

Abstract
The extracellular‐regulated kinase (ERK) signaling pathway plays important roles in regulating the malignant potential of cancer cells in vitro. However, the effect of ERK signaling on the prognosis of human tumors is not clearly understood. The present study examined the expression of phosphorylated ERK1/2 (p‐ERK1/2) as a hallmark of ERK activation, in relation to KRAS and BRAF mutations, in 63 endometrial cancer specimens with endometrioid‐subtype, in order to clarify the prognostic value of p‐ERK1/2 expression. Immmunohistochemical analysis revealed that 40 tumors (63%) expressed p‐ERK1/2, with varying levels of expression. Total ERK1/2 expression was also evaluated in a subset of tumors; most cases expressed ERK1/2 constitutively but no correlation was observed with p‐ERK expression, indicating that p‐ERK1/2 staining was not due to ERK overexpression but to hyperactivation of ERK1/2. There was no statistically significant correlation between p‐ERK1/2 expression and clinicopathological features, including patient age, International Federation of Gynecology and Obstetrics stage, pathological grade, myometrial invasion and lymph node metastasis. Sequencing analysis indicated that 23% of patients had a mutation in exon 1 of KRAS, whereas none of the patients had a mutation in exons 11 or 15 of BRAF, which are reportedly hot spots for mutation in many tumor types. There was no significant correlation between KRAS or BRAF status and p‐ERK1/2 expression. Unexpectedly, patients with low p‐ERK1/2 expression had significantly lower relapse‐free survival (P = 0.041) and overall survival (P = 0.020). Multivariate Cox regression analysis indicated that p‐ERK1/2 expression was an independent prognostic indicator for overall survival (P = 0.047). These findings suggest that ERK activation occurs in a KRAS‐ and BRAF‐independent manner in endometrial cancer, and is associated with favorable prognosis. (Cancer Sci 2007; 98: 652–658)
Adenocarcinoma of the endometrium is the most common gynecological malignancy in the USA. In Japan, it is the second most common gynecological cancer, but its frequency has increased dramatically in the last decade. Although there are well‐established surgical and chemotherapeutic treatments for endometrial cancer, the need for molecular‐target therapy has increased, especially for recurrent disease that has acquired radio‐ or chemoresistance; thus, there is a need for a better understanding of the molecular pathways of endometrial carcinogenesis. Several genetic alterations are reportedly associated with endometrial carcinogenesis, and the most frequent genetic abnormalities in cases of endometrial carcinogenesis are mutations in PTEN and KRAS. ( 1 , 2 , 3 , 4 , 5 ) A PTEN mutation activates the phosphatidylinositol‐3‐kinase (PI3K) pathway, and a KRAS mutation activates the extracellular‐regulated kinase (ERK)–mitogen‐activated protein kinase (MAPK) pathways; both mutations activate nuclear downstream targets and play major roles in most human carcinogenesis.( 6 ) The ERK–MAPK pathway is activated by mitogenic stimuli mediated by receptor tyrosine kinases and G protein‐coupled receptors, leading to sequential phosphorylation of Ras, Raf, MAP kinase‐ERK kinase (MEK) and ERK1/2. Phosphorylated ERK1/2 translocates to the nucleus and regulates a range of substrates (including the Ets and activator protein1/activating transcription factor (Ap1/ATF) families of transcription factors) that promote cell proliferation, motility, differentiation and survival.( 7 , 8 , 9 ) Thus, expression of phosphorylated ERK1/2 (p‐ERK1/2) is the hallmark of activated MAPK signaling. To our knowledge, there have been no reports of studies on the expression of p‐ERK1/2 in endometrial cancers.
A mutation in KRAS induces continuous activation of the ERK pathway, even in the absence of mitogenic stimuli, and is thought to play a critical role in carcinogenesis of various organs.( 6 ) Recently, we found that this KRAS mutation plays a causative role in endometrial carcinogenesis, using an in vitro model in which human endometrium‐derived immortalized cells were transfected with mutated KRAS alleles and were found to exhibit completely transformed phenotypes.( 10 , 11 )
In contrast to the results of those in vitro studies, genetic analyses by us and other researchers using clinical samples of endometrial cancer have identified the KRAS mutation only in 10–20% of patients.( 2 , 3 , 4 , 5 ) This discrepancy gives rise to several questions. Is MAPK signaling activated in only a small subset of endometrial cancers? Does the KRAS mutation inevitably confer activated MAPK signaling? Recent genetic studies indicate that frequent genetic mutations of BRAF in a wide variety of tumor types are etiological factors in activation of the MAPK pathway. However, the status of BRAF and the role of its mutation in activating MAPK pathways in endometrial cancer remain controversial.
