

Abstract
Epigenetic alterations of DNA methylation play an important role in the regulation of gene expression associated with chemosensitivity of gastric carcinomas. With the aim of improving the chemotherapeutic efficacy of gastric carcinoma, the effect of DNA methyltransferase inhibitor, 5‐aza‐CdR, on the chemosensitivity of five anticancer drugs was investigated. Human gastric cancer cell lines, OCUM‐2M and MKN‐74, and five anticancer drugs, 5‐FU, PTX, OXA, SN38, and GEM, were used. In both gastric cancer cell lines, a synergistic antiproliferative effect by a combination of 5‐aza‐CdR at 5 µM was found in SN38 and GEM. 5‐Aza‐CdR at 5 µM increased apoptosis induced by SN38 and GEM in both cell lines. 5‐Aza‐CdR increases the expression of DAPK‐2 and DAPK‐3, RASSF1, and THBS1 genes in both OCUM‐2M and MKN‐74 cells, but not that of hMLH1, p16, MGMT, E‐cadherin, and p53 genes. These findings suggest that 5‐aza‐CdR is a promising chemotherapeutical agent for gastric carcinomas, in combination with the anticancer drugs SN38 and GEM, in apoptosis signaling. The upregulation of DAPK‐2 and DAPK‐3, RASSF1, and THBS1 genes by 5‐aza‐CdR might be associated with the synergistic effect. (Cancer Sci 2006; 97: 938–944)
Abbreviations:
- 5‐aza‐CdR
5‐aza‐2′‐deoxycytidine
- 5‐FU
5‐fluorouracil
- DAPK
death‐associated protein kinases
- FITC
fluorescein‐isothiocyanate
- GAPDH
glyceraldehyde‐3‐phosphate dehydrogenase
- GEM
gemcitabine
- IC50
concentration causing 50% growth inhibition
- MGMT
O(6)‐methylguanine‐DNA methyltransferase
- MTT
3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide
- OXA
oxaliplatin
- PCR
polymerase chain reaction
- PI
propidium iodide
- PTX
paclitaxel
- RASSF1
Ras association domain family 1
- SN38
irinotecan
- TGFβ
transforming growth factor β; TGFBR1, transforming growth factor beta receptor 1
- THBS1
thrombospondin 1.
Hypermethylation of CpG islands at gene promoter regions on DNA might occur in both alleles or in combination with deletions or mutations of the other allele, resulting in gene inactivation of some tumor suppressor genes.( 1 ) These genes contain the apoptosis‐related gene DAPK, tumor suppressor genes p53, p16, RASSF1 and TGFBR1, DNA mismatch repair genes hMLH1 and MGMT, intracellular adhesion proteins E‐cadherin and angiogenesis inhibitor THBS1.( 2 , 3 , 4 , 5 , 6 ) Aberrant DNA methylation plays an important role in carcinogenesis and tumor apoptosis. The process of hypermethylation is carried out by DNA methyltransferase, which catalyzes the covalent addition of a methyl group from a donor S‐adenosylmethionine to the 5 position of cytosine, predominantly within the CpG dinucleotide.( 7 ) DNA methyltransferases are depleted by being bound to their inhibitors and are thereby unavailable for methylation, resulting in significant demethylation after repeated replication.( 8 ) The DNA methyltransferase inhibitor, 5‐aza‐CdR, has been shown to reverse the hypermethylation status of promoters, allowing re‐expression of silenced genes and ultimately inducing apoptosis and inhibiting tumor growth. Recently, several clinical trials of 5‐aza‐CdR have been reported, including a phase II study of 5‐aza‐CdR in patients with metastatic prostate cancer( 9 ) and a phase III study of decitabine in patients with myelodysplasia.( 10 ) Although 5‐aza‐CdR as a single agent is quite effective in acute or chronic leukemia and myelodysplasia syndrome( 11 , 12 ) the response rate of 5‐aza‐CdR in solid tumors is less than 10%.( 13 , 14 ) Kanda et al. have found that 5‐aza‐CdR sensitizes the antitumor effect of 5‐FU in chemoresistant hepatoma and pancreatic cancer cell lines.( 15 ) In this study, we explored the novel role of 5‐aza‐CdR as a chemo‐sensitizer in combination with chemotherapeutic agents for gastric carcinomas.
As one of the major cause of cancer death, gastric cancer remains threatening around the world( 16 ) and most patients in advanced stages need chemotherapy. Of the commonly used chemotherapeutical agents effective in gastric cancer, 5‐FU remains the primary one. Recently, however, several new drugs have emerged that provide better efficacy and prolonged survival for patients with advanced gastric cancer. These agents include taxanes such as PTX, the third‐generation platinum derivative OXA, the topoisomerase‐I inhibitor SN38, and the pyrimidine analog GEM. Even so, the response rate is low (20%−40%).( 17 , 18 , 19 , 20 ) Combination chemotherapy using anticancer drugs achieves a better response rate, exceeding the efficacy of single treatment, but carries a high risk of side‐effects. With the aim of improving the chemotherapeutic efficacy of gastric carcinoma, we investigated a novel combination regimen of anticancer drugs with DNA methyltransferase inhibitor 5‐aza‐CdR, and report that 5‐aza‐CdR interacts with anticancer drugs in a highly synergistic manner in gastric cancer cell lines to induce apoptosis.
