

Abstract
The receptor tyrosine kinase RET is expressed in a number of neuroblastoma tissues and cell lines, but its role in neuroblastoma remains to be determined. In this study, we examined the roles of RET protein in neuroblastoma by the RNA interference technique using the NB‐39‐nu neuroblastoma cell line. NB‐39‐nu neuroblastoma cells show high expression and elevated tyrosine phosphorylation of RET, although short interfering RNA against RET (RET siRNA) did not significantly inhibit cell proliferation or suppression of basal levels of phosphorylation of extracellular regulated kinase (ERK)1/2 or protein kinase B (AKT). By the addition of glial cell line‐derived neurotrophic factor (GDNF), both the expression and phosphorylation of RET and the phosphorylation of ERK1/2 and AKT were further increased, whereas cell proliferation was not stimulated under normal culture conditions. However, proliferation of cells cultured under non‐adherent conditions was significantly increased by GDNF. The increased proliferation was suppressed by RET siRNA, which also caused inhibition of the phosphorylation of ERK1/2 and AKT. These results suggest that RET signaling plays an important role in GDNF‐induced enhancement of non‐adherent proliferation of NB‐39‐nu cells, which might contribute to the metastasis of neuroblastoma. (Cancer Sci 2009; 100: 1034–1039)
RET is a receptor tyrosine kinase that is expressed in various neurons including central motor dopaminergic and noradrenergic neurons as well as peripheral enteric sensory and sympathetic neurons.( 1 , 2 ) The expression of RET has also been detected in the mesonephric duct and branching ureteric bud during embryogenesis of the kidney.( 1 , 2 ) It has been shown that RET is required for the development of the sympathetic, parasympathetic, and enteric nervous systems as well as the kidney and testis.( 1 , 3 ) Dysfunctions of RET, which are caused by mutations of the gene, lead to various neuroendocrine tumors or enteric disorders. Germline gain‐of‐function mutations of RET are responsible for the dominantly inherited cancer syndromes of multiple endocrine neoplasia types 2A and 2B and familial medullary thyroid carcinoma.( 4 , 5 , 6 ) In addition, somatic mutations or rearrangements of RET have also been found in sporadic medullary thyroid carcinoma and papillary thyroid carcinoma, respectively.( 7 , 8 , 9 , 10 ) Loss‐of‐function mutations in RET have been found in some cases of Hirchsprung's disease characterized by severe constipation and intestinal obstructions during childhood.( 11 , 12 )
Neuroblastoma is the most common extracranial tumor in children and is derived from sympathoadrenal lineage of the neural crest in which RET is expressed.( 13 ) Although RET is expressed in most neuroblastoma tissues and cell lines and its overexpression has been reported in some cell lines,( 14 , 15 , 16 , 17 ) its role in molecular pathogenesis in neuroblastoma remains to be determined.
Glial cell line‐derived neurotrophic factor (GDNF), a ligand of RET protein, was originally purified as a growth factor promoting survival of the embryonic dopaminergic neurons and exhibiting a potent trophic factor for spinal motor neurons and central noradrenergic neurons.( 18 , 19 , 20 ) GDNF induces dimerization of RET in the form of multicomponents with a coreceptor GDNF family receptor α 1 (GFRα‐1) and triggers autophosphorylation of RET in its intracellular tyrosine kinase domain. The phosphorylated tyrosines serve as docking sites for signaling molecules such as Shc, FRS2, IRS1/2 and Dok1/4/5.( 21 ) In neuroblastoma, it has been shown that extracellular regulated kinase (ERK), phosphoinositide 3‐kinase/protein kinase B (AKT), c‐Jun N‐terminal kinase, and p38 mitogen‐activated protein kinase (p38MAPK) pathways are activated mainly through tyrosine 1062,( 22 ) but their biological effects have not been fully elucidated.
In this study, we investigated the biological role of RET in neuroblastoma using the NB‐39‐nu neuroblastoma cell line, which shows extremely high expression and elevated phosphorylation of RET, by suppression of RET protein expression using RNA interference (RNAi).
Materials and Methods
Cell line and culture. NB‐39‐nu, Nagai,( 23 ) and YT‐nu( 24 ) were provided by the Carcinogenesis Division, National Cancer Center Research Institute (Tokyo, Japan). NB‐1 and TNB‐1 were obtained from the Human Science Research Resource Bank (Tokyo, Japan). SK‐N‐SH was obtained from Riken Cell Bank (Tsukuba, Japan) and TT was obtained from the American Type Culture Collection (Manassas, VA, USA). These cell lines were maintained in RPMI‐1640 medium with 10% heat‐inactivated fetal bovine serum, 100 units/mL penicillin, and 100 µg/mL streptomycin at 37°C with 5% CO2.
