

Abstract
Wiskott–Aldrich syndrome protein (WASP) and WASP family verprolin‐homologous protein (WAVE) family proteins activate cells’ major actin nucleating machinery, the actin‐related protein 2/3 (Arp2/3) complex, leading to the formation and remodeling of cortical actin filament networks. Cortical actin regulation is critical in many aspects of cell physiology including cell–cell adhesion and cell motility, whose dysregulation is directly associated with cancer invasion and metastasis. In line with this association, the WASP and WAVE family proteins have been reported to be involved in cancer malignancies. What is puzzling, however, is that they can act as either enhancers or suppressors of cancer malignancies depending on the type of cancer and its pathological stage. We are still far from understanding the roles of the WASP and WAVE family proteins in cancer progression. Here, we summarize the recent advances of studies of the WASP and WAVE family proteins with respect to cancer invasion and we offer a model that can account for the diverse outcomes originating from dysregulated WASP and WAVE family proteins in cancer development. (Cancer Sci 2010.)
Cancer cells spread by infiltrating adjacent tissues (invasion) and subsequently travelling to distant organs through the blood or lymph vessels (metastasis). Metastatic progression is a multistep process, known as a metastatic cascade. Although various cellular activities are altered during these steps, tumor cells, particularly those of epithelial origin, initially abrogate cadherin‐based cell–cell adhesion and become able to degrade the underlying basement membrane. They then gradually become motile within the stromal extracellular matrix (ECM) by increasing cell motility coupled with intensive proteolytic activities, resulting in the pathological state called local invasion. As tumor mortality results primarily from progression to the invasive and metastatic states, appreciating the mechanisms of invasive progression, the initiating step for metastasis, is critical to understanding cancer death.( 1 , 2 ) While tumor cells shift to the invasive state, the actin cytoskeleton plays a crucial role: its fundamental task is to provide mechanical underpinnings in cell–cell adhesion and control cell shape. Some benign tumors undergo epithelial‐to‐mesenchymal transition (EMT), in which junctional actin networks at cell–cell contacts are actively remodeled to loosen cell–cell adhesiveness,( 3 , 4 ) and actin polymerization at the leading edge of migrating cells provides force to push the plasma membrane forward.( 5 ) Recent findings suggest that invading cancer cells digest and rearrange the ECM at the specialized and focalized actin‐rich macrostructures called podosomes or invadopodia, which are thought to serve as scaffolds where proteolytic enzymes cluster.( 6 )
Wiskott–Aldrich syndrome protein (WASP) superfamily proteins are the major actin polymerizing factors in cells, and at least one WASP superfamily homologue has been found in all eukaryotic organisms examined thus far. In humans, the WASP superfamily consists of WASP, which is expressed exclusively in hematopoietic lineages, and its ubiquitous homologue Neural‐(N‐)WASP (WASP subfamily; WASPs in short hereafter); brain‐enriched WASP family verprolin‐homologous protein (WAVE)1 and WAVE3, and ubiquitous WAVE2 (WAVE/suppressor of cAR [SCAR] subfamily; WAVEs in short); and newly characterized members WASP and SCAR homologue (WASH), WASP homologue associated with actin, membranes, and microtubules (WHAMM), and junction‐mediating and regulatory protein (JMY).( 7 ) There is little information available regarding the biological roles of WASH, WHAMM, and JMY, so they are not described in detail here. WASPs and WAVEs commonly induce rapid actin polymerization beneath the biological membrane, which is necessary to carry out a broad spectrum of physiological and pathological processes including immune response, tissue morphogenesis, synaptogenesis, and pathogen infection, as well as cancer invasion and metastasis (for details, see reference 8 and accompanying references).( 8 ) Accumulating evidence suggests WASPs and WAVEs are profoundly involved in cancer progression.( 9 , 10 , 11 ) It was initially assumed WASPs and WAVEs would promote cancer invasion and metastasis because they positively regulate cell motility through actin polymerization; this was later revealed to be partially true( 10 , 12 ) but oversimplified. Recent reports indicate that WASPs and WAVEs can act as a suppressor or an enhancer for cancer malignancy, depending on the clinical or experimental setting (Table 1). These dual functions of WASPs and WAVEs in cancer likely arose from their multifaceted role in cells.( 7 , 8 ) To better understand invasive behavior and find more effective targets for anticancer drugs, it is necessary to discuss WASP superfamily proteins in an integrative context that covers their positive and negative effects on cancer invasion.
Table 1.
Aberrations of Wiskott–Aldrich syndrome proteins (WASPs) and WASP family verprolin‐homologous proteins (WAVEs) in cancers
| Gene | Aberration | Evidence | Correlation | Refs |
|---|---|---|---|---|
| WASPs | ||||
| WASP | Unknown | — | — | — |
| N‐WASP | Up (colon) | MA, RT | Met | [s1] |
| Up (esophagus) | MA | ND | [s2] | |
| Down (breast) | RT, WB, IHC | Met, bad prognosis | [s3] | |
| WIPs | ||||
| WIPF1/2/3 | Unknown | — | — | — |
| WAVEs | ||||
| WAVE1 | Up (prostate) | RT, IHC | Inv | [s4] |
| Up (leukemia) | RT, WB | Resistance to apoptosis | [s5] | |
| WAVE2 | Up (melanoma) | WB | Inv, met | (12) |
| Up (breast) | RT, IHC | Inv, bad prognosis | [s6, s7] | |
| Up (lung)† | IHC | Bad prognosis | [s8] | |
| Up (colon)† | IHC | Met | [s9] | |
| WAVE3 | TL (ganglioneuroblastoma) | MA, RT | Tumorigenesis | [s10] |
| Up (breast) | MA, RT, IHC | Inv | (99, 100) | |
| Up (prostate) | RT | Inv | [s11] | |
| ABIs | ||||
| Abi1 | Up (breast) | WB | Inv | [s12] |
| Down (stomach) | IHC | Met, bad prognosis | [s7] | |
| Abi2 | Unknown | — | — | — |
| Abi3 | Down (glioma) | WB | Met | [s13] |
| HSPC300 | ||||
| HSPC300 | Del (VHL kidney) | MAP | Suppression of tumorigenesis | [s14–s16] |
| Up (lung) | IHC | Met | [s17] | |
| CYFIPs | ||||
| CYFIP1 | Down (breast, colon, etc.) | IHC, RT | Inv | (95) |
| CYFIP2 | Unknown | — | — | — |
| NckAPs | ||||
| NckAP1/2 | Unknown | — | — | — |
References prefixed with “s” are provided as Supplementary Information. Abi, Abl‐interactor; Del, genomic deletion of HSPC300 accompanied by genetic loss of von Hippel‐Lindau (VHL) tumor suppressor. Loss of function of VHL gene is responsible for familial or sporadic VHL tumor syndrome; CYFIP, cytoplasmic FMR1 interacting protein 2; Down, gene product downregulated as indicated cancer advances; HSPC300, hematopoietic stem/progenitor cell protein 300; IHC, immunohistochemistry; Inv, invasion; MA, microarray; MAP, deletion mapping of chromosomes; Met, metastasis; NckAP, Nck‐associated protein; ND, not determined; N‐WASP, WASP homologue; RT, RT‐PCR; TL, genomic translocation found at the locus of the indicated gene; Up, gene product upregulated as indicated cancer advances; WB, Western blotting. —, not applicable; WIP, WASP‐interacting protein. †WAVE2 upregulation is seen with actin‐related protein 2 coexpression.
In this review, we focus on changes in the actin cytoskeleton during the stepwise invasive progression of cancer cells, with an emphasis on WASPs and WAVEs, and we summarize the roles of WASPs and WAVEs in different types of cancer and different stages of progression. Then, we propose a model that would account for the apparently inconsistent results for the roles of WASPs and WAVEs in cancer progression.
Molecular characteristics of WASP and WAVE family proteins
WASPs and WAVEs activate the actin‐related protein 2/3 (Arp2/3) complex downstream of Rho family GTPases. The WASP superfamily proteins can be defined by a characteristic domain architecture: a proline‐rich stretch followed by a conserved C‐terminal sequence called the VCA region (Fig. 1). The VCA region acts as a platform to simultaneously bind to both an actin monomer and the Arp2/3 complex, bringing them into close proximity. This spatial configuration activates the Arp2/3 complex to be an actin polymerization nucleus. The Arp2/3 complex, when activated by binding to the VCA region, binds mainly to the side of pre‐existing actin filaments, thereby creating a branch point and a new site for polymerization. Thus, all WASP superfamily proteins evoke Arp2/3 complex‐mediated actin polymerization, leading to a net growth of branched actin networks (see references 5, 13, and 14 for reviews).( 5 , 13 , 14 ) WASPs and WAVEs are known as effectors of Rho family small GTPases. Well‐characterized Rho family members Rac1 and Cdc42, when activated, induce characteristic actin‐based structures at the cell cortex, a flat sheet‐like plasma membrane extension known as the lamellipodium and a needle‐like plasma membrane protrusion known as the filopodium, respectively. RhoA, the founding member of the Rho family, induces stress fibers, contractile actin bundles formed on the ventral surface of cultured cells. N‐WASP was first identified to directly interact with activated Cdc42 and send signals toward the Arp2/3 complex, leading to the formation of filopodia (Fig. 1a), and later, WAVEs were identified as downstream targets of Rac1 to induce lamellipodia( 8 ) (Fig. 1b). Follow‐up studies showed that filopodia can be induced not only by WASPs but also by other actin modulators, such as enabled/vasodilator‐stimulated phosphoprotein (Ena/VASP) family proteins and formins.( 15 ) Apart from filopodia, early molecular and cell biological studies on WASPs revealed another important N‐WASP function, vesicle trafficking. N‐WASP is now known to mediate actin polymerization on endocytic vesicles and thereby mobilize them to be pinched off from the plasma membrane.( 8 )
Figure 1.