Materials and methods
Chemicals and anticancer drugs
A stock solution of 5‐aza‐CdR (Sigma, St Louis, MO, USA) at 10 mM in 99% ethanol, and five anticancer drugs, 5‐FU (Kyowa Hakko, Tokyo, Japan), PTX (Bristol‐Myers, Wallingford, Connecticut), OXA (Yakult, Tokyo, Japan), SN38 (Yakult) and GEM (Eli Lilly, Kobe, Japan), were used in this study. All reagents were formulated as recommended by their suppliers.
Cell culture and cell lines
The human gastric cancer cell lines OCUM‐2 M( 21 ) and MKN‐74( 22 ) were used in this study. MKN‐74 was derived from liver metastases of moderately differentiated tubular adenocarcinoma, and OCUM‐2M was derived from a primary tumor of poorly differentiated adenocarcinoma. OCUM‐2M was cultured in Dulbecco's modified Eagle's medium (Nikken Biomedical Laboratory, Kyoto, Japan), and MKN‐74 was cultured in RPMI‐1640 medium (Sigma, Tokyo, Japan). Both media were supplemented with 10% fetal bovine serum, 100 IU/mL penicillin (ICN Biomedical, Costa Mesa, CA, USA), 100 mg/mL streptomycin (ICN Biomedical), and 0.5 mM sodium pyruvate (Cambrex, Walkersville, MD, USA). The cells were cultured at 37°C in a humidified atmosphere of 5% CO2 in air.
Cell growth assays
Cancer cells (5 × 104) were placed in each well of a 96‐well plate. With or without the addition of 5‐aza‐CdR at 5 µM, and with or without anticancer drugs at the concentration of IC50, the plates were incubated for 72 h at 37°C. The suppression of cell proliferation was examined by MTT (Sigma, St Louis) colorimetric assay in which the formazan product of MTT was measured as absorbance at 550 nm using a microtiter plate reader (Model 550; Bio‐Rad Laboratories, Tokyo, Japan). The percentage of cell viability was determined as the ratio absorbance of the sample versus the control. The IC50 of each drug was determined as a drug concentration showing 50% cell growth inhibition as compared with the control cell growth. Six replicate wells were used for each drug concentration, and the testing was carried out independently three times. The potential synergy between the drugs and 5‐aza‐CdR was evaluated as follows, using Drewinko's fraction method.( 23 ) The synergistic, semiadditive, and antagonistic interactions were determined when the value was less than the expected value, more than the expected value but less than the drugs’ value, and more than the drugs’ value, respectively. The expected value was calculated by the combined effects (%) = the effects of the anticancer drug/control × the effects of the 5‐aza‐CdR/control × 100.
Flow cytometry
Apoptosis was detected using flow cytometry by staining cells with annexin V–FITC and PI (BD Pharmingen, San Diego, CA, USA) labeling. OCUM‐2M and MKN‐74 cells were seeded at a density of 1.0 × 105 cells/mL in a six‐well plate. With or without the addition of 5‐aza‐CdR (5 µM), and with or without anticancer drugs at the concentration of IC50, the plates were incubated for 72 h at 37°C. Cells were stained with 2.5 mL of Annexin V–FITC and/or 2.5 mL of PI (50 ng/mL) according to the instructions of the manufacturer, incubated for 15 min at room temperature in the dark, and immediately analyzed by FACScan flow cytometry (Becton Dickinson, Mounkain View, CA, USA). Viable cells do not stain with either dye, apoptotic cells only with annexin V–FITC, and secondary necrotic cells with both annexin V–FITC and PI.
Reverse transcription–PCR
We examined the expression at the mRNA level of genes, including DAPK‐1, DAPK‐2, DAPK‐3, p16, RASSF1, TGFBR1, p53, THBS, hMLH1, MGMT and E‐cadherin. The cells were seeded in a 100‐mm dish with the final concentration of 2.5 × 104 cells/mL, with or without 5‐aza‐CdR at 5 µM. After incubation for 48 h, total cellular RNA was extracted from OCUM‐2M and MKN‐74 using Trizol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. After the genomic DNA was removed by DNAse, cDNA was prepared from 1 mg of RNA with Maloney mouse leukemia virus reverse transcriptase (Invitrogen) using random primers (Invitrogen). The relevant cDNA was amplified by PCR using the primer pairs (Table 1) with Taq DNA polymerase (Invitrogen) in a thermal cycler. The PCR conditions were as follows: predenaturation at 94°C for 3 min, denaturation at 94°C for 30 s, annealing at 59°C for 30 s, extension at 72°C for 1 min with 35 cycles of the three repeated steps, and final incubation at 72°C for 10 min. The PCR products were applied to 2% agarose gel and electrophoresed. The mRNA level of the each gene was normalized by the internal control GAPDH.
Table 1.
