

Abstract
Lapatinib is a small molecule inhibitor of both HER2 and the epidermal growth factor receptor (EGFR). We investigated the effect of treatment with lapatinib alone or in combination with a fluoropyrimidine derivative S‐1 against pancreatic cancer. The HER2/EGFR expression in each of the four pancreatic cancer cell lines MiaPaca‐2, PANC‐1, Capan‐1 and Capan‐2 was measured by flow cytometry. The anti‐tumor effects of lapatinib (30 mg/kg) and/or S‐1 (10 mg/kg) were evaluated using female BALB/c nude mice xenografts generated using these four cell lines. Synergy between lapatinib and S‐1 was examined by median effect analysis in vitro. Resected pancreatic cancer tissues from 137 patients were immunohistochemically stained with anti‐human HER2 and EGFR antibodies. The administration of lapatinib as a single agent substantially suppressed tumor growth in vivo of all pancreatic cancer cell lines examined. A strong correlation was observed between HER2 expression and the anti‐tumor effect of lapatinib in vivo. Lapatinib synergized with S‐1 to inhibit the tumor growth of MiaPaca‐2 and PANC‐1 xenografts. When used as a single agent in vitro, lapatinib barely inhibit the cell growth of any cell line. However, lapatinib synergized with the anti‐tumor activity of the S‐1 components 5‐fluorouracil and 5‐chloro‐2,4‐dihydrogenase against all cell lines. Immunohistochemical staining demonstrated that 70% of the pancreatic cancers overexpressed HER2 and/or EGFR. Both lapatinib monotherapy and combined treatment with S‐1 may be promising treatments for patients with pancreatic cancers; the majority these cancers express lapatinib target molecules. (Cancer Sci 2009; 00: 000–000)
Pancreatic cancer is the fifth leading cause of cancer death in men and the sixth in women in Japan, and the disease population is similar to that in the USA and Europe. In Japan, the resectability rate of pancreatic cancer is approximately 40% and therefore, the majority of pancreatic cancers are unresectable. Despite recent advances in therapeutic strategies for pancreatic cancer, the 5‐year survival rate of International Union against Cancer (UICC) stage IV pancreatic cancer is only 4.0%.( 1 )
Chemotherapy of locally advanced or metastatic pancreatic cancer by intravenous administration of gemcitabine (GEM) modestly improves patient survival compared to treatment with continuous venous infusion of 5‐fluorouracil (5‐FU), as the median survival time (MST) is 5.65 versus 4.41 months, respectively.( 2 ) To date, two regimens that include GEM have been proved to result in a higher survival rate than GEM alone in phase III studies. One of these regimens is GEM together with erlotinib, an agent whose molecular target is the epidermal growth factor receptor (EGFR) (MST 6.24 vs 5.91 months for GEM alone).( 3 ) The second regimen is GEM together with capecitabine, an oral fluoropyrimidine that is enzymatically converted to 5‐FU within the tumor (MST 8.4 vs 7.2 months for GEM alone).( 4 ) In Japan, the oral fluorouracil derivative S‐1 has been clinically used for pancreatic cancer since the agent was approved by the Japanese health insurance industry in 2006. A phase II study of S‐1 efficacy against metastatic pancreatic cancer indicated that the MST of patients treated with S‐1 is 5.6 months.( 5 ) S‐1 contains tegafur (a masked compound of 5‐FU) and a reversible competitive dihydropyrimidine dehydrogenase (DPD) inhibitor, 5‐chloro‐2,4‐dihydrogenase (CDHP). Since DPD is a rate‐limiting enzyme that degrades 5‐FU, CDHP is expected to enhance the cytotoxicity of 5‐FU by prolonging high 5‐FU concentrations in blood and tumor tissues. Because S‐1 monotherapy does not have satisfactory efficacy against pancreatic cancer, many studies have tested if S‐1 is more efficient when combined with GEM. Two phase I and two phase II studies of this combination therapy showed promising efficacies (MST ranging 7.6–12.5 months).( 6 , 7 , 8 , 9 ) Since the general status of patients with advanced pancreatic cancer is usually unfavorable, weekly outpatient treatment of a GEM‐containing regimen may be a substantial physical burden for the patient. In this context, an effective oral anti‐cancer agent would be ideal for combination therapy with S‐1 for advanced pancreatic cancer. Since small molecule targeting agents are usually given orally, such an agent may be a candidate for combination therapy with S‐1.