In the present study, we analyzed expression of p‐ERK1/2 in relation to the mutation status of KRAS and BRAF in endometrial cancer. The present findings clearly indicate that the ERK–MAPK pathway was frequently activated, independent of the status of KRAS and BRAF. Furthermore, we obtained the novel finding that p‐ERK1/2 expression appears to have a favorable effect on patient survival.
Materials and Methods
Patient and tissue samples. A total of 63 patients with endometrioid‐type endometrial cancer (mean age, 57.5 years; range, 32–78 years) treated at the Department of Obstetrics and Gynecology, Kanazawa University Hospital, from January 1995 to December 2002, were enrolled for the study. All patients underwent a total abdominal or radical hysterectomy plus bilateral salpingo‐oophorectomy. Systemic retroperitoneal lymphadenectomy was carried out in approximately 70% of the patients. Staging was done for all patients using the International Federation of Gynecology and Obstetrics (FigO) surgical staging system: 46 tumors were classified as stage I (substages: Ia, 10 tumors; Ib, 28 tumors; Ic, eight tumors); six tumors were classified as stage II (substages: IIa, three tumors; IIb, three tumors); nine tumors were classified as stage III (substages: IIIa, three tumors; IIIc, six tumors); and two tumors were classified as stage IV. Based on histological examination, 34 tumors were classified as G1, 14 tumors were classified as G2, and 15 tumors were classified as G3. Patients with risk factors for recurrence (such as deep myometrial invasion, cervical involvement, special histology or positive peritoneal cytology) underwent external radiotherapy and/or 4–6 cycles of CAP chemotherapy (cisplatin 90 mg/m2; doxorubirin 50 mg/m2; cyclophosphamide 500 mg/m2) or TJ chemotherapy (paclitaxel, 180 mg/m2; carboplatin, according to Chatelut's formula [area under curve = 5 mg/ml/mL]) as a postoperative adjuvant therapy. Patient treatment was followed up with a gynecological examination, recording of laboratory data, transvaginal/abdominopelvic ultrasonography, and a radiologic examination. Data from regular follow‐up visits to the outpatient department were stored in a database specifically designed for endometrial carcinoma patients. A telephone survey to update the status of all surviving patients was made in August 2004. The exact date of disease recurrence was defined as the date when the apparent tumors were detected by ultrasonographic or radiologic examinations.
Tumor samples were collected at the time of surgery, with written informed consent and approval of the Ethics Committee of Kanazawa University. Half of each tissue sample was examined histologically by pathologists, and the remaining portion of each sample was frozen at −80°C until DNA extraction for mutation analysis.
Western blot analysis. To verify the validity of the antibody, an in vitro induction experiment for p‐ERK1/2 was carried out using immortalized endometrial epithelial cells (EM‐E6/E7/TERT) with or without oncogenic mutant KRAS alleles.( 10 , 11 ) The cells were incubated with or without epidermal growth factor (EGF), or were incubated with specific inhibitors of MEK (U0126), PI3K (wortmannin) and p38 (SB203580) for 15 min. After incubation, cell lysates were extracted and used for western blot analysis to visualize phosphorylated ERK. Whole‐cell lysates were prepared by resuspending cell pellets in buffer containing 50 mM Tris‐Cl (pH 7.4), 1% Nonidet P40 (Fluka), and 1 mM ethylenediaminetetracetic acid for 10 min at room temperature. The homogenate was centrifuged at 8000 g for 5 min. The supernatant was recovered, and its protein concentration was measured by Bradford assay. Then, 30 µg of extract was electrophoresed on a sodium dodecylsulfate–polyacrylamide gel with a gradient of 10–20% (Readygels J, 161‐J351, Bio‐Rad), and was transferred to a polyvinylidine difluoride membrane. Membranes were blocked with TBS‐T (150 mM NaCl, 20 mM Tris‐Cl [pH 7.5], 0.1% Tween) containing 5% non‐fat powdered milk, and were then incubated with specific primary antibody against p‐ERK1/2 or total ERK1/2, followed by incubation with horseradish peroxidase‐linked antirabbit IgG. Immunoreactive bands were visualized using the ECL detection system (Amersham), according to the manufacturer's instructions.
Immunohistochemistry. Immunohistochemical analysis of p‐ERK1/2 and total ERK1/2 was carried out using formalin‐fixed, paraffin‐embedded specimens of endometrial cancer tissue. The primary antibody for phosphorylated ERK1/2 was rabbit monoclonal phosphor‐p44/42 MAPK (Thr202/Tyr204) antibody (Cell Signaling Technology), which detects endogenous levels of human p42 and p44 MAP kinase (ERK1 and ERK2) only when they are phosphorylated at Thr202 and Tyr204, respectively. The antibody used for total ERK1/2 was rabbit polyclonal p44/42 MAPK antibody (Cell Signaling Technology), which detects endogenous levels of human p42 and p44 MAP kinase. Neither of these antibodies crossreacts with JNK/SAPK or p38 MAP kinase.