Primer sequences used in this study
| Gene | Sequence | Size of PCR products | |
|---|---|---|---|
| DAPK 1 | sense | TCTACCAGCCACGGGACTTC | 134 bp |
| antisense | GCTGGCCTGTGAGTAGACGT | ||
| DAPK 2 | sense | GCATCGTGTCCCTGTGCAAC | 121 bp |
| antisense | GCTTTCCTCCTGGCGATGTC | ||
| DAPK 3 | sense | CCCAACCCACGAATCAAGCTC | 236 bp |
| antisense | GCTGAGATGTTGGTGAGCGTC | ||
| p53 | sense | AGCGATGGTCTGGCCCCTCCT | 120 bp |
| antisense | CTCAGGCGGCTCATAGGGCAC | ||
| RASSF1 | sense | TTCACCTGCCACTACCGCTG | 292 bp |
| antisense | AGGGTGGCTTCTTGCTGGAG | ||
| TGFBR1 | sense | TCGAGTGCCAAATGAAGAGGAC | 278 bp |
| antisense | AAATCTCTGCCTCACGGAACCA | ||
| THBS1 | sense | GGGTTGTACGCCATCAGGGT | 258 bp |
| antisense | CAGAAAGGCCCGAGTATCCC | ||
| hMLH1 | sense | TCAGGCCAGCAGAGTGAAGT | 154 bp |
| antisense | GATCAGGCAGGTTAGCAAGC | ||
| p16 | sense | GAATAGTTACGGTCGGAGGCC | 304 bp |
| antisense | ATGGTTACTGCCTCTGGTGCC | ||
| MGMT | sense | CCAGCAAGAGTCGTTCACCAG | 134 bp |
| antisense | TCATTGCTCCTCCCACTGCTC | ||
| E‐cadherin | sense | TGATGCCCCCAATACCCCAG | 209 bp |
| antisense | CTGTGGAGGTGGTGAGAGAG | ||
| GAPDH | sense | ACCTGACCTGCCGTCTAGAA | 247 bp |
| antisense | TCCACCACCCTGTTGCTGTA |
Statistical methods
The quantitative ratios of different groups were compared using Student's t‐test. Probability values of P < 0.05 were regarded as statistically significant. All statistical tests were two‐sided.
Results
5‐Aza‐CdR increased the efficiency of anticancer drugs
Figure 1 shows the effects of 5‐aza‐CdR and/or anticancer drugs on the proliferation of gastric cancer cells. 5‐Aza‐CdR at 5 µM suppressed the proliferation of cancer cells in both cell lines, whereas the antiproliferation rates in response to 5‐aza‐CdR were low (90.4% and 95.3%) in OCUM‐2M and MKN‐74 cells, respectively. Anti‐cancer drugs were added to cancer cell cultures at the IC50 for each cell line (Table 2). The proliferation rate in response to combined‐exposure was evaluated by comparison with the expected additive effect. The proliferation rates for OCUM‐2M cells by 5‐FU, PTX, OXA, SN38, and GEM alone were 40.7%, 49.4%, 61.3%, 42.1%, and 60.2%, respectively. The proliferation rates for OCUM‐2M cells in response to the combination of 5‐aza‐CdR and 5‐FU, PTX, OXA, SN38, or GEM were 42.8%, 45.0%, 58.0%, 32.6%, and 49.2%, respectively. The proliferation effect for OCUM‐2M cells by combination of 5‐aza‐CdR was lower than the expected additive effect, evaluating that the combination of 5‐aza‐CdR with SN38 or GEM shows a synergistic effect. The 5‐aza‐CdR plus PTX or OXA shows a semiadditive effect. Although the combination with 5‐FU shows an antagonistic effect, the statistical data show no significance. The proliferation rates for MKN‐74 cells in response to 5‐FU, PTX, OXA, SN38, or GEM alone were 44.8%, 34.6%, 53.0%, 51.8%, and 42.6%, respectively. The proliferation rates for MKN‐74 cells in response to 5‐aza‐CdR plus 5‐FU, PTX, OXA, SN38, or GEM were 45.3%, 33.5%, 42.2%, 42.0%, and 36.5%, respectively. In MKN‐74 cells, the proliferation effect by combination of 5‐aza‐CdR was lower than the expected additive effect, evaluating that the combination of 5‐aza‐CdR with OXA, SN38, or GEM shows a synergistic effect. 5‐Aza‐CdR plus PTX shows a semiadditive effect. Similar to the OCUM‐2M cells, the combination with 5‐FU shows an antagonistic effect, but the statistical data show no significance. No morphologic change was found after treatment with 5‐aza‐CdR alone, whereas the numbers of large floating cells were increased by the treatment of the five anticancer drugs in OCUM‐2M and MKN‐74.
Figure 1.
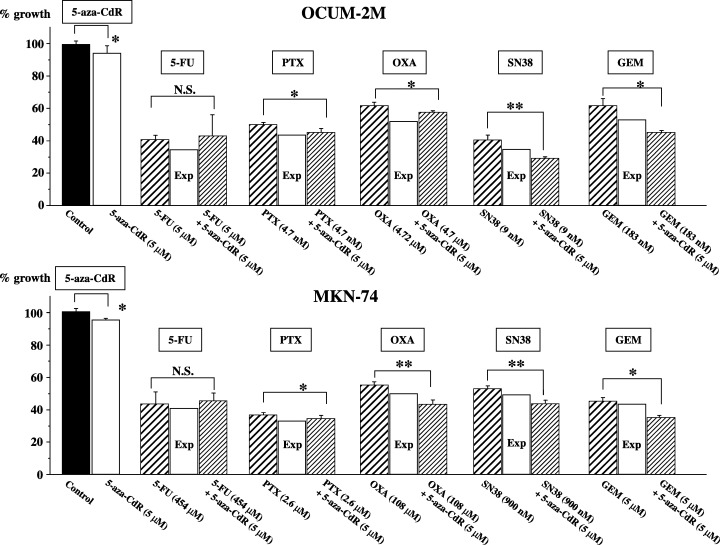
Synergistic or semiadditive effects of 5‐aza‐CdR with anticancer drugs in gastric cell lines. A synergistic antiproliferative effect in response to a combination of 5‐aza‐CdR with SN38 or GEM was observed in both OCUM‐2M and MKN‐74 cells. In MKN‐74 cells, the combination of 5‐aza‐CdR with OXA also showed a synergistic antiproliferative effect. A semiadditive antiproliferative effect was observed in response to a combination of 5‐aza‐CdR with OXA or PTX in OCUM‐2M, and with PTX in MKN‐74. No significant effect was observed in response to a combination of 5‐aza‐CdR with 5‐FU in either cell line. Each anticancer drug was added at the IC50 for each cell line. The synergistic, semiadditive, and antagonistic interactions were determined when the value was less than the expected value, more than the expected value but less than the drugs’ value, and more than the drugs’ value, respectively. The expected value of the combined effects (%) = effects of anticancer drug/control × effects of 5‐aza‐CdR/control × 100. The results are presented as the mean of three independent experiences, and the bars indicate the standard deviation. *P < 0.05 and **P < 0.01, compared with each anticancer drug alone. Exp, an expected additive value; N.S., not significant.