Short interfering RNA. Short interfering RNA against human RET (RET siRNA) was generated using a BLOCK‐iT RNAi TOPO Transcription Kit and BLOCK‐iT Complete Dicer RNAi Kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. In the generation of RET siRNA, 805 bp from the second nucleotide of the initiation codon of human RET was chosen as the target sequence, and amplified by polymerase chain reaction using human RET cDNA and the primers, 5′‐ tggcgaaggcgacgtccggt‐3′ and 5′‐cgagtcgtcctcgtcgtaca‐3′. The control LacZ siRNA was made using an expression plasmid containing the lacZ gene and polymerase chain reaction primers attached in the kit. Transfection was carried out with Lipofectamine 2000 (Invitrogen).
Antibodies and reagents. Anti‐ERK1/2 antibody, anti‐phospho‐ERK1/2 antibody (phospho‐p44/p42 mitogen‐activated protein kinase antibody), anti‐AKT antibody, and anti‐phospho‐AKT (Ser473) antibody were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti‐RET (C‐19) antibody and Chr‐A (chromogranin‐A) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti‐α‐tubulin (B‐5‐1‐2) antibodies were purchased from Sigma (St. Louis, MO, USA). Anti‐GAP‐43 was purchased from Zymed Laboratories (San Francisco, CA, USA). Anti‐phospho‐RET antibody was produced by immunizing rats with a synthetic phospho‐peptide corresponding to residues around Tyr1062 of a long form of human RET. Horseradish peroxidase‐conjugated anti‐mouse and anti‐rabbit antibodies were purchased from Amersham Pharmacia (Little Chalfont, UK). Human recombinant GDNF was purchased from Sigma.
Cell proliferation assay. Cell proliferation was analyzed by Tetra Color One (Seikagaku, Tokyo, Japan) according to the manufacturer's instructions. Briefly, 3 × 103 or 1 × 104 cells/well were seeded in normal or low‐cell‐binding 96‐well plates treated with 2‐methacryloxyethyl phosphorylcholine (MPC; Nalge Nunc International, Tokyo, Japan). The cells were cultured for 24 or 48 h with or without GDNF (50 ng/mL) then subjected to Tetra Color for 3 h, followed by measurement for proliferation. The absorbance of the samples was measured at 450 nm on a microplate reader model 550 (Bio‐Rad, Hercules, CA, USA).
Western blot analysis. The cell lysates were separated using sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and transferred to a polyvinylidene difluoride membrane (Immobilon‐P; Millipore, Bedford, MA, USA). After blocking of the membrane with blocking buffer (5% skim milk in Tris buffered saline containing 0.1% Tween‐20), the membrane was probed with antibodies for detection. The membrane was further probed with horseradish peroxidase‐conjugated anti‐rabbit or anti‐mouse IgG to visualize the reacted antibody.
Results
Expression of RET in various neuroblastoma cell lines. We first estimated the levels of the expression and phosphorylation of RET in various neuroblastoma cell lines and compared the estimated levels with those of the TT medullary thyroid carcinoma cell line, which carries C634W gain‐of‐function mutation of RET. Although RET was expressed in all six neuroblastoma cell lines, the levels of expression and phosphorylation of RET greatly varied among cell lines (Fig. 1a). Among the six neuroblastoma cell lines evaluated, two cell lines, NB‐39‐nu and Nagai, showed a high level of expression and phosphorylation of RET, which were comparable to or higher than those of TT.
Figure 1.