After specimens were deparaffinized in xylene and graded alcohols, epitope retrieval was done. The sections were heated in a microwave oven at 700 W for 10 min in 1 × Antigen Retrieval Solution (Biogenex). Then, endogenous peroxidase was blocked by immersing the sections in 0.3% H2O2 methanol for 30 min. After blocking with horse serum, the sections were incubated for 16 h at 4°C with the (above‐described) appropriate primary antibody at 1:100 dilution. The subsequent steps were carried out using the ABC‐elite kit (Vecter Laboratories), according to the manufacturer's instructions. Color development used the peroxidase substrate 3‐3′‐diaminobenzidine tetrahydrochloride (DAKO Cytomation). All slides were counterstained with GM hematoxylin stain solution (Muto Pure Chemical Co.).
As a positive control, we used sections of mouse tumors that arose from the endometrial cell line EM‐E6/E7/TERT/Ras with an oncogenic mutant KRAS allele.( 11 ) For negative controls, the primary antibody was omitted. The evaluation of immunohistochemistry results was carried out independently by two observers. Staining intensity was classified into four levels: no, weak, moderate and strong expression. Weak expression represented faint and/or focal expression, whereas moderate and strong expression represented significant expression at a rate of >5% positivity for expression among tumor cells in each section. The results of the staining were reviewed without knowledge of the clinicopathological data by two of the coauthors (S. K. and N. M.), who scored the percentage of positive nuclei and/or cytoplasm within tumors. The average staining score was registered, and there was no statistically significant difference in the scoring between observers. For the survival analysis, patients with no expression or weak expression were assigned to the low‐expression group, and those with moderate or strong expression were assigned to the high‐expression group.
Real‐time polymerase chain reaction. Total RNA was extracted from 22 surgical tissue specimens (14 samples with low p‐ERK expression and eight samples with high p‐ERK expression) using NucleoSpin RNA II (Marchery‐Nagel) and was reverse transcribed using SuperScript II Reverse Transcriptase and oligo (dT) primers (SuperScript First‐strand Synthesis System for RT‐PCR; Invitrogen). The mRNA expression levels of the ERK1 and ERK2 genes were measured by fluorescence‐based, TaqMan real‐time polymerase chain reaction (PCR), using the ABI PRISM 7000 Sequence Detection System (Applied Biosystems). Real‐time PCR was carried out with a 40‐µL reaction mixture containing 2 × TaqMan Universal PCR Master Mix (Applied Biosystems), 20 × primer–probe mix (MAPK1, Hs00177066‐m1; MAPK3, Hs00385075‐m1; Applied Biosystems). After 2 min at 50°C and 10 min at 95°C, amplification was carried out using 40 cycles of 95°C for 15 s and 60°C for 60 s. As the control, glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) mRNA expression was examined. To detect GAPDH mRNA expression, assays that measured the gene expression products of the GAPDH gene (Applied Biosystems) were used. The amplification plot, the graph of the increment of fluorescence reporter signal vs cycle number during PCR, was examined early in the reaction, at a point that represents the logarithmic phase of product accumulation.
KRAS and BRAF mutation analysis. We previously reported the presence of KRAS mutation in endometrial cancer. In the present study, we used a greater number of samples (44 cases) to assay for KRAS and BRAF mutation. Exon 1 of KRAS (including codons 12 and 13) and exons 11 and 15 of BRAF were amplified by PCR, using DNA extracted from endometrial cancer tissue as a template. The primer sets used for KRAS were as follows: forward, 5′‐GGTACTGGTGGAGTATTTGATAGTG‐3′; and reverse, 5′‐CATGAAAATGGTCAGAGAAACC‐3′. The primer sets used for BRAF were as follows: exon 11 forward, 5′‐TCCCTCTCAGGCATAAGGTAA‐3′; exon 11 reverse, 5′‐CGAACAGTGAATATTTCCTTTGAT‐3′; exon 15 forward, 5′‐TAGGAAAGCATCTCACCTCATCC‐3′; and exon 15 reverse, 5′‐CACTGATTTTTGTGAATACTGGGAAC‐3′. PCR products were purified using the Qiagen PCR purification kit (Qiagen), and direct sequencing was then carried out. PCR products with suspected mutations were reamplified and subsequently cloned into pGEM‐T Easy (Promega), and at least three of those clones were sequenced.
Statistical analysis. We used the χ2‐test for 2 × 2 tables to compare the categorical data. Life tables were computed using the Kaplan–Meier method, and the Log‐rank test was used to assess statistical significance. Cox proportional hazards analysis was used to determine the relative contribution of various factors to the risk of recurrence. A P‐value of <0.05 was considered to indicate statistical significance. All statistical analyses were done using the statistical package StatView version 5.0 (Abacus Concepts).
