

Abstract
Cholangiocarcinoma (CCA) is a major cause of cancer deaths in northeast Thailand. It is aggressive, highly metastatic, and responds poorly to traditional chemotherapy. We demonstrated the potential for Cepharanthine (CEP), a biscoclaurine alkaloid extracted from Stephania cepharantha, to treat CCA. CEP significantly inhibited growth of human CCA cell lines in a dose‐ and time‐dependent manner, regardless of the histologic type of tumor origin. Increasing cell apoptosis via caspase‐3 and capase‐9 activation was demonstrated in CEP‐treated cells. We found that CEP controlled the growth of CCA cells through nuclear factor‐kappa B (NF‐κB) inactivation by inhibiting nuclear translocation. CEP treatment effectively reduced tumor size in CCA‐inoculated mice without serious side effects. CEP also increased cell apoptosis in primary histocultures of CCA patients’ tissues; this was demonstrated by immunohistochemistry using TUNEL staining. Our results suggest that CEP possesses therapeutic potential against human CCA. (Cancer Sci 2010)
Cholangiocarcinoma (CCA) is an aggressive and lethal cancer arising from biliary epithelia within either the intrahepatic or extrahepatic biliary tracts. This cancer is rare worldwide but it is the most common liver cancer in northeast Thailand. Several conditions associated with chronic inflammation have been identified as risk factors for CCA. Infection with the liver fluke Opisthorchis viverrini is the most common risk factor for CCA in Thailand and in Southeast Asia; whereas Clonochis sinensis infection is the general risk for CCA in East Asia. For non‐liver fluke‐associated CCA, primary sclerosing cholangitis is the predisposing factor in Western countries.( 1 ) According to our research, there were no significant differences in the clinical features and biological behaviors of these three different etiologic CCAs; however, differences in the molecular signature of CCAs from different etiologic CCAs were recognized. A comparison of gene expression profiles for mass‐forming CCA (from Thai Opisthorchis viverrini‐related CCA) and Japanese (non‐liver fluke‐related CCA) revealed that the liver fluke‐related CCA exhibited increased expression of genes involved in xenobiotic metabolism, whereas those of non‐fluke‐related CCA showed enhanced genes related to growth factor signaling.( 2 )
Early detection of CCA is difficult since there are no specific symptoms during the early stages of tumor development. Consequently, the majority of CCA patients present with advanced incurable disease so these people are not good candidates for curative surgery. Even in those who have undergone complete surgical resection, recurrence is common and the 5‐year survival rate unfavorable.( 3 , 4 ) Novel treatment strategies directed against this malignancy are, therefore, urgently needed.
Cepharanthin (CEP), a biscoclaurine alkaloid extracted from the roots of Stephania cepharantha Hayata, is widely used in Japan for the treatment of various acute and chronic diseases without any serious side effects.( 5 ) It is known to have anti‐inflammatory, anti‐allergic, and immunomodulatory activities and hence is used for chemoprevention and treatment of many diseases. Regarding inflammation and shock, CEP affects antitumor activity,( 6 ) cell cycle arrest,( 7 ) anti‐proliferative, and pro‐apoptotic actions,( 8 , 9 ) as well as anti‐tumor invasion( 10 ) in many cancer cells. The potent enhancement of CEP on the sensitivity and restoration of anticancer drugs in cancer cells is also reported.( 7 )
As corroboration of the anti‐inflammatory and antitumor activity of CEP, we planned to demonstrate the antitumor activity of CEP in human CCA cell lines, in a CCA‐inoculated mouse model and in primary cultures of human CCA tissues, and to clarify the mechanism by which CEP acts on nuclear factor‐kappa B (NF‐κB) activity. To our knowledge, our findings provide the first evidence of the use of CEP to modulate the growth of CCA.
Materials and Methods
Cell lines. Human CCA cell lines were cultured in DMEM supplemented with 10% fetal calf serum, 1%l‐glutamine, and 100 U/mL penicillin and 100 μg/mL streptomycin at 37°C and 5% CO2.
Cell viability test. MTT assays were applied to test cell viability. In brief, 3 × 103 cells per well were seeded in a 96‐well dish and incubated with or without various concentration of CEP for 24, 48, and 72 h at 37°C, 5% CO2. Subsequently, 10 μL MTT (0.5 mg/mL final concentration) was added to each well. After 4 h of additional incubation, 100 μL of 0.01 N HCl in isopropanol was added to dissolve the crystals. Absorption at 570 nm for each sample was determined with an auto‐matic ELISA plate reader (Multiskan; Thermo Electron, Vantaa, Finland).
DNA fragmentation assay. Briefly, 106 cells were lysed in 100 μL of 10 mM Tris–HCl buffer (pH 7.4) containing 10 mM EDTA and 0.5% Triton X‐100. After centrifugation for 5 min at 20 000g, the supernatant was treated with 2 μL of 10 mg/mL RNase‐A and 2 μL of 10 mg/mL proteinase K. Subsequently, 20 μL of 5 M NaCl and 120 μL absolute isopropanol were added to the samples and kept at −20°C for 12 h. After centrifugation for 15 min at 20 000g, the pellets were dissolved in 20 μL of TE buffer (10 mM Tris–HCl and 1 mM EDTA) and loaded onto a 1.5% agarose gel in 1 × TBE buffer (89 mM Tris Base, 89 mM boric acid, and 2 mM EDTA) and electrophoresed at 100 volt for 30 min and stained with ethidium bromide.
