

Abstract
Invadopodia are ventral cell protrusions formed in invasive cancer cells. Because invadopodia have extracellular matrix (ECM) degradation activity, they are thought to function in cancer invasion. In this study, we examined the roles of phosphatidylinositol 4,5‐bisphosphate [PI(4,5)P2] and PI(4,5)P2‐producing enzymes in invadopodia formation in MDA‐MB‐231 human breast cancer cells. Immunofluorescence analysis showed that PI(4,5)P2 accumulates at invadopodia on the ventral cell surface. Injection of an anti‐PI(4,5)P2 antibody inhibited invadopodia formation along with gelatin degradation activity. Sequestering of PI(4,5)P2 by overexpression of the phospholipase C (PLC) δ1‐pleckstrin homology (PH) domain, a specific probe for PI(4,5)P2, also blocked invadopodia formation, while a mutated PLCδ1‐PH domain that lacks PI(4,5)P2‐binding activity had no effect. Cellular PI(4,5)P2 production is mainly mediated by type‐I phosphatidylinositol 4‐phosphate 5‐kinase (PIP5KI) family proteins, which include PIP5KIα, Iβ, and Iγ. Real‐time quantitative PCR analysis showed that PIP5KIα is a dominant isoform expressed in MDA‐MB‐231 cells. Knockdown of PIP5KIα by small‐interfering RNA (siRNA) inhibited invadopodia formation and gelatin degradation. Immunofluorescence analysis revealed that endogenous PIP5KIα protein localizes at invadopodia, which is corroborated by the observation that exogenously expressed green fluorescent protein (GFP)‐fused PIP5KIα protein also accumulates at gelatin degradation sites. These results indicate that localized production of PI(4,5)P2 by PIP5KIα is required for invadopodia formation and ECM degradation by human breast cancer cells. (Cancer Sci 2010)
Invadopodia are extracellular matrix (ECM)‐degrading protrusions formed at the ventral surface of invasive cancer cells.( 1 , 2 ) In the case of breast cancer cell lines, the ability to form invadopodia is closely related to their invasive and metastatic properties.( 3 , 4 ) Additionally, during intravasation, invadopodia‐like protrusions in tumor cells have been observed in vivo by intravital imaging.( 5 , 6 ) Therefore, invadopodia are proposed to function in local ECM degradation during cancer invasion and metastasis, although the in vivo relevance of invadopodia has yet to be determined. To date, many components of invadopodia have been reported, including proteins involved in the regulation of the actin cytoskeleton, cell signaling, cell–ECM adhesion, ECM degradation, and membrane remodeling.( 7 , 8 , 9 ) We and other researchers previously proposed that invadopodia formation occurs in several steps. Invadopodia precursors are assembled by an actin polymerization machinery containing neural Wiskott–Aldrich syndrome protein (N‐WASP), the Arp2/3 complex, and cortactin in response to extracellular stimuli.( 3 , 10 ) The invadopodia precursors are then stabilized by further actin polymerization induced by cofilin and Mena, and finally these structures gather matrix metalloproteinases to mature into functional invadopodia.( 3 , 11 , 12 ) However, it is unknown how these events occur at the restricted sites on the plasma membrane of invasive cancer cells.
The membrane phospholipid phosphatidylinositol 4,5‐bisphosphate [PI(4,5)P2] regulates diverse cellular processes, including signal transduction, endocytosis/phagocytosis, vesicular trafficking, and actin cytoskeletal reorganization.( 13 ) PI(4,5)P2 acts as a substrate for phospholipase C (PLC) and phosphatidylinositol 3‐kinase (PI3K), while it also directly regulates the activity and localization of target proteins that have a PI(4,5)P2 binding domain.( 14 ) PI(4,5)P2 production in mammalian cells is primarily mediated by type I phosphatidylinositol 4‐phosphate 5‐kinase (PIP5KI), which phosphorylates phosphatidylinositol 4‐phosphate at the D‐5 position of the inositol ring.( 15 ) Three isoforms of PIP5KI (PIP5KIα, Iβ, and Iγ in human nomenclature) and two alternatively spliced isoforms of PIP5KIγ (Iγ635 and Iγ661) have been identified in mammalian cells.( 16 ) Because the subcellular localization of PIP5KIs is unique to each isoform, they seem to have distinct cellular functions.( 17 , 18 )
Previous reports have identified that the localization and activity of several invadopodia components, such as N‐WASP, cofilin, and dynamin‐2, are regulated by PI(4,5)P2 ( 19 , 20 ) Additionally, Arf6, an essential regulator of invadopodia formation,( 21 , 22 ) is known to activate PIP5KIs and, therefore, stimulate PI(4,5)P2 synthesis.( 23 ) However, the importance of PI(4,5)P2 and PIP5KIs in invadopodia formation is yet to be determined.