Results
Expression of phosphorylated ERK1/2 in endometrial cancer. Figure 1 shows the results of western blot analysis to confirm the validity of the antibody used for p‐ERK1/2 immunohistochemistry. When the endometrial epithelium‐derived cell line (EM‐E6/E7/TERT) was treated with EGF, a band of p‐ERK1/2 (p‐p42/44) was clearly induced, whereas the amount of total ERK1/2 was not altered. We also examined the expression of p‐ERK1/2 in EM‐E6/E7/TERT cells with oncogenic mutant KRAS alleles. The expression of p‐ERK1/2 was significantly upregulated in these cells. This induction was partially cancelled by treatment with the specific MEK inhibitor U0126, but not by the p38 inhibitor SB203580 or the PI3K inhibitor wortmannin. These findings indicate that the main mechanism of ERK1/2 regulation upon mitogenic stimuli is its phosphorylation, and show that the antibody we used recognizes phosphorylated ERK1/2 with sufficient sensitivity and specificity.
Figure 1.

Validation of the phosphorylated extracellular‐regulated kinase (p‐ERK1/2) antibody using western blot analysis. The endometrial epithelium‐derived cell line EM‐E6/E7/TERT was incubated with or without epidermal growth factor (EGF) at 10 or 100 nM for 15 min, and protein extracts were prepared and subjected to western blot analysis to assay expression of p‐ERK1/2. EM‐E6/E7/TERT cells transfected with oncogenic mutant KRAS were used as a positive control of p‐ERK1/2 expression. Two bands (42 and 44 kDa) were found to be induced by treatment with EGF or transfection with oncogenic mutant KRAS, and this effect was specifically inhibited by treatment with a MAP kinase‐ERK kinase (MEK) inhibitor (U0126), but not by a p38 (SB203580) or phosphatidylinositol‐3‐kinase (PI3K) inhibitor (Wortmannin). The expression levels of total ERK1/2 were not altered by these treatments.
Expression levels of p‐ERK1/2 varied widely among samples (Fig. 2). We found that 18 samples had weak expression of p‐ERK1/2, 15 had moderate expression, and eight had strong expression. Thus, 40 of the 63 samples (63%) had p‐ERK1/2 expression. The p‐ERK1/2 signals were detected mainly in nuclei (Fig. 2c), but most samples with strong expression had both nucleic and cytoplasmic expression (Fig. 2e–f). We also examined the expression of total ERK1/2 in a subset of 26 tumor specimens. Homogeneous but varying levels of staining for total ERK1/2 were observed in all 26 samples, independent of the p‐ERK1/2 status (data not shown). Furthermore, we measured the expression levels of ERK1 or ERK2 mRNA by quantitative RT‐PCR in 22 patients whose RNA was available for the analysis, and compared them with the status of p‐ERK1/2 expression. As shown in Fig. 3, no significant correlation was observed between ERK1 or ERK2 mRNA expression and p‐ERK1/2 expression. These findings indicate that expression of p‐ERK1/2 is not the consequence of overexpression of ERK1/2 but the result of hyperactivation of ERK1/2.
Figure 2.
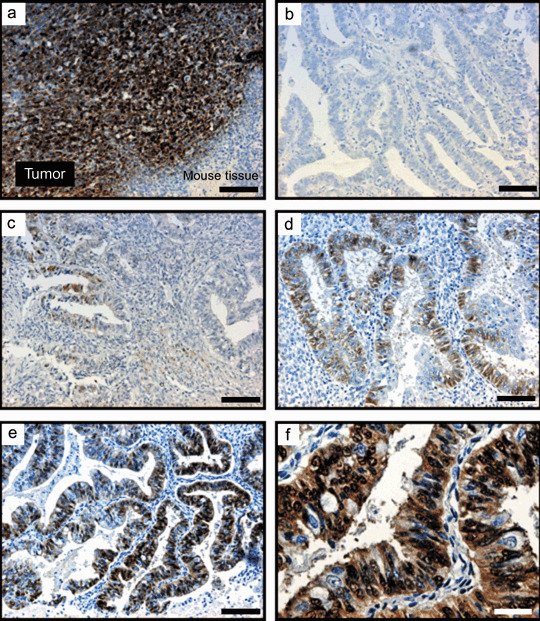
Immunohistochemical staining of phosphorylated extracellular‐regulated kinase (p‐ERK1/2) in endometrial cancer specimens. (a) Positive control consisted of sections of mouse tumor that arose from endometrial epithelial cells with an oncogenic mutant KRAS allele (EM‐E6/E7/TERT/Ras), and exhibited significant p‐ERK1/2 expression in tumor but not in surrounding mouse tissue. (b) A sample with negative staining. (c) A sample with weak, focal staining classified as weak expression. (d) A case with moderate expression. (e,f) A case with strong expression. (a–e) ×100; (f) ×400. Black bars = 100 µm. White bar = 25 µm.
Figure 3.
