of caspase‐8, caspase‐10, Fas and Bid. Data are shown as mean ± SD (n = 3). *P < 0.05, **P < 0.01 and ***P < 0.001 versus untreated controls.
Effect of caspase inhibitors on bufalin‐ or cinobufagin‐induced apoptosis in HepG2 cells. To further confirm the involvement of caspases in bufalin‐ and cinobufagin‐induced apoptosis, several caspase inhibitors such as caspase‐3 inhibitor Z‐DEVD‐FMK, caspase‐8 inhibitor Z‐IETD‐FMK, caspase‐9 inhibitor Z‐LEHD‐FMK and caspase‐10 inhibitor Z‐AEVD‐FMK were used. As shown in Figure 5, bufalin‐ and cinobufagin‐induced apoptosis was markedly blocked by these inhibitors and particularly by caspase‐10 inhibitor. As shown in Figure 6, the activity of caspase‐3, ‐9, ‐8 and ‐10 was reduced by these caspase inhibitors and particularly by caspase‐3 and ‐10 inhibitors. Furthermore, the roles of caspases in Fas‐ and mitochondria‐mediated pathways of apoptosis induced by bufalin and cinobufagin were determined using western blot. In the mitochondria‐mediated apoptotic pathway, activation of caspse‐3 and ‐9 and cleavage of PARP were obviously suppressed by caspase‐3 inhibitor (Fig. 7A) or caspase‐9 inhibitor (Fig. 7B). In the Fas‐mediated apoptotic pathway, activation of caspase‐8, ‐10, ‐9 and ‐3, cleavage of Bid, release of cytochrome c and cleavage of PARP induced by bufalin were markedly eliminated by caspase‐10 inhibitor (Fig. 8) and/or caspase‐8 inhibitor (Fig. 9). These results suggest that bufalin‐ and cinobufagin‐induced apoptosis in HepG2 cells were associated with both the Fas‐mediated caspase‐8/‐10‐dependent pathway and the mitochondria‐mediated caspase‐9‐dependent pathway, and the Fas‐mediated caspase‐10‐dependent pathway might play a more important role.
Figure 5.
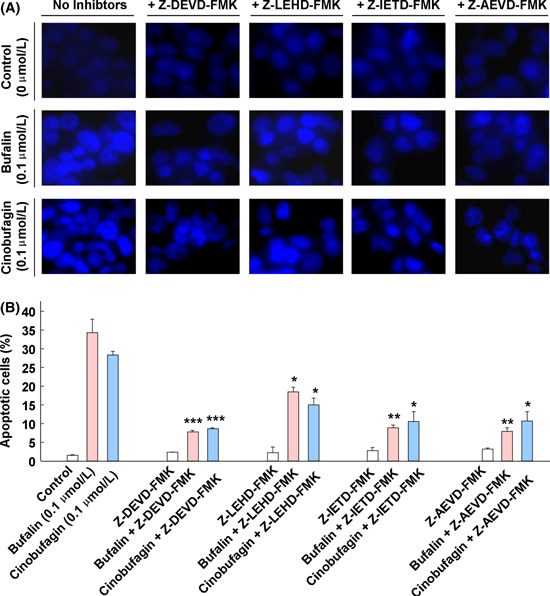
Effect of caspase inhibitors on the morphology and proportion of apoptotic cells induced by bufalin or cinobufagin in HepG2 cells. (A) Representative microphotographs of Hoechst 33258 staining (original magnification, ×400). (B) Proportion of apoptotic cells. The data represent mean ± SD (n = 3). *P < 0.05, **P < 0.01 and ***P < 0.001 versus bufalin or cinobufagin groups in the absence of caspase inhibitors.
Figure 6.