Here, we studied the roles of PI(4,5)P2 and PIP5KIs in invadopodia formation. We found that PI(4,5)P2 and PIP5KIα are essential regulators of invadopodia‐mediated ECM degradation by human breast cancer cells.
Materials and Methods
Cell culture. MDA‐MB‐231 human breast cancer cell line was obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured as described previously.( 4 )
Antibodies, reagents, and constructs. Alexa dyes, fluorescence‐labeled phalloidin, and secondary antibodies were purchased from Invitrogen (Carlsbad, CA, USA). Gelatin and other chemicals were purchased from Sigma (St. Louis, MO, USA). Anti‐PI(4,5)P2 antibody has been described previously.( 24 ) Anti‐PIP5KIα and anti‐β‐actin antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and Millipore (Billerica, MA, USA), respectively. Anti‐Akt and anti‐phospho‐Akt (Ser473) antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Green fluorescent protein (GFP)‐PIP5KIα constructs were kindly provided by Dr Kanaho (Tsukuba University, Ibaraki, Japan). For GFP‐PLCδ1‐pleckstrin homology (PH) domain constructs, cDNA encoding the mouse PLCδ1‐PH domain (amino acid 1–140) was subcloned into pEGFP‐C1 vector (Clontech, Mountain View, CA, USA). A PrimeSTAR Mutagenesis Basal Kit (Takara Bio, Shiga, Japan) was used to generate the R40D mutant of the PLCδ1‐PH domain.
Plasmid transfection. Cells were transfected with the indicated plasmids using Lipofectamine LTX (Invitrogen) according to the manufacturer’s instructions.
RNA interference. Stealth RNAi (Invitrogen) molecules were used for RNA interference (RNAi) experiments: Negative Control Medium GC Duplex #2, Stealth Select RNAi for PIP5KIα (HSS144742 and HSS144743) and for PIP5KIγ (HSS118703). Cells were transfected with 30 nm small‐interfering RNA (siRNA) using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s instructions. The cells were cultured for 48–72 h and subjected to invadopodia assay, reverse transcriptase–polymerase chain reaction (RT‐PCR) analysis, or immunoblotting.
RT‐PCR. Standard PCR amplification was performed as described previously.( 4 ) The sequences of primer pairs used were as follows: human cyclophilin B2 (5′‐GCACAGGAGGAAAGAGCATC‐3′ and 5′‐CTTCTCCACCTCGATCTTGC‐3′), human PIPK Iα (5′‐TGCCCATCAAGAAAATAGGC‐3′ and 5′‐GAAGTAGCGGAAGGCAACAG‐3′), human PIPK Iβ (5′‐AGACACAATGGGAGGCATTC‐3′ and 5′‐TCTCCTGTGAAGTGGCCTTT‐3′), and human PIPK Iγ (5′‐CAGGACTTCCGCTTCAAGAC‐3′ and 5′‐CACGCAGTACAGCCCATA‐GA‐3′). Quantitative RT‐PCR was performed with Thunderbird qPCR Mix (Toyobo, Osaka, Japan) in a CFX96 Real‐Time PCR Detection System (Bio‐Rad Laboratories, Hercules, CA, USA). The primer pairs used were as follows: human cyclophilin B2 (5′‐TTCTAGAGGGCATGGAGGTG‐3′ and 5′‐GCTTCTCCA‐CCTCGATCTTG‐3′), human PIPK Iα (5′‐CGGTCTGGCTC‐ATCTTTCTC‐3′ and 5′‐AAACATCAGGACGACCAAGG‐3′), human PIPK Iβ (5′‐AAGGATGAGAAGCGGGATTT‐3′ and 5′‐AATTGTGGTTGCCAAGGAAG‐3′), and human PIPK Iγ (5′‐AGAAGCCACTACAGCCTCCA‐3′ and 5′‐CCTGTCCA‐GACGACTGTGTG‐3′). These primer pairs have almost the same amplification efficiencies. Human mosaic cDNA template (Toyobo) was used as a positive control.
Epidermal growth factor (EGF) stimulation and immuno‐blotting. Cells were serum‐starved overnight in 0.35% bovine serum albumin/medium and stimulated with 50 ng/mL EGF for 10 min at 37°C. The cells were then lysed in lysis buffer containing 50 mm Tris–HCl, pH 7.5, 1% NP‐40, 2 mm EDTA, 100 mm NaCl, 1 mm sodium orthovanadate, and a protease inhibitor cocktail (Roche, Basel, Switzerland). The lysates were subjected to immunoblot analysis as described previously.( 3 ) To quantify the phospho‐Akt amount, densitometry was performed on scanned immunoblot images using the ImageJ 1.410 (National Institutes of Health, Bethesda, Maryland, USA, http://rsbwep.nih.gov/ij/) gel analysis tool.