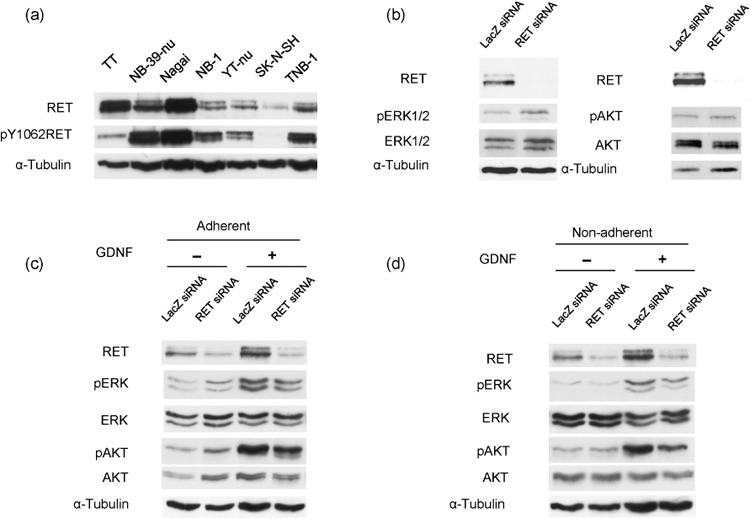
(a) Expression of RET in various neuroblastoma cell lines. Exponentially growing neuroblastoma cell lines NB‐39‐nu, Nagai, NB‐1, YT‐nu, SK‐N‐SH, TNB‐1, and a medullary thyroid carcinoma TT were harvested and the cell lysates subjected to Western blot analysis. (b) RET knockdown does not affect extracellular regulated kinase (ERK)1/2 or protein kinase B (AKT) signaling pathways in NB‐39‐nu cells. NB‐39‐nu cells (10 × 104/mL, 2 mL/well) were seeded in conventional 6‐well plates in 10% fetal bovine serum containing RPMI‐1640 medium, followed by treatment with LacZ short intefering RNA (siRNA) or RET siRNA at 0 h and 24 h, then harvested at 48 h for Western blot analysis. pAKT, phosphorylated AKT; pERK1/2, phosphorylated ERK1/2. (c, d) Glial cell line‐derived neurotrophic factor (GDNF) enhances ERK1/2 and AKT signaling pathways in NB‐39‐nu cells in a RET‐dependent manner. NB‐39‐nu cells (10 × 104/mL, 2 mL/well) were seeded in conventional 6‐well plates in 10% fetal bovine serum containing RPMI‐1640 medium, followed by treatment with LacZ siRNA or RET siRNA at 0 h and 24 h. After the cells were harvested at 48 h, the adherent cells were cultured with GDNF (50 ng/mL) for 24 h in conventional 6‐well plates (c), while the non‐adherent cells were cultured in 2‐methacryloxyethyl phosphorylcholine‐treated 6‐well plates (d). The treatment was terminated at 72 h by harvesting the cells for Western blot analysis.
GDNF enhances ERK1/2 and AKT signaling pathways in adherent NB‐39‐nu cells in a RET‐dependent manner. We next knocked down RET expression to see the effect of activated RET signaling in NB‐39‐nu cells, one of the two neuroblastoma cell lines that showed a remarkable level of RET expression. As shown in Figure 1(b), RET expression was dramatically suppressed by treatment using RET RNAi, whereas phosphorylation of ERK1/2 and AKT, which have been reported to be universal downstream targets of receptor tyrosine kinases including RET, were not significantly affected. This suggests that these downstream signaling pathways are not under the regulation of activated RET under normal culture conditions. However, phosphorylation of ERK1/2 and AKT were significantly enhanced by GDNF treatment in the existence of serum, as indicated in Figure 1(c), and this enhancement was inhibited by the knockdown of RET protein, indicating that ERK1/2 and AKT signals were activated by the stimulation with GDNF in a RET‐dependent manner in this cell line. Phosphorylation of AKT and ERK1/2 was also increased by stimulation of GDNF in the serum‐depleted condition after serum starvation, and the increased phosphorylation of AKT was inhibited by RET knockdown, whereas the inhibition of the increased phosphorylation of ERK1/2 by RET knockdown was not obvious (Fig. S1).
GDNF enhances cell proliferation of non‐adherent NB‐39‐nu cells but not of adherent cells. As RET downstream signaling was stimulated by the treatment of GDNF in NB‐39‐nu cells, we next examined the effect of GDNF on the proliferation of NB‐39‐nu cells. The growth of the cells cultured under adherent conditions was not significantly changed by the treatment with GDNF (Fig. 2a), but the growth of the cells under non‐adherent conditions was stimulated by the treatment with GDNF (Fig. 2b). In this study, we used the low attachment plate by Nunc (MPC plate as described in ‘Materials and Methods’).( 25 ) In the MPC plate, almost all cells grew under non‐adherent conditions, detached from the bottom of the plate (Fig. 4b, lower panels). The proliferation of NB‐39‐nu cells was also measured using Tetra Color One assay, as described in ‘Materials and Methods’, and it was confirmed that marked enhancement in the proliferation of the cells was specifically observed under non‐adherent conditions by the treatment with GDNF (Fig. 3). In addition, RET knockdown inhibited GDNF‐stimulated proliferation of NB‐39‐nu cells under non‐adherent conditions, suggesting that GDNF promotes non‐adherent proliferation of NB‐39‐nu cells in a RET‐dependent manner (Fig. 3). At the same time, knockdown of RET protein under non‐adherent conditions caused suppression of the amount of activated ERK1/2 and ATK (Fig. 1d).
Figure 2.