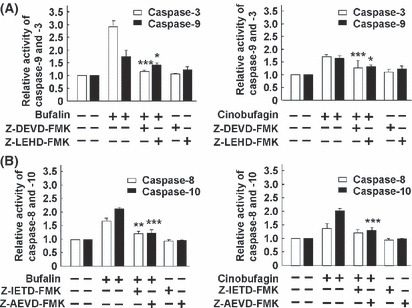
Effect of caspase inhibitors on caspase activities induced by bufalin or cinobufagin in HepG2 cells. (A) Relative activity of caspase‐9 and ‐3. (B) Relative activity of caspase‐8 and ‐10. The data represent mean ± SD (n = 3). *P < 0.05, **P < 0.01 and ***P < 0.001 versus bufalin or cinobufagin groups in the absence of caspase inhibitors.
Figure 7.
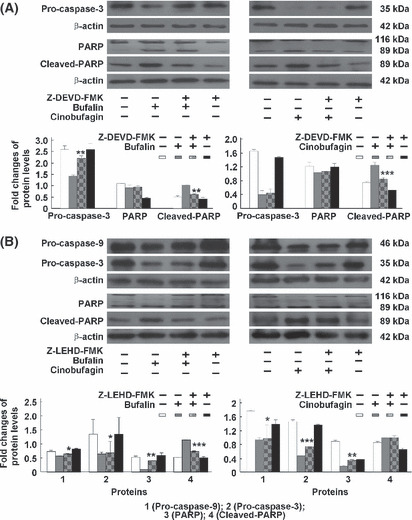
Effect of caspase‐9 and ‐3 inhibitors on caspase‐dependent apoptotic pathways induced by bufalin or cinobufagin in HepG2 cells. (A) Protein expression of pro‐caspase‐3, poly(ADP‐ribose)polymerase (PARP) and cleaved PARP. (B) Protein expression of pro‐caspase‐9, pro‐caspase‐3, PARP and cleaved PARP. The data represent mean ± SD (n = 3). *P < 0.05, **P < 0.01 and ***P < 0.001 versus bufalin or cinobufagin groups in the absence of caspase inhibitors.
Figure 8.
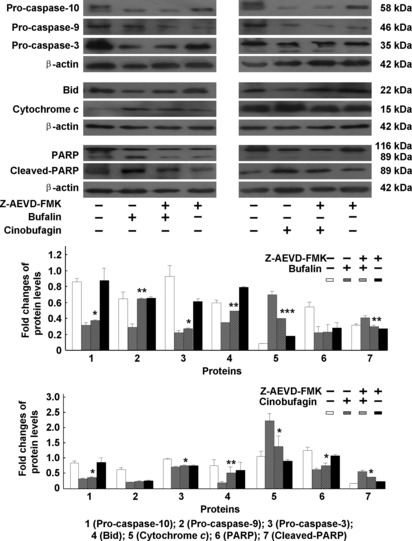
Effect of caspase‐10 inhibitor on caspase‐dependent apoptotic pathways induced by bufalin or cinobufagin in HepG2 cells. The protein expression of pro‐caspase‐10, ‐9, ‐3, Bid, cytochrome c, poly(ADP‐ribose)polymerase (PARP) and cleaved PARP was analyzed. The data represent mean ± SD (n = 3). *P < 0.05, **P < 0.01 and ***P < 0.001 versus bufalin or cinobufagin groups in the absence of caspase inhibitors.
Figure 9.
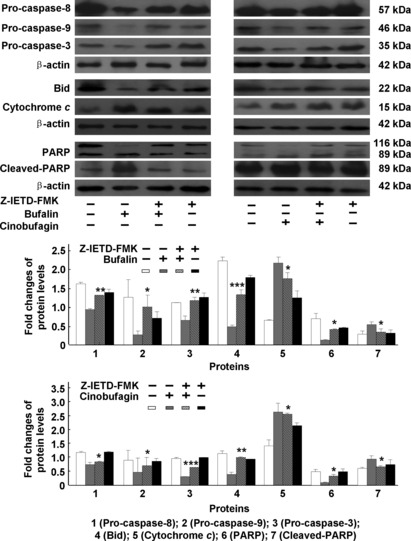
Effect of caspase‐8 inhibitor on caspase‐dependent apoptotic pathways induced by bufalin or cinobufagin in HepG2 cells. The protein expression of pro‐caspase‐8, ‐9, ‐3, Bid, cytochrome c, poly(ADP‐ribose)polymerase (PARP) and cleaved PARP was analyzed. The data represent mean ± SD (n = 3). *P < 0.05, **P < 0.01 and ***P < 0.001 versus bufalin or cinobufagin groups in the absence of caspase inhibitors.