Activation mechanisms for Wiskott–Aldrich syndrome proteins (WASPs) and WASP family verprolin‐homologous proteins (WAVEs). (a) Activation mechanisms for the WASP homologue, N‐WASP. It is proposed to be recruited to the plasma membrane through independent binding of Cdc42 and phosphatidylinositol‐(4,5)‐bisphosphate (PIP2) to the Cdc42/Rac‐interactive binding (CRIB/GBD) domain and basic amino acid motif (B), respectively. These interactions, as well as adaptor proteins (Nck or Grb2), cooperatively activate N‐WASP by stabilizing the VCA domain in an open conformation. Src family kinases (SFKs) phosphorylate and activate WASPs, representing another upstream input. (b) Activation mechanisms for WAVE2. Phosphatidylinositol‐(3,4,5)‐triphosphate (PIP3) binding to basic amino acids found in WAVEs and Rac‐GTP binding to complex subunit 140 kDa specifically Rac1‐associated protein (Sra1)/121F‐specific p53 inducible RNA (PIR121) are thought to recruit WAVEs to the membrane. For WAVE2, the Src‐homology 3 (SH3) domain of insulin receptor substrate (IRS) p53 can bind to the polyproline stretch of WAVE2 and this interaction is augmented in the presence of Rac‐GTP, thus indirectly contributing WAVE2 targeting to the membrane in a Rac‐dependent manner. All of these interactions seem to be involved in the activation of WAVE2, similar to the case for N‐WASP. Abi, Abl‐interactor; Arp2/3, actin‐related protein 2/3; CR16, corticosteroids and regional expression‐16; EVH1, Ena‐VASP homology 1; Hem1, hematopoietic protein 1, the NckAP1 paralogue; HSPC300, hematopoietic stem/progenitor cell protein 300; I‐BAR, inverse BAR; NckAP1, Nck‐associated protein 1; P, proline; pY, phosphotyrosine; RTK, receptor tyrosine kinase; SHD, SCAR‐homology domain; WH1, WASP homology 1; WHD, WAVE‐homology domain; WICH, WIP‐and CR 16‐homologous protein; WIP, WASP‐interacting protein.
Multiprotein complexes of WASPs and WAVEs. Endogenous WAVEs constitutively exist within a heterologous multiprotein complex, namely, the WAVE complex. When biochemically purified from bovine brain extracts, WAVE1 has been shown to form a stable heteropentameric protein complex with 121F‐specific p53 inducible RNA (PIR121; also known as cytoplasmic FMR1 interacting protein 2 [CYFIP2]), Nck‐associated protein 1 (NckAP1), Abl‐interactor 2 (Abi2), and hematopoietic stem/progenitor cell protein 300 (HSPC300; also known as Brick1) in a 1:1:1:1:1 molar ratio( 16 ) (Fig. 1b). This pioneering work led to the understanding that all mammalian WAVE isoforms tightly form a WAVE complex with the other four subunits or their paralogues.( 8 , 17 ) For a complete list of WAVE complex genes, see references 14 and 17).( 14 , 17 ) The N‐terminal WAVE‐homology domain (WHD)/SCAR‐homology domain (SHD), which is characteristic of WAVEs, serves as a docking site for Abi and HSPC300 to form a WAVE complex and is thus highly homologous between WAVE isoforms.( 7 ) Importantly, gel filtration chromatography or sucrose gradient separation, which were used to purify a WAVE complex, retrieved barely detectable levels of monomeric WAVEs, suggesting that most endogenous WAVEs are in the complexed state.( 16 , 18 , 19 )
Wiskott–Aldrich syndrome proteins in turn are complexed with WASP‐interacting protein (WIP) family proteins, consisting of WIP, corticosteroids and regional expression‐16 (CR 16), and WIP‐ and CR16‐homologous protein/WIP‐related protein (WICH/WIRE) forming the WASP/N‐WASP complex( 20 ) (Fig. 1a). WASPs share a characteristic domain located at their N‐terminus, the WASP homology 1/Ena‐VASP homology 1 domain, which directly interacts with conserved motifs found near the C‐terminus of WIPs. Similar to the WAVE complex, most endogenous WASPs appear to form a heterodimer with WIPs in a 1:1 molar ratio.( 20 , 21 )
In both WAVEs and WASPs, forming a multiprotein complex seems to protect each subunit from protease‐dependent degradation. For example, dsRNA‐mediated knockdown of any subunit in the WAVE complex in Drosophila S2 cells leads to degradation of SCAR (Drosophila homologue of human WAVEs) by way of the ubiquitin–proteasome pathway.( 22 ) Likewise, WASP protein levels in T cells from WIP knockout mice are dramatically decreased, and can be partially restored by MG132 proteasome inhibitor treatment.( 21 )
Regulation of WASP and WAVE activity by oncogenic signals. The activation mechanisms of WASPs and WAVEs have been extensively reviewed in recent years.( 8 , 14 , 17 ) Readers should consult these reviews for comprehensive understanding of the subject. Here we discuss how the activity of WASPs and WAVEs is affected by various oncogenic signals.
The WASPs are autoinhibited in a resting state, which is thought to preclude unintended actin polymerization deleterious to normal cellular function. Intramolecular interactions between Cdc42/Rac‐interactive binding domain (CRIB, also known as the GTPase‐binding domain [GBD]) and the VCA region mediate this autoinhibition. This “masked” VCA is released by competitive binding of various molecules to the CRIB/GBD domain or to the region nearby, allowing the VCA region to interact with and activate the Arp2/3 complex (Fig. 1a). Those molecules that can directly bind to and serve as activation inputs for N‐WASP are: (i) a GTP‐bound (active) form of Cdc42, which specifically binds to the CRIB/GBD domain; (ii) a negatively charged phospholipid species such as phosphatidylserine or phosphatidylinositol‐(4,5)‐bisphosphate (PI(4,5)P2);( 23 ) or (iii) Src‐homology 3 (SH3) domain‐containing proteins, such as SH2‐SH3 adaptor proteins Grb2 and Nck.( 24 , 25 ) Src family kinase (SFK)‐mediated phosphorylation at tyrosine 291/256 of WASP/N‐WASP also enhances their ability to induce Arp2/3‐dependent actin polymerization.( 26 , 27 ) These inputs cooperatively act on WASPs for their full activation. As Cdc42 and SFKs are membrane‐anchored proteins and SH2‐SH3 adaptor proteins are recruited to the plasma membrane under receptor tyrosine kinase (RTK) activation, WASPs are thought to be primarily activated at the membrane. Overexpression of RTKs, including the epidermal growth factor receptor family, is reported in a number of human cancers.( 28 ) Receptor tyrosine kinases not only recruit SH2‐SH3 adaptor proteins to the membrane, but also increase local Cdc42 activity, suggesting that WASPs are hyperactivated in cancers with enhanced RTK signaling. Src family kinases are also known as oncoproteins. Some cancerous phenotypes caused by aberrant SFK signals, including invadopodium formation, are potentially caused by hyperactivated WASPs (discussed later).
The WAVEs are activated downstream of Rac. However, WAVEs, unlike WASPs, do not possess the CRIB/GBD domain and thus cannot bind directly to Rac. Instead, insulin receptor substrate (IRS) p53 has been identified as a linker molecule connecting Rac1 and WAVEs (Fig. 1b). The N‐terminal domain of IRSp53, which is currently known as the inverse BAR (I‐BAR; also known as the Rac‐binding [RCB] or the IRSp53‐MIM‐homology domain [IMD]) domain, has been shown to specifically bind to active forms of Rac1. The C‐terminally located SH3 domain of IRSp53 in turn binds to the proline‐rich sequence of WAVEs and thereby enhances WAVE activity.( 19 , 29 ) In addition, IRSp53 recruits the WAVE2 complex to the plasma membrane, presumably because the I‐BAR domain of IRSp53 electrostatically interacts with negatively charged phosphoinositides including PI(4,5)P2 and phosphatidylinositol‐(3,4,5)‐triphosphate (PI(3,4,5)P3).( 19 , 30 ) Importantly, WAVE2 has much stronger affinity for IRSp53 than for WAVE1 or WAVE3.( 29 ) Therefore, the interaction with IRSp53 is likely to contribute primarily to activity regulation of WAVE2. An alternative, but not mutually exclusive, link between Rac and WAVEs is that WAVE complex subunit 140 kDa specifically Rac 1‐associated protein (Sra 1), the PIR121 paralogue, can interact specifically with GTP‐bound forms of Rac1.( 31 ) Although there has been a debate as to whether the WAVE complex is autoinhibited in a resting state, as is the case for WASPs, recent reports from two independent groups suggest a mechanism whereby the VCA region of WAVE1 and WAVE2 is inhibited by intracomplex interaction, probably between the VCA region and the Sra1‐NckAP1 subcomplex.( 32 , 33 ) The VCA region appears to be released through a conformational change that is triggered by binding of activated Rac to Sra1.( 32 ) Interestingly, Lebensohn and Kirschner( 33 ) provided evidence that a native WAVE2 complex purified from cellular extracts cannot be activated solely by Rac1/2‐GTP but is activated in the presence of both active Rac and negatively charged phospholipids, especially PI(3,4,5)P3. The activating mechanism of the WAVE2 complex by binding to phospholipids is unclear, but PI(3,4,5)P3 binding to the basic amino acid cluster found in WAVE2, which we have previously shown to be important for the recruitment of WAVE2 to the plasma membrane,( 34 ) might contribute to the activation of the WAVE2 complex. In a broad range of cancer types, signals that increase cellular levels of PI(3,4,5)P3 are aberrantly augmented by mutations in genes that constitute the phosphoinositide 3‐kinase (PI3K) pathway, such as activating mutations in PIK3CA (encoding a catalytic subunit of PI3K) and loss‐of‐function mutations in PTEN (encoding a lipid phosphatase for PI(3,4,5)P3).( 35 ) WAVEs can be directly activated by PI(3,4,5)P3 as discussed above. In addition, PI(3,4,5)P3 activates Rac through PI(3,4,5)P3‐responsive guanine‐nucleotide exchange factors for Rac (RacGEFs).( 36 , 37 ) Therefore, WAVEs are presumably hyperactivated in cancers with enhanced PI3K signaling and thus may contribute to cancer pathogenesis. Phosphorylation has also been reported for WAVE regulation: cyclin‐dependent kinase 5 serine/threonine kinase phosphorylates and inhibits WAVE1 activity,( 38 ) and conversely, c‐Abl tyrosine kinase activates WAVE2 through phosphorylation.( 39 ) c‐Abl is a proto‐oncogene product best studied in chronic myelogenous leukemia (CML), implicating the association between WAVE2 and CML.( 40 )
WAVEs in 2D cancer cell motility
Signals that orchestrate the leading edge formation in 2D cell motility. Although cancer cells in vivo migrate through three‐dimentional (3D) networks of ECM fibers and sometimes move collectively as a tumor mass (Fig. 2a), fundamental aspects of the molecular mechanism of cancer cell motility have been largely revealed in simple two‐dimentional (2D) environments such as a culture dish (Fig. 2b). We now know that cell migration is conceptualized as a cyclic process, as follows: (i) leading edge extension propelled by rapid actin assembly in the direction of movement; (ii) establishment of cell–substrate adhesion by integrins at the leading edge; and (iii) trailing edge contraction at the back, which is accomplished by myosin II motor protein‐driven contractile force generation (actomyosin contraction). Here we introduce a simplified view on the mechanism of the leading edge formation, which will help readers understand later discussion. More precise and global mechanisms of cell motility are elegantly summarized in previous reviews.( 5 , 11 , 13 , 41 )
Figure 2.