The EGFR and human epidermal growth factor receptor (HER2) tyrosine kinases play essential roles in ligand‐activated signaling pathways that regulate cell proliferation and differentiation.( 10 ) Many antibodies or small molecule inhibitors targeting HER2 and/or EGFR have already been approved for therapy in clinical trials. Lapatinib, a novel, orally administered, small‐molecule dual tyrosine kinase inhibitor of HER2 and EGFR, has been approved by the FDA for the treatment of patients with advanced or metastatic breast cancer whose tumors overexpress HER2. To date, lapatinib has not been approved as a therapeutic agent against pancreatic cancer in any country. Furthermore, there has been very little basic investigation of the potential anti‐tumor effect of lapatinib for pancreatic cancer.
The purpose of this study was to evaluate the anti‐tumor effect of lapatinib against pancreatic cancer when administered as a single agent, and to correlate lapatinib efficacy with tumor HER2 and EGFR expression. We further investigated potential synergy between lapatinib and S‐1 against human pancreatic cancer.
Materials and Methods
Cell culture. The pancreatic cancer cell lines MiaPaca‐2, PANC‐1, Capan‐1 and Capan‐2 were purchased from the American Type Culture Collection (Manassas, VA, USA). The cells were maintained at 37°C in a humidified atmosphere of 5% CO2 and were continuously grown in high‐glucose DMEM (Nikken Bio., Kyoto, Japan) supplemented with 10%FBS (Life Technologies, Inc., Grand Island, NY, USA), 100 IU/mL penicillin (ICN Biomedicals, Costa Mesa, CA, USA), 10 μg/mL streptomycin (ICN Biomedicals) and 0.5 mmol/L sodium pyruvate (Cambrex, Walkersville, MD, USA).
Compounds. For in vitro experiments, lapatinib was purchased from Toronto Research Chemicals, Inc. (North York, ON, Canada). For in vivo experiments, lapatinib (Tykerb) was purchased from GlaxoSmithKline (Brentford, UK). S‐1 and CDHP were kindly provided by Taiho Inc. (Tokyo, Japan), and 5‐FU, ethanol and DMSO were purchased from Wako Pure Chemical Industries, Ltd (Osaka, Japan).
Flow cytometric analysis. The expression level of HER2 and EGFR in pancreatic cancer cell lines was analyzed by flow cytometry using a FACScan (CellQuest FACSCalibur, Becton Dickinson, Mountain View, CA, USA). The cells were suspended at a density of 1 × 106 cells/mL in PBS supplemented with 4% FBS. The phycoerythrin‐labeled mouse anti‐human EGFR and HER2 mAbs (BD Biosciences, San Diego, CA, USA) were added as primary antibodies and the cells were incubated at 4°C for 30 min. After two washes with PBS supplemented with 4% FBS, the cells were re‐suspended in 1 mL of PBS supplemented with 4% FBS. Fluorescence was measured by flow cytometry using a FACScan analyzer (BD Biosciences). The percentage of EGFR and HER2‐positive cells and the mean fluorescence intensity (MFI) were calculated using the supplied FACScan software and were compared with those of isotype‐matched control‐stained cells. MFI data were presented as mean ± SD of three independent experiments.
Xenograft tumorigenicity studies. Female BALB/c nude mice (4 weeks old) were obtained from Nihon CLEA (Tokyo, Japan). The ethical standards of the experiment were approved by the Animal Research Committee of Osaka City University Graduate School of Medicine, and the animals were maintained in accordance with our institutional guidelines. The mice were acclimatized at the Animal Facility of Osaka City University Graduate School of Medicine for 1 week. To produce mouse tumors, pancreatic cancer cells (MiaPaca‐2, PANC‐1, Capan‐1, Capan‐2) were harvested from subconfluent cultures by treatment with 0.25% trypsin and 0.02% EDTA. Trypsinization was stopped using medium containing 10% FBS and the cells were washed once in serum‐free medium and resuspended in DMEM. For each cell line, 1 × 107 cells in 200 μL DMEM were inoculated s.c. into the left flank of the nude mice. Only single‐cell suspensions with >90% viability were used for injection. Treatment was initiated on the seventh day after cell injection, when the xenografts had reached a mean size of 25 to 50 mm3. Tumor‐bearing mice were randomly assigned to one of the following four treatment groups: (i) no treatment (n = 6); (ii) oral administration of S‐1 (10 mg/kg) five times a week for 6 weeks (n = 6); (iii) oral administration of lapatinib (30 mg/kg) five times a week for 6 weeks (n = 6); and (iv) oral administration of S‐1 (10 mg/kg) plus lapatinib (30 mg/kg) five times a week for 6 weeks (n = 6). The administration doses of S‐1 and lapatinib were determined by reference to previous reports.( 11 , 12 ) Tumors were measured weekly and tumor volume was determined using the formula: largest diameter × (smallest diameter)2 × 0.5. Tumor inhibition rate was calculated using the formula: (1 – [tumor volume after treatment]/[tumor volume of no treatment]) × 100 (%).