Discussion
Bufalin and cinobufagin have been reported to possess potent anticancer activity by triggering apoptosis in a wide spectrum of cancer cells.( 14 ) However, the exact mechanism and signaling pathways involved in bufalin‐ and cinobufagin‐induced apoptosis are not well established in HCC. The current study showed that bufalin and cinobufagin significantly induced apoptosis of HepG2 cells. Marked morphological changes indicative of cell apoptosis were clearly observed, and the apoptotic cell population increased in a dose‐dependent manner with bufalin and cinobufagin treatment. Moreover, the apoptotic rate induced by bufalin was higher than that induced by cinobufagin. Based on these findings, a series of experiments was performed to further determine potential apoptotic signaling pathways. Classical apoptosis can be initiated via two death‐signaling pathways, the intrinsic or mitochondria‐mediated pathway and the extrinsic or death receptor‐mediated pathway, which both result in the activation of caspases.( 27 ) The present study demonstrated that both the death receptor Fas‐ and mitochondria‐mediated pathways were found to be involved in apoptosis induced by bufalin and cinobufagin.
The Bcl‐2 family proteins Bax and Bcl‐2 play important roles in initiating the mitochondrial death cascade.( 21 ) Pro‐apoptotic protein Bax translocates to the mitochondria and integrates into the outer mitochondrial membrane, where it promotes the disruption of Δψm and the release of cytochrome c into the cytosol. In contrast, anti‐apoptotic protein Bcl‐2 prevents this process by preserving mitochondrial integrity. Thus, the ratio of Bax to Bcl‐2 is crucial to the sustenance of drug‐induced apoptosis in the mitochondria‐mediated apoptotic pathway.( 17 ) The present study showed that bufalin and cinobufagin upregulated the expression of Bax and downregulated the level of Bcl‐2, eventually leading to an increase in the ratio of Bax/Bcl‐2 protein levels. Following the disruption of Δψm and the release of cytochrome c, a caspase cascade including caspase‐9 and caspase‐3 is activated.( 23 ) Activated caspase‐3 subsequently cleaves intracellular protein PARP into cleaved PARP, which serves as an important marker of apoptosis.( 28 ) The present studies showed that a marked loss of Δψm and release of cytochrome c were observed in HepG2 cells with bufalin or cinobufagin treatment. Furthermore, the activation of caspase‐9 and ‐3 and the cleavage of PARP were detected, and these events were blocked markedly by caspase‐3 inhibitor and partially by caspase‐9 inhibitor. These results suggest that bufalin and cinobufagin induced apoptosis of HepG2 cells via a mitochondria‐mediated intrinsic apoptosis pathway (Fig. 10).
Figure 10.
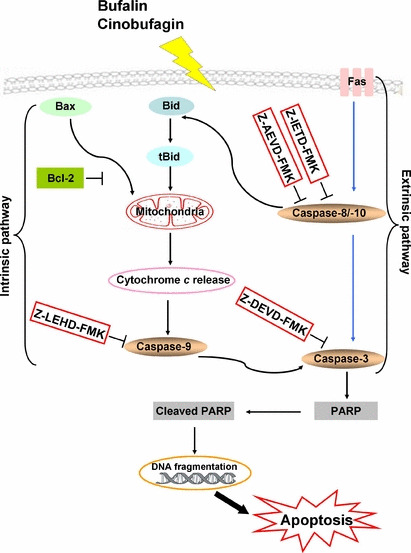
The Fas‐ and mitochondria‐mediated caspase‐dependent apoptotic pathways induced by bufalin and cinobufagin in HepG2 cells. In the intrinsic pathway, bufalin or cinobufagin upregulates Bax expression and downregulates Bcl‐2 expression, resulting in the disruption of Δψm and the release of cytochrome c. Its release leads to the activation of caspase‐9 and ‐3 and cleavage of the caspase‐3 substrate poly(ADP‐ribose)polymerase (PARP) and subsequently results in DNA fragmentation and apoptosis. In the extrinsic pathway, bufalin or cinobufagin upregulates Fas expression and then caspase‐8 and ‐10 are activated, subsequently inducing apoptotic cell death by activating caspase‐3 either directly or indirectly via the mitochondrial apoptotic pathway. Furthermore, these two apoptotic pathways can be inhibited by caspase‐3, ‐9, ‐8 and ‐10 inhibitors, indicating that these caspases play important roles in bufalin‐ and cinobufagin‐induced apoptosis.