Multiple modes of cancer cell motility in 3D. (a) Cancer progression is illustrated from the standpoint of cell motility mode. A primary tumor initially invades in a mode called collective migration. If cancer cells are released from a primary tumor mass and move as single cells, the cells can adopt two modes of invasion, round‐shape motility or elongated motility. EMT, epithelial‐to‐mesenchymal transition. (b) The actin cytoskeleton of a two‐dimensionally migrating cell. In 2D circumstances, characteristic actin structures (colored in red) are commonly seen in many cell types. They generally form lamellipodia, filopodia, and stress fibers. In malignant cancer cells, invadopodia are sometimes evident as large actin dots that show proteolytic activity around them. (c) The actin cytoskeleton in cells migrating in the elongated motility mode. The tip contains dense actin networks and its biogenesis is dependent on Wiskott–Aldrich syndrome protein family verprolin‐homologous protein and actin‐related protein 2/3 activities. (d) The actin cytoskeleton in cells migrating in the round‐shape motility mode. Protrusions used for migration are, in this case, membrane blebs. Both the formation and retraction of membrane blebs rely on actomyosin contractility.
Simply speaking, a signal to trigger actin polymerization at the leading edge starts with cell polarization in which PI3K asymmetrically accumulates toward the source of guidance cues, such as chemokines and growth factors.( 42 ) This PI3K accumulation triggers local increase in PI(3,4,5)P3 levels at the plasma membrane, leading to localized activation of Rac by RacGEFs.( 36 , 37 ) A positive feedback loop between Rac and PI(3,4,5)P3 is thought to be important to maintain polarized PI(3,4,5)P3 localization and persistent activation of Rac at the front of migrating cells.( 43 ) Cdc42 is also reported to be localized and activated at the leading edge of migrating neutrophils.( 44 ) The activated Rac and Cdc42 in turn recruit WAVEs and WASPs to the plasma membrane where they are activated, leading to the formation of lamellipodia and filopodia, respectively. Actin polymerization at the leading edge is prompted not only by WASPs and WAVEs, but also by actin depolymerizing factor (ADF)/cofilin that binds to and severs pre‐existing lamellipodial actin filaments, and thereby creates new filament ends that are compatible for polymerization. Actin depolymerizing factor/cofilin is inhibited when bound to PI(4,5)P2. Phospholipase C‐mediated hydrolysis of PI(4,5)P2 is thought to release ADF/cofilin from this inhibition, which is proposed to be an activating mechanism of ADF/cofilin at the leading edge in response to epidermal growth factor.( 45 ) However, cell motility, in practice, requires spatially and temporarily controlled cytoskeletal remodeling at the leading edge, which is coupled with cell polarization and cell–substrate adhesion. Thus, there should be a mechanism that coordinates the activities of Arp2/3 and ADF/cofilin. Coronin 1B has been proposed as a candidate molecule that links these two activities. Coronin 1B suppresses Arp2/3 activity by directly binding to the Arp2/3 complex, and at the same time, it dephosphorylates and thereby activates ADF/cofilin by recruiting the ADF/cofilin phosphatase Slingshot to the plasma membrane.( 46 ) In reality, there are hundreds of molecules other than the aforementioned proteins that orchestrate actin dynamics at the leading edge, and we need to continually refine our understanding of the mechanisms of leading edge formation.
WAVE2 regulates lamellipodium‐driven 2D cell motility. So, how much importance do WASPs and WAVEs have in 2D cell motility? It has been shown that WASPs have relatively compromised effects (when compared to those of WAVEs; see later) on protrusion formation and cell motility itself on 2D substrates in several cell types.( 47 , 48 , 49 ) Rather, it has been suggested that WASP and N‐WASP are required to control directionality of movement in macrophages( 47 ) and mammary carcinoma cells,( 49 ) respectively. This does not mean that filopodia are dispensable for cell motility, as filopodia are formed through several redundant signaling pathways, as described above. Recent evidence suggests that the filopodium serves as a precursor of integrin‐based adhesion by which cells sense mechanical properties of their exterior environment.( 50 , 51 ) Some of these filopodia grow to become mature focal adhesions, and in this sense, the filopodium affects cell motility. In contrast, it has been traditionally believed that the lamellipodium is the driving force in cell motility, and hence that cells’ ability to form lamellipodia critically influences cell motility. This notion is not merely an assumption, but was exemplified by a simple but thought‐provoking experiment carried out in 1984, in which a tiny fragment was excised from a broad lamellipodium that a fish keratinocyte usually forms when it randomly migrates on a culture dish. When it was observed under a video microscope, the fragment by itself was, surprisingly, found to migrate at a similar speed and persistence to an intact cell.( 52 ) This experiment strongly indicated that the lamellipodium per se is likely to give the protrusive force for cell motility, and that elementary units of cell motility correspond to the local chemical reactions within a restricted compartment of a lamellipodium. As described above, lamellipodial actin polymerization is regulated by WAVEs. In fact, lamellipodia are severely disrupted by genetic loss‐of‐function of WAVE2 in mouse endothelial cells( 53 ) and in mouse embryonic fibroblasts,( 54 ) or by RNAi‐mediated depletion of WAVE2 in B16F10 mouse melanoma cells( 12 ) and in mouse macrophages,( 55 ) all of which result in decreased 2D cell motility. Unexpectedly, however, in mammals, WAVE2 depletion predominantly prevents actin polymerization in the lamellipodium, suggesting non‐redundant functions between WAVE isoforms in lamellipodial actin regulation (Kurisu S, 2005, unpublished observations).( 12 , 54 ) This may be simply due to higher expression levels of WAVE2, compared to those of WAVE1 and WAVE3. Alternatively, it may be caused by the higher affinity of WAVE2 for IRSp53, which preferentially localizes and activates WAVE2 at the lamellipodial tips (Fig. 3a). Therefore, inhibiting WAVE2 activity can discourage cancer cells from showing a motile phenotype. Drugs that interfere with WAVE2 activation may be promising therapy against invasion and metastasis in cancer patients. Indeed, we have shown that invasion and metastasis are effectively suppressed when WAVE2 activity is inhibited in highly metastatic B16F10 cells.( 12 )
Figure 3.
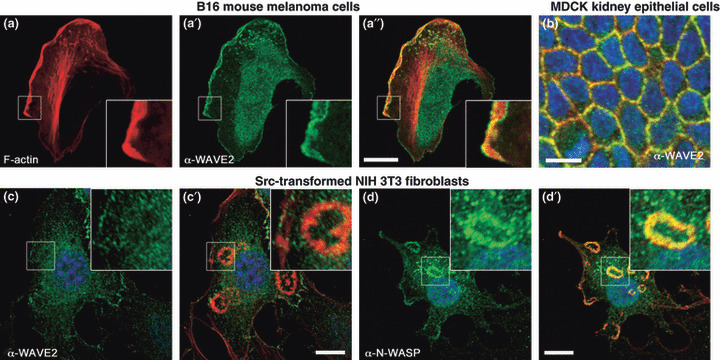
Characteristic subcellular localization of Wiskott–Aldrich syndrome protein (WASP) family verprolin‐homologous protein (WAVE)2 and WASP homologue, N‐WASP. (a–a″) B16 mouse melanoma cells form broad lamellipodia in the direction of movement. WAVE2 (shown in green) localizes in the tip of the lamellipodia. (b) In MDCK kidney epithelial cells cultured on the surface of type I collagen gel, WAVE2 accumulates at the cell–cell junction. (c,d) Src‐transformed NIH fibroblasts form invadopodial rosettes (magnified in insets of c and d). They are devoid of WAVE2 (c), whereas N‐WASP is strongly concentrated in invadopodia (d). Red and blue show phalloidin staining and Hoechst 34580 nuclear staining, respectively. Green in (a–c) and in (d) shows immunostaining for endogenous WAVE2 and N‐WASP, respectively. Bars, 10 μm.
WASPs and WAVEs in 3D cancer cell invasion
Plasticity of cancer cell motility within the 3D ECM. Recently, researchers have examined cancer motility in the 3D environment, which more closely mimics the in vivo cancer microenvironment than the 2D environment. One 3D observation strategy is to implant cancer cells in live animals and directly observe the cells under a microscope, which is sometimes called “intravital imaging”. Another technique is organotypic culture, in which cancer cells are usually embedded in type I collagen gel or in Matrigel, a mixture of ECM with similar constituents to those of the basement membrane. Using these techniques, combined with time‐lapse fluorescent confocal microscopy, it has been revealed that cancer cells show multiple “modes” of cell migration in 3D.( 56 , 57 ) There are at least three different modes of migration: collective motility; elongated motility; and round‐shape motility (Fig. 2a,c,d). Collective motility, which refers to cells moving in a group, is reported in organotypic cultures of breast cancer, fibrosarcoma, melanoma, and squamous cell carcinoma cells.( 58 , 59 ) From the histopathological resemblance between human tumor samples and cultured cancer cells in 3D, it is speculated that collective motility mimics the invasive front of human tumors, although direct evidence of this is missing. The elongated and round‐shape modes of motility describe solitary cells that have departed from the primary tumor mass through EMT or as a result of spontaneous cell–cell dissociation. Elongated motility is characterized by a bipolar spindle‐shaped morphology and dependence on protease activity for migration. This type of motility is usually seen in cancers of mesenchymal origin, such as glioma, fibrosarcoma, and melanoma, and in some aggressive carcinomas.( 57 , 60 , 61 ) Round‐shape motility, which is in contrast characterized by a short ellipsoid shape during cell movement and a relatively compromised dependence on protease activity for migration, is reported in some colon carcinoma and melanoma cells as well as in non‐neoplastic hematopoietic cells like lymphocytes and neutrophils.( 9 , 60 , 62 ) Importantly, some cancer cells can switch their migration mode, and this plasticity is proposed to explain why cancer cells continue to move even when one mode of motility has been suppressed by pharmacological intervention. Wolf et al. ( 57 ) first reported that a cocktail of protease inhibitors triggers mode‐shift from elongated to round‐shape motility in fibrosarcoma cells and in breast cancer cells, and they assumed this switching mechanism might underlie the escape pathway for cancers refractory to protease inhibitor‐based therapies.