Cell growth inhibition assay. Concentrations of 5‐FU/CDHP and lapatinib that induced IC50of each cell line were assessed using MTT (Sigma Chemicals, St. Louis, MO, USA). Briefly, cells were seeded in 96‐well tissue culture plates (Costar, Corning, NY, USA) at a density of 1 × 104 per 100 μL/well. Eight wells were assigned to each experimental treatment. The 5‐FU/CDHP mixture was used at a molar ratio of 1:1, which mimics human plasma pharmacokinetics after oral administration of S‐1.( 13 ) After 72‐h drug exposure, 10 μL of MTT solution (2 mg/mL) was added to each well and the cells were incubated for a further 2 h. The reaction was stopped by removal of MTT, and the formazan crystals that had formed were dissolved in DMSO (100 μL/well). The absorbance at 570 nm was recorded using a 96‐well microplate reader (Model 550, Bio‐Rad Laboratories, Hercules, CA, USA). The IC50 of each drug was determined using the dose–effect analysis model as previously described.( 14 )
Median effect analysis. Synergy between the drugs was assessed by carrying out median effect analysis as described by Chou and Talalay.( 15 ) The recommended experimental design for this analysis is to mix the two drugs at their IC50 concentration. However, in our experiments the inhibition of cell growth in each cell line by a high concentration of lapatinib only reached 5.0–8.0% inhibition. Therefore, instead of using the IC50 concentration of lapatinib for each cell line, a baseline dose of lapatinib was determined as 2.5 μm based on the IC50 determined for the inhibition of growth of other normal and tumor cell lines by lapatinib in previous reports.( 11 , 16 ) Lapatinib (2.5 μm) was mixed with 5‐FU or 5‐FU/CDHP at its IC50 value for each pancreatic cell line. The mixture was then serially diluted (4‐, 2‐, 1‐, 0.5‐, 0.25‐ and 0.125‐fold dilutions). The values obtained for growth inhibition of these different drug dilutions were then used to calculate the combination index (CI) by inserting the values as ‘A’ in Calcusyn software (Biosoft, Ferguson, MO, USA). Values of CI denote cytotoxicity: CI < 1 denotes synergistic cytotoxicity, CI = 1 addictive cytotoxicity and CI > 1 antagonistic cytotoxicity.
Immunohistochemical staining. Immunohistochemical staining was performed by the avidin–biotin–peroxidase complex method as described previously.( 17 ) Briefly, the expressions of HER2 and EGFR were assessed in pancreatic cancer tissue samples obtained from 137 patients who underwent macroscopically curative resection at the Department of Surgical Oncology, Osaka City University Hospital, from January 1983 to December 2008. The anti‐human HER2 rabbit polyclonal antibody (DAKO, Inc., Copenhagen, Denmark) and the anti‐human EGFR murine monoclonal antibody (Novocastra Laboratories Ltd, Newcastle, UK) were used as primary antibodies to detect EGFR and HER2 overexpression. All slides were examined by two of the authors (M.K. and B.N.). Final evaluations of ambiguous cases were decided after discussion between the two authors. Only the membrane staining intensity and pattern of HER2/EGFR protein immunoreactivity were evaluated according to the scale presented on the HercepTest™ Scoring Guidelines: Score 0, no staining is observed, or membrane staining is observed in less than 10% of the tumor cells; Score 1+, faint/barely perceptible membrane staining is detected in more than 10% of tumor cells (the cells exhibit incomplete membrane staining); Score 2+, weak or moderate complete membrane staining is observed in more than 10% of tumor cells; and Score 3+, strong complete membrane staining is observed in more than 10% of tumor cells.
Statistical analysis. The unpaired two‐tailed Student’s t‐test was used to compare mean values between two groups. Linear regression analysis was used to assess the relationship between the anti‐tumor effect of lapatinib and HER2/EGFR expression; P < 0.05 was considered statistically significant. Group data are presented as mean ± SD.