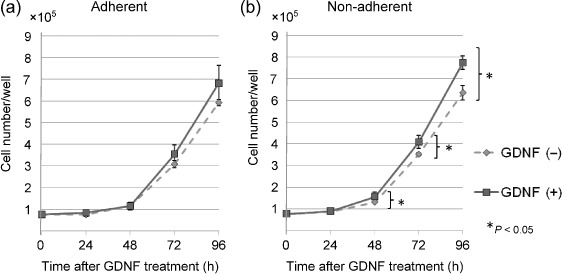
Glial cell line‐derived neurotrophic factor (GDNF) enhances the cell growth of non‐adherent NB‐39‐nu cells but not of adherent cells. Cells (3 × 104/mL, 2.5 mL/well) were seeded in conventional or 2‐methacryloxyethyl phosphorylcholine‐treated 6‐well plates, and cultured with GDNF (50 ng/mL) for 24, 48, 72, and 96 h. The cells were harvested by trypsinization at the indicated time points, then cell numbers were measured using a Coulter counter. Points, means of three replicates; bars, standard deviation.
Figure 4.
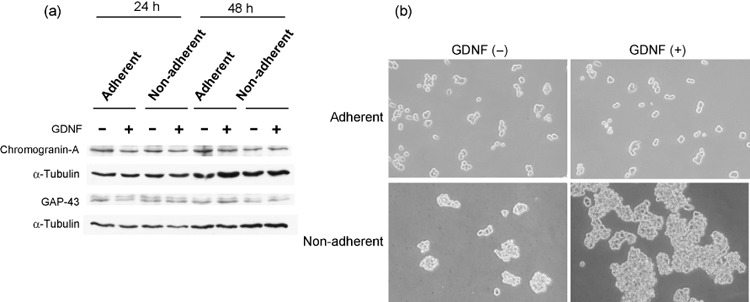
GDNF does not induce neuronal differentiation markers or morphological change in adherent NB‐39‐nu cells. NB‐39‐nu cells (10 × 104/mL, 2 mL/well) were seeded in conventional or 2‐methacryloxyethyl phosphorylcholine‐treated 6‐well plates in 10% fetal bovine serum containing RPMI‐1640 medium with or without glial cell line‐derived neurotrophic factor (GDNF; 50 ng/mL) for 24 or 48 h. (a) The cells were harvested for Western blot analysis at 24 h and 48 h. (b) Microscopic appearance of NB‐39‐nu cells cultured with or without GDNF under adherent or non‐adherent conditions at 48 h.
Figure 3.
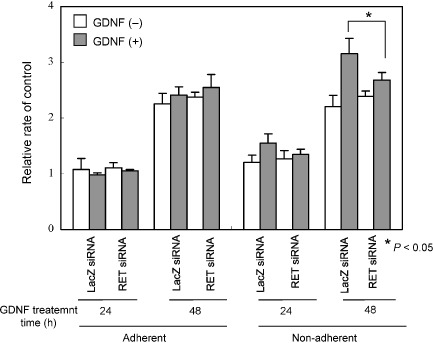
RET knockdown abrogates glial cell line‐derived neurotrophic factor (GDNF)‐stimulated proliferation of non‐adherent NB‐39‐nu neuroblastoma cells. NB‐39‐nu cells (10 × 104/mL, 2 mL/well) were seeded in conventional 6‐well plates in 10% fetal bovine serum containing RPMI‐1640 medium, followed by treatment with LacZ short interfering RNA (siRNA) or RET siRNA at 0 h and 24 h. At 48 h, the cells were harvested and seeded (3 × 103/mL, 0.1 mL/well) in conventional or 2‐methacryloxyethyl phosphorylcholine‐treated 96‐well plates with or without GDNF (50 ng/mL) for 24 or 48 h. Cell proliferations were determined at the indicated time points. Columns, means of three replicates; bars, standard deviation.
In addition to NB‐39‐nu cells, another neuroblastoma cell line, NB‐1, was evaluated for proliferation when treated with GDNF with or without of RET knockdown. Like NB‐39‐nu cells, the proliferation of NB‐1 cells was enhanced by GDNF under non‐adherent conditions, but not under adherent conditions, and the enhancement was inhibited by RET knockdown (Fig. S2).
GDNF does not induce neuronal differentiation markers in NB‐39‐nu cells. It has been shown that GDNF induces cell differentiation in most primary neuroblastoma tissues, which generally results in the inhibition of the proliferation of cancer cells, so we investigated whether differentiation was induced by GDNF treatment of NB‐39‐nu cells by monitoring two neuronal markers, GAP‐43 and chromogranin‐A. The treatment did not cause upregulation of these markers at a protein level, under either adherent or non‐adherent conditions (Fig. 4a). In addition, no obvious morphological changes, such as neurite outgrowth, were observed with the addition of GDNF under adherent conditions (Fig. 4b, upper panels). These findings suggest that the treatment of GDNF did not induce neuronal differentiation under the experimental conditions. Furthermore, the cells showed evidence of cluster formations under non‐adherent conditions, and the clusters of the cells treated with GDNF appeared to be significantly larger (Fig. 4b, lower panels).