Western blot analysis. Protein of whole cell or nuclear lysates were separated by 10% SDS‐polyacrylamide gel electrophoresis( 11 ) and blotted onto a PVDF membrane (GE Healthcare, Tokyo, Japan). Detection was performed using the Enhanced Chemiluminescence Western Blotting Detection System (ECL; GE Healthcare Bio‐Science, Buckinghamshire, UK). Primary antibodies used were: anti‐p65 (F‐6), anti‐p50 (NLS), anti‐p52 (C‐5), anti‐IκBα (C‐21), anti‐phospho (Ser32)‐IκBα (B‐9), anti‐IKKαβ (H‐470), anti‐phospho (Thr23)‐IKKαβ, anti‐Actin (C‐2), and anti‐Histone H1(N‐16) (Santa Cruz Biotechnology, Santa Cruz, CA, USA), caspase‐3 (9662), and caspase‐9 (9502) (Cell Signaling Technology, Danvers, MA, USA). Quantification of the western blots was performed using NIH Image software (NIH, Bethesda, MD, USA). Relative density was evaluated and normalized with actin or Histone H1.
Nuclear extraction. Cells were washed with PBS, lyzed in 1 mL hypotonic buffer (10 mM HEPES‐KOH [pH 7.9], 1.5 mM MgCl2, 10 mM KCl, 0.1% NP‐40, 0.5 mM DTT, and 0.5 mM PMSF) and incubated on ice for 15 min. Nuclei fraction was collected by centrifugation at 200g for 1 min, lysed with 100 μL of nuclear lysis buffer (50 mM HEPES‐KOH [pH 7.9], 0.1 mM EDTA and 10% glycerol, 420 mM KCl, 5 mM MgCl2, 0.1 mM DTT, 0.5 mM PMSF, and 2 μg/mL Aprotinin), and incubated on ice for 30 min. Nuclear lysate was obtained by centrifugation at 18 000g for 10 min.
Electrophoretic mobility shift assay. An electrophoretic mobility shift assay (EMSA) was performed using a 2nd generation DIG Gel Shift Kit (Roche Diagnostics, Mannheim, Germany). Double‐stranded oligonucleotide probes containing the mouse immunoglobulin kappa (Igκ) light‐chain NF‐κB consensus site were purchased from Promega (Madison, WI, USA). The oligonucleotide was 3′ end‐labeled with a digoxigenin‐11‐ddUTP. The nuclear extract (10 μg protein) was incubated with 1 μg of poly [d(I‐C)], 0.1 μg of poly‐L‐lysine, and DIG‐labeled oligonucleotide in binding buffer (20 mM HEPES [pH 7.6], 1 mM EDTA, 10 mM (NH4)2SO4, 0.2% Tween 20, and 30 mM KCl) for 15 min at 25°C. After the incubation, 5 × loading buffer (0.25 × TBE and 60% glycerol) was added, and the samples separated on 5% acrylamide gel in 0.5 × TBE buffer (89 mM Tris Base, 89 mM boric acid, and 2 mM EDTA). The oligonucleotide was electroblotted onto a positively charged nylon membrane and immunodetected using anti‐digoxigenin‐alkaline phosphatase. For analysis of the specific binding to κB sequence, a 200 × excess of the non‐biotin‐tagged NF‐κB oligonucleotides was added to the sample as a competitor.
In vivo assay. Eight‐ to 10‐week‐old male NOD/Scid/Jak3 deficient (NOJ) mice( 12 ) were housed and monitored in the animal research facility according to the institutional guidelines. All experimental protocols were approved by the Institutional Animal Care and Use Committee, Kumamoto University. Mice were subcutaneously injected with 4 × 106 cells of KKU‐M213 and KKU‐M214 at both flank sides, and were intraperitoneally injected with PBS or CEP (10 mg/kg/day) on the next day and every day for 17 days thereafter. On day 18, serum was collected from each animal and the tumors removed, weighted, and fixed in 4% paraformladehyde overnight for immunohistochemistry study.
Histoculture drug response assay (HDRA). Tumor tissues from patients with a papillary mass‐forming type of CCA (n = 6) and from patients with a non‐papillary type of CCA (n = 3), who underwent liver resection, were obtained from the Department of Surgery, Srinagarind Hospital, Faculty of Medicine, Khon Kaen University, Thailand. The average age of the CCA patients was 56.7 years (range, 52–69 years) and the male to female ratio 3.5:1. Informed consent was obtained from each subject, and the Khon Kaen University Ethics Committee for Human Research approved the research protocol (HE 471214).