Results
Flow cytometry. In order to correlate drug treatment data with tumor HER2 and EGFR expression we first quantified the expression of these growth factor receptors in four different pancreatic cancer cell lines by flow cytometry. The MFI values of HER2 in the cell lines MiaPaca‐2, PANC‐1, Capan‐1 and Capan‐2 were 86.8 ± 1.9, 33.2 ± 5.5, 10.9 ± 1.9 and 55.9 ± 9.5, respectively. The MFI values of EGFR in the same cell lines were 81.0 ± 4.4, 92.0 ± 3.6, 15.2 ± 14.4 and 64.1 ± 12.4, respectively (Fig. 1).
Figure 1.
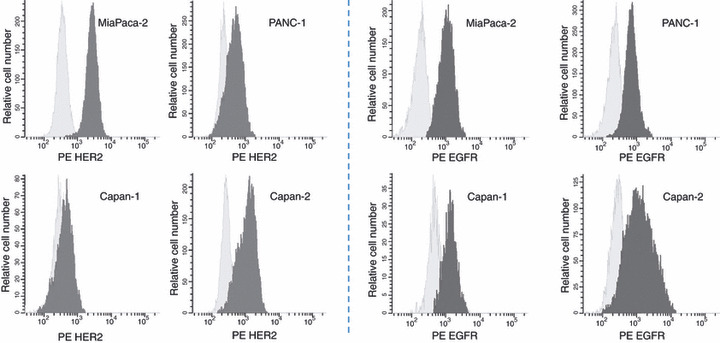
Expression of HER2 and EGFR on the four pancreatic cell lines MiaPaca‐2, PANC‐1, Capan‐1 and Capan‐2 was analyzed by flow cytometry using (left side four figures) phycoerythrin (PE)‐labeled mouse anti‐human HER2 and (right side four figures) EGFR antibody.
Xenograft tumorigenicity studies. The ability of the four pancreatic cell lines to form tumors in mice was next tested in xenograft tumorigenicity studies and the results are shown in Figure 2. The tumor inhibition rates of the xenografts after 6 weeks of treatment are summarized in Table 1. Synergistic effects were observed in MiaPaca‐2 and PANC‐1 (Table 1, Fig. 2).
Figure 2.
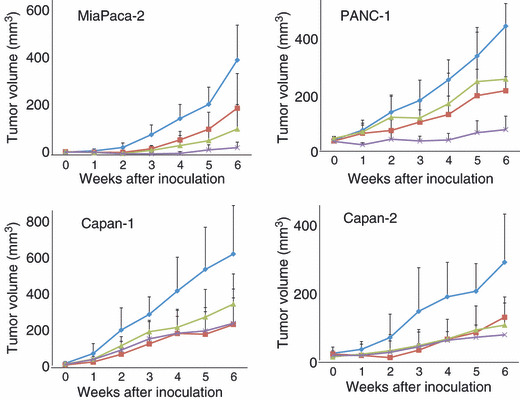
Anti‐tumor activity of lapatinib, S‐1 and lapatinib plus S‐1 against pancreatic cancer xenografts. BALB/c nude mice bearing the indicated tumor xenografts were treated for 6 weeks with no drug (◆), S‐1 (10 mg/kg) five times a week (), lapatinib (30 mg/kg) five times a week (), or S‐1 (10 mg/kg) plus lapatinib (30 mg/kg) five times a week (×). Error bars, ±SD.
Table 1.
Tumor inhibition rate at 6 weeks of treatment
| Cell line | Tumor inhibition rate (%) | P‐value | |||
|---|---|---|---|---|---|
| Lapatinib | S‐1 | Lapatinib + S‐1 | Lapatinib versus Lapatinib+S‐1 | S‐1 versus Lapatinib+S‐1 | |
| MiaPaca‐2 | 72.7 ± 25.8 | 50.7 ± 36.4 | 92.2 ± 5.8 | 0.003 | 0.034 |
| PANC‐1 | 42.3 ± 42.5 | 51.6 ± 11.1 | 82.7 ± 10.7 | 0.024 | 0.005 |
| Capan‐1 | 44.0 ± 31.2 | 62.0 ± 26.4 | 61.0 ± 22.3 | 0.057 | 0.927 |
| Capan‐2 | 62.8 ± 25.1 | 54.9 ± 14.2 | 72.4 ± 17.3 | 0.032 | 0.081 |
P‐value was analysed by Student’s t‐test.
Cell growth inhibition. We then assayed the ability of 5‐FU/CDHP and lapatinib to individually modulate growth inhibition of these cell lines. A dose‐dependent growth inhibition was observed following treatment of all cell lines with 5‐FU/CDHP for 72 h. The IC50 values of 5‐FU/CDHP were 7.9 μm for MiaPaca‐2, 5.7 μm for PANC‐1, 9.3 μm for Capan‐1 and 8.9 μm for Capan‐2. The percentage cell growth inhibition induced by lapatinib at a dose of 30 μm (the maximum dose in our experience) was 5.0% for MiaPaca‐2, 6.0% for PANC‐1, 5.0% for Capan‐1 and 8.0% for Capan‐2.