Discussion
It has been shown that RET is expressed in most neuroblastoma cell lines and tissues, although its role in neuroblastoma remains to be determined. In this study, we analyzed the role of RET protein in neuroblastoma using NB‐39‐nu cells in which especially high levels of expression and tyrosine phosphorylation of RET protein were observed. In two of the six cell lines examined, NB‐39‐nu and Nagai cells, the expression and phosphorylation levels of RET protein were comparable to or higher than those of TT cells. TT cells are derived from medullary thyroid carcinoma, and were found to harbor C634W gain‐of‐function mutation of RET. It was considered that activation of RET caused by gain‐of‐function mutation, which led to constitutive activation of downstream signaling, is responsible for oncogenesis in TT cells. Actually, it has been shown that the inhibition of RET signaling by expressing a dominant negative form of RET leads to suppression of ERK1/2 and AKT and the proliferation in TT cells, suggesting that the activated RET plays an important role in proliferation.( 26 , 27 ) Although RET protein shows high levels of tyrosine phosphorylation in NB‐39‐nu and Nagai cells without any known mutations, the proliferation and downstream signaling of RET were not inhibited by RET RNAi of NB‐39‐nu cells under normal culture conditions in this study.
However, ERK1/2 and AKT, downstream molecules of RET protein, were further activated upon the stimulation of its ligand, GDNF, in a RET‐dependent manner, suggesting a ligand‐dependent nature as a wild‐type RET protein. Nevertheless, the proliferation of the cells under adherent conditions was not stimulated by the treatment with GDNF. Interestingly, the proliferation of NB‐39‐nu cells was stimulated by GDNF treatment under non‐adherent conditions, and this was suppressed by RET siRNA. These results suggest that GDNF promotes non‐adherent proliferation of NB‐39‐nu cells in a RET‐dependent manner. Similar results of the effect of GDNF on proliferation were observed in another neuroblastoma cell line, NB‐1, suggesting that the phenomenon of RET‐dependent GDNF‐induced increase of proliferation under non‐adherent conditions was not exclusive to NB‐39‐nu cells.
We noticed that RET knockdown had an obvious inhibitory effect on the activation of both ERK1/2 and AKT in long‐term stimulation (24 h) by GDNF in the existence of serum, but it had little inhibitory effect on the activation of ERK1/2 in rapid stimulation (15 min) by GDNF after serum starvation, suggesting a special mechanism of long‐term activation of ERK1/2 by GDNF‐RET signaling.
When the NB‐39‐nu cells were cultured in non‐adherent conditions, they grew in cluster formations, as shown in Figure 4(b). It has been shown that intercellular adhesion under non‐adherent conditions generates the stimulations that support survival and proliferation in some cell lines.( 28 , 29 ) GDNF might enhance this intercellular stimulation generated by cluster formation of the cells, resulting in promotion of the proliferation of the cells. In the metastasis cascade, it can be considered to be a critical step that the metastasizing cancer cells become detached from the extracellular matrix and survive and proliferate in touch with adjacent cancer cells, suggesting the possibility that GDNF‐RET signaling might promote cancer metastasis by facilitating this step.( 30 , 31 )
It has been shown that GDNF induces differentiation in most neuroblastoma tissues by the primary culture system.( 17 ) Therefore, we initially suspected that the loss of the growth promotion effect of GDNF under adherent conditions was due to its induction of differentiation. However, GDNF stimulation did not cause either induction of neuronal markers, such as GAP‐43 and chromogranin‐A, nor morphological changes such as neurite outgrowth, suggesting that neuronal differentiation was not induced by the treatment of GDNF in NB‐39‐nu cells under adherent conditions.
We previously demonstrated that anaplastic lymphoma kinase (ALK) was overexpressed and activated by gene amplification in NB‐39‐nu cells, and found that the AKT and ERK1/2 signals were significantly blocked by the knockdown of ALK,( 32 , 33 ) suggesting that activation of ALK dominates the growth and survival signaling in this cell line. Thus, it is possible that the growth of NB‐39‐nu cells under GDNF‐free conditions, or under adherent conditions, is mainly sustained by activated ALK. This is also the case with NB‐1 overexpressing ALK. Furthermore, we observed that the proliferations in other neuroblastoma cell lines, TNB‐1 and YT‐nu, which are not overexpressing ALK were enhanced by GDNF under both adherent and non‐adherent conditions (data not shown), suggesting that the cell proliferation might be dominantly regulated by activated ALK which overcomes GDNF‐RET signaling under adherent conditions in NB‐39‐nu and NB‐1 cells. GDNF‐RET signaling might have a special role in non‐adherent growth that might not be supported by signals from other growth factor receptors, including ALK.