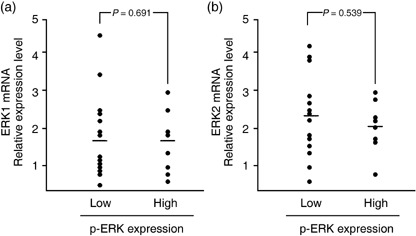
Relationship between extracellular‐regulated kinase (ERK1/2) mRNA and phosphorylated ERK (p‐ERK) expression in endometrial cancer specimens. Relative levels of (a) ERK1 and (b) ERK2 mRNA expression were measured by real‐time polymerase chain reaction in 22 endometrial cancer specimens and were compared between two groups with low and high p‐ERK expression. Bars represent mean values.
The patients were stratified into two groups depending on the expression levels of p‐ERK1/2. Patients with no or weak expression were assigned to the low‐expression group, and those with moderate or strong expression were assigned to the high‐expression group, and the relationships between p‐ERK1/2 expression and clinicopathological variables are shown in Table 1. There was no significant correlation between p‐ERK1/2 expression and clinicopathological characteristics including patient age, FigO staging, pathological grade, depth of myometrial invasion, pelvic lymph node metastasis, menopause or body mass index.
Table 1.
Associations between phosphorylated extracellular‐regulated kinase (p‐ERK1/2) expression and clinicopathological factors in patients with endometrial cancer
| Variables | No. patients | p‐ERK1/2 expression † | P‐value ‡ | |
|---|---|---|---|---|
| Low | High | |||
| Age (years) | ||||
| <60 | 39 | 18 | 21 | 0.06 |
| ≥60 | 24 | 5 | 19 | |
| FigO stage | ||||
| I | 46 | 18 | 28 | 0.56 |
| II‐IV | 17 | 5 | 12 | |
| Grade | ||||
| 1 | 34 | 16 | 18 | 0.07 |
| 2, 3 | 29 | 7 | 23 | |
| Myometrial invasion | ||||
| <1/2 | 47 | 20 | 27 | 0.13 |
| ≥1/2 | 16 | 3 | 13 | |
| Pelvic lymph node metastasis | ||||
| No | 58 | 21 | 37 | > 0.999 |
| Yes | 5 | 2 | 3 | |
| Menopause | ||||
| Premenopausal | 23 | 11 | 12 | 0.18 |
| Postmenopausal | 40 | 12 | 28 | |
| Body mass index | ||||
| <25 | 38 | 15 | 23 | 0.60 |
| ≥25 | 25 | 8 | 17 | |
High and low mean moderate/strong expression and no/weak expression, respectively.
‡ Fisher's exact method.
Identification of KRAS and BRAF mutation and correlation with p‐ERK expression. Of the 63 samples we examined, 44 were available for DNA sequencing. Of those 44 samples, 10 samples had a mutation in KRAS codon 12 and/or codon 13 (Table 2). We examined the correlation between KRAS mutation and p‐ERK1/2 expression. Seven of the 10 cancers with KRAS mutation (70%) had p‐ERK1/2 expression, whereas 24 of the 34 cancers without KRAS mutation (71%) had p‐ERK1/2 expression. Thus, there was no significant correlation between KRAS mutation and p‐ERK1/2 expression. Also, exons 11 and 15 of the BRAF gene were sequenced; no mutations were detected in any of the samples examined, and this was confirmed by repeated sequencing of both the upper and lower strands.
Table 2.
Correlation between KRAS/BRAF mutations and phosphorylated extracellular‐regulated kinase (p‐ERK1/2) expression in endometrial cancer
| p‐ERK1/2 expression | n | KRAS sequence | BRAF sequence | ||||
|---|---|---|---|---|---|---|---|
| Exon 1 | Exon 11 | Exon 15 | |||||
| Mutation | Wild type | Mutation | Wild type | Mutation | Wild type | ||
| None | 13 | 3 | 10 | 0 | 13 | 0 | 13 |
| Weak | 13 | 4 | 9 | 0 | 13 | 0 | 13 |
| Moderate | 12 | 1 | 11 | 0 | 12 | 0 | 12 |
| Strong | 6 | 2 | 4 | 0 | 6 | 0 | 6 |
| Total | 44 | 10 | 34 | 0 | 44 | 0 | 44 |
Effect of p‐ERK expression on prognosis of endometrial cancer. We next examined the prognostic effect of p‐ERK1/2 expression. The mean follow‐up for all patients was 4.08 years for relapse‐free survival (RFS; median, 3.51 years; range, 0.15–9.63 years) and 4.31 years for overall survival (OS; median, 3.75 years; range, 0.56–9.63 years). First, the effect of p‐ERK1/2 expression on RFS was analyzed. Among the 63 patients, 11 patients (17.5%) had suffered relapses of endometrial cancer at the time of the last follow‐up. Figure 4 illustrates univariate outcomes based on p‐ERK1/2 expression. The RFS for the high‐ and low‐expression groups were 95.7 and 75.0%, respectively (P = 0.0407). Next, the effect of p‐ERK1/2 on OS was analyzed. Among the 63 patients, 12 patients (19.0%) had died (eight from endometrial cancer, and four from other causes [breast cancer, melanoma, liver cancer and lung embolism]) at the time of the last follow‐up. The OS for the high and low p‐ERK1/2 expression groups were 95.7 and 72.5%, respectively (P = 0.0203). Thus, low p‐ERK1/2 expression negatively affected RFS and OS in univariate analysis. Among other clinicopathological factors, FigO stage ≥II (P < 0.0001), myometrial invasion ≥1/2 (P = 0.0009) and tumor grade ≥2 (P = 0.043) negatively affected RFS in univariate analysis. Similarly, FigO stage ≥II (P = 0.0170) and myometrial invasion ≥1/2 (P = 0.0111) negatively affected OS in univariate analysis (Table 3).