Immunofluorescence. Cells were stained with indicated antibodies and phalloidins as described previously.( 3 ) PI(4,5)P2 staining was also performed as described.( 25 ) Samples were observed with an Olympus IX81‐ZDC‐DSU confocal microscope (Olympus, Tokyo, Japan) equipped with a cooled charged‐coupled device (CCD) camera (ORCA‐ER; Hamamatsu, Shizuoka, Japan). Images were analyzed and processed with Metamorph (Universal Imaging, Buckinghamshire, UK), ImageJ 1.41o, and Adobe Photoshop software.
Invadopodia assay. Fluorescent gelatin‐coated dishes were prepared as described previously.( 26 ) In brief, gelatin was labeled with TRITC in borate buffer (40 mm NaCl, 50 mm Na2B4O7, pH 9.3) and extensively dialyzed against phosphate‐buffered saline (PBS). Coverslips (12 mm, circular) were coated with 25 mg/mL fluorescent gelatin and 20 mg/mL sucrose in PBS, and then crosslinked with 0.5% glutaraldehyde on ice for 15 min followed by 30 min at room temperature. After extensive washing with PBS, the coverslips were treated with 5 mg/mL sodium borohydrate for 3 min to quench autofluorescence of residual glutaraldehyde. The coverslips were then sterilized with 70% ethanol for 15 min and quenched in growth medium for 1 h before plating cells. Cells were cultured on the gelatin matrix for 3–7 h and then stained with the appropriate antibody and phalloidin to visualize invadopodia and gelatin degradation sites. To quantitate the degradation activity of invadopodia, the degradation area was calculated from images using ImageJ 1.41o software and normalized for the number of cells. In the cases of antibody microinjection and GFP‐PLCδ1 PH domain overexpression, the average values of control cells were set to 100% and relative degradation activities were shown. Data were obtained from three independent experiments. For siRNA knockdown experiments, at least 40 transfected cells were analyzed for each sample and the values of control cells were set to 100% in each experiment. Data shown are mean ± SE of four independent experiments.
Antibody microinjection. Cells were plated onto fluorescent gelatin, cultured for 2 h, and then microinjected with anti‐PI(4,5)P2 antibody or control mouse immunoglobulin G (IgG) using a microinjection system (Eppendorf, Hamburg, Germany). The cells were further cultured for 3 h to allow them to form invadopodia and degrade the gelatin matrix. Cells were then fixed and stained with Alexa Fluor 488‐conjugated antimouse IgG antibody and Alexa Fluor 633‐phalloidin to detect microinjected cells and invadopodia, respectively.
Quantification of the cellular PI(4,5)P2 amount. A PI(4,5)P2 Mass ELISA Kit (Echelon Biosciences, Salt Lake City, UT, USA) was used to determine the amount of cellular PI(4,5)P2 according to the manufacturer’s instructions.
Results
PI(4,5)P2 accumulates at invadopodia formed in invasive human breast cancer cells. Localization of PI(4,5)P2 in invadopodia was first determined by immunocytochemistry. MDA‐MB‐231 is a highly invasive human breast cancer cell line with a basal‐like phenotype, the most aggressive form of breast cancers.( 27 ) MDA‐MB‐231 cells were plated onto fluorescence‐labeled gelatin and then stained with an anti‐PI(4,5)P2 antibody, whose specificity was validated previously.( 24 ) MDA‐MB‐231 cells have been shown to form invadopodia, which are observed as dot‐like accumulations of actin filaments (F‐actin) associated with black areas formed by the degradation of fluorescent gelatin matrix. The PI(4,5)P2 signals were mainly observed at the plasma membrane (Fig. 1, left). On the ventral surface of the cells, the PI(4,5)P2 signals were observed as small vesicles and some of them were localized in close proximity to the degradation sites of gelatin formed by invadopodia (Fig. 1, right). The vesicular signals often surrounded the F‐actin structures of invadopodia (Fig. 1, right). These observations indicate that PI(4,5)P2 accumulates at invadopodia and associated gelatin degradation sites.
Figure 1.

PI(4,5)P2 accumulates at invadopodia formed in human breast cancer cells. (a) MDA‐MB‐231 cells were cultured on coverslips coated with fluorescent gelatin (red) and stained with anti‐PI(4,5)P2 antibody (green) and phalloidin (blue) as described in the Materials and Methods. Right panels are magnified images of the boxed region. Arrowheads denote accumulation of PI(4,5)P2 signals at invadopodia.