Stimulation of Fas has been shown to be highly efficient in the killing of tumor cells.( 29 ) At the cell surface, Fas receptor binding to its ligand leads to the formation of the death‐inducing signal complex, which contains the adaptor protein Fas‐associated death domain protein (FADD) and caspase‐8 and ‐10. FADD leads to the auto‐cleavage and activation of caspase‐8 and ‐10.( 30 , 31 ) A large amount of active caspase‐8 and ‐10 in turn activates downstream caspases such as caspase‐3, causing apoptosis. A small amount of active caspase‐8 and ‐10 cleaves the BH‐3‐only protein Bid into tBid, which subsequently triggers the release of cytochrome c and activation of the mitochondria‐mediated apoptosis pathways.( 32 , 33 ) The present study showed that the protein expression of Fas was upregulated and caspase‐8 and ‐10 were activated with bufalin or cinobufagin treatment. The caspase‐3, ‐8 and ‐10 activation and PARP cleavage during apoptosis induced by bufalin and cinobufagin were blocked markedly by caspase‐10 inhibitor and partially by caspase‐8 inhibitor. These results suggest that a caspase‐10‐dependent Fas‐mediated extrinsic pathway might play a critical role in bufalin‐ and cinobufagin‐induced apoptosis in HepG2 cells (Fig. 10).
Moreover, a noteworthy finding was that Bid was cleaved with bufalin or cinobufagin treatment, leading to a decrease in Bid protein levels. Bid is considered to be a molecular linker bridging the death receptor pathway and mitochondria pathway.( 24 ) Once Bid is cleaved by active caspase‐8 and caspase‐10 as described above, tBid translocates to mitochondria and induces mitochondrial damage and release of cytochrome c.( 24 ) The current data indicate that the release of cytochrome c was blocked markedly by caspase‐10 inhibitor and partially by caspase‐8 inhibitor. The process of Bid cleavage induced by bufalin and cinobufagin was reversed by caspase‐10 inhibitor. Our results suggest that the cross‐talk between Fas‐ and mitochondria‐mediated pathways might exist in bufalin‐ and cinobufagin‐induced apoptosis (Fig. 10).
In conclusion, the present study demonstrated that bufalin and cinobufagin induced apoptosis of HepG2 cells via both Fas‐ and mitochondria‐mediated apoptotic pathways (Fig. 10). Caspase‐3, ‐8, ‐9 and ‐10 all took part in regulation of these two pathways and a caspase‐10‐dependent Fas‐mediated pathway might play a critical role. These findings should provide important clues for further evaluating the potential potency of bufalin and cinobufagin for use in HCC therapy.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgment
This project was supported by Grants‐in‐Aid from the Ministry of Education, Science, Sports, and Culture of Japan and JSPS.
