

Abstract
Interferons‐α/β, which are produced upon viral infection, are key soluble factors for the establishment of an antiviral state, but are also produced at low levels in the absence of infection. Herein, we demonstrate that a weak signal by these constitutively produced IFN‐α/β show a preventive role in cellular transformation. Ifnar1‐deficient (Ifnar1−/– ) MEF, which are devoid of IFN‐α/β signal, undergo a spontaneous transformation during long‐term cell culture. Similar to Irf1−/– MEF, primary Ifnar1−/– MEF become tumorigenic in nude mice by the expression of activated c‐Ha‐Ras oncoprotein. However, Ifnar1−/– MEF do not show any abnormal growth properties. A similar observation is made in Ifnb−/– MEF that fail to produce constitutive IFN‐α/β, whereas such a transforming property is not found in MEF that lack any of the IFN receptor downstream molecules including Stat1, IRF9 and IRF1. Furthermore, Ifnar1−/– mice develop chemically‐induced skin papilloma more severely than wild‐type mice. In addition, the expression levels of IFNAR1 mRNA are significantly decreased in human gastric cancer tissues. These results suggest a cell‐intrinsic role of the weak signal by constitutively produced IFN‐α/β to prevent cells from transformation, which may be mediated by a hitherto‐unknown pathway(s) downstream of the IFN‐α/β receptor. (Cancer Sci 2009; 100: 449–456)
- Abbreviations: AAF
IFN‐α‐activated factor
- DMBA
7,12‐dimethylbenz(a)anthracene
- DMEM
Dulbecco's modified Eagle medium
- FCS
fetal calf serum
- IFN
interferon
- IFNAR
IFN‐α/β receptor
- IRF
IFN regulatory factor
- IL‐6
interleukin‐6
- ISG
IFN‐stimulated genes
- ISGF3
IFN‐stimulated gene factor 3
- ISRE
IFN‐stimulated responsive element
- Jak
Janus kinase
- MEF
mouse embryonic fibroblasts
- OAS
2′‐5′oligoadenylate synthetase
- PBS
phosphate‐buffered saline
- PKR
ds‐RNA‐dependent protein kinase
- RT‐PCR
reverse‐transcription polymerase chain reaction
- SNP
single‐nucleotide polymorphisms
- Stat
signal transducer and activator of transcription
- TPA
12‐O‐tetradecanoylphorbol 13‐acetate
- TRAIL
tumor necrosis factor‐related apoptosis inducing ligand.
Interferons‐α/β are massively produced upon viral infection, and function for antiviral defense in the innate immune system. The IFN function is based upon several signaling pathways induced by the activation of the IFNAR complex, which consists of at least two subunits, IFNAR1 and IFNAR2.( 1 , 2 , 3 ) Binding of IFN‐α/β to IFNAR results in the activation of the Jak–Stat pathway, which leads to formation of two transcriptional activator complexes, AAF( 4 ) and ISGF3( 5 , 6 , 7 ). AAF is a homodimer of tyrosine‐phosphorylated Stat1, whereas ISGF3 is a heterotrimeric complex of tyrosine‐phosphorylated Stat1, Stat2 and another transcription factor family member, IRF9/p48/ISGF3γ.( 5 , 8 ) The nuclear translocation of these transcription complexes is followed by the transcriptional induction of a large number of target genes (ISG) to evoke versatile activities in the host defense system. Other biological activities induced by treatment with large amounts of IFN‐α/β have also been well documented including antiproliferative and antitumor activities. Recently, we found that IFN‐α/β induce the mRNA of p53, a tumor suppressor, in an ISGF3‐dependent manner, which contributes to the enhancement of apoptosis triggered by DNA damage.( 9 )
In contrast to the above roles of massive IFN‐α/β that are induced by viral infection, there have been intriguing observations that the constitutive expression of IFN‐α/β occurs in the absence of viral infection, albeit at very low levels both in vitro and in vivo.( 10 , 11 , 12 , 13 ) In fact, we previously found that low levels of IFN‐α/β mRNA can be detected in MEF and in other murine cells/tissues by RT‐PCR analysis,( 14 , 15 , 16 ) and that a weak signal by these constitutively produced IFN‐α/β, transmitted independently of viral infection, is critical for priming cells to enhance their response to other stimuli (reviewed in( 8 )). In particular, it has been shown that this constitutive, weak IFN‐α/β signal plays an essential role for eliciting robust responses of cells to IFN‐γ and IL‐6, which are key cytokines to regulate adaptive immune responses.( 16 , 17 ) Evidence has also been provided for its amplifying effect on IFN‐α/β production upon viral infection by sustaining the constitutive expression level of IRF7,( 14 ) which is an IFN‐inducible transcriptional factor crucial for virus‐induced IFN‐α/β gene induction.( 18 , 19 ) Furthermore, the constitutive IFN‐α/β signaling is also crucial for enhancing the activation of CD8+ T cells, but it needs to be properly attenuated to maintain homeostatic CD8+ T‐cell responses.( 15 , 20 ) Given these enhancing effects of the constitutive IFN‐α/β signaling in immune responses, we propose that there is a role for constitutively produced IFN‐α/β in tumor prevention.