It was previously reported that the proliferation of SH‐SY5Y neuroblastoma cell line was enhanced by GDNF with the activation of the ERK1/2 and AKT signaling pathways under conventional conditions.( 34 ) Conversely, as we have shown, GDNF did not stimulate the proliferation of NB‐39‐nu cells under adherent conditions, but it did enhance the signaling pathway of ERK1/2 and AKT. Although the mechanism by which the differential ability of GDNF to proliferate the cells between these two cell lines remains to be elucidated, the dominant influence of the activated ALK pathway in NB‐39‐nu cells is naturally suspected. It could be postulated that other pathways such as the signal from NCAM,( 35 ) also a receptor for GNDF, might contribute differently to the signaling pathway for proliferation between these two cell lines growing under adherent conditions.
Various neuroblastoma cell lines secrete GDNF under the controls of various cytokines and growth factors that are also generated from neuroblastoma.( 17 , 36 ) Thus, the fact that GDNF promotes the non‐adherent growth of NB‐39‐nu neuroblastoma cells might suggest that GDNF generated from neuroblastoma cells could contribute to tumor growth, progression, and metastasis in an autocrine and paracrine fashion, at least in some neuroblastoma.
Supporting information
Fig. S1. Glial cell line‐derived neurotrophic factor (GDNF)‐induced enhancement of extracellular regulated kinase (ERK)1/2 and protein kinase B (AKT) signaling in serum‐depleted conditions in NB‐39‐nu neuroblastoma cells. NB‐39‐nu cells (10 × 104/mL, 2 mL/well) were seeded in conventional 6‐well plates in 10% fetal bovine serum containing RPMI‐1640 medium, followed by treatment with LacZ short interfering RNA (siRNA) or RET siRNA at 0 h and 24 h, then harvested at 48 h. After culturing the cells in serum‐depleted condition for 24 h under adherent conditions, the cells were treated with GDNF (50 ng/mL) in serum‐depleted conditions for 15 min, then harvested for Western blot analysis.
Fig. S2. RET knockdown abrogates glial cell line‐derived neurotrophic factor (GDNF)‐stimulated proliferation of non‐adherent NB‐1 neuroblastoma cells. NB‐1 cells (10 × 104/mL, 2 mL/well) were seeded in conventional 6‐well plates in 10% fetal bovine serum containing RPMI‐1640 medium, followed by treatment with LacZ short interfering RNA (siRNA) or RET siRNA at 0 h and 24 h. At the 48 h time point, the cells were harvested and seeded (1 × 104/mL, 0.1 mL/well) in conventional or 2‐methacryloxyethyl phosphorylcholine‐treated 96‐well plates in 10% fetal bovine serum containing RPMI‐1640 medium with or without GDNF (50 ng/mL) for 24 or 48 h. RET knockdown was confirmed for the cells harvested at 48 h by Western blot analysis. Cell proliferations were determined at the indicated time points by Tetra Color One assay as described in Materials and Methods. Columns, means of three replicates; bars, standard deviation.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Acknowledgments
This work was supported by a Grant‐in Aid for Cancer Research by the Ministry of Education, Culture, Sports, Science and Technology of Japan and in part by a Grant‐in‐Aid from the Ministry of Health, Labor and Welfare of Japan for the 3rd‐term Comprehensive 10‐year Strategy for Cancer Control.
References
- 1. Pachnis V, Mankoo B, Costantini F. Expression of the c‐ret proto‐oncogene during mouse embryogenesis. Development 1993; 119: 1005–17. [DOI] [PubMed] [Google Scholar]
- 2. Tsuzuki T, Takahashi M, Asai N, Iwashita T, Matsuyama M, Asai J. Spatial and temporal expression of the ret proto‐oncogene product in embryonic, infant and adult rat tissues. Oncogene 1995; 10: 191–8. [PubMed] [Google Scholar]
- 3. Jain S, Naughton CK, Yang M et al . Mice expressing a dominant‐negative Ret mutation phenocopy human Hirschsprung disease and delineate a direct role of Ret in spermatogenesis. Development 2004; 131: 5503–13. [DOI] [PubMed] [Google Scholar]
- 4. Donis KH, Dou S, Chi D et al . Mutations in the RET proto‐oncogene are associated with MEN 2A and FMTC. Hum Mol Genet 1993; 2: 851–6. [DOI] [PubMed] [Google Scholar]
