

Abstract
The size of anticancer agent‐incorporating micelles can be controlled within the diameter range of 20–100 nm to ensure that they do not penetrate normal vessel walls. With this development, it is expected that the incidence of drug‐induced side‐effects may be decreased owing to the reduced drug distribution in normal tissue. Micelle systems can also evade non‐specific capture by the reticuloendothelial system because the outer shell of a micelle is covered with polyethylene glycol. Consequently, a polymer micelle carrier can be delivered selectively to a tumor by utilizing the enhanced permeability and retention effect. Moreover, a water‐insoluble drug can be incorporated into polymer micelles. Presently, several anticancer agent‐incorporating micelle carrier systems are under preclinical and clinical evaluation. Furthermore, nucleic acid‐incorporating micelle carrier systems are also being developed. (Cancer Sci 2009; 100: 572–579)
Nanotechnology is one of the fast‐moving technologies and is presently contributing significantly to the progress of medical science. Drugs categorized under the drug delivery system (DDS) are made primarily by utilizing nanotechnology. In the field of oncology, DDS drugs have been produced and evaluated in preclinical or clinical trials, with some already approved for clinical use (Table 1). More specifically, DDS can be used for active or passive targeting of tumor tissues. Passive targeting refers to the development of monoclonal antibodies directed against tumor‐related molecules, allowing targeting of a tumor from the specific binding of antibodies with respective antigens. However, the application of DDS using monoclonal antibodies is restricted to tumors expressing high levels of related antigens.
Table 1.
Examples of drug delivery systems in oncology and their stage of development
|
Passive targeting | |||
|---|---|---|---|
| Name | Platform | Compound | Clinical stage |
| NK105 | Micelles | Paclitaxel | P2 |
| NC‐6004 | Micelles | Cisplatin | P1/2 |
| NK012 | Micelles | SN‐38 | P2 |
| Smancs | Polymer conjugate | Neocarzinostatin | Approved |
| Doxil | Liposome | Doxorubicin | Approved |
| Abraxane | Albumin particle | Paclitaxel | Approved |
| Xyota,˜ | Polymer conjugate | Paclitaxel | P3 |
| CT‐2106 | Polymer conjugate | Camptothecin | P2 |
| EndoTAG | Cationic Liposome | Paclitaxel | P2 |
| MAG‐CPT | Polymer conjugate | Camptothecin | P1 |
| LE‐SN‐38 | Liposome | SN‐38 | P2 |
| PK1 | Polymer conjugate | Doxorubicin | P2 |
| ,hT‐101 | Polymer conjugate | Camptothecin | P2 |
| SP1049C | Micelles | Doxorubicin | P3 |
| CPX‐1 | Liposome | CPT‐11,floxuridine | P2 |
|
Active targeting | |||
|---|---|---|---|
| Name | Platform | Compound | Clinical Stage |
| Mylotarg | Anti‐CD33‐Ab | Calicheamicin | Approved |
| Zevalin | Anti‐CD20‐Ab | 90Y | Approved |
| Bexxar | Anti‐CD20‐Ab | 131 I | Approved |
| PK2 | Galactose‐Polymer | Doxorubicin | P1 |
| MCC465 | Ab‐liposome | Doxorubicin | P1 |
| MBP‐426 | Transferrin‐liposome | Oxaliplatin | P1 |
| CALAA‐01 | Transferrin‐polymer | siRNA | P1 |
| T‐DM1 | Anti‐HER2‐Ab | DM1 | P1 |
Passive targeting can be achieved by utilizing the enhanced permeability and retention (EPR) effect.( 1 , 2 ) This effect is based on the pathophysiological characteristics of solid tumor tissues, namely, hypervascularity, incomplete vascular architecture, secretion of vascular permeability factors stimulating extravasation within cancer tissue, and absence of effective lymphatic drainage from tumors that impedes the efficient clearance of macromolecules accumulated in solid tumor tissues (Fig. 1A,B).
Figure 1.

(A) A diagram of normal and tumor tissue, demonstrating the presence of a lymphatic duct in normal tissue (upper) but the absence of any lymphatic duct in tumor tissue (lower). Small molecules easily leak from normal vessels in the body, which gives small molecules a short plasma half life. On the other hand, macromolecules have a long plasma half life because they are too large to pass through the normal vessel walls, unless they are trapped by the reticuloendothelial system in various organs. In the solid tumor tissues shown in the lower panel, it was found that solid tumors generally possess several pathophysiological characteristics: hypervasculature, secretion of vascular permeability factors stimulating extravasation of macromolecules within the cancer, and absence of effective lymphatic drainage from tumors that impedes the efficient clearance of macromolecules accumulated in solid tumor tissues. These characteristics of solid tumors are the basis of the enhanced permeability and retention (EPR) effect. Summarizing these findings, conventional low‐molecular‐weight anticancer agents disappear before reaching the tumor tissues and exerting their cell‐killing effect. On the other hand, macromolecules and nanoparticles including micelle carriers should have time to reach the tumor, extravasate from the tumor capillaries, and stay for a long time in the tumor tissue, by means of the EPR effect. (B) Example of the EPR effect. This in vivo imaging demonstrates that both control whole immunoglobulin G (IgG) and the specific whole monoclonal antibody (mAb) accumulated selectively in the tumor tissue on day 1 after invravenous injection. On day 7, a greater degree of retention of the specific whole mAb as compared to the control IgG was noted. On the other hand, the F(ab) region of the specific mAb with a molecular weight of 45 000 accumulated in the tumor to the same extent as the control whole IgG. Interestingly, fluorescence of the F(ab) could also be detected in both the kidneys, which implied that the antigen binding fragment F(ab) could easily pass through the kidney glomerulus. This accumulation of the control IgG in the tumor represents the EPR effect. The findings suggest that not only the specific affinity of the mAb, but also the size of the molecules and the stability of the molecules in blood are important for tumor‐selective targeting.