Figure 4.

Kaplan–Meier plots of (a) relapse‐free and (b) overall survival of groups defined by phosphorylated extracellular‐regulated kinase (p‐ERK1/2) immunohistochemistry The tumors were divided into two groups: high and low expression of p‐ERK1/2.
Table 3.
Prognostic analysis for overall survival
| Variable | Risk factor | Univariate (Kaplan–Meier) Log rank test (P‐value) | Multivariate (Cox regression model) Hazard ratio (95% confidence interval) | P‐value |
|---|---|---|---|---|
| FigO stage | ≥II | 0.0170 | 3.492 (1.012–12.049) | 0.0478 |
| Myometrial invasion | ≥1/2 | 0.0111 | 2.341 (0.713–7.693) | 0.1610 |
| p‐ERK1/2 expression | Low | 0.0203 | 8.202 (1.026–65.550) | 0.0472 |
Low means no or weak expression. p‐ERK1/2, phosphorylated extracellular‐regulated kinase.
Finally, when the above prognostic factors that affected RFS or OS evaluated in the univariate analyses were included in a Cox proportional‐hazard analysis, only FigO stage was an independent predictive factor for RFS (Hazard ratio 6.21, P = 0.01), and both FigO stage (Hazard ratio 3.492, P = 0.0478) and low p‐ERK1/2 expression (Hazard ratio 8.202, P = 0.0472) were independent predictive factors of OS (Table 3).
Discussion
In the present study, we demonstrated that the ERK–MAPK pathway was frequently activated, independent of the status of KRAS and BRAF, in endometrioid‐type endometrial cancer. We found that 65% of the endometrial cancers expressed p‐ERK1/2, whereas the KRAS mutation was detected in 22% of the patients, and there was no significant correlation between KRAS mutation and p‐ERK1/2 expression. Thus, the present results indicate that KRAS mutation is not a major determinant of ERK activation in endometrial cancer. Alternate pathways may therefore exist for ERK activation, such as via crosstalk with the PI3K pathway.( 12 ) It is likely that activation of PI3K signaling by the PTEN mutation (which was detected in 30–40% of patients in a previous study)( 5 ) contributes to increased p‐ERK1/2 expression via crosstalk. However, we found that three of the 10 patients with the KRAS mutation lacked p‐ERK1/2 expression. The reason for the lack of p‐ERK1/2 expression despite KRAS mutation remains unclear. Recently, several negative regulators of the MAPK signaling pathway upstream of ERK were identified, including Sprouty and Spred.( 13 , 14 , 15 , 16 ) Spred inhibits MAPK signaling at the level of Raf by binding to Ras and Raf, whereas Sprouty is thought to target Grb, Sos and Raf. Activation of these negative regulators inhibits phosphorylation of ERK1/2, even in the presence of the KRAS mutation. We are currently analyzing the status of expression of these negative regulators in relation to the KRAS mutation and p‐ERK1/2 expression levels. Overall, our data indicate that complex regulatory pathways are involved in ERK phosphorylation in endometrial cancer, which does not necessarily involve the KRAS mutation.
Another interesting finding of the present study is the complete lack of BRAF mutation in endometrial cancer. Although an increasing number of studies have identified mutations of BRAF in a wide variety of cancer types,( 17 ) there is controversy as to whether BRAF mutations are associated with endometrial cancer. To our knowledge, there have been four reported studies of BRAF mutations in endometrial cancer. In three of those four studies, no BRAF mutations or a very low frequency of BRAF mutations was detected: one of 146 cases,( 18 ) one of 48 cases,( 19 ) and none of 67 cases.( 20 ) In the remaining study, which was conducted in Japan, 20 of 97 endometrial cancer patients (21%) had a BRAF mutation.( 21 ) This discrepancy raises confusion regarding the involvement of BRAF mutations in endometrial carcinogenesis. The question may thus arise whether BRAF is population‐specific, especially with Japanese. However, the present results make this unlikely, and are consistent with the three above‐described non‐Japanese reports. The available evidence indicates that BRAF mutations are not major constituents of MAPK signaling in endometrial cancer, whereas they play a major role in other epithelial tumors.