Median effect analysis. Potential synergy of lapatinib with 5‐FU or lapatinib with 5‐FU/CDHP on cell growth inhibition was than assayed by median effect analysis. This analysis revealed that a combined treatment of lapatinib and 5‐FU/CDHP generated strong synergistic effects on cell growth inhibition for all cell lines examined (Fig. 3). The CI values of lapatinib with 5‐FU/CDHP were smaller than those of lapatinib with 5‐FU alone (Table 2).
Figure 3.
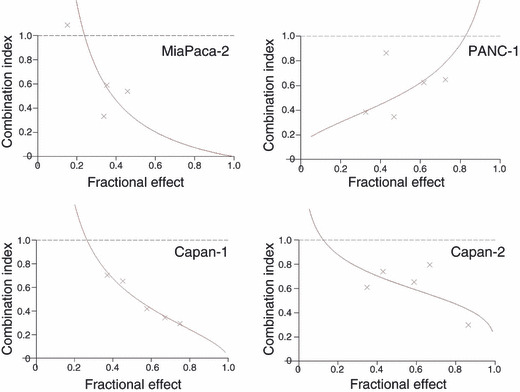
Synergistic anti‐tumor activity of lapatinib and 5‐FU/CDHP for the four pancreatic cancer cell lines was evaluated by median effect analysis and subsequent calculation of the combination index (CI). CI < 1 denotes synergistic cytotoxicity.
Table 2.
Comparion of combination index of lapatinib plus 5‐FU with/without CDHP in pancreatic cancer cells
| Cell line | Combination index | |
|---|---|---|
| Lapatinib + 5‐FU | Lapatinib + 5‐FU/CDHP | |
| MiaPaca‐2 | 0.630 | 0.310 |
| PANC‐1 | 0.795 | 0.529 |
| Capan‐1 | 0.865 | 0.533 |
| Capan‐2 | 1.014 | 0.643 |
Association between HER2/EGFR expression and anti‐tumor effect of lapatinib. To determine if lapatinib anti‐tumor activity against pancreatic cancer xenografts depended on tumor expression of HER2 or EGFR, linear regression analysis of HER2/EGFR expression (MFI) and the relative tumor volume of pancreatic cancer xenografts after 6 weeks of lapatinib treatment was carried out (Fig. 4). This analysis suggested a strong correlation between HER2 expression and the anti‐tumor effect of lapatinib (r = 0.935, P = 0.065). In contrast, there appeared to be no relationship between EGFR expression and the anti‐tumor effect of lapatinib (r = 0.315, P = 0.685).
Figure 4.
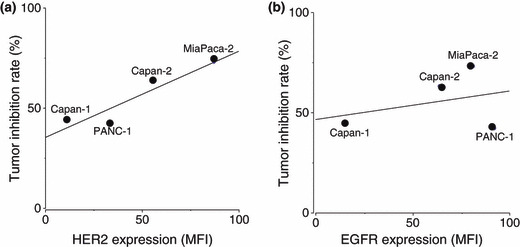
Linear regression analysis of (a) HER2 and (b) EGFR expression versus tumor inhibition rate following lapatinib treatment of tumor xenografts. HER2/EGFR expression of the indicated tumor xenografts was evaluated as the mean fluorescence intensity (MFI) that was measured by flow cytometry.
Immunohistochemistry. Representative positive staining of HER2/EGFR in cancer cells is shown in Figure 5. HER2 was not stained in 22 cases. Of the HER2 positively stained cases, 29 cases had a score of 1+, 42 cases had a score of 2+ and 44 cases had a score of 3+. Therefore, HER2 overexpression (a score of 2+ or 3+) was observed in 86 cases (62.8%). Regarding EGFR expression, 30 cases had a score of 0, 49 cases had of a score of 1+, 53 cases had a score of 2+ and 5 cases had a score of 3+. EGFR overexpression (a score of 2+ or 3+) was observed in 58 cases (42.3%). Seventy percent of cases had the overexpression of HER2 and/or EGFR.
Figure 5.

Representative positive immunohistochemical staining of (a) HER2 and (b) EGFR in pancreatic cancer tissue. Tissue obtained from human pancreatic tumors was immunohistochemically stained using specific anti‐HER2 and ‐EGFR antibodies. Original magnification × 400.