Several techniques have been developed to maximally utilize the EPR effect such as modification of drug structures and development of drug carriers. Polymeric micelle‐based anticancer drugs were originally developed by Kataoka et al. in the late 1980s or early 1990s.( 3 , 4 , 5 ) Polymeric micelles were expected to increase the accumulation of drugs in tumor tissues by utilizing the EPR effect as well as to incorporate various kinds of drugs into their inner core with relatively high stability by chemical conjugation or physical entrapment. Also, the size of micelles can be controlled within the diameter range of 20–100 nm to ensure that they do not penetrate normal vessel walls. With this development, it is expected that the incidence of drug‐induced side‐effects may be decreased owing to the reduced drug distribution in normal tissues.
In this paper, we review recent developments in polymeric micelle systems presently being evaluated in clinical trials and introduce new micellar formulations.
Anticancer agent‐incorporating micelle carrier systems under clinical evaluation
NK105: paclitaxel (PTX)‐incorporating micellar nanoparticle
Background. PTX is one of the most useful anticancer agents against various cancers, including ovarian, breast and lung cancers.( 6 , 7 ) However, it produces serious adverse effects such as neutropenia and peripheral sensory neuropathy. In addition, anaphylaxis and other severe hypersensitive reactions have been reported in 2–4% of patients receiving the drug even after premedication with anti‐allergic agents; these adverse reactions have been attributed to the mixture of Cremophor EL and ethanol used for solubilizing PTX.( 8 , 9 ) Of the adverse reactions, neutropenia can be prevented or managed effectively by administering a granulocyte colony‐stimulating factor. On the other hand, there are no effective therapies to prevent or reduce nerve damage associated with PTX‐induced peripheral neuropathy. Thus, neurotoxicity constitutes a significant dose‐limiting toxicity of the drug.( 10 , 11 )
Preparation and characterization of NK105. To construct NK105 micellar nanoparticles (Fig. 2A), block copolymers consisting of polyethylene glycol (PEG) and polyaspartate,( 3 , 4 , 5 ) were used. PTX was incorporated into polymeric micelles formed by physical entrapment utilizing hydrophobic interactions between PTX and the block copolymer polyaspartate chain. NK105 was prepared by facilitating the self association of NK105 polymers and PTX. NK105 was obtained as a freeze‐dried formulation and contained about 23% (w/w) of PTX The weight‐average diameter of the nanoparticles was approximately 85 nm with a narrow size distribution.( 12 )
Figure 2.
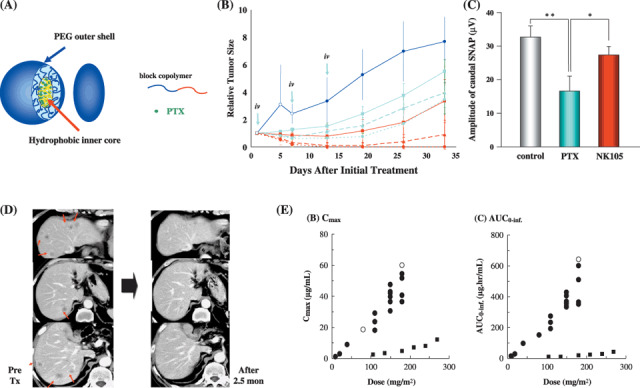
(A) The schematic structure of NK105, a micelle‐forming polymeric drug, that is a polyethylene glycol (PEG)‐poly (aspartic acid) block copolymer conjugated with paclitaxel (PTX). NK105 was obtained as a freeze‐dried formulation and contained approximately 23% (w/w) of PTX. The weight‐average diameter of the nanoparticles was approximately 85 nm with a narrow size distribution. (B) Antitumor activity of PTX or NK105 on HT‐29 human colon cancer xenografts.Each drug was administered intravenously weekly for 3 times at a PTX equivalent dose of 25 (¦?¦), 50 (mg/kg), or 100 mg/kg (mg/kg). Light blue lines show the treatment with PTX. Red lines show the treatment with NK105. NK105 exhibited significant superior antitumor activity as compared with PTX at respective dose levels (P < 0.001). Especially, tumors disappeared after the first dosing to mice treated with NK105 at 100 mg/kg. Dark blue line (control). Each point: mean ± SE. (C) The amplitude of caudal sensory nerve action potential after the drugs administration for 2 months. The amplitude was significantly smaller in the PTX group as compared to control or NK105 administration group. (D) Serial computed tomography scans. A 60‐year‐old man with pancreatic cancer who was treated with NK105 at a dose level of 150 mg/m2. Baseline scan (left) showing multiple metastasis in the liver. Partial response, characterized by a more than 90% decrease in the size of the liver metastasis (right) compared with the baseline scan. The antitumor response was maintained for nearly 1 year. (E) Correlations between dosage and maximum contentration (Cmax) and area under the curve (AUC) for both PTX ( ) d NK105 (
) d NK105 ( ). Both Cmax and AUC of NK105 were significantly higher than those of PTX. Different from PTX, the Cmax and AUC of NK105 increased linearly at doses between 10 and 15 mg/m2. The AUC of NK105 at the recommended dose was 10–30‐fold higher than that of PTX at a conventional dose, 210 mg/m2.
). Both Cmax and AUC of NK105 were significantly higher than those of PTX. Different from PTX, the Cmax and AUC of NK105 increased linearly at doses between 10 and 15 mg/m2. The AUC of NK105 at the recommended dose was 10–30‐fold higher than that of PTX at a conventional dose, 210 mg/m2.
Preclinical studies. In an in vivo pharmacokinetics study using colon 26 tumor‐bearing CDF1 mice, the plasma concentration at 5 min (C5min) and the area under the curve (AUC) of NK105 were 11‐ to 20‐fold and 50‐ to 86‐fold higher than those of PTX. The maximum contentration (Cmax) and AUC of NK105 in colon 26 tumors were approximately 3 and 25 times higher than those of PTX. NK105 continued to accumulate in the tumors until 72 h postinjection (data not shown).
In BALB/c mice bearing subcutaneous HT‐29 colon cancer tumors, NK105 exhibited superior antitumor activity compared with PTX (P < 0.001). Tumor suppression by NK105 increased in a dose‐dependent manner (Fig. 2B).