The primary tissue culture was prepared for a drug response assay in a 24‐well microplate.( 13 ) Fresh cancerous tissues from CCA patients were made into approximately 10‐mg slices and placed on a collagen sponge gel (Sumitomo Pharma, Osaka, Japan), which was submerged in RPMI‐1640 medium containing 20% fetal calf serum, in the presence or absence of 10 and 20 μg/mL CEP. Plates were incubated for 4 days at 37°C, 5% CO2, and then the tumor tissues were processed for immunohistochemistry staining using TUNEL assay for apoptotic cells analysis.
Terminal deoxynucleotide transferase‐mediated dUTP nick‐end labeling (TUNEL) assay. Histologic analysis of DNA fragmentation was used to identify apoptotic cells in the paraffin sections of the HDRA. In situ TUNEL was done using the DeadEnd Colorimetric TUNEL System (Promega). TUNEL‐positive cells were quantified in at least four high‐power fields (×40) of randomly selected tissue sections.
Statistical analysis. The results were presented as a mean ± SD for at least three separate experiments. Statistical significance was determined using the Student’s t‐test and P < 0.05 was required for statistical significance.
Results
Cepharanthin inhibits growth of CCA cells by induction of apoptosis. The effects of CEP on the growth of CCA cells were observed in seven human CCA cell lines established from primary tumors of liver fluke‐associated CCA patients with different histological types; namely, KKU‐M055 and KKU‐100 (poorly differentiated CCA), KKU‐M139 (adeno‐squamous CCA), KKU‐M156 (moderately differentiated CCA), KKU‐M214, KKU‐OCA17 (well‐differentiated CCA), and KKU‐M213 (mixed papillary and non‐papillary CCA). As shown in Figure 1(a), CEP inhibited the growth of all CCA cell lines examined. The effective doses were varied; however, CEP at 20 μg/mL effectively suppressed growth of all cell lines to <40% of the control. CEP significantly inhibited the growth of KKU‐M213 and KKU‐M214 (P < 0.001) in a dose‐ and time‐dependent manner (Fig. 1b,c).
Figure 1.
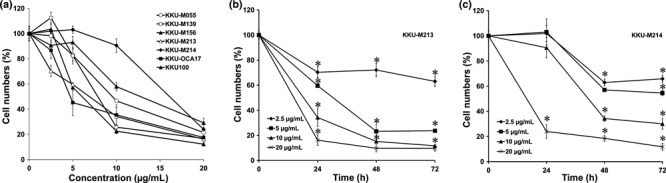
Anti‐proliferative activity of cepharanthin (CEP) in cholangiocarcinoma (CCA) cell lines. (a) Seven human CCA cell lines were treated with 2.5, 5, 10, and 20 μg/mL of CEP for 24 h; (b) KKU‐M213 and (c) KKU‐M214 were treated with various concentrations of CEP for 24, 48, and 72 h. Cell growth was analyzed by MTT assay. Percentages of cell viability relative to control cells (without CEP) are shown as mean ± SD from three separate experiments. *P < 0.001.
Cepharanthin induces apoptosis in CCA cell lines via the caspase pathway. DNA fragmentation was examined for CEP‐induced cell death in CCA cell lines. CEP‐treated cells showed a significant increase of DNA ladders over time and with greater concentrations of CEP. CCA cells treated with 10 and 20 μg/mL of CEP for 24 and 48 h showed a gradual increase in apoptotic cells, as determined by DNA fragmentation (Fig. 2a).
Figure 2.

Cepharanthin (CEP) induces cell apoptosis via activation of caspase‐3 and ‐9. (a) KKU‐M213 and KKU‐M214 cells were treated with 20 μg/mL CEP for 24 and 48 h. DNA fragmentation was analyzed by agarose gel electrophoresis. (b) Caspase‐3 and ‐9 were activated by CEP treatment. Expression of pro‐ and active‐caspase 3 and ‐9 were determined by western blot analysis.
Expression of caspase‐3 and ‐9 are hallmarks of cells undergoing apoptosis. Both enzymes express in cells as inactive precursors and are cleaved to active caspase‐3 and ‐9 which initiate and execute the apoptotic cascade. To investigate the mechanism by which CEP induced cell apoptosis, we first examined the activation of caspase‐3 and ‐9 in CCA cells in the presence of 20 μg/mL of CEP, at various times. Western blot analyses showed that pro‐caspase‐3 and ‐9 gradually decreased with time in CCA cells treated with CEP, whereas the activated forms progressively increased (Fig. 2b). Thus, CEP‐induced apoptosis in CCA cells is associated with activation of the caspase pathway.
Cepharanthin suppresses the endogenous activity of NF‐κB. Since a number of studies have reported the significance of NF‐κB for cell survival and proliferation, we investigated further whether CEP exerted its effect on inhibiting NF‐κB activity. We performed EMSA and western blot analysis to assess the effects of CEP on NF‐κB DNA‐binding activity and on the translocation of NF‐κB subunits from cytosol to the nuclei. CEP significantly decreased the NF‐κB DNA‐binding activity in KKU‐M213 cells in a time‐dependent manner over untreated cells (Fig. 3a). CEP effectively inhibited NF‐κB DNA binding within 6 h and completely at 24 h after treatment.