PI(4,5)P2 is required for invadopodia formation. The role of PI(4,5)P2 in invadopodia formation was then examined. Microinjection of an anti‐PI(4,5)P2 antibody was previously shown to inhibit the cellular functions of PI(4,5)P2.( 24 ) MDA‐MB‐231 cells were plated onto fluorescent gelatin and microinjected with control IgG or anti‐PI(4,5)P2 antibody. The cells were further cultured to allow them to form invadopodia and degrade the gelatin matrix. Cells injected with anti‐PI(4,5)P2 antibody showed a decreased ability to form invadopodia and degrade the gelatin matrix compared to the uninjected and control IgG‐injected cells (Fig. 2a,b). It was also observed that microinjection of anti‐PI(4,5)P2 antibody tend to increase cortical F‐actin (Fig. 2a). However, some of the cells injected with anti‐PI(4,5)P2 antibody showed impaired invadopodia formation without such changes in the actin cytoskeleton, excluding the possibility that the loss of invadopodia is secondarily caused by aberrant accumulation of F‐actin. The role of PI(4,5)P2 in invadopodia formation was also determined using a PLCδ1‐PH domain fused to GFP (GFP‐PLCδ1‐PH). GFP‐PLCδ1‐PH specifically binds to PI(4,5)P2 and therefore sequesters PI(4,5)P2 upon overexpression.( 28 ) Compared to control GFP‐transfected cells, invadopodia formation and gelatin degradation were significantly impaired in cells overexpressing GFP‐PLCδ1‐PH (Fig. 2c,d). This effect was not observed when cells were transfected with the R40D mutant of GFP‐PLCδ1‐PH, which possesses a point mutation in an amino acid essential for PI(4,5)P2 binding( 29 ) (Fig. 2c,d). Although the GFP‐PLCδ1‐PH signals were almost exclusively observed at the plasma membrane, signals in the R40D mutant were mainly detected in the cytosol and the nucleus similar to the control GFP signals (Fig. 2c). These results demonstrate that PI(4,5)P2 function is necessary for invadopodia formation and ECM degradation.
Figure 2.

PI(4,5)P2 is necessary for invadopodia formation. (a) MDA‐MB‐231 cells were plated onto fluorescent gelatin‐coated coverslips for 2 h and microinjected with mouse anti‐PI(4,5)P2 antibody. Cells were further cultured for 3 h to allow them to form invadopodia and then stained with phalloidin and antimouse IgG secondary antibody to detect injected cells. The arrowhead denotes the gelatin degradation sites. (b) Quantification of invadopodia activity. The degradation area of the gelatin matrix was calculated as described in the Materials and Methods and shown as a percentage of uninjected cells. Data are mean ± SEM (n = 95, 47, and 100 cells for uninjected, control (Ctrl) IgG, and anti‐PI(4,5)P2 antibody injected cells, respectively). *P < 0.04, Student’s t‐test. (c) MDA‐MB‐231 cells transfected with GFP or GFP‐PLCδ1‐PH constructs (WT and R40D) were cultured on fluorescent gelatin‐coated coverslips. Representative microscopy images of the cells are shown. The arrowhead denotes the gelatin degradation sites. (d) The degradation area of the gelatin was quantified among cells transfected with the indicated constructs. Data are mean ± SEM (n = 51, 51, and 48 cells for control, WT, and R40D, respectively). **P < 0.02, Student’s t‐test.
PIP5KIα is an essential regulator of invadopodia formation. PI(4,5)P2 in mammalian cells is mainly produced by the PIP5KI family, which consists of three isoforms, PIP5KIα, Iβ, and Iγ. We next determined whether the PIP5KI family is involved in invadopodia formation. The expression of the PIP5KI family in MDA‐MB‐231 cells was examined by real‐time quantitative PCR analysis. PIP5KIα was abundantly expressed in these cells, while the expression level of PIP5KIγ was only about 7% of that of Iα (Fig. 3a). The expression of Iβ was undetectable in this analysis (Fig. 3a), although the primer set used could efficiently detect Iβ expression when human cDNA mixture was used as a template (data not shown). These data indicated that PIP5KIα is the most abundantly expressed isoform in MDA‐MB‐231 cells.
Figure 3.

PIP5KIα is an essential regulator of invadopodia formation. (a) Expression of PIP5KIs in MDA‐MB‐231 cells was analyzed by real‐time quantitative PCR. Relative expression of PIP5KIs normalized by cyclophilin B expression is shown. Data are mean ± SEM of three independent experiments. (b) MDA‐MB‐231 cells were transfected with non‐silencing control (Ctrl), PIP5KIα, or Iγ siRNA, and subjected to RT‐PCR (left) and immunoblot (right) analyses. Cyclophilin B (Cycl) and actin were used as controls. (c) Cells transfected with siRNAs were cultured on fluorescent gelatin‐coated coverslips and stained with phalloidin to visualize invadopodia. (d) The degradation area of the gelatin was quantified among cells transfected with the indicated siRNAs. Data are mean ± SEM of four independent determinations. In each determination, at least 40 cells were analyzed. *P < 0.02 and **P < 0.0003, Student’s t‐test.