Elongated motility versus round‐shape motility from an actin perspective. The actin morphology of migrating cells in 3D differs strikingly from that in 2D. Cells that have resumed elongated motility no longer form broad lamellipodia at the leading edge. Instead, they usually extend long finger‐like protrusions rich in F‐actin, which here we call “pseudopodia” (2, 3). The pseudopodium can be structurally divided into two compartments: a shaft, which is supported by cortical thick actin bundles, and a tip, which shows dense phalloidin staining with filopodium‐like spikes inserted into the pericellular ECM. The tip seemingly contains a miniature of the lamellipodium found in 2D, implied by the shape of the dense actin in the tip. The tip is formed by the activities of WAVE2 and Arp2/3 (Fig. 4d,e), and its oscillatory motion of extension and retraction resembles the motion of lamellipodia when the tip is observed under a video microscope.( 12 , 61 ) The tip contains a substratum anchorage site where β1‐integrin concentrates and constitutes a functional adhesive receptor for the ECM.( 62 , 63 ) Thus, the force required for membrane extension at the tip is likely to be provided by Arp2/3‐dependent actin polymerization, similar to the case of lamellipodium in 2D. A striking difference between 2D and 3D, however, is that focal adhesions, characteristic of cell–ECM interaction in 2D cell culture, are not evident in the 3D environment. Accordingly, stress fibers in 2D, contractile actin bundles with each side anchored to the ECM by the focal adhesions, are not detectable in 3D.( 64 ) Stress fibers in 2D are bundled by non‐muscle myosin II motor proteins and generate actomyosin‐based traction force to pull the rear of migrating cells. In 3D, migrating cells form longitudinal (i.e. parallel to the direction of movement) actin bundles at the cell cortex, as seen in the shaft of pseudopodia. These cortical bundles are also colocalized with myosin II and thus are likely to generate traction force, suggesting that they could be the 3D analogues of stress fibers. It is not known whether cortical bundles are connected to the ECM at their ends, as is the case in 2D stress fibers, as focal adhesions in 3D are not clearly visible. As a whole, the mechanism of 3D elongated motility is similar to that of 2D motility, in that cells use WAVE2‐ and Arp2/3‐dependent protrusive forces, despite cell morphologies of 2D and 3D motility appearing quite different.
Figure 4.
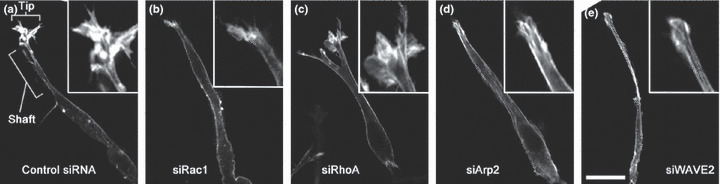
Pseudopodial F‐actin in cells showing elongated motility in 3D. (a) Control HT1080 cells form well‐developed tip and shaft structures. (b–e) HT1080 cells treated with Rac1, RhoA, actin‐related protein (Arp)2, and Wiskott–Aldrich syndrome protein family verprolin‐homologous protein (WAVE)2 siRNA, respectively. Note that the tip structures are lost in Rac1, Arp2, and WAVE2 siRNA treated cells. Cells are embedded in type I collagen gels. F‐actin is visualized by staining with fluorescently labeled phalloidin. Insets show magnification of the tip structure. Details are described in reference( 61 ). Bar, 20 μm.
Round‐shape motility uses completely different mechanisms. Cells rapidly inflate their plasma membrane in the direction of movement in a balloon‐like shape called a membrane bleb, and actin networks gradually fill the cortex of the bleb (Fig. 2d). How actin is supplied to the bleb cortex is unknown, but Gadea et al. ( 65 ) raises the hypothesis that N‐WASP is responsible for actin nucleation at the bleb cortex from the fact that N‐WASP knockdown suppresses round‐shape motility. The newly formed cortical actin mesh within a bleb then generates contractile force by way of myosin II activity, by which the bleb retracts to the cell body.( 66 ) This traction is one of the candidates that generate the force required to move forward, although controversy still exists on the issue of force generation in round‐shape motility.( 67 ) It has been proposed that membrane blebs form from a local rise in hydrostatic pressure beneath the plasma membrane, leading to either a rupture in cortical actin networks or a local detachment of the plasma membrane from the cortical cytoskeleton.( 68 ) In both cases, the hydrostatic pressure gradient gives rise to a cytoplasmic flow, resulting in inflation of the membrane to form a bleb. Local rise in hydrostatic pressure is somehow caused by the local increase in myosin II activity, and thus both protrusive and contractile forces of round‐shape motility are myosin II‐dependent. In concordance with this, invasive migration of colon carcinoma cells that show round‐shape motility is strongly suppressed by inhibition of Rho kinase I/II (ROCKI/II), a direct kinase that phosphorylates the myosin regulatory light chain (MLC) leading to actomyosin contraction.( 61 )
The mechanisms of collective migration have recently been excellently reviewed,( 58 ) so we will not describe them in detail. Briefly, in many cases of collective migration seen in cancer invasion, as well as in morphogenic movement during development, a group of cells that jointly moves within the ECM is frequently guided by a subpopulation of “tip” or “leader” cells. In the majority of described cases of collective migration, leader cells assume elongated morphology, and thus we speculate that collective migration is basically governed by similar mechanisms in the leader cells as those mechanisms governing elongated motility.
WAVE2 controls conversion between elongated and round‐shape modes of motility. Both elongated and round‐shape modes of motility can be seen even in a clonal population of a cultured cancer cell line. Many cancer cell lines have a unique population bias toward one of these motility traits, by which we categorize them simply as elongated or round‐shaped. Which mode each cell in the population adopts seems to be stochastically decided by a combination of genetic matters of the cell line and epigenetic variations stemming from the extracellular microenvironment in which each cell is deposited. Environmental control of motility has been implicated by several previous studies that showed cancer cells treated with protease inhibitors convert their shape from elongated to rounded and keep moving within the ECM.( 57 , 60 , 61 ) More direct evidence is that ECM mechanical rigidity strongly influences motility modes of glioma cells.( 69 ) Genetic and cell‐intrinsic attributes that affect mode control of motility are still unclear, but Rac and Rho signals are key to understanding the mechanism.
As mentioned above, depletion of WAVE2, a Rac effector, suppressed elongated motility by inhibiting Arp2/3‐mediated actin polymerization at the tip of pseudopodia, but unexpectedly it also increased the population of cells showing round‐shape motility in some cell types, such as HT1080 cells( 61 ) and A375M2 melanoma cells.( 60 , 70 ) Thus, WAVE2 not only promotes pseudopod extension, but also seems to suppress actomyosin contractility. Sanz‐Moreno et al. ( 70 ) showed that MLC2 phosphorylation, a measure of myosin contractility, is enhanced in Rac1‐ or WAVE2‐depleted melanoma cells. Also, we quantified active RhoA levels in WAVE2‐ or Arp3‐depleted HT1080 cells, but could not detect any change in RhoA activity compared to non‐depleted cells.( 61 ) Therefore, WAVE2 is likely to somehow suppress actomyosin contraction through formation of Arp2/3‐dependent actin mesh without affecting RhoA activity. How Rac1‐WAVE2‐Arp2/3 signals suppress MLC2 phosphorylation remains to be determined, although a mutual antagonism between Rac and Rho activities has been reported in a number of studies. The molecules mediating this antagonism are currently supposed to be GTPase activating proteins for Rho family (RhoGAPs). The human genome encodes more than 70 RhoGAPs, but their functions in cells are largely unknown. Among them, p190RhoGAP, which is activated by active Rac, has been shown to inactivate Rho in a Rac‐dependent manner, although its association with elongated motility is yet to be investigated.( 71 ) In a reciprocal way, suppressing the activities of Rho or its downstream targets such as ROCK and myosins reduces the proportion of cells of round‐shape motility and simultaneously increases the proportion of cells of elongated motility.( 60 , 61 , 70 ) This conversion from round‐shape to elongated mode seems to be regulated by RhoGAPs, as evidenced in the study of melanoma cells by Sanz‐Moreno et al. that showed GTPase activating protein for Aplysia Ras‐related homologue (ARHGAP)22 downregulates Rac1 activity and suppresses elongated motility.( 70 ) However, we cannot predict which mode a given case of cancer prefers, and how cancer cells make the initial decision of which motility mode to use remains an open question.