Paclitaxel treatment has been shown to cause cumulative sensory‐dominant peripheral neurotoxicity in humans characterized clinically by numbness and/or paraesthesia of the extremities. Pathologically, axonal swelling, vesicular degeneration, and demyelination have also been observed. We therefore examined the pathologic effects of free PTX and NK105 using both electrophysiological and morphological methods. After drug administration for 2 months, the amplitude of the caudal sensory nerve action potential in the control group increased in association with rat maturation. The amplitude was significantly smaller in the PTX group than in the control group (P < 0.01); it was significantly larger in the NK105 group than in the PTX group (P < 0.05); and it was comparable between the NK105 group and the control group (Fig. 2C).
Clinical study. A phase I study was designed to determine the maximum tolerated dose (MTD), dose‐limiting toxicities (DLTs), and recommended dose (RD) of NK105 for phase II, as well as its pharmacokinetics.( 13 )
NK105 was administered by intravenous infusion for 1 h every 3 weeks without anti‐allergic premedication. The starting dose was 10 mg PTX equivalent/m2, and the dose escalated according to the accelerated titration method. DLTs were observed in two patients at 180 mg/m2 (grade 4 neutropenia lasting for more than 5 days), which was determined as MTD. Allergic reactions were not observed in any of the patients except in one. The RD was 150 mg/m2. A partial response was observed in one pancreatic cancer patient who received more than 12 courses of NK105 (Fig. 2D). Colon and gastric cancer patients experienced stable disease lasting for ten and seven courses, respectively. Despite long‐term administration, only grade 1 or 2 neuropathy was observed when the dose or period of drug administration was modified. The Cmax and AUC of NK105 showed dose‐dependent characteristics. The plasma AUC of NK105 at 150 mg/m2 was approximately 30‐fold higher than that of the commonly used PTX formulation (Fig. 2E).
Dose‐limiting toxicity was Grade 4 neutropenia. NK105 facilitates prolonged systemic PTX exposure in plasma. Tri‐weekly 1‐h infusion of NK105 was feasible and well tolerated, with antitumor activity. A phase II study of NK105 against advanced stomach cancer as a second‐line therapy is currently underway.
NC‐6004: cisplatin‐incorporating micellar nanoparticle
Background. Cisplatin (cis‐dichlorodiammineplatinum [II]: CDDP] is a key drug in the chemotherapy of various cancers, including lung, gastrointestinal and genitourinary cancers.( 14 , 15 ) However, it is often necessary to discontinue CDDP treatment because of its adverse reactions (e.g. nephrotoxicity and neurotoxicity) despite its persisting effects.( 16 ) To date, platinum analogs (e.g. carboplatin and oxaliplatin)( 17 ) have been developed to overcome these CDDP‐related disadvantages. Consequently, these analogs have become the standard drugs for ovarian( 18 ) and colon( 19 ) cancers. On the other hand, these regimens including CDDP constitute the standard treatment for lung, gastric, testicular( 20 ) and urothelial( 21 ) cancers. Therefore, the development of DDS technology is anticipated, which would enable better selective CDDP accumulation in solid tumors while lessening its distribution in normal tissue.
Preparation and characterization of NC‐6004. NC‐6004 was prepared according to a slightly modified procedure reported by Nishiyama et al.( 22 ) (Fig. 3A). NC‐6004 consists of PEG, a hydrophilic chain constituting the outer shell of micelles, and the coordinate complex of poly(glutamic acid) and CDDP, a polymer‐metal complex‐forming chain constituting the inner core of micelles. A narrowly distributed size of polymeric micelles (20 nm) was confirmed by dynamic light‐scattering measurement. Also, static light‐scattering measurement revealed that the CDDP‐loaded micelles showed no dissociation upon dilution and the critical micellar concentration was <5 × 10−7, suggesting remarkable stability compared with typical micelles from amphiphilic block copolymers.( 22 )
Figure 3.

(A) Cisplatin‐incorporating polymeric micelles (NC‐6004) consists of polyethylene glycol (PEG), a hydrophilic chain which constitutes the outer shell of the micelles, and the coordinate complex of Poly(Glu) and cis‐dichlorodiammineplatinum (II) (CDDP) constitutes the inner core of the micelles. The mean particle size of NC‐6004 was approximately 20 nm. (B) Nephrotoxicity of CDDP and NC‐6004 in rats. The CDDP 10 mg/kg administration group showed significantly higher concentrations of blood urea nitrogen (BUN) and creatinine as compared with the control group and with the NC6004 10 mg/kg administration group. (C) Neurotoxicity of CDDP and NC‐6004 in rats. Rats were given 2 mg/kg of CDDP () or NC‐6004 ( ) twice a week, 11 administrations in total. Animals given NC‐6004 showed no delay in sensory nerve conductive velocity (SNCV) as compared with the control (¦). On the other hand, animals given CDDP showed a significant delay in SNCV as compared with animals given NC‐6004 (A). The analysis by inductively‐coupled plasma–mass spectrometer revealed that sciatic nerve concentrations of platinum (Pt) in mice given CDDP were more than 2‐fold higher than NC‐6004 (b). *P < 0.05 NS: not significant.
) twice a week, 11 administrations in total. Animals given NC‐6004 showed no delay in sensory nerve conductive velocity (SNCV) as compared with the control (¦). On the other hand, animals given CDDP showed a significant delay in SNCV as compared with animals given NC‐6004 (A). The analysis by inductively‐coupled plasma–mass spectrometer revealed that sciatic nerve concentrations of platinum (Pt) in mice given CDDP were more than 2‐fold higher than NC‐6004 (b). *P < 0.05 NS: not significant.
Preclinical study. NC‐6004 showed a very long blood retention profile compared with CDDP. The AUC0‐t and Cmax values were significantly higher in animals given NC‐6004 than in animals given CDDP, namely, 65‐fold and 8‐fold, respectively (P < 0.001 and P < 0.001, respectively). Regarding platinum accumulation in the tumor, platinum concentrations peaked at 10 min following CDDP administration and at 48 h following NC‐6004 administration. The Cmax in the tumor was 2.5‐fold higher for NC‐6004 than for CDDP (P < 0.001). Furthermore, the tumor AUC was 3.6‐fold higher for NC‐6004 than for CDDP (81.2 µg/mL/h and 22.6 µg/mL/h, respectively).( 23 )
In nude mice implanted with the human gastric cancer cell line MKN‐45, NC‐6004 administration groups (5 mg/kg of CDDP) showed no significant difference in tumor growth rate compared with CDDP administration groups. Regarding time‐course changes in body weight change rate, the CDDP (5 mg/kg) administration group showed a significant decrease (P < 0.001) in body weight compared with the control group. On the other hand, the NC‐6004 administration group showed no decrease in body weight compared with the control group (data not shown).