Table 2.
IC50 of OCUM‐2M and MKN‐74 cells to five anticancer drugs
| IC50 | ||
|---|---|---|
| OCUM‐2M | MKN‐74 | |
| 5‐FU | 5.1 ± 1.9 µM | 0.6 ± 0.1 mM |
| PTX | 4.7 ± 1.9 nM | 2.6 ± 0.1 µM |
| OXA | 4.7 ± 1.3 µM | 108.3 ± 10.5 µM |
| SN38 | 9.0 ± 1.2 nM | 0.9 ± 0.1 µM |
| GEM | 183.3 ± 12.9 nM | 5.0 ± 1.6 µM |
The values correspond to the results of three independent studies expressed as the mean ± standard deviation.
5‐Aza‐CdR increased apoptosis induced by anticancer drugs in gastric cancer cell lines
Figure 2 shows the rates of apoptosis induced by 5‐aza‐CdR and/or anticancer drugs in gastric cancer cell lines. Anticancer drugs were added to the cancer cell cultures at the IC50 for each cell line. In OCUM‐2M cells, 5‐aza‐CdR induced apoptosis at a rate of 4.7%, compared with the control of 2.4%. The apoptosis rates induced by 5‐FU, PTX, OXA, SN38, and GEM alone were 7.0%, 5.1%, 17.0%, 8.8%, and 12.7%, respectively. In addition, the apoptosis rates induced by combined exposure of 5‐aza‐CdR with anticancer drugs, 5‐FU, PTX, OXA, SN38, and GEM were 6.0%, 8.7%, 20.5%, 21.4%, and 24.5%, respectively. In MKN‐74 cells, 5‐aza‐CdR induced apoptosis at a rate of 3.7%, compared with the control rate of 5.7%. The apoptosis rates induced by 5‐FU, PTX, OXA, SN38, and GEM alone were 22.2%, 16.7%, 23.4%, 15.8%, and 14.4%, respectively. and the apoptosis rates induced by combined exposure of 5‐aza‐CdR with 5‐FU, PTX, OXA, SN38, and GEM were 24.5%, 38.9%, 38.4%, 26.5%, and 21.7%, respectively. 5‐Aza‐CdR at 5 µM increased apoptosis induced by SN38 and GEM in both cell lines, and increased the apoptosis rate induced by PTX and OXA in MKN‐74 cells.
Figure 2.
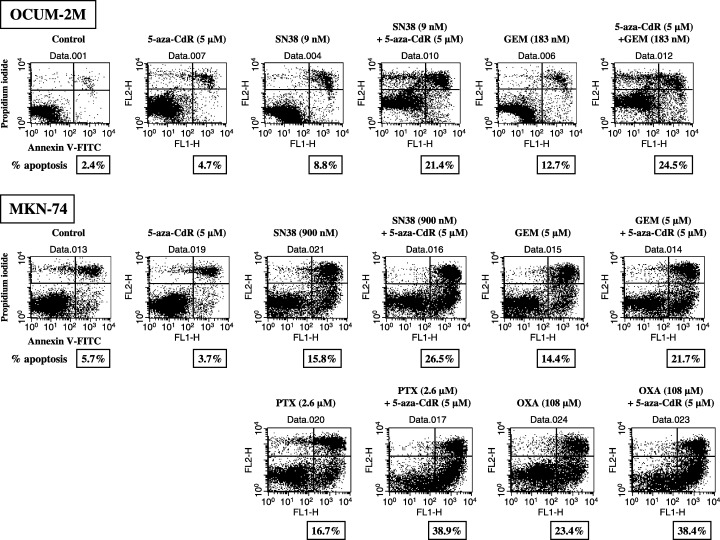
Apoptosis induction by DNA methyltransferase inhibitor. To clarify the induction of apoptosis during the growth suppression of OCUM‐2M and MKN‐74 cell lines by combined exposure of 5‐aza‐CdR with anticancer drugs, cells were double stained with Annexin V–FITC and PI. Cells staining annexin V positive and PI negative were considered to be apoptotic. Anticancer drugs were added to the cancer cell cultures at the IC50 for each cell line. In OCUM‐2M, 5‐aza‐CdR induced apoptosis at a rate of 4.7%, compared with the control of 2.4%. The apoptosis rates induced by SN38, and GEM alone were 8.8%, and 12.7%, respectively. In addition, the apoptosis rates induced by combined exposure of 5‐aza‐CdR with anticancer drugs, SN38, and GEM were 21.4%, and 24.5%, respectively. In MKN‐74, 5‐aza‐CdR induced apoptosis at a rate of 3.7%, compared with the control rate of 5.7%. The apoptosis rates induced by SN38, GEM, PTX and OXA alone were 15.8%, 14.4%, 16.7% and 23.4%, respectively. The apoptosis rates induced by combined exposure of 5‐aza‐CdR with SN38, GEM, PTX and OXA were 26.5%, 21.7%, 38.9% and 38.4%, respectively. 5‐Aza‐CdR at 5 µM increased apoptosis induced by SN38 and GEM in both cell lines, and increased the apoptosis rate induced by PTX and OXA in MKN‐74 cells.