N‐WASP is essential to form podosomes/invadopodia, centers of ECM degradation. To invade 3D ECM, cancer cells need to not only change their shape for movement (i.e. cell elongation or bleb formation), but also enzymatically degrade and sever ECM fibers to remodel the surrounding matrix. Pseudopodia of cells invading in an elongated shape are active zones for ECM proteolysis where proteolytic enzymes, such as matrix metalloproteases, are concentrated.( 72 ) In recent years, another class of actin‐based cellular protrusion specialized for ECM proteolysis has been the subject of intense 3D cell motility studies. This class is called podosomes or invadopodia, and they are usually found on the ventral surface of 2D cultured cells (Fig. 2b). Monocyte‐derived lineages, including macrophages, lymphocytes, and osteoclasts, form these protrusions under physiological conditions, and the name “podosome” is preferentially used in this situation. In contrast, the “invadopodium” usually refers to the protrusions that resemble podosomes but are abused by cancer cells for invasion.( 10 ) We can easily observe invadopodia in Src‐transformed fibroblasts and in some malignant cancer cells.( 6 , 10 , 73 ) On 2D substrates, podosomes/invadopodia appear as microscopically discernible actin dots; in some cases, these dots coagulate to form ring‐like structures (called rosettes) at cell–substrate contact sites (Fig. 3c,d). Importantly, there is experimental evidence of extensive ECM proteolysis by podosomes/invadopodia.( 74 , 75 ) Various proteases, including matrix metalloproteases,( 76 ) cathepsins,( 77 ) and ADAM (a disintegrin and metalloprotease) family proteases,( 78 ) are known to be concentrated in podosomes/invadopodia. This proteolytic activity of podosomes/invadopodia plays an important role for some cell types’ migration in 3D; for example, inhibiting the formation of podosomes impairs macrophage migration in 3D ECM but not on 2D glass surfaces.( 79 )
The actin‐rich core structure of podosomes/invadopodia is essential for their maintenance. This structure contains a variety of actin regulatory proteins involved in actin polymerization, cross‐linking, and filament turnover. Filamentous actin at the core is mainly supplied by (N‐)WASP/WIP and Arp2/3 actin polymerization machinery but not by WAVE complexes.( 73 , 80 , 81 , 82 ) N‐WASP knockdown almost completely disrupts podosome/invadopodium formation( 73 , 82 ) and N‐WASP, but not WAVEs, intensely localizes in podosomes/invadopodia (Fig. 3c,d). Cross‐linking proteins such as α‐actinin( 81 ) and fascin( 83 ) are thought to strengthen the actin core structure, and proteins that affect filament elongation and stability, such as formins, Ena/VASP family, and ADF/Cofilin, induce rapid turnover of actin in podosomes/invadopodia( 10 , 84 ) Interestingly, the list of molecules involved in the development of podosomes/invadopodia is like that of focal adhesions. However, podosomes/invadopodia are believed to be distinct from what we call focal adhesions. A striking difference is that podosomes/invadopodia stably contain actin polymerizing factors, (N‐)WASP and the Arp2/3 complex, whereas focal adhesions do not.( 73 , 81 ) This difference has an important implication for the functionality of podosomes/invadopodia: locally restricted rapid actin polymerization generates protrusive force at the cell–substrate interface causing cells to form a unique protrusive structure that is different from the above‐mentioned pseudopodia or membrane blebs. Some cancer cells use invadopodia to invade 3D ECM, but how invadopodia are used (or not used) in conjunction with elongated and round‐shape motility needs to be clarified. Intriguingly, recent evidence suggests that invadopodia are likely to facilitate elongated motility but not round‐shape motility.( 83 )
We previously found that PI(3,4)P2 accumulation at the plasma membrane triggers localization signals where podosomes/invadopodia will form. Usually restricted to extremely low levels of abundance at the plasma membrane, PI(3,4)P2 was found to be enriched in podosomes/invadopodia, and PI(3,4)P2 production was revealed to be essential for their formation.( 73 ) Surprisingly, even after blocking actin polymerization by treating cells with latrunculin A, rosette‐like plasma membrane structures rich in PI(3,4)P2 were still seen, indicating that PI(3,4)P2 production precedes actin assembly in podosome/invadopodium formation. Phosphatidylinositol‐(3,4)‐bisphosphate recruits a scaffolding protein, tyrosine kinase substrate 5/five SH3 domains (Tks5/FISH), which has a PI(3,4)P2‐binding domain, along with its binding proteins, such as N‐WASP, adaptor protein Grb2,( 73 ) and ADAM family proteases.( 78 ) These molecular interactions seem to be responsible for organizing the functional podosome/invadopodium macrostructure. Originally found as an Src‐family kinase substrate, Tks5/FISH has been shown to be essential for podosome formation.( 85 ) It is a unique molecule in that it has five consecutive SH3 domains, all of which can bind to N‐WASP.( 73 ) Since many SH3 domain‐containing proteins, when bound to the N‐WASP proline‐rich region, are known to activate N‐WASP,( 8 ) Tks5/FISH presumably unlocks and boosts N‐WASP activities just at the site podosomes/invadopodia are formed, and evokes actin polymerization through Arp2/3 complex activation, leading to the formation of mature podosomes/invadopodia. This sequential event emanating from PI(3,4)P2 production may differentiate focal adhesions into podosomes/invadopodia, as the PI(3,4)P2‐rich plasma membrane rings always preferentially appear near focal adhesions.( 73 )
WAVEs regulate actin dynamics at cell–cell junctions
Dynamic actin turnover at cell–cell junctions. Tight control of cell–cell adhesion is instrumental to normal development and maintenance of the shape of multicellular organs. Cancer cells that originate from epithelia often weaken or abrogate cell–cell adhesion prior to being locomotive as a single cell, and it has been established that loss of cell–cell adhesion is one of the hallmarks of cancer progression.( 1 , 3 , 4 ) The epithelial cell–cell junction is a polarized structure, in which cells are connected to each other near the apical end of the lateral cell interface through specialized adhesive structures called adherens junctions (AJs). The core component of the AJ is the transmembrane adhesive receptor E‐cadherin, whose ectodomain homophilically binds to other E‐cadherin ectodomains, and whose cytoplasmic tail is indirectly connected to the perijunctional actin cytoskeleton. These linkages are believed to be the central mechanism for generating an adhesive force and the mechanical strength of AJs. The cytoplasmic tail of E‐cadherin binds to β‐catenin, and β‐catenin in turn associates with α‐catenin. α‐Catenin can directly bind to F‐actin, suggesting that α‐catenin functions as a molecular bridge that physically connects E‐cadherin and F‐actin. Thus, it had been traditionally believed that E‐cadherin at mature AJs maintains stationary connection to perijunctional actin cytoskeletons through α/β‐catenins.( 86 ) However, this view was challenged in 2005 by the finding that reconstituted E‐cadherin–catenin complex cannot bind directly to F‐actin.( 87 , 88 ) Instead, these studies suggested that F‐actin shows a steady‐state turnover at the junctions, and that E‐cadherin is likely to keep continuous but transient contact with this dynamic actin network. Which molecule and what mechanism coordinate these dynamic cadherin–actin interactions are still unclear and under investigation.
WAVE2 dysfunction leads to loss of cell–cell adhesion integrity. Perijunctional actin should be continuously polymerized to sustain its steady‐state turnover. The Rac‐WAVE‐Arp2/3 pathway( 89 ) and the Rho‐formin pathway( 90 ) are proposed to mediate the actin nucleation at cell–cell contacts. We previously showed that the development of AJs is markedly delayed by knockdown of WAVE2 (and, to a lesser extent, WAVE1) in canine kidney epithelial cells.( 89 ) A previous study showed that E‐cadherin ligation triggers Arp2/3‐dependent actin polymerization at nascent adhesions.( 91 ) Thus, the WAVE2‐Arp2/3 pathway represents important signaling branches for de novo actin polymerization at developing cell–cell contacts. Of course, it should be realized that the development and maturation process of cell–cell contacts might rely on mechanisms distinct from those for maintenance and/or remodeling of cell–cell adhesion. However, we speculate that WAVE2 is also involved in the maintenance and remodeling of AJs for several reasons. First, WAVE2 strongly localizes to the lateral wall of contacting epithelial cells even after they form mature AJs (Fig. 3b). Second, Tiam1, a Rac‐specific RhoGEF, localizes to a similar area as WAVE2, and Tiam1 is required for maintenance of cell–cell adhesion.( 92 ) Finally, constitutively active Rac1 can drastically increase the number of actin filaments at mature cell–cell junctions.( 93 ) These observations suggest that activated Rac and high concentrations of WAVE2 coexist at mature cell–cell junctions, thereby continuously stimulating Arp2/3 complex‐mediated actin polymerization at the junction. In contrast to the Arp2/3 complex, formins can induce unbranched actin fibers. Thus, branched and unbranched actin populations are likely to intermingle and constitute dense actin networks at cell–cell contacts. Although the relative contribution of the Arp2/3 complex and formins for actin supply at AJs is unclear, these two different actin nucleators seem to cooperatively maintain steady‐state actin turnover for cell–cell adhesion.
Actin regulation at AJs has significant impacts for cancer dissemination. Of note, while homophilic cadherin ligation can induce actin assembly at the perijunctional space,( 91 ) the integrity of the actin cytoskeleton is also required to establish proper cadherin‐based cell–cell adhesion.( 94 ) Thus, not only loss‐of‐function of E‐cadherin, but also dysfunction in actin regulators at AJs, could potentially accelerate metastatic progression. Recently, Silva and colleagues( 95 ) found that reduced expression of the WAVE complex subunit Sra1/CYFIP1 causes loss of epithelial cell adhesion and promotes tumor progression, which implicates Sra1 as a suppressor of invasion in epithelial cancers. Silencing other WAVE complex components causes the same phenotype as Sra1 knockdown in MCF10A mammary epithelial cells, suggesting that Sra1 regulates epithelial morphology as part of the WAVE complex. Thus, WAVE dysfunction is likely to underlie the reduced cell adhesiveness, which contributes to abnormal epithelial cell morphology and promotes the invasive potential of epithelial cells.
Multiple levels of regulation of cancer invasion by WASPs and WAVEs
As discussed above, WASPs are required for actin assembly in invadopodium formation and possibly for bleb formation in round‐shape movement in 3D ECM. Invadopodia act not just as protrusive cellular structures, but also as storage of proteases and their secreting machinery for cancer cell invasion into the ECM, suggesting that WASPs are multimodal promoters in cancer cell invasion. However, WAVE2 is required for formation and stabilization of cell–cell adhesion in epithelial cells, presumably blocking the transition from epithelial to mesenchymal phenotype. Thus, WAVE2 appears to act as a tumor suppressor in epithelial‐like benign tumors. However, WAVEs, especially WAVE2, induce actin meshwork at the leading edge in single elongated cells and enhance the invasive capacity of cancer cells. Therefore, WAVE2 works as an invasion promoter once cancer cells start to invade as a single elongated cell in a later stage of cancer progression.