- 5. Mulligan LM, Kwok JB, Healey CS et al . Germ‐line mutations of the RET proto‐oncogene in multiple endocrine neoplasia type 2A. Nature 1993; 363: 458–60. [DOI] [PubMed] [Google Scholar]
- 6. Hofstra RM, Landsvater RM, Ceccherini I et al . A mutation in the RET proto‐oncogene associated with multiple endocrine neoplasia type 2B and sporadic medullary thyroid carcinoma. Nature 1994; 367: 375–6. [DOI] [PubMed] [Google Scholar]
- 7. Eng C, Mulligan LM, Smith DP et al . Mutation of the RET protooncogene in sporadic medullary thyroid carcinoma. Genes Chromosomes Cancer 1995; 12: 209–12. [DOI] [PubMed] [Google Scholar]
- 8. Eng C, Smith DP, Mulligan LM et al . A novel point mutation in the tyrosine kinase domain of the RET proto‐oncogene in sporadic medullary thyroid carcinoma and in a family with FMTC. Oncogene 1995; 10: 509–13. [PubMed] [Google Scholar]
- 9. Grieco M, Santoro M, Berlingieri MT et al . PTC is a novel rearranged form of the ret proto‐oncogene and is frequently detected in vivo in human thyroid papillary carcinomas. Cell 1990; 60: 557–63. [DOI] [PubMed] [Google Scholar]
- 10. Bongarzone I, Monzini N, Borrello MG et al . Molecular characterization of a thyroid tumor‐specific transforming sequence formed by the fusion of ret tyrosine kinase and the regulatory subunit RIα of cyclic AMP‐dependent protein kinase A. Mol Cell Biol 1993; 13: 358–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Edery P, Lyonnet S, Mulligan LM et al . Mutations of the RET proto‐oncogene in Hirschsprung's disease. Nature 1994; 367: 378–80. [DOI] [PubMed] [Google Scholar]
- 12. Romeo G, Ronchetto P, Luo Y et al . Point mutations affecting the tyrosine kinase domain of the RET proto‐oncogene in Hirschsprung's disease. Nature 1994; 367: 377–8. [DOI] [PubMed] [Google Scholar]
- 13. Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer 2003; 3: 203–16. [DOI] [PubMed] [Google Scholar]
- 14. Nagao M, Ishizaka Y, Nakagawara A et al . Expression of ret proto‐oncogene in human neuroblastomas. Jpn J Cancer Res 1990; 81: 309–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ikeda I, Ishizaka Y, Tahira T et al . Specific expression of the ret proto‐oncogene in human neuroblastoma cell lines. Oncogene 1990; 5: 1291–6. [PubMed] [Google Scholar]
- 16. Borrello MG, Bongarzone I, Pierotti MA et al . trk and ret proto‐oncogene expression in human neuroblastoma specimens: high frequency of trk expression in non‐advanced stages. Int J Cancer 1993; 54: 540–5. [DOI] [PubMed] [Google Scholar]
- 17. Hishiki T, Nimura Y, Isogai E et al . Glial cell line‐derived neurotrophic factor/neurturin‐induced differentiation and its enhancement by retinoic acid in primary human neuroblastomas expressing c‐Ret, GFRα‐1, and GFRα‐2. Cancer Res 1998; 58: 2158–65. [PubMed] [Google Scholar]
- 18. Lin LF, Doherty DH, Lile JD, Bektesh S, Collins F. GDNF: a glial cell line‐derived neurotrophic factor for midbrain dopaminergic neurons. Science 1993; 260: 1130–2. [DOI] [PubMed] [Google Scholar]
- 19. Henderson CE, Phillips HS, Pollock RA et al . GDNF: a potent survival factor for motoneurons present in peripheral nerve and muscle. Science 1994; 266: 1062–4. [DOI] [PubMed] [Google Scholar]
- 20. Arenas E, Trupp M, Akerud P, Ibanez CF. GDNF prevents degeneration and promotes the phenotype of brain noradrenergic neurons in vivo . Neuron 1995; 15: 1465–73. [DOI] [PubMed] [Google Scholar]
- 21. Arighi E, Borrello MG, Sariola H. RET tyrosine kinase signaling in development and cancer. Cytokine Growth Factor Rev 2005; 16: 441–67. [DOI] [PubMed] [Google Scholar]
- 22. Hayashi H, Ichihara M, Iwashita T et al . Characterization of intracellular signals via tyrosine 1062 in RET activated by glial cell line‐derived neurotrophic factor. Oncogene 2000; 19: 4469–75. [DOI] [PubMed] [Google Scholar]
- 23. Ishikawa S, Ohshima Y, Suzuki T, Oboshi S. Primitive neuroectodermal tumor (neuroepithelioma) of spinal nerve root – Report of an adult case and establishment of a cell line. Acta Pathol Jpn 1979; 29: 289–301. [DOI] [PubMed] [Google Scholar]
- 24. Kuga N, Yoshida K, Seido T et al . Heterotransplantation of cultured human cancer cells and human cancer tissues into nude mice. Gann 1975; 66: 547–60. [PubMed] [Google Scholar]