Effects of 5‐aza‐CdR on gene expression in gastric cancer cells
5‐Aza‐CdR treatment increased the expression of DAPK‐2, DAPK‐3, RASSF1, and THBS1 genes in both OCUM‐2M and MKN‐74 cells. 5‐Aza‐CdR increased the expression of DAPK1 and TGFBR1 gene in MKN‐74 cells, but not in OCUM‐2M cells. In addition, the expression of hMLH1, p16, MGMT, E‐cadherin, and p53 genes showed no alteration in response to 5‐aza‐CdR in OCUM‐2M cells, nor in MKN‐74 cells (Fig. 3).
Figure 3.
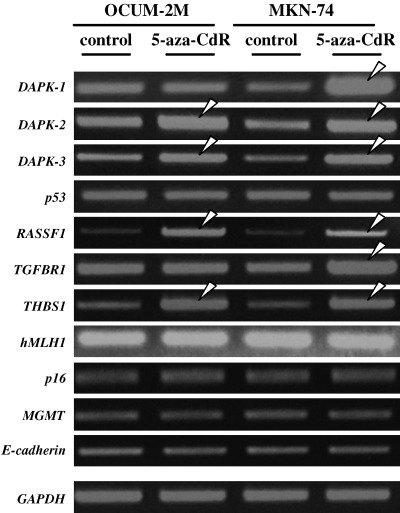
Effects of 5‐aza‐CdR on gene expression by reverse transcription–PCR. Expression of DAPK‐2, DAPK‐3, RASSF1 and THBS1 increased in OCUM‐2M and MKN‐74 cells. The expression of DAPK1 and TGFBR1 increased in MKN‐74, but not in OCUM‐2M. The expression of hMLH1, p16, MGMT, E‐cadherin and p53 genes did not change in either cell line.
Discussion
In this study, DNA methyltransferase inhibitor, 5‐aza‐CdR, showed synergism or semiaddition when given simultaneously with four anticancer drugs, PTX, OXA, SN38, and GEM, in human gastric cancer cells. In both OCUM‐2M and MKN‐74 cells, a synergistic antiproliferative effect was observed in response to a combination of 5‐aza‐CdR with SN38 or GEM, and a semiadditive effect was observed with PTX. The combination of 5‐aza‐CdR with OXA showed a synergistic antiproliferative effect in MKN‐74 cells, but a semiadditive effect in OCUM‐2M cells. These four anticancer drugs have different mechanisms of antitumor effect. These findings suggest that 5‐aza‐CdR could have wide chemotherapeutic efficacy with various types of anticancer drugs in gastric carcinoma. Although 5‐aza‐CdR as a single agent has demonstrated little activity in solid tumors( 13 , 14 ) the present findings open a new window to further consider the use of 5‐aza‐CdR in solid tumors, including gastric cancer.
This study indicated that a low dose of 5‐aza‐CdR showed a synergistic or semiadditive antiproliferative effect in combination with the anticancer drugs. Different concentrations of 5‐aza‐CdR, ranging from 0.1 to 30 µM, have been applied in previous studies. Timmermann et al.( 24 ) reported that 3 µM is not sufficient to re‐activate the expression of silenced genes, and Yang et al.( 25 ) reported that 5 µM of 5‐aza‐CdR is enough to re‐activate the expression of silenced genes. We then used 2 µM, 5 µM and 10 µM of 5‐aza‐CdR. In our preliminary assay, 2 µM of 5‐aza‐CdR did not show synergistic or additive effects with anticancer drugs, whereas 5 µM and 10 µM increased the chemosensitivity of anticancer drugs. Then we used 5 µM of 5‐aza‐CdR in our experiment. Six cell lines, OCUM‐2M, MKN74, OCUM‐8, OCUM‐2MD3, MKN‐7, and MKN‐45 were used in our preliminary study. In OCUM‐8, OCUM‐2MD3, MKN‐7, and MKN‐45 lines, the combination effects of 5‐aza‐CdR with five anticancer drugs were generally similar to that in OCUM‐2M and MKN74 cells (data not shown). The major toxicity produced by 5‐aza‐CdR is myelosuppression.( 26 , 27 ) The optimal dose‐schedule for 5‐aza‐CdR with a novel mechanism of action remains to be determined. The above four anticancer drugs are used clinically either weekly or biweekly in gastric cancers. The epigenetic alteration of mRNA by DNA methyltransferase inhibitor is reversible.( 28 ) In humans, 5‐aza‐CdR has a short half‐life of 15–25 min due to rapid inactivation by liver cytidine deaminase.( 26 ) The low‐dose prolonged exposure 5‐aza‐CdR schedule was well tolerated and delivered on an outpatient basis, and low doses are as, or more, effective than higher doses.( 29 ) The exposure of 5‐aza‐CdR for 48 h enhanced the expression of genes associated with chemosensitivity. These findings suggested that a low dose of 5‐aza‐CdR 48 h ahead might be used effectively and safely with these anticancer drugs.
In contrast, no significant effect was observed in response to a combination of 5‐aza‐CdR with 5‐FU, and an antiproliferative effect was observed in combination with GEM. Although both 5‐FU and GEM are pyrimidine antimetabolites that interfere with the synthesis of DNA, GEM further acts as an effective inhibitor of DNA repair, which was specifically demonstrated with regard to DNA damage induced by other drugs. This might be one of the reasons for the different antiproliferative effects of combining 5‐aza‐CdR and GEM or 5‐FU.