In order to examine the roles of PIP5KIα and Iγ in invadopodia formation, cells were transfected with siRNAs targeting these enzymes. Efficient knockdown of each isoform was confirmed by RT‐PCR analysis (Fig. 3b, left), and immunoblotting in the case of PIP5KIα (Fig. 3b, right). Cells with a reduced level of PIP5KIα showed a marked decrease in invadopodia formation and gelatin degradation activity (Fig. 3c,d). Similar results were obtained with another siRNA targeting a different region of the PIP5KIα gene (Fig. 3b,d). In contrast, such effects were not observed in cells transfected with PIP5KIγ siRNA (Fig. 3c,d), which should target both Iγ635 and Iγ661. Additionally, PIP5KIβ siRNA also had no effect on invadopodia formation (data not shown). These results indicate that PIP5KIα, among the PIP5KI family members, is specifically involved in invadopodia formation.
PIP5KIα knockdown does not affect PI3K signaling. PI3K activities are necessary for invadopodia formation in human melanoma cells.( 30 ) Because PI(4,5)P2 serves as a substrate of PI3K for generating PI(3,4,5)P3, it is possible that suppression of invadopodia formation by PIP5KIα knockdown is due to the inhibition of PI3K signaling. To test this possibility, the effects of PIP5KIα knockdown on the activation of PI3K signaling were examined. EGF stimulation is known to activate PI3K signaling, which results in the phosphorylation and activation of the downstream target Akt. As expected, EGF stimulation markedly increased the amount of phospho‐Akt in control cells (Fig. 4a,b). The level of phospho‐Akt in PIP5KIα knockdown cells was comparable to that in control cells (Fig. 4a,b). Total cellular content of PI(4,5)P2 in PIP5KIα knockdown cells was also examined using an enzyme‐linked immunosorbent assay (ELISA)‐based method. This analysis showed that the total amount of PI(4,5)P2 in PIP5KIα knockdown cells decreased to 61% of that in control cells (Fig. 4c). Because the PI(3,4,5)P3 level is considerably lower than that of PI(4,5)P2,( 31 ) the reduction of PI(4,5)P2 by PIP5KIα knockdown may have minimal effect on PI3K signaling.
Figure 4.

PIP5KIα knockdown does not affect PI3K signaling. (a) MDA‐MB‐231 cells were transfected with non‐silencing control (Ctrl) or PIP5KIα siRNA. The cells were serum‐starved and stimulated with 50 ng/mL of EGF for 10 min. Cell lysates were then prepared and subjected to immunoblotting with antibodies against total and phospho‐Akt. (b) The relative amount of phospho‐Akt was calculated from the immunoblots. Data are mean ± SEM of four independent experiments. (c) Relative amount of cellular PI(4,5)P2 was determined as described in the Materials and Methods. Data are mean ± SEM of four independent determinations. *P < 0.002, Student’s t‐test.
PIP5KIα localizes at invadopodia. Localization of endogenous PIP5KIα at invadopodia was examined by immunocytochemistry. MDA‐MB‐231 cells were cultured on gelatin matrix and stained with anti‐PIP5KIα antibody and phalloidin. PIP5KIα mainly localized at the plasma membrane (Fig. 5a). At the ventral membrane of the cells, the PIP5KIα signals accumulated at some invadopodia, but not all (Fig. 5a). To confirm the localization of PIP5KIα at invadopodia, cells were transfected with a GFP‐PIP5KIα construct. The signals for GFP‐PIP5KIα also accumulated at the gelatin degradation sites on the ventral cell membrane (Fig. 5b). These observations indicate that PIP5KIα localizes and functions at invadopodia.
Figure 5.
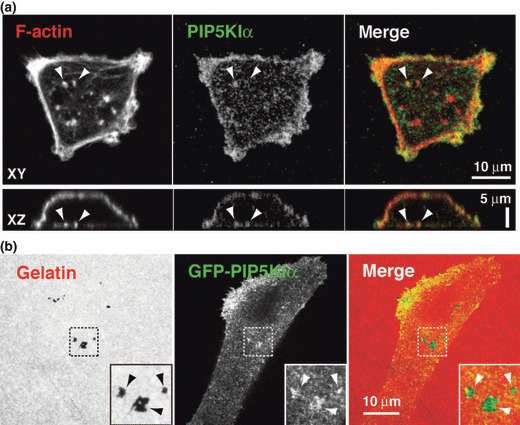
PIP5KIα localizes at invadopodia. (a) MDA‐MB‐231 cells plated onto gelatin‐coated coverslips were stained with anti‐PIP5KIα antibody and phalloidin. Upper and lower images were XY and XZ sections obtained by confocal microscopy. Arrowheads denote invadopodia associated with PIP5KIα signals. (b) Cells transfected with the GFP‐PIP5KIα construct were cultured on fluorescent gelatin and observed by confocal microscopy. Inserts are magnified images of the boxed regions. Arrowheads denote accumulation of GFP‐PIP5KIα signals at the sites of gelatin degradation.