Regarding renal function, the CDDP (10 mg/kg) administration group showed significantly higher plasma concentrations of BUN and creatinine than the control group (P < 0.05 and P < 0.001, respectively) and NC‐6004 (10 mg/kg) administration group (P < 0.05 and P < 0.001, respectively) (Fig. 3B).
In neurological examination, rats given NC‐6004 showed no delay in sensory nerve conductive velocity (SNCV) compared with animals given 5% glucose. On the other hand, rats given CDDP showed a significant delay (P < 0.05) in SNCV compared with animals given NC‐6004. Sciatic nerve concentrations of platinum were significantly (P < 0.05) lower in rats given NC‐6004 (Fig. 3C). This finding is believed to be a factor that reduced neurotoxicity following NC‐6004 administration compared with CDDP administration.
Clinical study. A phase I clinical trial of NC‐6004 has recently been completed in the UK.( 24 ) The starting dose of NC‐6004 was 10 mg/m2. NC‐6004 was administered once every 3 weeks with only 1000 mL water loading at the day of administration. Administration of doses up to 120 mg/m2 was performed without inducing significant nephrotoxicity. Although nausea and vomiting are typical CDDP adverse effects, those caused by NC‐6004 were generally mild. However, hypersensitivity reactions caused by NC‐6004 occurred more frequently than those caused by CDDP. A phase II study will soon be started involving sufficient anti‐allergic premedication.
NK012: SN‐38‐incorporating micellar nanoparticle
Background. Irinotecan hydrochloride (CPT‐11) has recently been demonstrated to be active against colorectal, lung, and ovarian cancers.( 25 , 26 , 27 , 28 , 29 ) CPT‐11 is a prodrug and is converted to 7‐ethyl‐10‐hydroxy‐CPT (SN‐38), a biologically active metabolite of CPT‐11, by carboxylesterases (CEs). SN‐38 (Fig. 4A) is an analog of the plant alkaloid camptothecin which targets DNA topoisomerase I. SN‐38 exhibits up to 1000‐fold more potent cytotoxic activity against various cancer cells in vitro than CPT‐11.( 30 ) Although CPT‐11 is converted to SN‐38 in the liver and tumors, the metabolic conversion rate is less than 10% of the original volume of CPT‐11.( 31 , 32 ) Moreover, the conversion of CPT‐11 to SN‐38 depends on the genetic interindividual variability of CE activity.( 33 ) Thus, further efficient use of SN‐38 might be of great advantage and may be attractive for cancer treatment. The progress of the manufacturing technology of ‘micellar nanoparticles’ may make it possible to use SN‐38 for in vivo experiments and further clinical use.
Figure 4.
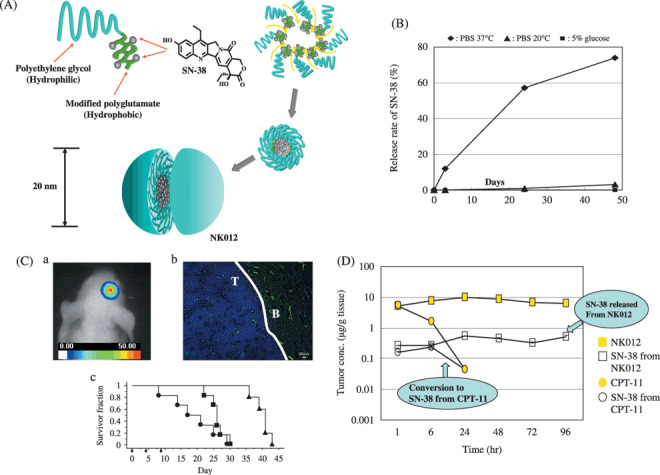
(A) Schematic structure of NK012. A polymeric micelle carrier of NK012 consists of a block copolymer of polyethylene glycol (PEG) (molecular weight of ≈5000) and partially modified polyglutamate (≈ 20 units). Polyethylene glycol (hydrophilic) is believed to be the outer shell and SN‐38 was incorporated into the inner core of the micelle. (B) The releasing rates of 7‐ethyl‐10‐hydroxy‐CPT (SN‐38) from NK012. The data suggested that NK012 is stable in 5% glucose solution before administration and starts to release SN‐38, gradually, under physiological conditions after administration. (C) (a) An orthotopic glioma model. Twenty days after U87MG/Luc inoculation. (b) The orthotopic tumor was visualized using a photon imager. The maximum tolerated dose (MTD) of NK012 (30 mg/kg) or Irinotecan hydrochloride (CPT‐11) (66.7 mg/kg) was injected intravenously into the tail vein of mice. 24 h after NK012 injection, mice were also administered with fluorescein Lycopersicon esculentum lectin (100 µL/mouse) to visualize tumor blood vessels (green). NK012 (blue) was accumulated selectively in tumor tissue. T: tumor, B: normal brain (c) NK012 (30 mg/kg/day), CPT‐11 (66.7 mg/kg/day) () and saline () were intravenously given on days 0 (20 days after tumor inoculation), 4, and 8 ( ). Kaplan‐Meier analysis was performed to determine the effect of drugs on time to morbidity, and statistical differences were ranked according to the Mantel‐Cox log‐rank test using StatView 5.0. (D) Intra‐tumor distribution of CPT‐11, NK012 (or polymer bound SN‐38), and free SN‐38 after administration of NK012 and CPT‐11 to mice bearing Capan1 xenografts. The time profiles of polymer‐bound SN‐38 (), free SN‐38 released from NK012 (), CPT‐11 (), and free SN‐38 converted from CPT‐11 (
). Kaplan‐Meier analysis was performed to determine the effect of drugs on time to morbidity, and statistical differences were ranked according to the Mantel‐Cox log‐rank test using StatView 5.0. (D) Intra‐tumor distribution of CPT‐11, NK012 (or polymer bound SN‐38), and free SN‐38 after administration of NK012 and CPT‐11 to mice bearing Capan1 xenografts. The time profiles of polymer‐bound SN‐38 (), free SN‐38 released from NK012 (), CPT‐11 (), and free SN‐38 converted from CPT‐11 ( ) were obtained by high performance liquid chromatography (HPLC) analysis. The time‐points examined were 1, 6, 24, 48, 72, and 96 h after the administration of CPT‐11 or NK012.