How can we reconcile these opposing effects of WAVE2 in cancer invasion? It is generally believed that invasion and metastasis of carcinoma are prompted by EMT, in which specialized transcription factors known as EMT inducers downregulate epithelial genes, such as E‐cadherin, and at the same time upregulate mesenchymal genes to enhance single cell motility and subsequent dissemination of tumor cells.( 3 ) Recent reports showed that the EMT‐inducer zinc finger E‐box binding homeobox1 (ZEB1) is a crucial promoter of metastasis and inhibits expression of the microRNA‐200 (miR‐200) family, whose members are strong inducers of the epithelial phenotype.( 96 ) Interestingly, ZEB1 not only represses epithelial genes but also increases the expression of stem cell factors such as Sox2 and Klf4 by downregulating the stemness‐inhibiting miR‐203.( 97 ) As described above, there are three WAVE isoforms, WAVE1, WAVE2, and WAVE3. A role for WAVE3 is as yet unclear. However, it has been reported that there is a significant correlation between the expression levels of WAVE3 and advanced stages of breast cancer, suggesting the function of WAVE3 is a metastasis‐promoting protein.( 98 , 99 ) Surprisingly, it was found that expression of WAVE3, but not of WAVE1 or WAVE2, is controlled under the miR‐200 family as is the case for other EMT regulating factors.( 100 ) The miR‐200‐mediated downregulation of WAVE3 leads to a significant reduction in the invasive potential of breast cancer cells. Loss of WAVE3 expression downstream of miR‐200 also results in a dramatic change in cell morphology resembling that seen in the mesenchymal‐to‐epithelial transition, a reverse process of EMT.( 100 ) These data indicate that EMT inducers, by repressing specific classes of miRNAs (miR‐200 family and miR‐203), convert cancer cells to the the undifferentiated and stem cell‐like state, and WAVE3 may participate in increasing the mobility of those stem cell‐like cancer cells. We hypothesize that during EMT, WAVE3, whose expression is repressed by the miR‐200 family, will be transiently expressed to antagonize WAVE2 at cell–cell junctions and counteract cell–cell junction maintenance. We postulate that the activities of WAVE isoforms are not equal in lamellipodium induction and cell–cell adhesion formation, which may be corroborated by the existence of isoform‐specific regulatory proteins, such as IRSp53 and cyclin‐dependent kinase 5 (see above). Indeed, ectopic expression of WAVE3 in T47D breast cancer cells induces EMT‐like changes, as seen in Figure 5, showing disintegrating E‐cadherin‐based adhesion in WAVE3‐expressing cells (Fig. 5b, arrowheads). This antagonism between WAVE2 and WAVE3 can be accomplished by the following mechanism. Because WAVE isoforms share components of the WAVE complex, WAVE3 overexpression would lead to a reduction in the amount of WAVE2 and a change in the ratio of WAVE isoforms expressed. The expression level of total WAVEs is adjusted or “capped” by the level of the least expressed WAVE complex component, and excess WAVEs that fail to form a complex are degraded in proteasomes. We call this mutual relation the “isoform sequestration model” (Fig. 6a). If Abi is least expressed and is later overexpressed, the expression level of total WAVEs is increased to that of the second least expressed HSPC300 (far right panel in Fig. 6a). Depletion of Sra1, as reported in some carcinomas,( 95 ) results in reduction of the expression level of total WAVE isoforms to that of the Sra1 homologue PIR121/CYFIP2. Again, when WAVE3 expression is increased, WAVE1 and WAVE2 are reduced to meet the levels of other proteins that comprise the complex. This quantitative regulation of WAVE expression levels may underlie the dual function of WAVE2 in invasion. In the earlier stage of cancer progression, cancer cells inhibit WAVE2 (possibly through WAVE3 overproduction or through deletion of Sra1/CYFIP1) to acquire dissociating abilities from E‐cadherin‐mediated cell–cell adhesion. Later, some cancer cells may restore WAVE2 activity (possibly through re‐repression of WAVE3 or through overexpression of Abi) to increase cancer cell motility, which leads to metastatic progression. We summarize the inter‐relation of WASP and WAVE family proteins in cancer cell invasion and metastasis in Figure 6b.
Figure 5.
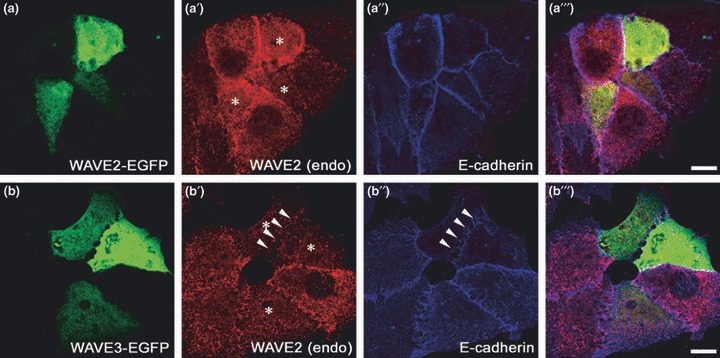
Forced expression of Wiskott–Aldrich syndrome protein family verprolin‐homologous protein (WAVE)3 in T47D breast cancer cells leads to disruption of E‐cadherin‐based cell–cell adhesion, indicating that WAVE3 may function as an invasion promoter in some primary benign tumors. Enhanced green fluorescent protein (EGFP)‐tagged WAVE2 (a) or WAVE3 (b) are ectopically expressed in T47D cells. Cells were fixed 48 h after transfection, immunostained with anti‐WAVE2 antibody (a′ and b′) and anti‐E‐cadherin antibody (a″ and b″), and observed by confocal microscopy. Cells expressing EGFP‐WAVEs (green) are denoted by asterisks (*) in a′ and b′. Note that the cell–cell junction between neighboring EGFP‐WAVE3 overexpressing cells is disrupted, judged from the disappearance of E‐cadherin staining (arrowheads in b″). In concordance, WAVE2 is no longer accumulated at the dissociating cell–cell junction. These cells also show decreased endogenous levels of WAVE2 expression (red in b′). Bars, 10 μm.
Figure 6.
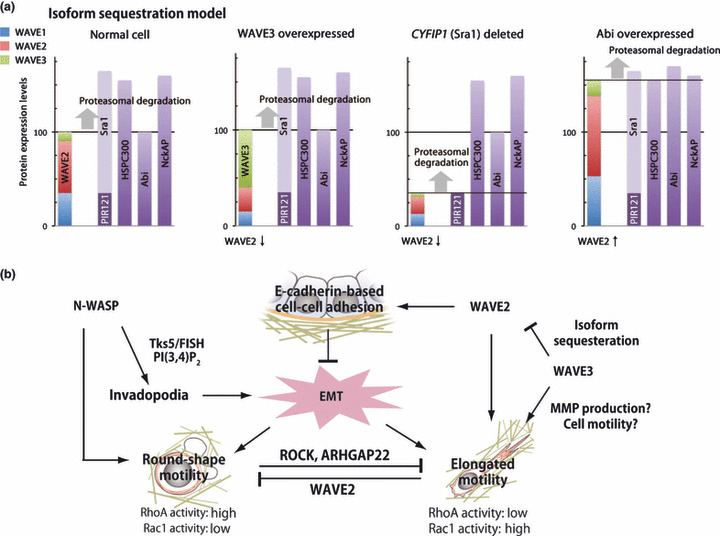
(a) The isoform sequestration model. Wiskott‐Aldrich syndrome protein (WASP) family verprolin‐homologous protein (WAVE) isoform expression levels can be altered by three possible mechanisms. If WAVE3 is overexpressed (left), the amounts of WAVE1 and WAVE2 that can successfully form the WAVE complex is decreased, and thus expression levels of WAVE1 and WAVE2 are reduced. If CYFIP1/Sra1 is deleted from the genome (centre), the absolute amount of WAVEs is capped to the level of CYFIP2/PIR121. If least‐expressed Abi is increased (right), for example via chromosomal amplification, the total amount of WAVE isoforms will be increased to the level of another component of the WAVE complex. Y‐axis of bar charts corresponds to the amount of the indicated protein. (b) Functional associations of WASPs and WAVEs in cancer progression. The WASP homologue N‐WASP affects 3D cell motility through invadopodium formation. WAVE2 has dual effects on cancer invasion: in the early phase of cancer progression, WAVE2 helps to form cell–cell adhesion and thus suppresses cancer cell invasion. However, at the later stage of progression, WAVE2 enhances elongated cell motility and acts as an invasion/metastasis promoter. Inhibiting WAVE2 activity in cancer cells invading in the elongated motility fashion induces a mode‐shift from elongated to round‐shape motility. Abi, Abl‐interactor; ARHGAP22, GTPase activating protein for Aplysia Ras‐related homologue 22; EMT, epithelial‐to‐mesenchymal transition; HSPC300, hematopoietic stem/progenitor cell protein 300; MMP, matrix metalloproteases; NckAP, Nck‐associated protein; PI(3,4)P2, phosphatidylinositol‐(3,4)‐bisphosphate; PIR121, 121F‐specific p53 inducible RNA; ROCK, Rho kinase; Sra1, 140 kDa specifically Rac1‐associated protein; Tks5/FISH, tyrosine kinase substrate 5/five SH3 domains. [Correction added after online publication 27 September 2010: Round‐shape motility RhoA activity is corrected to high, Rac1 activity is corrected to low.]
Concluding remarks
During the last decade, we have seen dramatic advancements in our understanding of WASPs and WAVEs in cancer cells. However, we still do not have a clear‐cut answer to the question of which molecule in WASP and WAVE signaling pathways could be targeted for anticancer drug development. It is of note that ROCK inhibitor treatment shifts single disseminating cancer cells toward the WAVE2‐dependent motility mode (Fig. 6b), suggesting that WAVE2 and its associated molecules, such as IRSp53, would be an attractive target for anti‐invasion drug development. The combined use of ROCK inhibitors and drugs that block WAVE2 activity would be effective against invasion of a broad spectrum of cancer cell types. Alternatively, N‐WASP inhibitors may block invasion of some cancer types that rely mostly on invadopodia to invade. However, to further validate WASP and WAVE family proteins as anticancer targets, it is urgent to determine exactly what contribution WASPs and WAVEs make in the various modes of cancer invasion.
Acknowledgments
We would like to thank Dr. Daisuke Yamazaki (Kobe University, Kobe, Japan) for providing insightful comments on this review and photographs of 3D invading cells used in Figure 4. We also thank Dr. Tsukasa Oikawa (Keio University, Tokyo, Japan) for helpful discussions regarding invadopodia. The writing of this review was supported by Grants‐in‐Aid for Scientic Research on Priority Areas “Cancer Cell Adhesion and Motility” from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (MEXT) to Tadaomi Takenawa.