Figure 3.
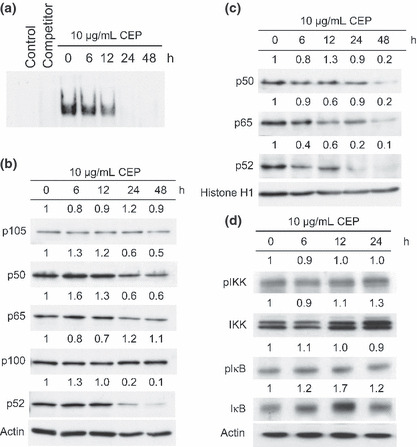
Cepharanthin (CEP) inhibited DNA‐binding activity, nuclear translocation, and expression of nuclear factor‐kappa B (NF‐κB) in CCA cells. KKU‐M213 cells were treated with or without 10 μg/mL of CEP for 6, 12, 24, and 48 h, and the effects of CEP on NF‐κB activity determined. (a) Inhibitions of constitutive NF‐κB binding activity were examined in nuclear extracts by electrophoretic mobility shift assay (EMSA). Reaction without cell lysate was used as control and reaction with unlabeling oligonucleotide (× 200 of labeling probe) was used as a competitor. (b) CEP inhibited expression of NF‐κB p50/p105, p52/p100, and p65 in whole cell lysate of KKU‐M213, as determined by western blotting. (c) CEP reduced nuclear translocations of NF‐κB p50, p52, and p65. (d) Amount of IKK, IκB, and their phosphorylated forms after CEP treatment. Intensity of each band was normalized with actin or Histone H1, and compared relative to those without CEP (=1), as indicated by the number shown on the top of each band.
We further investigated the mechanism by which CEP suppressed NF‐κB activity. Western blot analysis of NF‐κB proteins from KKU‐M213 cells treated with 10 μg/mL CEP revealed that CEP treatment had no effect on the expression of p100 or p105 (Fig. 3b). By contrast, CEP reduced the amount of constitutive p50, p65, and p52 in whole cell lysates (Fig. 3b) and nuclear fractions in a time‐dependent manner (Fig. 3c). These results indicate that CEP suppressed NF‐κB DNA‐binding activity by inhibiting NF‐κB activation, thereby reducing the amounts of NF‐κB p50, p65, and p52 translocated into the nucleus.
Since the inhibitors of NF‐κB (IκB) and IκB kinase (IKK) are upstream regulators of NF‐κB activation, we investigated whether these two factors are involved in the suppression of NF‐κB activation by CEP. As shown in Figure 3(d), untreated CCA cells constitutively expressed IκB, IKK, and their phosphorylated forms – pIκB and pIKK. CEP treatment for 24 h slightly increased the amounts of IκB and IKK and to some extent reduced the amounts of pIκB but not pIKK. Thus, the direct effects of CEP on IκB and IKK are unclear. These results indicated that CEP inhibited the activation of NF‐κB families by an IKK‐independent mechanism.
Antitumor effect of CEP in CCA‐inoculated mice. We further examined whether CEP suppressed the growth of tumors in CCA‐inoculated mice. CCA cells were injected subcutaneously into the flank of NOJ mice which were injected intraperitoneally with either PBS (as the control) or 10 mg/kg CEP in PBS for 17 days. Mice treated with CEP were healthy and had similar body weights as the control mice (Fig. 4a). No side effects were observed during CEP treatment. Supplementation of CEP significantly reduced tumor sizes (Fig. 4b) and tumor weights (Fig. 4c) in the inoculated KKU‐M213 (P < 0.05) and KKU‐M214 (P < 0.001) mice as compared to the controls that received PBS alone.
Figure 4.
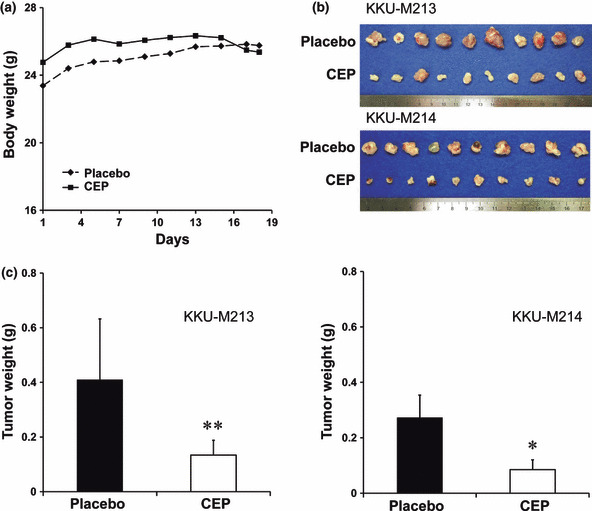
Antitumor activity of cepharanthin (CEP) in CCA‐inoculated NOD/Scid/Jak3 KO mice. Mice were inoculated with 4 × 106 cells of KKU‐M213 and KKU‐M214 in each flank and intraperitoneally injected with PBS or CEP every day for 17 days thereafter. (a) Average body weights of the mice in each group were compared. (b) CCA tumor tissues were obtained from placebo and CEP‐treated mice. (c) Comparison of tumor weights were obtained from placebo and CEP‐treated mice. **P < 0.05; *P < 0.001 independent‐sample t‐test compared to the placebo group.