) were obtained by high performance liquid chromatography (HPLC) analysis. The time‐points examined were 1, 6, 24, 48, 72, and 96 h after the administration of CPT‐11 or NK012.
Preparation and characterization of NK012. NK012 is an SN‐38‐loaded polymeric micelle constructed in an aqueous milieu by the self‐assembly of an amphiphilic block copolymer, PEG‐PGlu(SN‐38).( 34 ) NK012 was obtained as a freeze‐dried formulation and contained about 20% (w/w) of SN‐38 (Fig. 4A). The mean particle size of NK012 is 20 nm in diameter with a relatively narrow range (Fig. 4A). The releasing rates of SN‐38 from NK012 in phosphate buffered saline at 37°C were 57% and 74% at 24 h and 48 h, and those in 5% glucose solution at the same temperature and times were 1% and 3%, respectively (Fig. 4B). These results indicate that NK012 can release SN‐38 under neutral conditions even without a hydrolytic enzyme, and is stable in 5% glucose solution. Thus, NK012 is suggested to be stable before administration and starts to release SN‐38 gradually under physiological conditions following administration.
Preclinical study. Following CPT‐11 injection, the plasma concentrations of CPT‐11 and SN‐38 rapidly decreased with time in a log‐linear fashion. On the other hand, NK012 (polymer‐bound SN‐38) exhibited slower clearance. In tumor xenografts, NK012 clearance was significantly slower and free SN‐38 concentration was maintained for a long time following injection.( 34 , 35 ) Interestingly, there was no significant difference in the kinetic characteristics of free SN‐38 in the small intestine between mice treated with NK012 and CPT‐11.
Deviating from the ordinary experimental tumor model, tumors were allowed to grow until they became very large (approximately 1.5 cm), and then treatment was initiated. NK012 showed potent antitumor activity against bulky small cell lung cancer (SCLC) line, SBC‐3/Neo, tumors compared with CPT‐11. A striking antitumor activity was observed in mice treated with NK012 when we compared its antitumor activity with CPT‐11 using SBC‐3/VEGF (vascular endothelial growth factor) cells. In the clinical setting, CPT‐11/5‐FU (fluorouracil) combination therapy is now a standard regimen for colorectal cancer.( 25 , 26 ) Therefore, it was speculated that the use of NK012 in place of CPT‐11 in combination with 5‐FU may yield superior results. As expected, the therapeutic effect of NK012/5‐FU was significantly superior to that of CPT‐11/5‐FU against HT‐29 xenografts (P = 0.0004)( 36 ) (data not shown). In other tumors such as renal cancer,( 37 ) glioma,( 38 ) gastric cancer( 39 ) and pancreatic cancer,( 35 ) NK012 exerted significant superior antitumor activity and induced longer survival compared with CPT‐11. In a glioma orthotopic xenograft model, both NK012 and CPT‐11 appeared to be able to effectively extravasate from the blood–brain tumor barrier but not from normal brain vessels (Fig. 4C).( 38 ) In a pancreatic cancer cell line, Capan‐1 tumor xenograft as a hypovascular tumor model, it was also demonstrated that NK012 showed significantly more potent antitumor activity than CPT‐11. Pharmacological examination revealed that only a slight conversion of SN‐38 from CPT‐11 was observed from 1 h to 24 h, and no SN‐38 was detected thereafter. On the other hand, SN‐38 released from NK012 continued to be detected from 1 h to 96 h following NK012 injection( 35 ) (Fig. 4D).
Thus, NK012, which combines enhanced distribution with prolonged sustained release of SN‐38 within tumors, is ideal for the treatment of hypovascular tumors since the antitumor activity of SN‐38 is time‐dependent.
Clinical study. Two independent phase I clinical trials have been conducted in the National Cancer Center in Japan( 40 ) and the Sarah Canon Cancer Center in the US( 41 ) in patients with advanced solid tumors to define the MTD, DLT, and recommended phase II dose. NK012 is infused intravenously over 60 min every 21 days until disease progression or unacceptable toxicity occurs. The MTD and RD were 37 mg/m2 and 28 mg/m2, respectively. DLT was neutropenia, and diarrhea was mild. The pharmacokinetics (PK) profile in the US study was similar to that in the Japanese study. Antitumor activity was also promising. Partial responses (PR)s were obtained in three patients with triple negative breast cancer, one patient with esophageal cancer and one patient with SCLC. A phase II study in patients with triple negative breast and colorectal cancers will soon be started in the US and Japan.