Supporting information
Data S1. References.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
References
- 1. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100: 57–70. [DOI] [PubMed] [Google Scholar]
- 2. Eccles SA, Welch DR. Metastasis: recent discoveries and novel treatment strategies. Lancet 2007; 369: 1742–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Thiery JP, Sleeman JP. Complex networks orchestrate epithelial‐mesenchymal transitions. Nat Rev Mol Cell Biol 2006; 7: 131–42. [DOI] [PubMed] [Google Scholar]
- 4. Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev 2009; 28: 15–33. [DOI] [PubMed] [Google Scholar]
- 5. Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell 2003; 112: 453–65. [DOI] [PubMed] [Google Scholar]
- 6. Poincloux R, Lizarraga F, Chavrier P. Matrix invasion by tumour cells: a focus on MT1‐MMP trafficking to invadopodia. J Cell Sci 2009; 122: 3015–24. [DOI] [PubMed] [Google Scholar]
- 7. Kurisu S, Takenawa T. The WASP and WAVE family proteins. Genome Biol 2009; 10: 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Takenawa T, Suetsugu S. The WASP‐WAVE protein network: connecting the membrane to the cytoskeleton. Nat Rev Mol Cell Biol 2007; 8: 37–48. [DOI] [PubMed] [Google Scholar]
- 9. Yamazaki D, Kurisu S, Takenawa T. Regulation of cancer cell motility through actin reorganization. Cancer Sci 2005; 96: 379–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yamaguchi H, Condeelis J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim Biophys Acta 2007; 1773: 642–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Olson MF, Sahai E. The actin cytoskeleton in cancer cell motility. Clin Exp Metastasis 2009; 26: 273–87. [DOI] [PubMed] [Google Scholar]
- 12. Kurisu S, Suetsugu S, Yamazaki D, Yamaguchi H, Takenawa T. Rac‐WAVE2 signaling is involved in the invasive and metastatic phenotypes of murine melanoma cells. Oncogene 2005; 24: 1309–19. [DOI] [PubMed] [Google Scholar]
- 13. Le Clainche C, Carlier MF. Regulation of actin assembly associated with protrusion and adhesion in cell migration. Physiol Rev 2008; 88: 489–513. [DOI] [PubMed] [Google Scholar]
- 14. Insall RH, Machesky LM. Actin dynamics at the leading edge: from simple machinery to complex networks. Dev Cell 2009; 17: 310–22. [DOI] [PubMed] [Google Scholar]
- 15. Mattila PK, Lappalainen P. Filopodia: molecular architecture and cellular functions. Nat Rev Mol Cell Biol 2008; 9: 446–54. [DOI] [PubMed] [Google Scholar]
- 16. Eden S, Rohatgi R, Podtelejnikov AV, Mann M, Kirschner MW. Mechanism of regulation of WAVE1‐induced actin nucleation by Rac1 and Nck. Nature 2002; 418: 790–3. [DOI] [PubMed] [Google Scholar]
- 17. Derivery E, Gautreau A. Generation of branched actin networks: assembly and regulation of the N‐WASP and WAVE molecular machines. Bioessays 2010; 32: 119–31. [DOI] [PubMed] [Google Scholar]
- 18. Gautreau A, Ho HY, Li J, Steen H, Gygi SP, Kirschner MW. Purification and architecture of the ubiquitous Wave complex. Proc Natl Acad Sci U S A 2004; 101: 4379–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Suetsugu S, Kurisu S, Oikawa T, Yamazaki D, Oda A, Takenawa T. Optimization of WAVE2 complex‐induced actin polymerization by membrane‐bound IRSp53, PIP(3), and Rac. J Cell Biol 2006; 173: 571–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ho HY, Rohatgi R, Ma L, Kirschner MW. CR16 forms a complex with N‐WASP in brain and is a novel member of a conserved proline‐rich actin‐binding protein family. Proc Natl Acad Sci U S A 2001; 98: 11306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. De La Fuente MA, Sasahara Y, Calamito M et al. WIP is a chaperone for Wiskott‐Aldrich syndrome protein (WASP). Proc Natl Acad Sci U S A 2007; 104: 926–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kunda P, Craig G, Dominguez V, Baum B. Abi, Sra1, and Kette control the stability and localization of SCAR/WAVE to regulate the formation of actin‐based protrusions. Curr Biol 2003; 13: 1867–75. [DOI] [PubMed] [Google Scholar]
- 23. Takano K, Toyooka K, Suetsugu S. EFC/F‐BAR proteins and the N‐WASP‐WIP complex induce membrane curvature‐dependent actin polymerization. EMBO J 2008; 27: 2817–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Carlier MF, Nioche P, Broutin‐L’Hermite I et al. GRB2 links signaling to actin assembly by enhancing interaction of neural Wiskott‐Aldrich syndrome protein (N‐WASp) with actin‐related protein (ARP2/3) complex. J Biol Chem 2000; 275: 21946–52. [DOI] [PubMed] [Google Scholar]
- 25. Rohatgi R, Nollau P, Ho HY, Kirschner MW, Mayer BJ. Nck and phosphatidylinositol 4,5‐bisphosphate synergistically activate actin polymerization through the N‐WASP‐Arp2/3 pathway. J Biol Chem 2001; 276: 26448–52. [DOI] [PubMed] [Google Scholar]
- 26. Cory GO, Cramer R, Blanchoin L, Ridley AJ. Phosphorylation of the WASP‐VCA domain increases its affinity for the Arp2/3 complex and enhances actin polymerization by WASP. Mol Cell 2003; 11: 1229–39. [DOI] [PubMed] [Google Scholar]
- 27. Suetsugu S, Hattori M, Miki H et al. Sustained activation of N‐WASP through phosphorylation is essential for neurite extension. Dev Cell 2002; 3: 645–58. [DOI] [PubMed] [Google Scholar]
- 28. Baselga J, Swain SM. Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer 2009; 9: 463–75. [DOI] [PubMed] [Google Scholar]
- 29. Miki H, Yamaguchi H, Suetsugu S, Takenawa T. IRSp53 is an essential intermediate between Rac and WAVE in the regulation of membrane ruffling. Nature 2000; 408: 732–5. [DOI] [PubMed] [Google Scholar]
- 30. Mattila PK, Pykalainen A, Saarikangas J et al. Missing‐in‐metastasis and IRSp53 deform PI(4,5)P2‐rich membranes by an inverse BAR domain‐like mechanism. J Cell Biol 2007; 176: 953–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kobayashi K, Kuroda S, Fukata M et al. p140Sra‐1 (specifically Rac1‐associated protein) is a novel specific target for Rac1 small GTPase. J Biol Chem 1998; 273: 291–5. [DOI] [PubMed] [Google Scholar]
- 32. Ismail AM, Padrick SB, Chen B, Umetani J, Rosen MK. The WAVE regulatory complex is inhibited. Nat Struct Mol Biol 2009; 16: 561–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lebensohn AM, Kirschner MW. Activation of the WAVE complex by coincident signals controls actin assembly. Mol Cell 2009; 36: 512–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Oikawa T, Yamaguchi H, Itoh T et al. PtdIns(3,4,5)P3 binding is necessary for WAVE2‐induced formation of lamellipodia. Nat Cell Biol 2004; 6: 420–6. [DOI] [PubMed] [Google Scholar]
- 35. Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene 2008; 27: 5497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide‐exchange factors. Nat Rev Mol Cell Biol 2005; 6: 167–80. [DOI] [PubMed] [Google Scholar]
- 37. Cote JF, Vuori K. GEF what? Dock180 and related proteins help Rac to polarize cells in new ways. Trends Cell Biol 2007; 17: 383–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim Y, Sung JY, Ceglia I et al. Phosphorylation of WAVE1 regulates actin polymerization and dendritic spine morphology. Nature 2006; 442: 814–7. [DOI] [PubMed] [Google Scholar]
- 39. Leng Y, Zhang J, Badour K et al. Abelson‐interactor‐1 promotes WAVE2 membrane translocation and Abelson‐mediated tyrosine phosphorylation required for WAVE2 activation. Proc Natl Acad Sci U S A 2005; 102: 1098–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li Y, Clough N, Sun X et al. Bcr‐Abl induces abnormal cytoskeleton remodeling, beta1 integrin clustering and increased cell adhesion to fibronectin through the Abl interactor 1 pathway. J Cell Sci 2007; 120: 1436–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ridley AJ, Schwartz MA, Burridge K et al. Cell migration: integrating signals from front to back. Science 2003; 302: 1704–9. [DOI] [PubMed] [Google Scholar]
- 42. Kolsch V, Charest PG, Firtel RA. The regulation of cell motility and chemotaxis by phospholipid signaling. J Cell Sci 2008; 121: 551–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Weiner OD, Neilsen PO, Prestwich GD, Kirschner MW, Cantley LC, Bourne HR. A PtdInsP(3)‐ and Rho GTPase‐mediated positive feedback loop regulates neutrophil polarity. Nat Cell Biol 2002; 4: 509–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li Z, Hannigan M, Mo Z et al. Directional sensing requires G beta gamma‐mediated PAK1 and PIX alpha‐dependent activation of Cdc42. Cell 2003; 114: 215–27. [DOI] [PubMed] [Google Scholar]
- 45. Van Rheenen J, Song X, Van Roosmalen W et al. EGF‐induced PIP2 hydrolysis releases and activates cofilin locally in carcinoma cells. J Cell Biol 2007; 179: 1247–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cai L, Marshall TW, Uetrecht AC, Schafer DA, Bear JE. Coronin 1B coordinates Arp2/3 complex and cofilin activities at the leading edge. Cell 2007; 128: 915–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zicha D, Allen WE, Brickell PM et al. Chemotaxis of macrophages is abolished in the Wiskott‐Aldrich syndrome. Br J Haematol 1998; 101: 659–65. [DOI] [PubMed] [Google Scholar]
- 48. Snapper SB, Takeshima F, Anton I et al. N‐WASP deficiency reveals distinct pathways for cell surface projections and microbial actin‐based motility. Nat Cell Biol 2001; 3: 897–904. [DOI] [PubMed] [Google Scholar]
- 49. Desmarais V, Yamaguchi H, Oser M et al. N‐WASP and cortactin are involved in invadopodium‐dependent chemotaxis to EGF in breast tumor cells. Cell Motil Cytoskeleton 2009; 66: 303–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Galbraith CG, Yamada KM, Galbraith JA. Polymerizing actin fibers position integrins primed to probe for adhesion sites. Science 2007; 315: 992–5. [DOI] [PubMed] [Google Scholar]
- 51. Nemethova M, Auinger S, Small JV. Building the actin cytoskeleton: filopodia contribute to the construction of contractile bundles in the lamella. J Cell Biol 2008; 180: 1233–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Euteneuer U, Schliwa M. Persistent, directional motility of cells and cytoplasmic fragments in the absence of microtubules. Nature 1984; 310: 58–61. [DOI] [PubMed] [Google Scholar]