The rate of apoptosis was increased by 5‐aza‐CdR with SN38 or GEM in both OCUM‐2M and MKN‐74 cells, and with PTX and OXA in MKN‐74. A synergistic antiproliferative effect in response to a combination of 5‐aza‐CdR with SN38 or GEM was observed in both cell lines, and with PTX and OXA in MKN‐74. Apoptosis induction in response to a combination of 5‐aza‐CdR is closely associated with the antiproliferative effect. These findings suggested that one of the mechanisms responsible for the synergistic antiproliferative effect of 5‐aza‐CdR might be explained by strengthened apoptosis.
It is well known that the proapoptotic DAPK gene families can be epigenetically regulated by DNA methyltransferase inhibitors.( 30 ) In our study, DAPK‐2 and DAPK‐3 were upregulated by 5‐aza‐CdR in both gastric cancer cells, although DAPK‐1 was only increased in MKN‐74. DAPK‐1 and DAPK‐2 are responsible for the induction of apoptosis.( 31 ) Loss of DAPK expression has been documented in many cancer types as a result of aberrant methylation in promoters, and restored or enhanced expression of DAPK has been shown to increase the cell's apoptotic sensitivity.( 32 ) These findings suggest that the induction of apoptosis by the DAPK family, and especially DAPK‐2 and DAPK‐3, might participate in the synergism of cotreatment of DNA methyltransferase inhibitor with anticancer drugs.
Transcription of RASSF1 is observed to be increased through the demethylation pathway in a variety of human cancers, including gastric cancer cell lines and primary tumors.( 33 ) Acting as an effecter molecule of the Ras growth inhibition pathway, RASSF1 has been demonstrated to be involved in apoptotic signaling, microtubule stabilization, and mitotic progression.( 34 ) In our study showing that 5‐aza‐CdR increases chemosensitivity in gastric cell lines, the treatment of 5‐aza‐CdR also promoted the expression of RASSF1 in both cell lines. RASSF1 might therefore also explain the mechanism of synergism or semiaddition of 5‐aza‐CdR in apoptotic signaling.
Hypermethylation of TGFBRI has been suggested as a cause of abnormal expression and has been found to be associated with resistance to TGFβ in gastric cancer. Increased expression of TGFBR1 can be achieved by demethylation of 5‐aza‐CdR.( 35 ) In our study, 5‐aza‐CdR upregulated the transcription of TGFBR1 in MKN‐74 cells, but not in OCUM‐2M cells, indicating that TGFBR1 might play a role in the synergism of semiaddition by a combination of 5‐aza‐CdR with anticancer drugs in some, although not all, cell lines. The expression of THBS1 was increased in these two gastric cancer cell lines. The TGFβ pathway has also been reported to be involved in sensitivity to chemotherapy. THBS1 is shown to be able to convert the latent TGFβ to its active state before TGFβ binding with its receptors.( 36 ) Thus, the ability of THBS1 to activate TGFβ provides additional mechanisms for inhibition of tumor growth.( 37 , 38 )
The treatment with 5‐aza‐CdR did not change the expression of p53 in either cell line. It was reported that absence of functional p53 was critical for cells to undergo apoptosis( 39 ) however, Jackson‐Grusby et al. pointed out that demethylation could lead to p53‐mediated cell death.( 40 ) In our case, both OCUM‐2M and MKN‐74 have a wild‐type p53. Although the 5‐aza‐CdR did not affect p53 expression, apoptosis induction by 5‐aza‐dC with anticancer drugs might be dependent in part on the p53 pathway. The function of p53 in the combination of 5‐aza‐dC with anticancer drugs still needs to be examined in future study.( 41 , 42 )
In contrast, no significant change of expression was found in hMLH1, p16, MGMT, and E‐cadherin genes, but these genes have been reported to show hypermethylation in gastric cancers.( 4 , 5 , 6 ) Although it is possible that sufficient demethylation does not occur in some genes depending on dose and incubation time of 5‐aza‐CdR on cell lines, DNA demethylation might not alter the transcription of hMLH1, p16, MGMT and E‐cadherin, and the synergistic and semiadditive effect of 5‐aza‐CdR on chemotherapeutical agents is not likely to be related to these genes.
In conclusion, our data suggest that 5‐aza‐CdR can increase the chemosensitivity of PTX, OXA, SN38, and GEM in gastric cancer cells in apoptotic signaling. The synergistic or semiadditive effect of 5‐aza‐CdR might be mediated by enhanced expression of the DAPK family, RASSF1, and THBS1 genes. Our results might provide a rationale for combination chemotherapy of DNA methyltransferase inhibitors with traditional anticancer drugs in gastric cancers.
Acknowledgments
This study was partially funded by the Japan–China Sasagawa Medical Scholarship, and by a Grant‐in Aid from the Osaka City University Medical Research Foundation.