Future micelle formulations
The development of smart polymeric micelles that dynamically change their properties owing to their sensitivity to chemical or physical stimuli is the most promising trend, leading to the development of targeting therapy with high efficacy and ensured safety.( 42 ) In this way, such micelles can respond to pathological or physiological endogenous stimuli already present in the body or to externally applied stimuli such as temperature, light or ultrasound. One sophisticated and rational formulation approach is to increase the hydrophobicity of the core of the polymeric micelles loaded with a platinum‐based drug. This is to modulate the drug‐releasing profile induced by chloride ion in the body fluid so as to have a sufficient induction period, and thus avoiding systemic drug leakage and achieving selective and sustained drug release at the site of a solid tumor.( 43 ) Based on this strategy, dichloro (trans‐l‐1,2‐diaminocyclohexane) platinum (II) (DachPt)‐loaded micelles have recently been developed showing longer life in blood circulation and improved anticancer effects compared with cisplatin‐loaded polymeric micelles (Fig. 5A).( 44 ) A pH‐triggered system also shows great promise in the treatment of intractable cancers. Here, the therapeutic agent should be stably associated with the hydrophobic core, and drug release is expected to occur with the destabilization of the micelle structure in response to an acidic pH of tumor tissue as well as with disruption of intracellular compartments such as the endosome and lysosome.( 45 ) Notable antitumor efficacy against hypovascular cancer, including pancreatic and diffuse‐type gastric cancers of doxorubicin‐incorporating polymeric micelles with a pH‐responding property was demonstrated to emphasize the promising application of DDS for the treatment of intractable cancers.( 46 ) Photodynamic therapy, which involves the systemic administration of photosensitizers (PSs) and the subsequent photoirradiation of the diseased sites, is a promising physical approach for cancer treatment. However, PSs readily form aggregates, resulting in a significant reduction in singlet oxygen production. To prevent the self‐quenching of PSs, ionic dendritic PSs (DPs) have been prepared and incorporated into polymeric micelles (Fig. 5B).( 47 ) Eventually, the DP‐loaded micelles showed a significant increase in photocytotoxicity compared with free DPs, achieving a remarkable photodynamic efficacy against a subcutaneous tumor model in experimental animals by systemic injection.
Figure 5.
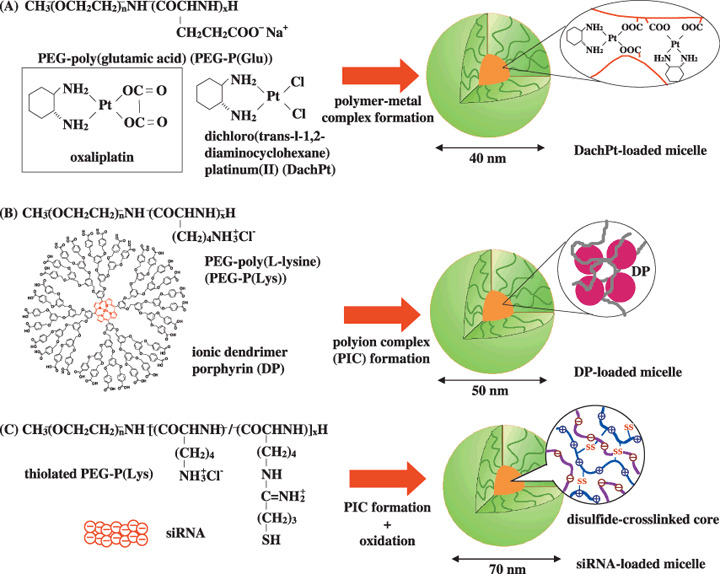
Formation of polymeric micelles incorporating dichloro (trans‐l‐1,2‐diaminocyclohexane) platinum (II) (DachPt) (A), ionic dendritic photosensitizers (B) and siRNA (C).
Success in gene and nucleic acid delivery indeed relies on the development of safe and effective carriers. In this regard, polyion complex (PIC) micelles, which are formed between nucleic acid and PEG‐polycation block copolymers, have received much attention owing to their small size (~100 nm) and excellent biocompatibility.( 48 ) Recently, an siRNA‐incorporating PIC micelle has been prepared from the PEG‐poly(lysine) block copolymer with sulthydryl (SH) groups in the side chain (Fig. 5C).( 49 ) This PIC micelle is expected to release the loaded siRNA selectively in the cytoplasm by cleavage of disulfide cross‐linking in response to a reductive intracellular environment, leading to the effective silencing of target genes related to oncogenesis. On the other hand, the disulfide cross‐linking is sufficiently stable in the blood compartment, enabling prolonged circulation because of the oxidized atmosphere in the body. Furthermore, facilitated endosomal escape occurs in the PIC micelle having an intermediated layer between the shell and the core phase to exert selective destablization of the endosomal membrane through the proton sponge effect as well as direct perturbation of the membrane structure.( 50 ) The evaluation of these PIC micelles loaded with plasmid DNA or siRNA will soon be performed in vivo for future molecular therapy.
Conclusion
Presently, the phrase EPR proposed by Maeda et al. has become a fundamental principle in the field of DDS. Until recently, the EPR effect has not been recognized in the field of oncology; however, some oncologists have now become more acquainted with this effect since some drugs such as doxil, abraxane and several PEGylated proteinacious agents formulated based on the EPR effect have been approved in the field of oncology. Micelle carrier systems described here are obviously categorized as DDS based on the EPR effect. We believe that some anticancer agent‐incorporating micelle nanoparticles may soon be approved for clinical use.
Acknowledgments
This work was supported by Third Term Comprehensive Control Research for Cancer from the Ministry of Health, Labour and Welfare of Japan, a Grant‐in‐Aid for Scientific Research or Priority Areas from the Ministry of Education, Culture, Sports, Science and Technology, and Japanese Foundation for Multidisciplinary Treatment of Cancer. We thank all researchers involved in this work and Ms. K Shiina for her secretarial support.