Antitumor activity of CEP on CCA patient tissues. The effect of CEP on the growth of CCA was investigated in nine cases of CCA patient tissues by histoculture drug response assay. CCA tissue slices were incubated in a medium with or without CEP for 4 days. Immunohistochemistry using TUNEL staining revealed a significantly higher number of apoptotic cells in tissue sections treated with 10 or 20 μg/mL of CEP (Fig. 5b,c) than controls incubated in a medium without CEP (Fig. 5a). CEP dose‐dependently induced cell apoptosis in CCA tissues (Fig. 5d).
Figure 5.
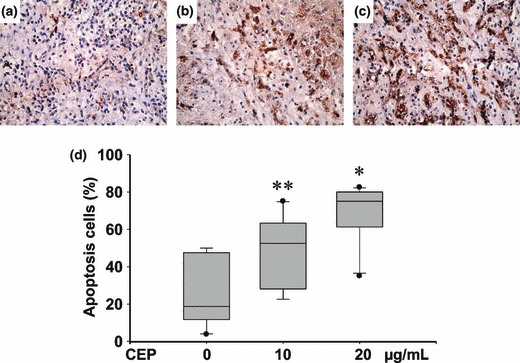
Cepharanthin (CEP) increased cell apoptosis in primary histoculture of cholangiocarcinoma (CCA) patient tissues. Fresh cancerous tissue slices from CCA patients were cultured on a collagen sponge gel submerged in RPMI‐1640 with 20% fetal calf serum, in the absence (a) or presence of (b) 10 μg/mL CEP or (c) 20 μg/mL CEP for 4 days. Apoptotic cells were determined using immunohistochemistry staining with TUNEL assay. (d) CEP significantly increased the number of apoptotic cells in a dose‐dependent manner compared with the control group. **P < 0.05; *P < 0.001 independent‐sample t‐test compared with the control group. (magnification of a–c: × 40).
Discussion
Cepharanthine (CEP) – alkaloids from the root extract of Stephania cepharantha Hayata – has been widely used in Japan with a variety of biological and pharmacological effects for acute and chronic diseases including cancer.( 14 ) Recently, it was demonstrated that CEP acts via inhibition of NF‐κB activation.( 15 , 16 ) We demonstrated that biscoclaurine alkaloid CEP effectively limits growth of CCA in vitro and in vivo via inactivation of NF‐κB action. CEP exhibited potent pro‐apoptotic effects on CCA cells resulting in induction of cell death and reduction of tumor growth in NOD/Scid/Jak‐3‐deficient mice without any observed side effects. Moreover, CEP effectively reduced tumor growth of patient tissues, as determined by histoculture drug response assay.
CEP inhibited growth of CCA cell lines of KKU‐M213 and KKU‐M214 at concentrations ranging between 2.5 and 20 μg/mL (4.12–32.96 μM), which are clinically achievable,( 17 , 18 ) suggesting the clinical efficacy of CEP for CCA. Several mechanisms for CEP‐mediated inhibition of tumor cell survival have been proposed.( 14 ) Our results clearly showed that CEP suppressed tumor growth by inducing cell apoptosis in CCA cells as evidenced by DNA ladder formation, a hallmark feature of apoptosis. The apoptosis‐inducing effects of CEP have been demonstrated in a variety of cancer cells.( 9 , 19 , 20 ) Production of reactive oxygen species and induction of p27 were evident in CEP‐treated neutrophils( 21 ) and squamous cell carcinoma,( 16 ) respectively.