References
- 1. Villanueva A, Minguez B, Forner A, Reig M, Llovet JM. Hepatocellular carcinoma: novel molecular approaches for diagnosis, prognosis, and therapy. Annu Rev Med 2010; 61: 317–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aravalli RN, Steer CJ, Cressman EN. Molecular mechanisms of hepatocellular carcinoma. Hepatology 2008; 48: 2047–63. [DOI] [PubMed] [Google Scholar]
- 3. El‐Serag HB, Marrero JA, Rudolph L, Reddy KR. Diagnosis and treatment of hepatocellular carcinoma. Gastroenterology 2008; 134: 1752–63. [DOI] [PubMed] [Google Scholar]
- 4. Thomas M. Molecular targeted therapy for hepatocellular carcinoma. J Gastroenterol 2009; 44: 136–41. [DOI] [PubMed] [Google Scholar]
- 5. Tsuchiya M, Kono H, Matsuda M, Fujii H, Rusyn I. Protective effect of Juzen‐taiho‐to hepatocarcinogenesis is mediated through the inhibition of kupffer cell‐induced oxidative stress. Int J Cancer 2008; 123: 2503–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang Y, Tang X, Liu X, Li F, Lin X. Simultaneous determination of three bufadienolides in rat plasma after intravenous administration of bufadienolides extract by ultra performance liquid chromatography electrospray ionization tandem mass spectrometry. Anal Chim Acta 2008; 610: 224–31. [DOI] [PubMed] [Google Scholar]
- 7. Wang Z, Wen J, Zhang J, Ye M, Guo D. Simultaneous determination of four bufadienolides in human liver by high‐performance liquid chromatography. Biomed Chromatogr 2004; 18: 318–22. [DOI] [PubMed] [Google Scholar]
- 8. Chen A, Yu J, Zhang L et al. Microarray and biochemical analysis of bufalin‐induced apoptosis of HL‐60 cells. Biotechnol Lett 2009; 31: 487–94. [DOI] [PubMed] [Google Scholar]
- 9. Kawazoe N, Watabe M, Masuda Y, Nakajo S, Nakaya K. Tiam1 is involved in the regulation of bufalin‐induced apoptosis in human leukemia cells. Oncogene 1999; 18: 2413–21. [DOI] [PubMed] [Google Scholar]
- 10. Watabe M, Ito K, Masuda Y, Nakajo S, Nakaya K. Activation of AP‐1 is required for bufalin‐induced apoptosis in human leukemia U937 cells. Oncogene 1998; 16: 779–87. [DOI] [PubMed] [Google Scholar]
- 11. Masuda Y, Kawazoe N, Nakajo S, Yoshida T, Kuroiwa Y, Nakaya K. Bufalin induces apoptosis and influences the expression of apoptosis‐related genes in human leukemia cells. Leuk Res 1995; 19: 549–56. [DOI] [PubMed] [Google Scholar]
- 12. Yu CH, Kan SF, Pu HF, Jea Chien E, Wang PS. Apoptotic signaling in bufalin‐ and cinobufagin‐treated androgen‐dependent and ‐independent human prostate cancer cells. Cancer Sci 2008; 99: 2467–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Han KQ, Huang G, Gu W, Su YH, Huang XQ, Ling CQ. Anti‐tumor activities and apoptosis‐regulated mechanisms of bufalin on the orthotopic transplantation tumor model of human hepatocellular carcinoma in nude mice. World J Gastroenterol 2007; 13: 3374–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Meng Z, Yang P, Shen Y et al. Pilot study of huachansu in patients with hepatocellular carcinoma, non‐small‐cell lung cancer, or pancreatic cancer. Cancer 2009; 115: 5309–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Qi F, Li A, Inagaki Y et al. Antitumor activity of extracts and compounds from the skin of the toad Bufo bufo gargarizans Cantor. Int Immunopharmacol 2010. doi: 10.1016/j.intimp.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 16. Qi FH, Li AY, Lv H et al. Apoptosis‐inducing effect of cinobufacini, Bufo bufo gargarizans Cantor skin extract, on human hepatoma cell line BEL‐7402. Drug Discov Ther 2008; 2: 339–43. [PubMed] [Google Scholar]