References
- 1. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res 1986; 46: 6387–92. [PubMed] [Google Scholar]
- 2. Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Contro Rel 2000; 65: 271–84. [DOI] [PubMed] [Google Scholar]
- 3. Kataoka K, Kwon GS, Yokoyama M, Okano T, Sakurai Y. Block copolymer micelles as vehicles for drug delivery. J Controlled Release 1993; 24: 119–32. [Google Scholar]
- 4. Yokoyama M, Miyauchi M, Yamada N et al . Polymer micelles as novel drug carrier: Adriamycin‐conjugted poly (ethylene glycol)‐poly (aspartic acid) block copolymer. J Controlled Release 1990; 11: 269–78. [Google Scholar]
- 5. Yokoyama M, Okano T, Sakurai Y, Ekimoto H, Shibazaki C, Kataoka K. Toxicity and antitumor activity against solid tumors of micelle‐forming polymeric anticancer drug and its extremely long circulation in blood. Cancer Res 1991; 51: 3229–36. [PubMed] [Google Scholar]
- 6. Khayat D, Antoine EC, Coeffic D. Taxol in the management of cancers of the breast and the ovary. Cancer Invest 2000; 18: 242–60. [DOI] [PubMed] [Google Scholar]
- 7. Carney DN. Chemotherapy in the management of patients with inoperable non‐small cell lung cancer. Semin Oncol 1996; 23: 71–5. [PubMed] [Google Scholar]
- 8. Weiss RB, Donehower RC, Wiernik PH et al . Hypersensitivity reactions from taxol. J Clin Oncol 1990; 8: 1263–8. [DOI] [PubMed] [Google Scholar]
- 9. Rowinsky EK, Donehower RC. Paclitaxel (taxol). N Engl J Med 1995; 332: 1004–14. [DOI] [PubMed] [Google Scholar]
- 10. Rowinsky EK, Chaudhry V, Forastiere AA et al . Phase I and pharmacologic study of paclitaxel and cisplatin with granulocyte colony‐stimulating factor: neuromuscular toxicity is dose‐limiting. J Clin Oncol 1993; 11: 2010–20. [DOI] [PubMed] [Google Scholar]
- 11. Wasserheit C, Frazein A, Oratz R et al . Phase II trial of paclitaxel and cisplatin in women with advanced breast cancer: an active regimen with limiting neurotoxicity. J Clin Oncol 1996; 14: 1993–9. [DOI] [PubMed] [Google Scholar]
- 12. Hamaguchi T, Matsumura Y, Suzuki M et al . NK105, a paclitaxel‐incorporating micellar nanoparticle formulation, can extend in vivo antitumour activity and reduce the neurotoxicity of paclitaxel. Br J Cancer 2005; 92: 1240–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hamaguchi T, Kato K, Matsumura Y et al . A Phase I and Pharmacokinetic Study of NK105, a Paclitaxel‐incorporating Micellar Nanoparticle Formulation. Br J Cancer 2007; 97: 170–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Horwich A, Sleijfer DT, Fossa SD et al . Randomized trial of bleomycin, etoposide, and cisplatin compared with bleomycin, etoposide, and carboplatin in good‐prognosis metastatic nonseminomatous germ cell cancer: a Multiinstitutional Medical Research Council/European Organization for Research and Treatment of Cancer Trial. J Clin Oncol 1997; 15: 1844–52. [DOI] [PubMed] [Google Scholar]
- 15. Roth BJ. Chemotherapy for advanced bladder cancer. Semin Oncol 1996; 23: 633–44. [PubMed] [Google Scholar]
- 16. Pinzani V, Bressolle F, Haug IJ, Galtier M, Blayac JP, Balmes P. Cisplatin‐induced renal toxicity and toxicity‐modulating strategies: a review. Cancer Chemother Pharmacol 1994; 35: 1–9. [DOI] [PubMed] [Google Scholar]
- 17. Cleare MJ, Hydes PC, Malerbi BW, Watkins DM. Anti‐tumor platinum complexes. relationships between chemical properties and activity. Biochimie 1978; 60: 835–50. [Google Scholar]
- 18. Du Bois A, Luck HJ, Meier W et al . A randomized clinical trial of cisplatin/paclitaxel versus carboplatin/paclitaxel as first‐line treatment of ovarian cancer. J Natl Cancer Inst 2003; 95: 1320–9. [DOI] [PubMed] [Google Scholar]
- 19. Cassidy J, Tabernero J, Twelves C et al . XELOX (capecitabine plus oxaliplatin): active first‐line therapy for patients with metastatic colorectal cancer. J Clin Oncol 2004; 22: 2084–91. [DOI] [PubMed] [Google Scholar]
- 20. Horwich A, Sleijfer DT, Fossa SD et al . Randomized trial of bleomycin, etoposide, and cisplatin compared with bleomycin, etoposide, and carboplatin in good‐prognosis metastatic nonseminomatous germ cell cancer: a Multiinstitutional Medical Research Council/European Organization for Research and Treatment of Cancer Trial. J Clin Oncol 1997; 15: 1844–52. [DOI] [PubMed] [Google Scholar]
- 21. Bellmunt J, Ribas A, Eres N et al . Carboplatin‐based versus cisplatin‐based chemotherapy in the treatment of surgically incurable advanced bladder carcinoma. Cancer 1997; 80: 1966–72. [DOI] [PubMed] [Google Scholar]
- 22. Nishiyama N, Okazaki S, Matsumura Y et al . Novel cisplatin‐incorporated polymeric micelles can eradicate solid tumors in mice. Cancer Res 2003; 63: 8977–83. [PubMed] [Google Scholar]
- 23. Uchino H, Matsumura Y, Negishi T et al . Cisplatin‐incorporating polymeric micelles (NC‐6004) can reduce nephrotoxicity and neurotoxicity of cisplatin in rats. Br J Cancer 2005; 93: 678–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wilson RH, Plummer R, Adam J et al . Phase I and pharmacokinetic study of NC‐6004, a new platinum entity of cisplatin‐conjugated polymer forming micelles. Am Soc Clin Oncol 2008; (Abstract# 2573).