References
- 1. Jones PA, Laird PW. Cancer epigenetics comes of age. Nat Genet 1999; 21: 163–7. [DOI] [PubMed] [Google Scholar]
- 2. Harada K, Toyooka S, Maitra A et al. Aberrant promoter methylation and silencing of the RASSF1A gene in pediatric tumors and cell lines. Oncogene 2002; 21: 4345–9. [DOI] [PubMed] [Google Scholar]
- 3. Zhang Q, Rubenstein JN, Jang TL et al. Insensitivity to transforming growth factor‐beta results from promoter methylation of cognate receptors in human prostate cancer cells (LNCaP). Mol Endocrinol 2005; 19: 2390–9. [DOI] [PubMed] [Google Scholar]
- 4. Oue N, Mitani Y, Motoshita J et al. Accumulation of DNA methylation is associated with tumor stage in gastric cancer. Cancer 2006; 106: 1250–9. [DOI] [PubMed] [Google Scholar]
- 5. Schildhaus HU, Krockel I, Lippert H, Malfertheiner P, Roessner A, Schneider‐Stock R. Promoter hypermethylation of p16INK4a, E‐cadherin, O6‐MGMT, DAPK and FHIT in adenocarcinomas of the esophagus, esophagogastric junction and proximal stomach. Int J Oncol 2005; 26: 1493–500. [PubMed] [Google Scholar]
- 6. Oue N, Matsumura S, Nakayama H et al. Reduced expression of the TSP1 gene and its association with promoter hypermethylation in gastric carcinoma. Oncology 2003; 64: 423–9. [DOI] [PubMed] [Google Scholar]
- 7. Robertson KD. DNA methylation, methyltransferases, and cancer. Oncogene 2001; 20: 3139–55. [DOI] [PubMed] [Google Scholar]
- 8. Santini V, Kantarjian HM, Issa JP. Changes in DNA methylation in neoplasia: pathophysiology and therapeutic implications. Ann Intern Med 2001; 134: 573–86. [DOI] [PubMed] [Google Scholar]
- 9. Thibault A, Figg WD, Bergan RC et al. A phase II study of 5‐aza‐2′deoxycytidine (decitabine) in hormone independent metastatic (D2) prostate cancer. Tumori 1998; 84: 87–9. [DOI] [PubMed] [Google Scholar]
- 10. Schaefer HE, Lubbert M. The hematopathological basis for studying effects of the demethylating agent 5‐aza‐2′‐deoxycytidine (decitabine) in myelodysplasia. Ann Hematol 2005; 84 (Suppl. 13): 67–79. [DOI] [PubMed] [Google Scholar]
- 11. Kantarjian HM, O’Brien SM, Keating M et al. Results of decitabine therapy in the accelerated and blastic phases of chronic myelogenous leukemia. Leukemia 1997; 11: 1617–20. [DOI] [PubMed] [Google Scholar]
- 12. Momparler RL, Rivard GE, Gyger M. Clinical trial on 5‐aza‐2′‐deoxycytidine in patients with acute leukemia. Pharmacol Ther 1985; 30: 277–86. [DOI] [PubMed] [Google Scholar]
- 13. Van Groeningen CJ, Leyva A, O’Brien AM, Gall HE, Pinedo HM. Phase I and pharmacokinetic study of 5‐aza‐2′‐deoxycytidine (NSC 127716) in cancer patients. Cancer Res 1986; 46: 4831–6. [PubMed] [Google Scholar]
- 14. Abele R, Clavel M, Dodion P et al. The EORTC Early Clinical Trials Cooperative Group experience with 5‐aza‐2′‐deoxycytidine (NSC 127716) in patients with colo‐rectal, head and neck, renal carcinomas and malignant melanomas. Eur J Cancer Clin Oncol 1987; 23: 1921–4. [DOI] [PubMed] [Google Scholar]
- 15. Kanda T, Tada M, Imazeki F, Yokosuka O, Nagao K, Saisho H. 5‐aza‐2′‐deoxycytidine sensitizes hepatoma and pancreatic cancer cell lines. Oncol Rep 2005; 14: 975–9. [PubMed] [Google Scholar]
- 16. Jemal A, Thomas A, Murray T, Thun M. Cancer statistics, 2002. CA Cancer J Clin 2002; 52: 23–47. [DOI] [PubMed] [Google Scholar]
- 17. Ajani JA, Fairweather J, Pisters PW, Charnsangavej C. Irinotecan and cisplatin in advanced gastric or gastroesophageal junction carcinoma. Oncology (Williston Park) 2000; 14: 19–21. [PubMed] [Google Scholar]
- 18. Correale P, Fulfaro F, Marsili S et al. Gemcitabine (GEM) plus oxaliplatin, folinic acid, and 5‐fluorouracil (FOLFOX‐4) in patients with advanced gastric cancer. Cancer Chemother Pharmacol 2005; 56: 563–8. [DOI] [PubMed] [Google Scholar]
- 19. Louvet C, Andre T, Tigaud JM et al. Phase II study of oxaliplatin, fluorouracil, and folinic acid in locally advanced or metastatic gastric cancer patients. J Clin Oncol 2002; 20: 4543–8. [DOI] [PubMed] [Google Scholar]
- 20. Murad AM, Petroianu A, Guimaraes RC, Aragao BC, Cabral LO, Scalabrini‐Neto AO. Phase II trial of the combination of paclitaxel and 5‐fluorouracil in the treatment of advanced gastric cancer: a novel, safe, and effective regimen. Am J Clin Oncol 1999; 22: 580–6. [DOI] [PubMed] [Google Scholar]
- 21. Yashiro M, Chung YS, Nishimura S, Inoue T, Sowa M. Establishment of two new scirrhous gastric cancer cell lines: analysis of factors associated with disseminated metastasis. Br J Cancer 1995; 72: 1200–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Niigata HHJ. Establishment of cultured cell lines of human stomach cancer origin and their morphological characteristics. Exp Med 1977; 91: 737–63. [Google Scholar]