Discussion
EGFR, HER2, HER3 and HER4 are members of the EGFR superfamily and play an important role in cancer development and progression. Upon ligand binding, the extracellular domain undergoes a conformational change, allowing the formation of homodimers or heterodimers with other members of the EGFR family. Dimerization induces tyrosine phosphorylation of specific residues in the intracellular domain that serve as docking sites for adaptor proteins and downstream effectors. As a result, two important signaling pathways, the phosphatidyl inositol 3‐kinase (PI3K) and MAPK pathways, leading to cell proliferation and survival.( 18 , 19 , 20 ) Lapatinib reversibly inhibits the tyrosine kinase activities of both HER2 and the EGFR and thereby inhibits downstream PI3K and MAPK pathways.( 21 )
To date, there have only been two reports concerning the clinical use of lapatinib as a treatment for pancreatic cancer. One study is an ongoing phase II study of the use of lapatinib for unresectable pancreatic cancer that is being carried out in the USA (Clinical Trials Government Identifier NCT00881621). The second study is a phase I study also in the USA aimed at evaluation of the clinically relevant dose of lapatinib in combination with GEM and GEM/oxaliplatin (GEMOX) for patients with advanced pancreaticobiliary cancer.( 22 ) In this phase I study, a dose of lapatinib of 1500 mg/day was administered together with weekly administration of GEM. The maximum tolerated dose of lapatinib was 1000 mg/day when combined with GEMOX administration. To the best of our knowledge, there have been few basic studies on the potential of lapatinib for the treatment of pancreatic cancer.
Our results showed that treatment with lapatinib as a single agent reduced the volume of tumor xenografts generated from four different pancreatic tumor cell lines by 42.3–72.7% of the volume of the corresponding control tumor that received no treatment (Table 1). Moreover, the anti‐tumor effect of lapatinib increased with the tumor expression of HER2 (Fig. 4). A strong correlation (r = 0.935) was observed between HER2 expression and the anti‐tumor effect of lapatinib, although a significant correlation could not be demonstrated due to the small number of cell lines tested (P = 0.065). We failed to demonstrate the correlation between EGFR expression and anti‐tumor effect of lapatinib in four cell lines tested. However, a positive correlation between the two parameters was suggested in the cell lines except PANC‐1. The anti‐tumor effect of lapatinib may be also expected through the EGFR inhibition. At least, these data suggested that either inhibition of HER2 activation or of HER2 downstream signaling pathways may be the main mechanism underlying the anti‐tumor effect of lapatinib in pancreatic cancer cells.
In our in vivo study, tumor growth inhibition in the lapatinib plus S‐1‐treated group was significantly higher than that in the lapatinib mono‐treated group for tumors generated from the MiaPaca‐2, PANC‐1 and Capan‐2 cell lines (Table 1). Furthermore, synergy between lapatinib and 5‐FU/CDHP for tumor inhibition was shown for all four cell lines by median effect analysis (Fig. 3). Lapatinib and 5‐FU alone also demonstrated the synergistic effect in three cell lines (MiaPaca‐2, PANC‐1, Capan‐1) and additive effect in Capan‐2 (Table 2). The smaller CI values of lapatinib with 5‐FU/CDHP compared to those of lapatinib with 5‐FU alone may indicate that CDHP may enhance the synergistic effect of lapatinib with 5‐FU.
A synergistic anti‐tumor effect has been previously shown for S‐1 and gefitinib, an epidermal growth factor receptor inhibitor, in non‐small cell lung cancer cell lines.( 12 ) In that study, it was demonstrated that thymidylate synthase (TS) downregulation was induced by gefitinib within 48 h, leading to the speculation that TS downregulation by gefitinib may be responsible for the synergistic anti‐tumor effect of the combined treatment with S‐1 and gefitinib. The possibility that inhibition of TS by lapatinib underlies the synergy between lapatinib and S‐1 should therefore be investigated in future studies. In contrast to the anti‐tumor activity of lapatinib that we observed in our in vivo study, little anti‐tumor activity of lapatinib was observed when it was administered as a single agent in vitro. These apparently contradictory data may be partly explained by the fact that the cytostatic effect of lapatinib may not be observed after a short exposure to lapatinib (72 h), which was the time used for the MTT assay in vitro.