- 25. Saltz LB, Cox JV, Blanke C et al . Irinotecan plus fluorouracil and leucovorin for metastatic colorectal cancer. Irinotecan Study Group N Engl J Med 2000; 343: 905–14. [DOI] [PubMed] [Google Scholar]
- 26. Douillard JY, Cunningham D, Roth AD et al . Irinotecan combined with fluorouracil compared with fluorouracil alone as first‐line treatment for metastatic colorectal cancer: a multicentre randomised trial. Lancet 2000; 355: 1041–7. [DOI] [PubMed] [Google Scholar]
- 27. Noda K, Nishiwaki Y, Kawahara M et al . Irinotecan plus cisplatin compared with etoposide plus cisplatin for extensive small‐cell lung cancer. N Engl J Med 2002; 346: 85–91. [DOI] [PubMed] [Google Scholar]
- 28. Negoro S, Masuda N, Takada Y et al . CPT‐11 Lung Cancer Study Group West . Randomised phase III trial of irinotecan combined with cisplatin for advanced non‐small‐cell lung cancer. Br J Cancer 2003; 88: 335–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bodurka DC, Levenback C, Wolf JK et al . Phase II trial of irinotecan in patients with metastatic epithelial ovarian cancer or peritoneal cancer. J Clin Oncol 2003; 21: 291–7. [DOI] [PubMed] [Google Scholar]
- 30. Takimoto CH, Arbuck SG. Topoisomerase I targeting agents: the camptothecins. In: Chabner BA, Lango DL, eds. Cancer Chemotherapy and Biotherapy: Principal and Practice, 3rd edn. Philadelphia (PA): Lippincott Williams & Wilkins, 2001: 579–646. [Google Scholar]
- 31. Slatter JG, Schaaf LJ, Sams JP et al . Pharmacokinetics, metabolism, and excretion of irinotecan (CPT‐11) following I.V. infusion of [(14) C]CPT‐11 in cancer patients. Drug Metab Dispos 2000; 28: 423–33. [PubMed] [Google Scholar]
- 32. Rothenberg ML, Kuhn JG, Burris HA 3rd et al . Phase I and pharmacokinetic trial of weekly CPT‐11. J Clin Oncol 1993; 11: 2194–204. [DOI] [PubMed] [Google Scholar]
- 33. Guichard S, Terret C, Hennebelle I et al . CPT‐11 converting carboxylesterase and topoisomerase activities in tumor and normal colon and liver tissues. Br J Cancer 1999; 80: 364–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Koizumi F, Kitagawa M, Negishi T et al . Novel SN‐38‐incorporating polymeric micelles, NK012, eradicate vascular endothelial growth factor‐secreting bulky tumors. Cancer Res 2006; 66: 10048–56. [DOI] [PubMed] [Google Scholar]
- 35. Saito Y, Yasunaga M, Kuroda J, Koga Y, Matsumura Y. Enhanced distribution of NK012 and prolonged sustained‐release of SN‐38 within tumors are the key strategic point for a hypovascular tumor. Caner Sci 2008; 99: 1258–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nakajima T, Yasunaga Matsumura Y et al . Syneragistic antitumor activity of the novel SN‐38 incorporating polymeric micelles, NK012, combined with 5‐fluorouracil in a mouse model of colorectal cancer, as compared with that of irinotecan plus 5‐fluorouracil. Int J Cancer 2008; 122: 22148–53. [DOI] [PubMed] [Google Scholar]
- 37. Sumitomo M, Koizumi F, Asano T et al . Novel SN‐38‐incorparated polymeric micelles, NK012, strongly supperss renal cancer progression. Cancer Res 2008, 2008; 122: 2148–53. [DOI] [PubMed] [Google Scholar]
- 38. Kuroda J, Kuratsu J, Yasunaga M, Koga Y, Matsumura Y. Potent antitumor effect of SN‐38‐incorporating polymeric micelle, NK012, against malignant glioma. Int J Cancer 2009; in press. [DOI] [PubMed] [Google Scholar]
- 39. Nakajima‐Eguchi T, Yanagihara K et al . Antitumor effect of SN‐38‐releasing polymeric micelles, NK012, on spontaneous peritoneal metastases from orthotopic gastoric cancer in mice sompared with irinotecan. Caner Res 2008; in press. [DOI] [PubMed] [Google Scholar]
- 40. Kato K, Hamaguchi T, Shirao K et al . PhaseI study of NK012, polymer micelle SN‐38, in patients with advanced cancer. Proc Ams Soc Clin Oncol GI 2008; (Abstract#485).
- 41. Burris IIIHA, Infante JR, Spigel DR et al . A phaseI dose‐escalation study of NK012. Proc Am Soc Clin Oncol 2008; (Abstract#2538).
- 42. Nishiyama N, Kataoka K. Current state, achievements, and future prospects of polymeric micelles as nanocarriers for drug and gene delivery. Pharmacol Ther 2006; 112 (3): 630–48. [DOI] [PubMed] [Google Scholar]
- 43. Cabral H, Nishiyama N, Okazaki S, Koyama H, Kataoka K. Preparation and biological properties of dichloro (1,2‐diaminocyclohexane) platinum (II) (DACHPt)‐loaded polymeric micelles. J Control Release 2005; 101 (1–3): 223–32. [DOI] [PubMed] [Google Scholar]
- 44. Cabral H, Nishiyama N, Kataoka K. Optimization of (1,2‐diamino‐cyclohexane) platinum (II)‐loaded polymeric micelles directed to improved tumor targeting and enhanced antitumor activity. J Control Release 2007; 121 (3): 146–55. [DOI] [PubMed] [Google Scholar]
- 45. Bae Y, Fukushima S, Harada A, Kataoka K. Design of environment‐sensitive supramolecular assemblies for intracellular drug delivery: polymeric micelles that are responsive to intracellular pH change. Angew Chem Int Ed 2003; 42 (38): 4640–3. [DOI] [PubMed] [Google Scholar]
- 46. Kano MR, Bae Y, Iwata C et al . Improvement of cancer‐targeting therapy, using nanocarriers for intractable solid tumors by inhibition of TGF‐beta signaling. P Natl Acad Sci USA 2007; 104 (9): 3460–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stapert HR, Nishiyama N, Jiang D‐L, Aida T, Kataoka K. Polyion complex micelles encapsulating light‐harvesting ionic dendrimer zinc porphyrins. Langmuir 2000; 16 (21): 8182–8. [Google Scholar]
- 48. Kataoka K, Togawa H, Harada A, Yasugi K, Matsumoto T, Katayose S. Spontaneous formation of polyion complex micelles with narrow distribution from antisense oligonucleotide and cationic block copolymer in physiological saline. Macromolecules 1996; 29 (26): 8556–7. [Google Scholar]
- 49. Matsumoto S, Christie RJ, Nishiyama N et al . Environment‐responsive block copolymer micelles with a disulfide cross‐linked core for enhanced siRNA delivery. Biomaromolecules in press. [DOI] [PubMed] [Google Scholar]
- 50. Miyata K, Oba M, Kano MR et al . Polyplex micelles from triblock copolymers composed of tandemly aligned segments with biocompatible, endosomal escaping, and DNA‐condensing functions for systemic gene delivery to pancreatic tumor. Pharm. Res in press. [DOI] [PubMed] [Google Scholar]