- 53. Yamazaki D, Suetsugu S, Miki H et al. WAVE2 is required for directed cell migration and cardiovascular development. Nature 2003; 424: 452–6. [DOI] [PubMed] [Google Scholar]
- 54. Suetsugu S, Yamazaki D, Kurisu S, Takenawa T. Differential roles of WAVE1 and WAVE2 in dorsal and peripheral ruffle formation for fibroblast cell migration. Dev Cell 2003; 5: 595–609. [DOI] [PubMed] [Google Scholar]
- 55. Kheir WA, Gevrey JC, Yamaguchi H, Isaac B, Cox D. A WAVE2‐Abi1 complex mediates CSF‐1‐induced F‐actin‐rich membrane protrusions and migration in macrophages. J Cell Sci 2005; 118: 5369–79. [DOI] [PubMed] [Google Scholar]
- 56. Sahai E. Illuminating the metastatic process. Nat Rev Cancer 2007; 7: 737–49. [DOI] [PubMed] [Google Scholar]
- 57. Wolf K, Mazo I, Leung H et al. Compensation mechanism in tumor cell migration: mesenchymal‐amoeboid transition after blocking of pericellular proteolysis. J Cell Biol 2003; 160: 267–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Friedl P, Gilmour D. Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol 2009; 10: 445–57. [DOI] [PubMed] [Google Scholar]
- 59. Gaggioli C, Hooper S, Hidalgo‐Carcedo C et al. Fibroblast‐led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol 2007; 9: 1392–400. [DOI] [PubMed] [Google Scholar]
- 60. Sahai E, Marshall CJ. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat Cell Biol 2003; 5: 711–9. [DOI] [PubMed] [Google Scholar]
- 61. Yamazaki D, Kurisu S, Takenawa T. Involvement of Rac and Rho signaling in cancer cell motility in 3D substrates. Oncogene 2009; 28: 1570–83. [DOI] [PubMed] [Google Scholar]
- 62. Friedl P, Wolf K. Tumour‐cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer 2003; 3: 362–74. [DOI] [PubMed] [Google Scholar]
- 63. Hegerfeldt Y, Tusch M, Brocker EB, Friedl P. Collective cell movement in primary melanoma explants: plasticity of cell‐cell interaction, beta1‐integrin function, and migration strategies. Cancer Res 2002; 62: 2125–30. [PubMed] [Google Scholar]
- 64. Cukierman E, Pankov R, Stevens DR, Yamada KM. Taking cell‐matrix adhesions to the third dimension. Science 2001; 294: 1708–12. [DOI] [PubMed] [Google Scholar]
- 65. Gadea G, Sanz‐Moreno V, Self A, Godi A, Marshall CJ. DOCK10‐mediated Cdc42 activation is necessary for amoeboid invasion of melanoma cells. Curr Biol 2008; 18: 1456–65. [DOI] [PubMed] [Google Scholar]
- 66. Charras GT, Hu CK, Coughlin M, Mitchison TJ. Reassembly of contractile actin cortex in cell blebs. J Cell Biol 2006; 175: 477–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lammermann T, Sixt M. Mechanical modes of ‘amoeboid’ cell migration. Curr Opin Cell Biol 2009; 21: 636–44. [DOI] [PubMed] [Google Scholar]
- 68. Charras GT, Yarrow JC, Horton MA, Mahadevan L, Mitchison TJ. Non‐equilibration of hydrostatic pressure in blebbing cells. Nature 2005; 435: 365–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ulrich TA, De Juan Pardo EM, Kumar S. The mechanical rigidity of the extracellular matrix regulates the structure, motility, and proliferation of glioma cells. Cancer Res 2009; 69: 4167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sanz‐Moreno V, Gadea G, Ahn J et al. Rac activation and inactivation control plasticity of tumor cell movement. Cell 2008; 135: 510–23. [DOI] [PubMed] [Google Scholar]
- 71. Nimnual AS, Taylor LJ, Bar‐Sagi D. Redox‐dependent downregulation of Rho by Rac. Nat Cell Biol 2003; 5: 236–41. [DOI] [PubMed] [Google Scholar]
- 72. Friedl P, Wolf K. Proteolytic interstitial cell migration: a five‐step process. Cancer Metastasis Rev 2009; 28: 129–35. [DOI] [PubMed] [Google Scholar]
- 73. Oikawa T, Itoh T, Takenawa T. Sequential signals toward podosome formation in NIH‐src cells. J Cell Biol 2008; 182: 157–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chen WT. Proteolytic activity of specialized surface protrusions formed at rosette contact sites of transformed cells. J Exp Zool 1989; 251: 167–85. [DOI] [PubMed] [Google Scholar]
- 75. Bowden ET, Barth M, Thomas D, Glazer RI, Mueller SC. An invasion‐related complex of cortactin, paxillin and PKCmu associates with invadopodia at sites of extracellular matrix degradation. Oncogene 1999; 18: 4440–9. [DOI] [PubMed] [Google Scholar]
- 76. Monsky WL, Kelly T, Lin CY et al. Binding and localization of M(r) 72,000 matrix metalloproteinase at cell surface invadopodia. Cancer Res 1993; 53: 3159–64. [PubMed] [Google Scholar]
- 77. Tu C, Ortega‐Cava CF, Chen G et al. Lysosomal cathepsin B participates in the podosome‐mediated extracellular matrix degradation and invasion via secreted lysosomes in v‐Src fibroblasts. Cancer Res 2008; 68: 9147–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Abram CL, Seals DF, Pass I et al. The adaptor protein fish associates with members of the ADAMs family and localizes to podosomes of Src‐transformed cells. J Biol Chem 2003; 278: 16844–51. [DOI] [PubMed] [Google Scholar]
- 79. Cougoule C, Le Cabec V, Poincloux R et al. Three‐dimensional migration of macrophages requires Hck for podosome organization and extracellular matrix proteolysis. Blood 2010; 115: 1444–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Mizutani K, Miki H, He H, Maruta H, Takenawa T. Essential role of neural Wiskott‐Aldrich syndrome protein in podosome formation and degradation of extracellular matrix in src‐transformed fibroblasts. Cancer Res 2002; 62: 669–74. [PubMed] [Google Scholar]
- 81. Kaverina I, Stradal TE, Gimona M. Podosome formation in cultured A7r5 vascular smooth muscle cells requires Arp2/3‐dependent de‐novo actin polymerization at discrete microdomains. J Cell Sci 2003; 116: 4915–24. [DOI] [PubMed] [Google Scholar]
- 82. Yamaguchi H, Lorenz M, Kempiak S et al. Molecular mechanisms of invadopodium formation: the role of the N‐WASP‐Arp2/3 complex pathway and cofilin. J Cell Biol 2005; 168: 441–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Li A, Dawson JC, Forero‐Vargas M et al. The actin‐bundling protein fascin stabilizes actin in invadopodia and potentiates protrusive invasion. Curr Biol 2010; 20: 339–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Albiges‐Rizo C, Destaing O, Fourcade B, Planus E, Block MR. Actin machinery and mechanosensitivity in invadopodia, podosomes and focal adhesions. J Cell Sci 2009; 122: 3037–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Seals DF, Azucena EF Jr, Pass I et al. The adaptor protein Tks5/Fish is required for podosome formation and function, and for the protease‐driven invasion of cancer cells. Cancer Cell 2005; 7: 155–65. [DOI] [PubMed] [Google Scholar]
- 86. Gates J, Peifer M. Can 1000 reviews be wrong? Actin, alpha‐Catenin, and adherens junctions. Cell 2005; 123: 769–72. [DOI] [PubMed] [Google Scholar]
- 87. Yamada S, Pokutta S, Drees F, Weis WI, Nelson WJ. Deconstructing the cadherin‐catenin‐actin complex. Cell 2005; 123: 889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Drees F, Pokutta S, Yamada S, Nelson WJ, Weis WI. Alpha‐catenin is a molecular switch that binds E‐cadherin‐beta‐catenin and regulates actin‐filament assembly. Cell 2005; 123: 903–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Yamazaki D, Oikawa T, Takenawa T. Rac‐WAVE‐mediated actin reorganization is required for organization and maintenance of cell‐cell adhesion. J Cell Sci 2007; 120: 86–100. [DOI] [PubMed] [Google Scholar]
- 90. Sahai E, Marshall CJ. ROCK and Dia have opposing effects on adherens junctions downstream of Rho. Nat Cell Biol 2002; 4: 408–15. [DOI] [PubMed] [Google Scholar]
- 91. Kovacs EM, Goodwin M, Ali RG, Paterson AD, Yap AS. Cadherin‐directed actin assembly: E‐cadherin physically associates with the Arp2/3 complex to direct actin assembly in nascent adhesive contacts. Curr Biol 2002; 12: 379–82. [DOI] [PubMed] [Google Scholar]
- 92. Malliri A, Van Es S, Huveneers S, Collard JG. The Rac exchange factor Tiam1 is required for the establishment and maintenance of cadherin‐based adhesions. J Biol Chem 2004; 279: 30092–8. [DOI] [PubMed] [Google Scholar]
- 93. Takaishi K, Sasaki T, Kotani H, Nishioka H, Takai Y. Regulation of cell‐cell adhesion by rac and rho small G proteins in MDCK cells. J Cell Biol 1997; 139: 1047–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Angres B, Barth A, Nelson WJ. Mechanism for transition from initial to stable cell‐cell adhesion: kinetic analysis of E‐cadherin‐mediated adhesion using a quantitative adhesion assay. J Cell Biol 1996; 134: 549–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Silva JM, Ezhkova E, Silva J et al. Cyfip1 is a putative invasion suppressor in epithelial cancers. Cell 2009; 137: 1047–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Burk U, Schubert J, Wellner U et al. A reciprocal repression between ZEB1 and members of the miR‐200 family promotes EMT and invasion in cancer cells. EMBO Rep 2008; 9: 582–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Wellner U, Schubert J, Burk UC et al. The EMT‐activator ZEB1 promotes tumorigenicity by repressing stemness‐inhibiting microRNAs. Nat Cell Biol 2009; 11: 1487–95. [DOI] [PubMed] [Google Scholar]
- 98. Wang W, Goswami S, Lapidus K et al. Identification and testing of a gene expression signature of invasive carcinoma cells within primary mammary tumors. Cancer Res 2004; 64: 8585–94. [DOI] [PubMed] [Google Scholar]
- 99. Sossey‐Alaoui K, Safina A, Li X et al. Down‐regulation of WAVE3, a metastasis promoter gene, inhibits invasion and metastasis of breast cancer cells. Am J Pathol 2007; 170: 2112–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Sossey‐Alaoui K, Bialkowska K, Plow EF. The miR200 family of microRNAs regulates WAVE3‐dependent cancer cell invasion. J Biol Chem 2009; 284: 33019–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. References.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item