- 23. Drewinko B, Loo TL, Brown B, Gottlieb JA, Freireich EJ. Combination chemotherapy in vitro with adriamycin. Observations of additive, antagonistic, and synergistic effects when used in two‐drug combinations on cultured human lymphoma cells. Cancer Biochem Biophys 1976; 1: 187–95. [PubMed] [Google Scholar]
- 24. Timmermann S, Hinds PW, Munger K. Re‐expression of endogenous p16ink4a in oral squamous cell carcinoma lines by 5‐aza‐2′‐deoxycytidine treatment induces a senescence‐like state. Oncogene 1998; 17: 3445–53. [DOI] [PubMed] [Google Scholar]
- 25. Yang QW, Liu S, Tian Y et al. Methylation‐associated silencing of the thrombospondin‐1 gene in human neuroblastoma. Cancer Res 2003; 63: 6299–310. [PubMed] [Google Scholar]
- 26. Momparler RL. Pharmacology of 5‐Aza‐2′‐deoxycytidine (decitabine). Semin Hematol 2005; 42: S9–16. [DOI] [PubMed] [Google Scholar]
- 27. Issa JP, Gharibyan V, Cortes J et al. Phase II study of low‐dose decitabine in patients with chronic myelogenous leukemia resistant to imatinib mesylate. J Clin Oncol 2005; 23: 3948–56. [DOI] [PubMed] [Google Scholar]
- 28. Samlowski WE, Leachman SA, Wade M et al. Evaluation of a 7‐day continuous intravenous infusion of decitabine: inhibition of promoter‐specific and global genomic DNA methylation. J Clin Oncol 2005; 23: 3897–905. [DOI] [PubMed] [Google Scholar]
- 29. Issa JP, Garcia‐Manero G, Giles FJ et al. Phase 1 study of low‐dose prolonged exposure schedules of the hypomethylating agent 5‐aza‐2′‐deoxycytidine (decitabine) in hematopoietic malignancies. Blood 2004; 103: 1635–40. [DOI] [PubMed] [Google Scholar]
- 30. Zhang S, Kong WJ, Wang YJ, Han YC, Zhang D. [Inhibitory effect of 5‐Aza‐2′‐deoxycytidine on human nasopharyngeal carcinoma xenograft in nude mice]. Ai Zheng 2005; 24: 1201–5. [PubMed] [Google Scholar]
- 31. Kogel D, Prehn JH, Scheidtmann KH. The DAP kinase family of pro‐apoptotic proteins: novel players in the apoptotic game. Bioessays 2001; 23: 352–8. [DOI] [PubMed] [Google Scholar]
- 32. Katzenellenbogen RA, Baylin SB, Herman JG. Hypermethylation of the DAP‐kinase CpG island is a common alteration in B‐cell malignancies. Blood 1999; 93: 4347–53. [PubMed] [Google Scholar]
- 33. Byun DS, Lee MG, Chae KS, Ryu BG, Chi SG. Frequent epigenetic inactivation of RASSF1A by aberrant promoter hypermethylation in human gastric adenocarcinoma. Cancer Res 2001; 61: 7034–8. [PubMed] [Google Scholar]
- 34. Agathanggelou A, Cooper WN, Latif F. Role of the Ras association domain family 1 tumor suppressor gene in human cancers. Cancer Res 2005; 65: 3497–508. [DOI] [PubMed] [Google Scholar]
- 35. Kang SH, Bang YJ, Im YH et al. Transcriptional repression of the transforming growth factor‐beta type I receptor gene by DNA methylation results in the development of TGF‐beta resistance in human gastric cancer. Oncogene 1999; 18: 7280–6. [DOI] [PubMed] [Google Scholar]
- 36. Yung S, Lee CY, Zhang Q, Lau SK, Tsang RC, Chan TM. Elevated glucose induction of thrombospondin‐1 up‐regulates fibronectin synthesis in proximal renal tubular epithelial cells through TGF‐{beta}1 dependent and TGF‐{beta}1 independent pathways. Nephrol Dial Transplant 2006. (in press). [DOI] [PubMed]
- 37. Murphy‐Ullrich JE, Poczatek M. Activation of latent TGF‐beta by thrombospondin‐1: mechanisms and physiology. Cytokine Growth Factor Rev 2000; 11: 59–69. [DOI] [PubMed] [Google Scholar]
- 38. Yee KO, Streit M, Hawighorst T, Detmar M, Lawler J. Expression of the type‐1 repeats of thrombospondin‐1 inhibits tumor growth through activation of transforming growth factor‐beta. Am J Pathol 2004; 165: 541–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nieto M, Samper E, Fraga MF, Gonzalez de Buitrago G, Esteller M, Serrano M. The absence of p53 is critical for the induction of apoptosis by 5‐aza‐2′‐deoxycytidine. Oncogene 2004; 23: 735–43. [DOI] [PubMed] [Google Scholar]
- 40. Jackson‐Grusby L, Beard C, Possemato R et al. Loss of genomic methylation causes p53‐dependent apoptosis and epigenetic deregulation. Nat Genet 2001; 27: 31–9. [DOI] [PubMed] [Google Scholar]
- 41. Yamamoto M, Maehara Y, Oda S, Ichiyoshi Y, Kusumoto T, Sugimachi K. The p53 tumor suppressor gene in anticancer agent‐induced apoptosis and chemosensitivity of human gastrointestinal cancer cell lines. Cancer Chemother Pharmacol 1999; 43: 43–9. [DOI] [PubMed] [Google Scholar]
- 42. Yukimoto K, Nakata B, Muguruma K et al. Apoptosis and thymidylate synthase inductions by 5‐fluorouracil in gastric cancer cells with or without p53 mutation. Int J Oncol 2001; 19: 373–8. [DOI] [PubMed] [Google Scholar]