- 25. Uekita T, Jia L, Narisawa‐Saito M, Yokota J, Kiyono T, Sakai R. CUB domain‐containing protein 1 is a novel regulator of anoikis resistance in lung adenocarcinoma. Mol Cell Biol 2007; 27: 7649–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Drosten M, Stiewe T, Putzer BM. Antitumor capacity of a dominant‐negative RET proto‐oncogene mutant in a medullary thyroid carcinoma model. Hum Gene Ther 2003; 14: 971–82. [DOI] [PubMed] [Google Scholar]
- 27. Drosten M, Hilken G, Bockmann M et al . Role of MEN2A‐derived RET in maintenance and proliferation of medullary thyroid carcinoma. J Natl Cancer Inst 2004; 96: 1231–9. [DOI] [PubMed] [Google Scholar]
- 28. Bates RC, Edwards NS, Yates JD. Spheroids and cell survival. Crit Rev Oncol Hematol 2000; 36: 61–74. [DOI] [PubMed] [Google Scholar]
- 29. Lawlor ER, Scheel C, Irving J, Sorensen PH. Anchorage‐independent multi‐cellular spheroids as an in vitro model of growth signaling in Ewing tumors. Oncogene 2002; 21: 307–18. [DOI] [PubMed] [Google Scholar]
- 30. Volk T, Geiger B, Raz A. Motility and adhesive properties of high‐ and low‐metastatic murine neoplastic cells. Cancer Res 1984; 44: 811–24. [PubMed] [Google Scholar]
- 31. Nabi IR, Raz A. Loss of metastatic responsiveness to cell shape modulation in a newly characterized B16 melanoma adhesive cell variant. Cancer Res 1988; 48: 1258–64. [PubMed] [Google Scholar]
- 32. Miyake I, Hakomori Y, Misu Y et al . Domain‐specific function of ShcC docking protein in neuroblastoma cells. Oncogene 2005; 24: 3206–15. [DOI] [PubMed] [Google Scholar]
- 33. Osajima‐Hakomori Y, Miyake I, Ohira M, Nakagawara A, Nakagawa A, Sakai R. Biological role of anaplastic lymphoma kinase in neuroblastoma. Am J Pathol 2005; 167: 213–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hirata Y, Kiuchi K. Mitogenic effect of glial cell line‐derived neurotrophic factor is dependent on the activation of p70S6 kinase, but independent of the activation of ERK and up‐regulation of Ret in SH‐SY5Y cells. Brain Res 2003; 983: 1–12. [DOI] [PubMed] [Google Scholar]
- 35. Paratcha G, Ledda F, Ibanez CF. The neural cell adhesion molecule NCAM is an alternative signaling receptor for GDNF family ligands. Cell 2003; 113: 867–79. [DOI] [PubMed] [Google Scholar]
- 36. Verity AN, Wyatt TL, Lee W et al . Differential regulation of glial cell line‐derived neurotrophic factor (GDNF) expression in human neuroblastoma and glioblastoma cell lines. J Neurosci Res 1999; 55: 187–97. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Glial cell line‐derived neurotrophic factor (GDNF)‐induced enhancement of extracellular regulated kinase (ERK)1/2 and protein kinase B (AKT) signaling in serum‐depleted conditions in NB‐39‐nu neuroblastoma cells. NB‐39‐nu cells (10 × 104/mL, 2 mL/well) were seeded in conventional 6‐well plates in 10% fetal bovine serum containing RPMI‐1640 medium, followed by treatment with LacZ short interfering RNA (siRNA) or RET siRNA at 0 h and 24 h, then harvested at 48 h. After culturing the cells in serum‐depleted condition for 24 h under adherent conditions, the cells were treated with GDNF (50 ng/mL) in serum‐depleted conditions for 15 min, then harvested for Western blot analysis.
Fig. S2. RET knockdown abrogates glial cell line‐derived neurotrophic factor (GDNF)‐stimulated proliferation of non‐adherent NB‐1 neuroblastoma cells. NB‐1 cells (10 × 104/mL, 2 mL/well) were seeded in conventional 6‐well plates in 10% fetal bovine serum containing RPMI‐1640 medium, followed by treatment with LacZ short interfering RNA (siRNA) or RET siRNA at 0 h and 24 h. At the 48 h time point, the cells were harvested and seeded (1 × 104/mL, 0.1 mL/well) in conventional or 2‐methacryloxyethyl phosphorylcholine‐treated 96‐well plates in 10% fetal bovine serum containing RPMI‐1640 medium with or without GDNF (50 ng/mL) for 24 or 48 h. RET knockdown was confirmed for the cells harvested at 48 h by Western blot analysis. Cell proliferations were determined at the indicated time points by Tetra Color One assay as described in Materials and Methods. Columns, means of three replicates; bars, standard deviation.
Please note: Wiley‐Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item