The novel finding we obtained in the present study is that p‐ERK1/2 expression appears to have a favorable effect on patient survival. This is in contrast to previous in vitro data showing that the MAPK pathway is involved in cellular proliferation and motility. Limited investigations indicate that p‐ERK expression has prognostic significance in various tumor types, but the results of theses studies are controversial. In salivary gland cancer, high levels of expression of p‐ERK were associated with early progression of the tumor.( 22 ) In breast cancer, high levels of MAPK activity in the cytosol coincided with positive lymph node involvement and higher risk of relapse within 40 months of follow‐up, but there was no significant correlation.( 23 ) In contrast, a recent study demonstrated that high p‐ERK expression in breast cancer correlated with a low frequency of recurrence and infrequent fatal outcome, and that high p‐ERK expression was an independent indicator of long relapse‐free survival and overall survival in multivariate analysis.( 24 ) Another study of breast cancer indicates that p‐ERK expression is associated with small tumors with better survival.( 19 ) In a study of ovarian tumors, patients with high ERK and p‐ERK expression in pleural effusions had better overall survival than patients with low ERK or p‐ERK values.( 25 ) Why do such discrepancies arise among studies or tumor types? One possible explanation is different assay conditions of immunohistochemistry or western blotting. For evaluation of p‐ERK expression, the type of antibody used is critical, and validation of the antibody should be carried out carefully. Differences between methods of lysate extraction in western blotting may cause differences in results. ERK is located in both the nucleus and cytosol, but is found mainly in the nucleus. In whole‐cell lysate extraction, the ratio of cytoplasmic and nuclear fractions depends strongly on the method of extraction, which affects evaluation of expression levels. In addition, differences in the ratio of epithelial and stromal components in each tumor sample cause differences in the results of ERK expression assays, which is a limitation and shortcoming of western blot analysis.
The physiological importance of MAPK signaling is due to the fact that it transduces signals from membrane receptors to transcriptional machinery in the nucleus. MAPK signaling plays pivotal roles in physiological proliferation of normal cells. In fact, normal endometrium expresses p‐ERK more frequently than endometrial cancer, and proliferation of normal endometrium appears to be highly dependent on MAPK signaling (Kyo S, Hashimoto M unpublished data), indicating that p‐ERK expression is required for proliferation of normal endometrium, rather than progression of cancer cells. Cancer cells usually obtain their own pathway for progression and may not highly depend on such physiological signals. For example, during tumorigenesis in estrogen‐dependent organs, cancer cells are likely to lose expression of estrogen receptors and achieve hormone‐independent growth, leading to more aggressive phenotypes, in which genetic rearrangements such as mutation, deletion and translocation of genes more seriously contribute to the malignant potential of cells. This theory may be applied to p‐ERK expression in endometrial cancer.
In summary, the present results indicate that p‐ERK1/2 is frequently expressed in endometrial cancer, independent of KRAS and BRAF status, indicating that RAS/RAF mutation is not the main cause of MAPK signaling in this tumor type. The expression level of p‐ERK1/2 is an independent prognostic indicator, and may be useful as a clinical parameter for prediction of patient survival. The present data raise the question of whether molecular therapies that target the MEK–ERK pathway are effective against endometrial cancer.
Acknowledgments
We wish to thank members of the Department of Pathology, Kanazawa University Hospital, for tissue preparation for immunohistochemistry. This study was supported by a Grant‐in‐Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS) and the Megumi Medical Foundation of Kanazawa University.