In our study, CEP‐induced apoptosis in CCA cells is partly via activations of caspase‐3 and caspase‐9, both enzymes known to play a key role in apoptosis regardless of the stimuli.( 22 ) CEP at high doses can induce excessive production of oxygen reactive species in cancer cells( 20 ) and consequently acts directly on the mitochondrial membrane, resulting in the release of cytochrome C and activating caspase‐9 and caspase‐3.( 9 )
Nuclear factor‐kappa B (NF‐κB) is an extensively anti‐apoptotic transcription factor whose DNA binding is potently and rapidly induced by tumor nectrosis factor‐α (TNF‐α), infrared radiation, and other stimuli, especially chemotherapeutic drugs in almost all cells. Constitutive NF‐κB activation has been observed in a wide variety of cancers including those of the prostate,( 23 ) colon,( 24 ) lung,( 25 ) and CCA.( 26 ) It is also associated with a resistance to apoptosis, since many of its target genes coded for anti‐apoptotic molecules such as cIAPs, c‐FLIP, A20, and BclXL.( 27 ) Many findings indicate that the blockade of NF‐κB signaling pathways produces a new and effective strategy for anticancer therapy.( 27 , 28 )
Because of the pivotal role that NF‐κB plays in promoting tumor cell proliferation, survival, migration, and angiogenesis, it is a likely target for experimental and/or pharmacological intervention for the treatment of cancer. CEP has been shown to exert its effect via inhibition of NF‐κB activation. In general, the IκB form a complex with NF‐κB which prevents the translocation of NF‐κB from the cytoplasm into the nucleus. The specific phosphorylation of IκBα by an IKK complex causes degradation of IκB and allows the translocation of NF‐κB to the nucleus. It has been shown that treatment of primary effusion lymphoma cells with CEP abrogated NF‐κB activity by blocking the phosphorylation of p65.( 15 )
In our study, CEP clearly reduced the nuclear translocations of NF‐κB (p50, p52, and p65) and, as a result, abolished NF‐κB–DNA complex formations, as shown by the EMSA assay. The mechanism by which CEP inactivates NF‐κB activity is not well understood; however, inactivation of NF‐κB by inhibiting nuclear translocation seems to be the general mechanism of CEP regardless to cancer origin. CEP effectively reduced growth of human primary effusion lymphoma cell lines, in vitro and in vivo, via decreasing protein expression of p65 and inhibition of nuclear translocation.( 15 ) In addition to the present study, decreases in the expression of NF‐κB p50, p65 and inhibition of NF‐κB nuclear translocation by caffeic acid phenethyl ester also effectively suppressed growth of extrahepatic CCA‐derived cell lines.( 29 ) From these perspective, activation of the NF‐κB pathway may play a central role in oncogenesis of CCA and drugs or chemicals targeting inhibition of NF‐κB action may efficiently inhibit growth of CCA.
CEP, a biscoclaurine alcakloid, is known to possess various biological and pharmacological activities.( 14 ) Apart from the anti‐inflammatory function via NF‐kB inhibition, CEP also exerts various activities which suppress tumor growth, such as: (i) inhibiting the activity of P‐glycoprotein;( 30 , 31 ) (ii) inducing cell cycle arrest by down‐regulation of cyclin E and up‐regulation of p27;( 32 ) (iii) inducing apoptosis effects through the activation of caspase‐9 and caspase‐3; and (iv) regulating the expression of Bcl‐2 and Bax protein.( 8 , 33 ) From this perspective, utilizing CEP as an antitumor agent in CCA should have more of an advantage than merely using it is an anti‐inflammatory agent.
In our study, the effect of CEP on suppression of tumor growth was demonstrated in CCA‐inoculated mice without any side effects. Similar effects and the safety of CEP have been reported for studies on leukemia cells,( 34 ) hepatocellular carcinoma,( 30 ) and primary effusion lymphoma.( 15 ) The effect of CEP on patients’ tissues determined by the histoculture drug response assay was first demonstrated in our study. The number of apoptotic cells as indicated by positive TUNEL staining was significantly higher in tissues cultured in the presence of CEP in a dose‐dependent manner. This three‐dimensional, native state, histoculture assay simulates the tumor architecture in the body and may reflect the CEP‐responsiveness of tumors if CEP is given as a supplement to CCA patients.
CEP effectively inhibited growth of CCA cell lines regardless of histologic type. As shown in this study, growth of seven CCA cell lines established from intrahepatic CCA with different histologic types was significantly suppressed by CEP treatment. A similar effect was also observed when human tissues from different histological types of CCA were treated with CEP. These results imply that CEP may efficiently inhibit growth of CCA regardless of the type of tumor origin.
In this study, we used a relatively high amount of CEP (10–20 μg/mL for in vitro study and 10 mg/kg for in vivo study). As for the mice study, as high as 10–100 mg/kg of CEP has been effectively used in several studies without obvious adverse effects.( 14 , 15 ) Moreover, a dose of 40–100 mg/body/day of CEP has been used in several clinical applications such as idiopathic thrombocytopenic purpura (ITP) and in cancer patients without any side effects.( 17 , 35 ) These findings suggest that CEP is an efficient alternative anticancer agent and that higher dose of CEP can be safely used for cancer patients.
In conclusion, we demonstrated that CEP exhibits antitumor activity against human CCA cells by enhancing apoptosis through the inhibition of NF‐κB activation. The antitumor activity of CEP was shown in CCA cell lines, as demonstrated in CCA‐inoculated mice and confirmed in the primary histoculture of patients’ tissues. For the first time, it has been demonstrated that CEP abrogates NF‐κB activity by inhibiting NF‐κB activation, leading to the inhibition of NF‐κB nuclear translocation and DNA‐binding activity. Since CEP exhibits diverse pharmacological effects, there may be other pathways apart from NF‐κB pathway undergirding the action of CEP. In view of the fact that CEP is a safe drug, which has been used in Japan for several decades( 14 ) and effectively suppresses the growth of CCA cells in culture, patient tissues, and tumor‐inoculated mice without obvious side effects, we propose that CEP be approved as a novel drug of choice for the treatment of CCA patients.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
This project was supported by the Research Team Strengthening Grant, National Genetic Engineering and Biotechnology Center, National Science and Technology Development Agency, Thailand, and the Royal Golden Jubilee – PhD Program for W. Seubwai and S. Wongkham (PHD/0034/2549). We thank Mr. Bryan Roderick Hamman and Mrs. Janice Loewen Hamman for assistance with the English‐language presentation of the manuscript.