Discussion
Here, we showed that PI(4,5)P2 accumulates at invadopodia and that inhibition of PI(4,5)P2 function by antibody microinjection or overexpression of PLCδ1‐PH, a PI(4,5)P2‐binding domain, blocks invadopodia formation and ECM degradation by human breast cancer cells. Consistent with these results, a gelsolin peptide, which binds to and sequesters PI(4,5)P2 in a similar manner to PLCδ1‐PH, introduced into osteoclasts inhibits the formation of podosomes, structures closely related to invadopodia.( 32 ) Moreover, Ziel et al. ( 33 ) recently reported that anchor cells form F‐actin and PI(4,5)P2‐rich invasive protrusions of the ventral plasma membrane to invade the basement membrane in C. elegans. Although the functional relationship between these structures and invadopodia needs further investigation, PI(4,5)P2 may have a conserved function in the formation of invasive membrane structures in a broad range of cell types.
The staining of PI(4,5)P2 were observed at vesicular structures surrounding invadopodia. These structures morphologically resemble the internalized lipid raft membranes that accumulate and traffic around invadopodia.( 4 ) As disruption of lipid rafts inhibits invadopodia formation, these vesicular structures likely have an important role, such as trafficking of invadopodia components, in invadopodia biogenesis.( 4 ) Previous studies have shown that PI(4,5)P2 is enriched in lipid raft fractions, and promotes recruitment and activation of specific signaling components.( 34 ) Therefore, PI(4,5)P2 may regulate transport and activation of invadopodia components in these lipid raft‐enriched vesicular structures.
PIP5KIs generate PI(4,5)P2 and have been implicated in several cellular processes that require dynamic actin cytoskeleton, such as cell migration, endocytosis, phagocytosis, and neurite remodeling.( 35 ) In this study, we found that PIP5KIα is an essential regulator of invadopodia formation and localizes at invadopodia; however, it is worth noting that PIP5KIα did not localize at all invadopodia. This observation suggests that PIP5KIα is not a structural component of invadopodia but rather has regulatory functions in invadopodia formation. It is possible that PIP5KIα is transiently recruited to invadopodia at specific stages of invadopodia formation.
Interestingly, PIP5KIα knockdown did not completely deplete cellular PI(4,5)P2 and had little effect on the PI3K signaling pathway. This suggests that a pool of PI(4,5)P2 locally produced by PIP5KIα directly regulates invadopodia formation rather than acting as a precursor for PI(3,4,5)P3 formation. Because PI(4,5)P2 directly binds to several components of invadopodia, such as N‐WASP, cofilin, and dynamin‐2,( 19 , 20 ) it may control localization and/or activation of these proteins at invadopodia assembly sites.
Synaptojanin‐2, which dephosphorylates PI(4,5)P2 at the D‐5 position on the inositol ring, localizes at invadopodia.( 36 ) Given that PIP5KIα and synaptojanin‐2 both localize and function at invadopodia, PI(4,5)P2 turnover may play an important role in invadopodia formation. Supporting this idea, overexpression of PIP5KIα in MDA‐MB‐231 cells, which should interfere with the PI(4,5)P2 turnover, inhibited invadopodia formation (data not shown). In the endocytic process, local PI(4,5)P2 turnover mediated by PIP5KI and synaptojanin is necessary for proper formation and internalization of clathrin‐coated pits.( 37 ) Therefore, it would be important to determine if similar PI(4,5)P2 turnover occurs at invadopodia.
Arf6 is a small GTPase that has been implicated in the regulation of the actin cytoskeleton and membrane trafficking. These functions of Arf6 are at least partly mediated by the direct activation of PIP5KIs.( 23 , 38 ) Recently, Arf6 has been reported to function in invadopodia‐mediated cancer cell invasion.( 21 , 22 ) Therefore, PIP5KIα may act as a downstream target of Arf6 for invadopodia formation. Further studies are needed to elucidate this possibility.
In conclusion, our results provide evidence that PI(4,5)P2 and PIP5KIα are essential regulators of invadopodia formation. These findings identify a novel role for PI(4,5)P2 in cancer cell invasion and should provide new insights into the molecular mechanisms of invadopodia formation. This will contribute to the development of new therapeutic strategies for cancer invasion and metastasis.