The effect of trastuzumab, an antibody that blocks HER2 function, on pancreatic cancer with a high level of HER2 expression has been examined both in vitro and in vivo. ( 23 ) In the in vitro experiment, trastuzumab treatment had no anti‐proliferative effect against 16 pancreatic cell lines. In experiments with tumor xenografts in nude mice that were generated by inoculation of three pancreatic cell lines, only tumors generated from the Capan‐1 cell line, which demonstrates high HER2 expression, were suppressed by trastuzumab. It was suggested that this anti‐tumor effect might have been induced by an antibody‐dependent cell‐mediated cytotoxic mechanism. However, a phase II clinical trial of trastuzumab in combination with GEM for patients with metastatic pancreatic cancer showed only a 6% response rate, a median survival time of 7 months, and a 1‐year survival of 19%.( 24 ) In contrast, in our study, lapatinib inhibited the xenograft tumorigenicity of all four of the pancreatic cell lines examined, indicating a potential advantage of lapatinib over trastuzumab for anti‐tumor therapy.
As described previously, the overall survival of patients with pancreatic cancer was improved by treatment with a combination of erlotinib and GEM compared to treatment with GEM alone.( 3 ) It has been reported that lapatinib may bind an inactive kinase conformation of the EGFR, whereas erlotinib may bind an active kinase conformation. Binding to an inactive form of a kinase may provide an advantage for receptor inhibition by reducing the rate of inhibitor dissociation, thereby allowing for prolonged effects in biological systems even after the total concentration of inhibitor has dropped. Indeed, after treatment with lapatinib, EGFR receptor autophosphorylation recovers much more slowly than following treatment with erlotinib.( 25 ) Consequently, we speculate that the anti‐tumor effect of lapatinib will be as good as, or better than, that of erlotinib.
Our immunohistochemical results demonstrated that 70% of the pancreatic cancer cases that we examined overexpress HER2 and/or EGFR. Therefore, lapatinib is expected to have an anti‐tumor effect on the tumors of a majority of pancreatic cancer patients.
In conclusion, our results indicate that lapatinib has a strong anti‐tumor effect on pancreatic cancer. Furthermore, this effect is potentiated in combination with S‐1. Both lapatinib and S‐1 are oral agents and have mild side‐effects.( 5 , 26 ) A prospective clinical study is necessary to confirm the mild side‐effects by the combination therapy of lapatinib plus S‐1. At least, combination therapy of lapatinib plus S‐1 may provide a favorable quality of life for patients with pancreatic cancer by avoiding the frequent outpatient‐based treatment that is required for intravenous infusion.
Disclosure Statement
All authors have no conflict of interests.
Acknowledgment
This study was supported by a Ministry of Education, Culture, Sports, Science and Technology of Japan‐Grant‐in‐aid for Scientific Research (C) 19590317.
References
- 1. Matsuno S, Egawa S, Fukuyama S et al. Pancreatic cancer registry in Japan. Pancreas 2004; 28: 219–30. [DOI] [PubMed] [Google Scholar]
- 2. Burris HA, Moore MJ, Andersen J et al. Improvements in survival and clinical benefit with gemcitabine as first‐line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 1997; 15: 2403–13. [DOI] [PubMed] [Google Scholar]
- 3. Moore MJ, Goldstein D, Hamm J et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 2007; 25: 1960–6. [DOI] [PubMed] [Google Scholar]
- 4. Herrmann R, Bodoky G, Ruhstaller T et al. Gemcitabine plus capecitabine compared with gemcitabine alone in advanced pancreatic cancer: a randomized, multicenter, phase III trial of the Swiss Group for Clinical Cancer Research and the Central European Cooperative Oncology Group. J Clin Oncol 2007; 25: 2212–7. [DOI] [PubMed] [Google Scholar]
- 5. Ueno H, Okusaka T, Ikeda M, Takezako Y, Morizane C. An early phase II study of S‐1 in patients with metastatic pancreatic cancer. Oncology 2005; 68: 171–8. [DOI] [PubMed] [Google Scholar]
- 6. Nakamura K, Yamaguchi T, Ishihara T et al. Phase I trial of S‐1 combined with gemcitabine in metastatic pancreatic cancer. Br J Cancer 2005; 92: 2134–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ueno H, Okusaka T, Ikeda M et al. A phase I study of combination chemotherapy with gemcitabine and oral S‐1 for advanced pancreatic cancer. Oncology 2005; 69: 421–7. [DOI] [PubMed] [Google Scholar]