References
- 1. Burak K, Angulo P, Pasha TM, Egan K, Petz J, Lindor KD. Incidence and risk factors for cholangiocarcinoma in primary sclerosing cholangitis. Am J Gastroenterol 2004; 99(3): 523–6. [DOI] [PubMed] [Google Scholar]
- 2. Jinawath N, Chamgramol Y, Furukawa Y et al. Comparison of gene expression profiles between Opisthorchis viverrini and non‐Opisthorchis viverrini associated human intrahepatic cholangiocarcinoma. Hepatology 2006; 44(4): 1025–38. [DOI] [PubMed] [Google Scholar]
- 3. Anderson CD, Pinson CW, Berlin J, Chari RS. Diagnosis and treatment of cholangiocarcinoma. Oncologist 2004; 9(1): 43–57. [DOI] [PubMed] [Google Scholar]
- 4. Gores GJ. Cholangiocarcinoma: current concepts and insights. Hepatology 2003; 37(5): 961–9. [DOI] [PubMed] [Google Scholar]
- 5. Kimoto T, Suemitsu K, Nakayama H, Komori E, Ohtani M, Ando S. Therapeutic experience of venomous snakebites by the Japanese viper (Agkistrodon halys Blomhoffii) with low dose of antivenin: report of 43 consecutive cases. Gan To Kagaku Ryoho 1997; 66(2): 71–7. [PubMed] [Google Scholar]
- 6. Bun SS, Laget M, Chea A, Bun H, Ollivier E, Elias R. Cytotoxic activity of alkaloids isolated from Stephania rotunda. Phytother Res 2009; 23(4): 587–90. [DOI] [PubMed] [Google Scholar]
- 7. Harada K, Ferdous T, Itashiki Y et al. Effects of cepharanthine alone and in combination with fluoropyrimidine anticancer agent, S‐1, on tumor growth of human oral squamous cell carcinoma xenografts in nude mice. Anticancer Res 2009; 29(4): 1263–70. [PubMed] [Google Scholar]
- 8. Biswas KK, Tancharoen S, Sarker KP, Kawahara K, Hashiguchi T, Maruyama I. Cepharanthine triggers apoptosis in a human hepatocellular carcinoma cell line (HuH‐7) through the activation of JNK1/2 and the downregulation of Akt. FEBS Lett 2006; 580(2): 703–10. [DOI] [PubMed] [Google Scholar]
- 9. Wu J, Suzuki H, Akhand AA, Zhou YW, Hossain K, Nakashima I. Modes of activation of mitogen‐activated protein kinases and their roles in cepharanthine‐induced apoptosis in human leukemia cells. Cell Signal 2002; 14(6): 509–15. [DOI] [PubMed] [Google Scholar]
- 10. Ebina T. [Intratumoral administration of biological preparations – recommendation for integrative medicine]. Gan To Kagaku Ryoho 2001; 28(11): 1515–8. [PubMed] [Google Scholar]
- 11. Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970; 227(5259): 680–5. [DOI] [PubMed] [Google Scholar]
- 12. Okada S, Harada H, Ito T, Saito T, Suzu S. Early development of human hematopoietic and acquired immune systems in new born NOD/Scid/Jak3null mice intrahepatic engrafted with cord blood‐derived CD34+ cells. Int J Hematol 2008; 88(5): 476–82. [DOI] [PubMed] [Google Scholar]
- 13. Furukawa T, Kubota T, Watanabe M et al. Chemosensitivity testing of clinical gastrointestinal cancers using histoculture and the MTT end‐point. Anticancer Res 1992; 12(5): 1377–82. [PubMed] [Google Scholar]
- 14. Furusawa S, Wu J. The effects of biscoclaurine alkaloid cepharanthine on mammalian cells: implications for cancer, shock, and inflammatory diseases. Life Sci 2007; 80(12): 1073–9. [DOI] [PubMed] [Google Scholar]
- 15. Takahashi‐Makise N, Suzu S, Hiyoshi M et al. Biscoclaurine alkaloid cepharanthine inhibits the growth of primary effusion lymphoma in vitro and in vivo and induces apoptosis via suppression of the NF‐kappaB pathway. Int J Cancer 2009; 125(6): 1464–72. [DOI] [PubMed] [Google Scholar]
- 16. Azuma M, Aota K, Tamatani T et al. Suppression of tumor necrosis factor alpha‐induced matrix metalloproteinase 9 production in human salivary gland acinar cells by cepharanthine occurs via down‐regulation of nuclear factor kappaB: a possible therapeutic agent for preventing the destruction of the acinar structure in the salivary glands of Sjogren’s syndrome patients. Arthritis Rheum 2002; 46(6): 1585–94. [DOI] [PubMed] [Google Scholar]
- 17. Suzuki S, Abe R, Nihei M, Kimijima I, Tsuchiya A, Nomizu T. [Efficacy of Cepharanthin for preventing leukopenia and thrombocytopenia induced by chemotherapy in breast cancer patient—prospective randomized study]. Gan To Kagaku Ryoho 1990; 17(6): 1195–200. [PubMed] [Google Scholar]