Acknowledgments
We are grateful to Dr Yasunori Kanaho for kindly providing plasmids. We thank Yumiko Konko, Keiko Takayama, Yukiko Takeo, and Juri Yamada for their technical assistance. This work was supported by Grants‐in‐Aid for Scientific Research (B), for Young Scientists (B), and for Cancer Research by the Ministry of Education, Culture, Sports, Science and Technology of Japan, and in part by a Grant‐in‐Aid from the Ministry of Health, Labour and Welfare of Japan for the 3rd‐term Comprehensive 10‐year Strategy for Cancer Control. This work was also supported in part by the Mochida Memorial Foundation for Medical and Pharmaceutical Research, ONO Medical Research Foundation, and Takeda Science Foundation.
References
- 1. Buccione R, Caldieri G, Ayala I. Invadopodia: specialized tumor cell structures for the focal degradation of the extracellular matrix. Cancer Metastasis Rev 2009; 28: 137–49. [DOI] [PubMed] [Google Scholar]
- 2. Weaver AM. Invadopodia: specialized cell structures for cancer invasion. Clin Exp Metastasis 2006; 23: 97–105. [DOI] [PubMed] [Google Scholar]
- 3. Yamaguchi H, Lorenz M, Kempiak S et al. Molecular mechanisms of invadopodium formation: the role of the N‐WASP‐Arp2/3 complex pathway and cofilin. J Cell Biol 2005; 168: 441–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yamaguchi H, Takeo Y, Yoshida S, Kouchi Z, Nakamura Y, Fukami K. Lipid rafts and caveolin‐1 are required for invadopodia formation and extracellular matrix degradation by human breast cancer cells. Cancer Res 2009; 69: 8594–602. [DOI] [PubMed] [Google Scholar]
- 5. Sidani M, Wyckoff J, Xue C, Segall JE, Condeelis J. Probing the microenvironment of mammary tumors using multiphoton microscopy. J Mammary Gland Biol Neoplasia 2006; 11: 151–63. [DOI] [PubMed] [Google Scholar]
- 6. Yamaguchi H, Wyckoff J, Condeelis J. Cell migration in tumors. Curr Opin Cell Biol 2005; 17: 559–64. [DOI] [PubMed] [Google Scholar]
- 7. Caldieri G, Buccione R. Aiming for invadopodia: organizing polarized delivery at sites of invasion. Trends Cell Biol 2010; 20: 64–70. [DOI] [PubMed] [Google Scholar]
- 8. Gimona M, Buccione R, Courtneidge SA, Linder S. Assembly and biological role of podosomes and invadopodia. Curr Opin Cell Biol 2008; 20: 235–41. [DOI] [PubMed] [Google Scholar]
- 9. Linder S. The matrix corroded: podosomes and invadopodia in extracellular matrix degradation. Trends Cell Biol 2007; 17: 107–17. [DOI] [PubMed] [Google Scholar]
- 10. Oser M, Yamaguchi H, Mader CC et al. Cortactin regulates cofilin and N‐WASp activities to control the stages of invadopodium assembly and maturation. J Cell Biol 2009; 186: 571–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Artym VV, Zhang Y, Seillier‐Moiseiwitsch F, Yamada KM, Mueller SC. Dynamic interactions of cortactin and membrane type 1 matrix metalloproteinase at invadopodia: defining the stages of invadopodia formation and function. Cancer Res 2006; 66: 3034–43. [DOI] [PubMed] [Google Scholar]
- 12. Philippar U, Roussos ET, Oser M et al. A Mena invasion isoform potentiates EGF‐induced carcinoma cell invasion and metastasis. Dev Cell 2008; 15: 813–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Di Paolo G, DeCamilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature 2006; 443: 651–7. [DOI] [PubMed] [Google Scholar]
- 14. Lemmon MA. Membrane recognition by phospholipid‐binding domains. Nat Rev Mol Cell Biol 2008; 9: 99–111. [DOI] [PubMed] [Google Scholar]
- 15. Fruman DA, Meyers RE, Cantley LC. Phosphoinositide kinases. Annu Rev Biochem 1998; 67: 481–507. [DOI] [PubMed] [Google Scholar]
- 16. Anderson RA, Boronenkov IV, Doughman SD, Kunz J, Loijens JC. Phosphatidylinositol phosphate kinases, a multifaceted family of signaling enzymes. J Biol Chem 1999; 274: 9907–10. [DOI] [PubMed] [Google Scholar]
- 17. Balla A, Balla T. Phosphatidylinositol 4‐kinases: old enzymes with emerging functions. Trends Cell Biol 2006; 16: 351–61. [DOI] [PubMed] [Google Scholar]
- 18. Doughman RL, Firestone AJ, Anderson RA. Phosphatidylinositol phosphate kinases put PI4,5P(2) in its place. J Membr Biol 2003; 194: 77–89. [DOI] [PubMed] [Google Scholar]