- 8. Nakamura K, Yamaguchi T, Ishihara T, Sudo K, Kato H, Saisho H. Phase II trial of oral S‐1 combined with gemcitabine in metastatic pancreatic cancer. Br J Cancer 2006; 94: 1575–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ueno H, Okusaka T, Furuse J et al. A multicenter phase II study of gemcitabine and S‐1 combination therapy (GS therapy) in patients with metastatic pancreatic cancer. J Clin Oncol 2007; 25: 4550. [DOI] [PubMed] [Google Scholar]
- 10. Sibilia M, Wagner EF. Strain‐dependent epithelial defects in mice lacking the EGF receptor. Science 1995; 269: 234–8. [DOI] [PubMed] [Google Scholar]
- 11. Konecny GE, Pegram MD, Venkatesan N et al. Activity of the dual kinase inhibitor lapatinib (GW572016) against HER‐2‐overexpressing and trastuzumab‐treated breast cancer cells. Cancer Res 2006; 66: 1630–9. [DOI] [PubMed] [Google Scholar]
- 12. Okabe T, Okamoto I, Tsukioka S et al. Synergistic antitumor effect of S‐1 and the epidermal growth factor receptor inhibitor gefitinib in non‐small cell lung cancer cell lines: role of gefitinib‐induced down‐regulation of thymidylase synthase. Mol Cancer Ther 2008; 7: 599–606. [DOI] [PubMed] [Google Scholar]
- 13. Kim WY, Nakata B, Hirakawa K. Alternative pharmacokinetics of S‐1 components, 5‐fluorouracil, dihydrofluorouracil and α‐fluoro‐β‐alanine after oral administration of S‐1 following total gastrectomy. Cancer Sci 2007; 98: 1604–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chou TC. Derivation and properties of Michaelis‐Menten type and Hill type equations for reference ligands. J Theor Biol 1976; 59: 253–76. [DOI] [PubMed] [Google Scholar]
- 15. Chou TC, Talalay P. Quantitative analysis of dose–effect relationship: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984; 22: 27–55. [DOI] [PubMed] [Google Scholar]
- 16. Rusnak DW, Alligood KJ, Mullin RJ et al. Assessment of epidermal growth factor receptor (EGFR, ErbB1) and HER2 (ErbB2) protein expression levels and response to lapatinib (Tykerb, GW572016) in an expanded panel of human normal and tumour cell lines. Cell Prolif 2007; 40: 580–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Komoto M, Nakata B, Amano R et al. HER2 overexpression correlates with survival after curative resection of pancreatic cancer. Cancer Sci 2009; 100: 1243–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yarden Y, Slimkowski MX. Untangling the ErbB signaling network. Nat Rev Mol Cell Biol 2001; 2: 127–37. [DOI] [PubMed] [Google Scholar]
- 19. Graus‐Porta D, Beerli PR, Daly JM, Hynes NE. ErbB‐2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J 1997; 16: 1647–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Karunagaran D, Tzahar E, Beerli PR et al. ErbB‐2 is a common auxiliary submit of NDF and EGF receptors: implications for breast cancer. EMBO J 1996; 15: 254–64. [PMC free article] [PubMed] [Google Scholar]
- 21. Xia W, Mullin RJ, Keith BR et al. Antitumor activity of GW572016:a dual tyrosine kinase inhibitor blocks EGF activation of EGFR/erbB2 and downstream Erk1/2 and AKT pathways. Oncogene 2002; 21: 6255–63. [DOI] [PubMed] [Google Scholar]
- 22. Safran H, Miner T, Resnick T et al. Lapatinib/gemcitabine and lapatinib/gemcitabine/oxaliplatin. Am J Clin Oncol 2008; 31: 140–4. 18391597 [Google Scholar]
- 23. Kimura K, Sawada T, Komatsu M et al. Antitumor effect of trastuzumab for pancreatic cancer with high HER‐2 expression and enhancement of effect by combined therapy with gemcitabine. Clin Cancer Res 2006; 12: 4925–32. [DOI] [PubMed] [Google Scholar]
- 24. Safran H, Iannitti D, Ramanathan R et al. Herceptin and gemcitabine for metastatic pancreatic cancers that overexpress HER‐2/neu. Cancer Invest 2004; 22: 706–12. [DOI] [PubMed] [Google Scholar]
- 25. Wood ER, Truesdale AT, Octerlonely B et al. A unique structure for epidermal growth factor receptor bound to GW572016 (lapatinib): relationship among protein conformation, inhibitor off‐rate, and receptor activity in tumor cells. Cancer Res 2004; 64: 6652–9. [DOI] [PubMed] [Google Scholar]
- 26. Geyer CE, Forster J, Lindquist D et al. Lapatinib plus capecitabine for HER‐2‐positive advanced breast cancer. N Engl J Med 2006; 355: 2733–43. [DOI] [PubMed] [Google Scholar]