- 18. Suzuki R, Hara M, Shindoh J et al. [Effects of cepharanthin on leukopenia and thrombocytopenia induced by chemotherapy in lung cancer patients]. Gan To Kagaku Ryoho 1992; 19(5): 647–52. [PubMed] [Google Scholar]
- 19. Harada K, Ohira S, Isse K et al. Lipopolysaccharide activates nuclear factor‐kappaB through toll‐like receptors and related molecules in cultured biliary epithelial cells. Lab Invest 2003; 83(11): 1657–67. [DOI] [PubMed] [Google Scholar]
- 20. Furusawa S, Wu J, Fujimura T et al. Cepharanthine inhibits proliferation of cancer cells by inducing apoptosis. Methods Find Exp Clin Pharmacol 1998; 20(2): 87–97. [DOI] [PubMed] [Google Scholar]
- 21. Akamatsu H, Komura J, Asada Y, Niwa Y. Effects of cepharanthin on neutrophil chemotaxis, phagocytosis, and reactive oxygen species generation. J Dermatol 1991; 18(11): 643–8. [DOI] [PubMed] [Google Scholar]
- 22. Nicholson DW, Thornberry NA. Caspases: killer proteases. Trends Biochem Sci 1997; 22(8): 299–306. [DOI] [PubMed] [Google Scholar]
- 23. Suh J, Payvandi F, Edelstein LC et al. Mechanisms of constitutive NF‐kappaB activation in human prostate cancer cells. Prostate 2002; 52(3): 183–200. [DOI] [PubMed] [Google Scholar]
- 24. Kojima M, Morisaki T, Sasaki N et al. Increased nuclear factor‐kB activation in human colorectal carcinoma and its correlation with tumor progression. Anticancer Res 2004; 24(2B): 675–81. [PubMed] [Google Scholar]
- 25. Mukhopadhyay T, Roth JA, Maxwell SA. Altered expression of the p50 subunit of the NF‐kappa B transcription factor complex in non‐small cell lung carcinoma. Oncogene 1995; 11(5): 999–1003. [PubMed] [Google Scholar]
- 26. Pinlaor S, Hiraku Y, Yongvanit P et al. iNOS‐dependent DNA damage via NF‐kappaB expression in hamsters infected with Opisthorchis viverrini and its suppression by the antihelminthic drug praziquantel. Int J Cancer 2006; 119(5): 1067–72. [DOI] [PubMed] [Google Scholar]
- 27. Karin M, Cao Y, Greten FR, Li ZW. NF‐kappaB in cancer: from innocent bystander to major culprit. Nat Rev 2002; 2(4): 301–10. [DOI] [PubMed] [Google Scholar]
- 28. Tamatani T, Azuma M, Motegi K, Takamaru N, Kawashima Y, Bando T. Cepharanthin‐enhanced radiosensitivity through the inhibition of radiation‐induced nuclear factor‐kappaB activity in human oral squamous cell carcinoma cells. Int J Oncol 2007; 31(4): 761–8. [PubMed] [Google Scholar]
- 29. Onori P, DeMorrow S, Gaudio E et al. Caffeic acid phenethyl ester decreases cholangiocarcinoma growth by inhibition of NF‐kappaB and induction of apoptosis. Int J Cancer 2009; 125: 565–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nakajima A, Yamamoto Y, Taura K et al. Beneficial effect of cepharanthine on overcoming drug‐resistance of hepatocellular carcinoma. Int J Oncol 2004; 24(3): 635–45. [PubMed] [Google Scholar]
- 31. Ikeda R, Che XF, Yamaguchi T et al. Cepharanthine potently enhances the sensitivity of anticancer agents in K562 cells. Cancer Sci 2005; 96(6): 372–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Harada K, Supriatno ????, Yamamoto S, Kawaguchi S, Yoshida H, Sato M. Cepharanthine exerts antitumor activity on oral squamous cell carcinoma cell lines by induction of p27Kip1. Anticancer Res 2003; 23(2B): 1441–8. [PubMed] [Google Scholar]
- 33. Kikukawa Y, Okuno Y, Tatetsu H et al. Induction of cell cycle arrest and apoptosis in myeloma cells by cepharanthine, a biscoclaurine alkaloid. Int J Oncol 2008; 33(4): 807–14. [PubMed] [Google Scholar]
- 34. Braun T, Carvalho G, Fabre C, Grosjean J, Fenaux P, Kroemer G. Targeting NF‐kappaB in hematologic malignancies. Cell Death Differ 2006; 13(5): 748–58. [DOI] [PubMed] [Google Scholar]
- 35. Tsukikawa S, Oikawa H, Satoh T et al. [The effect of cepharanthin on adjuvant chemotherapy induced bone marrow suppression in patients with breast cancer]. Gan To Kagaku Ryoho 1990; 17 (4 Pt 1): 645–8. [PubMed] [Google Scholar]