- 17. Qi F, Li A, Zhao L et al. Cinobufacini, an aqueous extract from Bufo bufo gargarizans Cantor, induces apoptosis through a mitochondria‐mediated pathway in human hepatocellular carcinoma cells. J Ethnopharmacol 2010; 128: 654–61. [DOI] [PubMed] [Google Scholar]
- 18. Wang DL, Qi FH, Xu HL et al. Apoptosis‐inducing activity of compounds screened and characterized from cinobufacini by bioassay‐guided isolation. Mol Med Report 2010; 3: 717–22. [DOI] [PubMed] [Google Scholar]
- 19. Inagaki Y, Tang W, Zhang L, Du GH, Xu WF, Kokudo N. Novel aminopeptidase N (APN/CD13) inhibitor 24F can suppress invasion of hepatocellular carcinoma cells as well as angiogenesis. Biosci Trends 2010; 4: 56–60. [PubMed] [Google Scholar]
- 20. Yan G, Duan RH, Yin K et al. Inhibition of survivin expression to induce the apoptosis of hepatocarcinoma cells by adenovirus‐mediated siRNA. Biosci Trends 2008; 2: 88–93. [PubMed] [Google Scholar]
- 21. Gross A, McDonnell JM, Korsmeyer SJ. BCL‐2 family members and the mitochondria in apoptosis. Genes Dev 1999; 13: 1899–911. [DOI] [PubMed] [Google Scholar]
- 22. Hellbrand EE, Varbiro G. Development of mitochondrial permeability transition inhibitory agents: a novel drug target. Drug Discov Ther 2010; 4: 54–61. [PubMed] [Google Scholar]
- 23. Haga N, Fujita N, Tsuruo T. Involvement of mitochondrial aggregation in arsenic trioxide (As2O3)‐induced apoptosis in human glioblastoma cells. Cancer Sci 2005; 96: 825–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yin XM. Bid, a BH3‐only multi‐functional molecule, is at the cross road of life and death. Gene 2006; 369: 7–19. [DOI] [PubMed] [Google Scholar]
- 25. Xian M, Ito K, Nakazato T et al. Zerumbone, a bioactive sesquiterpene, induces G2/M cell cycle arrest and apoptosis in leukemia cells via a Fas‐ and mitochondria‐mediated pathway. Cancer Sci 2007; 98: 118–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Debatin KM, Krammer PH. Death receptors in chemotherapy and cancer. Oncogene 2004; 23: 2950–66. [DOI] [PubMed] [Google Scholar]
- 27. Green DR. Apoptotic pathways: ten minutes to dead. Cell 2005; 121: 671–4. [DOI] [PubMed] [Google Scholar]
- 28. Scovassi AI, Poirier GG. Poly (ADP‐ribosylation) and apoptosis. Mol Cell Biochem 1999; 199: 125–37. [DOI] [PubMed] [Google Scholar]
- 29. Goto M. Elevation of soluble Fas (APO‐1, CD95) ligand in natural aging and Werner syndrome. Biosci Trends 2008; 2: 124–7. [PubMed] [Google Scholar]
- 30. Wajant H. The Fas signaling pathway: more than a paradigm. Science 2002; 296: 1635–6. [DOI] [PubMed] [Google Scholar]
- 31. Abd El‐Ghany RM, Sharaf NM, Kassem LA, Mahran LG, Heikal OA. Thymoquinone triggers anti‐apoptotic signaling targeting death ligand and apoptotic regulators in a model of hepatic ischemia reperfusion injury. Drug Discov Ther 2009; 3: 296–306. [PubMed] [Google Scholar]
- 32. Milhas D, Cuvillier O, Therville N et al. Caspase‐10 triggers Bid cleavage and caspase cascade activation in FasL‐induced apoptosis. J Bio Chem 2006; 280: 19836–42. [DOI] [PubMed] [Google Scholar]
- 33. Roucou X, Montessuit S, Antonsson B, Martinou JC. Bax oligomerization in mitochondrial membranes requires tBid (caspase‐8‐cleaved Bid) and a mitochondrial protein. Biochem J 2002; 368: 915–21. [DOI] [PMC free article] [PubMed] [Google Scholar]