- 19. Ling K, Schill NJ, Wagoner MP, Sun Y, Anderson RA. Movin’ on up: the role of PtdIns(4,5)P(2) in cell migration. Trends Cell Biol 2006; 16: 276–84. [DOI] [PubMed] [Google Scholar]
- 20. Takenawa T, Itoh T. Phosphoinositides, key molecules for regulation of actin cytoskeletal organization and membrane traffic from the plasma membrane. Biochim Biophys Acta 2001; 1533: 190–206. [DOI] [PubMed] [Google Scholar]
- 21. Hashimoto S, Onodera Y, Hashimoto A et al. Requirement for Arf6 in breast cancer invasive activities. Proc Natl Acad Sci U S A 2004; 101: 6647–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tague SE, Muralidharan V, D’Souza‐Schorey C. ADP‐ribosylation factor 6 regulates tumor cell invasion through the activation of the MEK/ERK signaling pathway. Proc Natl Acad Sci U S A 2004; 101: 9671–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Funakoshi Y, Hasegawa H, Kanaho Y. Activation mechanisms of PIP5K isozymes by the small GTPase ARF6. Adv Enzyme Regul. 2009. doi: 10.1016/j.advenzreg.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 24. Fukami K, Matsuoka K, Nakanishi O, Yamakawa A, Kawai S, Takenawa T. Antibody to phosphatidylinositol 4,5‐bisphosphate inhibits oncogene‐induced mitogenesis. Proc Natl Acad Sci U S A 1988; 85: 9057–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Van Rheenen J, Song X, Van Roosmalen W et al. EGF‐induced PIP2 hydrolysis releases and activates cofilin locally in carcinoma cells. J Cell Biol 2007; 179: 1247–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bowden ET, Coopman PJ, Mueller SC. Invadopodia: unique methods for measurement of extracellular matrix degradation in vitro. Methods Cell Biol 2001; 63: 613–27. [DOI] [PubMed] [Google Scholar]
- 27. Neve RM, Chin K, Fridlyand J et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006; 10: 515–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Balla T. Imaging and manipulating phosphoinositides in living cells. J Physiol 2007; 582: 927–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yagisawa H, Sakuma K, Paterson HF et al. Replacements of single basic amino acids in the pleckstrin homology domain of phospholipase C‐delta1 alter the ligand binding, phospholipase activity, and interaction with the plasma membrane. J Biol Chem 1998; 273: 417–24. [DOI] [PubMed] [Google Scholar]
- 30. Nakahara H, Otani T, Sasaki T, Miura Y, Takai Y, Kogo M. Involvement of Cdc42 and Rac small G proteins in invadopodia formation of RPMI7951 cells. Genes Cells 2003; 8: 1019–27. [DOI] [PubMed] [Google Scholar]
- 31. Pendaries C, Tronchere H, Plantavid M, Payrastre B. Phosphoinositide signaling disorders in human diseases. FEBS Lett 2003; 546: 25–31. [DOI] [PubMed] [Google Scholar]
- 32. Biswas RS, Baker D, Hruska KA, Chellaiah MA. Polyphosphoinositides‐dependent regulation of the osteoclast actin cytoskeleton and bone resorption. BMC Cell Biol 2004; 5: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ziel JW, Hagedorn EJ, Audhya A, Sherwood DR. UNC‐6 (netrin) orients the invasive membrane of the anchor cell in C. elegans. Nat Cell Biol 2009; 11: 183–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Caroni P. Actin cytoskeleton regulation through modulation of PI(4,5)P2 rafts. EMBO J 2001; 20: 4332–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mao YS, Yin HL. Regulation of the actin cytoskeleton by phosphatidylinositol 4‐phosphate 5 kinases. Pflugers Arch 2007; 455: 5–18. [DOI] [PubMed] [Google Scholar]
- 36. Chuang YY, Tran NL, Rusk N, Nakada M, Berens ME, Symons M. Role of synaptojanin 2 in glioma cell migration and invasion. Cancer Res 2004; 64: 8271–5. [DOI] [PubMed] [Google Scholar]
- 37. Sun Y, Carroll S, Kaksonen M, Toshima JY, Drubin DG. PtdIns(4,5)P2 turnover is required for multiple stages during clathrin‐ and actin‐dependent endocytic internalization. J Cell Biol 2007; 177: 355–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Aikawa Y, Martin TF. ARF6 regulates a plasma membrane pool of phosphatidylinositol(4,5)bisphosphate required for regulated exocytosis. J Cell Biol 2003; 162: 647–59. [DOI] [PMC free article] [PubMed] [Google Scholar]