References
- 1. Tashiro H, Blazes MS, Wu R et al. Mutations in PTEN are frequent in endometrial carcinoma but rare in other common gynecological malignancies. Cancer Res 1997; 57: 3935–40. [PubMed] [Google Scholar]
- 2. Mizuuchi H, Nasim S, Kudo R, Silverberg SG, Greenhouse S, Garrett CT. Clinical implications of K‐ras mutations in malignant epithelial tumors of the endometrium. Cancer Res 1992; 52: 2777–81. [PubMed] [Google Scholar]
- 3. Imamura T, Arima T, Kato H, Miyamoto S, Sasazuki T, Wake N. Chromosomal deletions and K‐ras gene mutations in human endometrial carcinomas. Int J Cancer 1992; 51: 47–52. [DOI] [PubMed] [Google Scholar]
- 4. Enomoto T, Fujita M, Inoue M et al. Alterations of the p53 tumor suppressor gene and its association with activation of the c‐K‐ras‐2 protooncogene in premalignant and malignant lesions of the human uterine endometrium. Cancer Res 1993; 53: 1883–8. [PubMed] [Google Scholar]
- 5. Kanaya T, Kyo S, Sakaguchi J et al. Association of mismatch repair deficiency with PTEN frameshift mutations in endometrial cancers and the precursors in a Japanese population. Am J Clin Pathol 2005; 124: 1–8. [DOI] [PubMed] [Google Scholar]
- 6. Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006; 441: 424–30. [DOI] [PubMed] [Google Scholar]
- 7. Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal‐regulated kinase activation. Cell 1999; 80: 179–85. [DOI] [PubMed] [Google Scholar]
- 8. Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature 2001; 410: 37–40. [DOI] [PubMed] [Google Scholar]
- 9. Pearson G, Robinson F, Beers Gibson T et al. Mitogen‐activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev 2001; 22: 153–83. [DOI] [PubMed] [Google Scholar]
- 10. Kyo S, Nakamura M, Kiyono T et al. Successful immortalization of endometrial glandular cells with normal structural and functional characteristics. Am J Pathol 2003; 163: 2259–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mizumoto Y, Kyo S, Ohno S et al. Creation of tumorigenic human endometrial epithelial cells with intact chromosomes by introducing defined genetic elements. Oncogene 2006; 25: 5673–82. [DOI] [PubMed] [Google Scholar]
- 12. Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer 2006; 6: 184–92. [DOI] [PubMed] [Google Scholar]
- 13. Hacohen N, Kramer S, Sutherland D, Hiromi Y, Krasnow MA. Sprouty encodes a novel antagonist of FGF signaling that patterns apical branching of the Drosophila airways. Cell 1998; 92: 253–63. [DOI] [PubMed] [Google Scholar]
- 14. Wakioka T, Sasaki A, Kato R et al. Spred is a Sprouty‐related suppressor of Ras signalling. Nature 2001; 412: 647–51. [DOI] [PubMed] [Google Scholar]
- 15. Furthauer M, Lin W, Ang SL, Thisse B, Thisse C. Sef is a feedback‐induced antagonist of Ras/MAPK‐mediated FGF signalling. Nat Cell Biol 2002; 4: 170–4. [DOI] [PubMed] [Google Scholar]
- 16. Tsang M, Friesel R, Kudoh T, Dawid IB. Identification of Sef, a novel modulator of FGF signalling. Nat Cell Biol 2002; 4: 165–9. [DOI] [PubMed] [Google Scholar]
- 17. Davies H, Bignell GR, Cox C et al. Mutations of the BRAF gene in human cancer. Nature 2002; 417: 949–54. [DOI] [PubMed] [Google Scholar]
- 18. Mutch DG, Powell MA, Mallon MA, Goodfellow PJ. RAS/RAF mutation and defective DNA mismatch repair in endometrial cancers. Am J Obstet Gynecol 2004; 190: 935–42. [DOI] [PubMed] [Google Scholar]
- 19. Svensson S, Jirstrom K, Ryden L et al. ERK phosphorylation is linked to VEGFR2 expression and Ets‐2 phosphorylation in breast cancer and is associated with tamoxifen treatment resistance and small tumours with good prognosis. Oncogene 2005; 24: 4370–9. [DOI] [PubMed] [Google Scholar]
- 20. Pappa KI, Choleza M, Markaki S et al. Consistent absence of BRAF mutations in cervical and endometrial cancer despite KRAS mutation status. Gynecol Oncol 2006; 100: 596–600. [DOI] [PubMed] [Google Scholar]
- 21. Feng YZ, Shiozawa T, Miyamoto T et al. BRAF mutation in endometrial carcinoma and hyperplasia: correlation with KRAS and p53 mutations and mismatch repair protein expression. Clin Cancer Res 2005; 11: 6133–8. [DOI] [PubMed] [Google Scholar]
- 22. Handra‐Luca A, Bilal H, Bertrand JC, Fouret P. Extra‐cellular signal‐regulated ERK‐1/ERK‐2 pathway activation in human salivary gland mucoepidermoid carcinoma: association to aggressive tumor behavior and tumor cell proliferation. Am J Pathol 2003; 163: 957–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mueller H, Flury N, Eppenberger‐Castori S, Kueng W, David F, Eppenberger U. Potential prognostic value of mitogen‐activated protein kinase activity for disease‐free survival of primary breast cancer patients. Int J Cancer 2000; 89: 384–8. [DOI] [PubMed] [Google Scholar]
- 24. Milde‐Langosch K, Bamberger AM, Rieck G et al. Expression and prognostic relevance of activated extracellular‐regulated kinases (ERK1/2) in breast cancer. Br J Cancer 2005; 92: 2206–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Givant‐Horwitz V, Davidson B, Lazarovici P et al. Mitogen‐activated protein kinases (MAPK) as predictors of clinical outcome in serous ovarian carcinoma in effusions. Gynecol Oncol 2003; 91: 160–72. [DOI] [PubMed] [Google Scholar]