In this study, we have found a novel role for the weak signaling induced by constitutively produced IFN‐α/β in the regulation of oncogenesis. Long‐term cell cultures of Ifnar1−/– MEF and Ifnb−/– MEF, which show undetectable or hardly detectable signaling by the constitutively produced IFN‐α/β, respectively, resulted in the formation of transformed foci, although at a low frequency, which are tumor‐forming when transplanted in nude mice. On the other hand, such a transforming property was not found in MEF lacking Stat1, IRF9 or IRF1, all of which are essential mediators in the downstream signaling pathways activated by the IFNAR. Moreover, we found that Ifnar1−/– mice were predisposed to chemically induced skin papilloma formation, as compared with wild‐type mice. Notably, the expression level of IFNAR1 mRNA was significantly decreased in human gastric cancer tissues. These findings may address a novel biological significance of the weak signal by constitutively produced IFN‐α/β, which is mediated by a non‐canonical pathway independent of ISGF3 or AAF. The implication of these present findings will be discussed in the context of a homeostatic regulatory role of the constitutive, weak IFN‐α/β signaling in tumor prevention.
Materials and Methods
Cell culture. Primary mouse embryonic fibroblasts derived from wild‐type, Ifnar1−/– , Ifnb−/– , Stat1−/– , Irf9−/– , Irf1−/– and Trp53−/– at day 12–15 embryos were prepared as described previously.( 16 ) All cultures were maintained in DMEM (Gibco) supplemented with 10% FCS (Sigma).
Retroviral vectors and gene transfer. Oncogenic Ras (H‐RasV12)( 21 ) was expressed using retroviral gene transfer.( 22 ) Similar expression levels of Ras oncoprotein in each infected cell populations were confirmed by western blot analysis with anti‐Ras antibody (Oncogene). Mouse IFNAR1 was retrovirally expressed with the pBabe‐puro/mIFNAR1 construct in MEF. Infected cell populations were selected by culture in puromycin (2.5 µg/mL, 3 days) to eliminate uninfected cells as previously described.( 16 )
Analysis of cell proliferation. For (3H)thymidine incorporation assay, 1.0 × 105 cells/well in flat‐bottomed 96‐well plates were plated in triplicate, and cultured with DMEM supplemented with 10% FCS. Cells were pulsed with 0.5 µCi/well of (3H)thymidine for the last 16 h of the 48‐culture period, and the (3H)thymidine uptake levels were measured by scintillation counting.
Spontaneous transformation of Ifnar1 −/– MEF. 1.0 × 106 cells were plated in 10‐cm dishes, and cultured with DMEM supplemented with 10% FCS. The medium was routinely changed once weekly until transformed foci were observed.
Methylcellulose gel assay and tumorigenicity in nude mice. Methylcellulose gel assay was carried out as previously described.( 22 ) For in vivo tumorigenicity assays, the method was followed as previously described.( 22 ) Briefly, 1.0 × 106 cells/100 µL of PBS were injected s.c. into 6‐week‐old male athymic nude mice (CLEA Japan). Each animal received two injections, one on each rear flank. MEF from at least two different embryo preparations were used for each genotype, and at least eight animals were examined for each line of MEF. Mice were monitored for 6 weeks to evaluate tumor formation.
Chemically induced tumor development. The procedure was followed as previously described.( 23 ) The backs of 7‐week‐old mice were shaved, and treated with DMBA (20 mg in 200 mL acetone; Sigma). Mice were subsequently treated twice a week with TPA (200 µL of 1.0 × 10−4 M in acetone) for 15 weeks, and then monitored for 20 weeks. All mice were housed and treated in accordance with protocols approved by the Institutional Animal Care and Use Committee of University of Tokyo.
Cell culture for senescence property. Cells were routinely passaged every 3 or 4 days in 60‐mm dishes with DMEM containing 10% FCS. When reaching confluence, cells were reseeded at a density of 3 × 105 cells/dish. Accumulative cell numbers and growth rates were calculated at each passage.
Immunoprecipitation and immunoblotting. Immunoprecipitation and immunoblotting were carried out as described.( 16 ) Polyclonal antibodies against IFNAR1 (R‐100; sc‐845) were purchased from Santa Cruz Biotechnology.
Quantitative RT‐PCR analysis with clinical specimens. Surgically resected from 14 primary gastric cancer tissues and the adjacent non‐cancerous tissues were obtained from the patients treated at Sapporo Medical University Hospital. Informed consent was obtained from all patients before collection of the specimens. To evaluate the expression levels of human IFNAR1 and IFNAR2 mRNA in gastric tissues, total RNA was extracted from these clinical specimens using Sepasol‐RNA I (Nacalai Tesque) and quantitative real‐time RT‐PCR analysis was performed by using a LightCycler and SYBRGreen system (Roche). Data were normalized by the level of β‐actin expression in each sample. For the amplification of IFNAR1 mRNA and IFNAR2 mRNA, the following primers were used: IFNAR1 forward primer, 5′‐TCC GCG TAC AAG CAT CTG AT‐3′ and reverse primer, 5′‐GAC TGT TTT GGA GCA CCG ATA‐3′; and IFNAR2 forward primer, 5′‐GGC CAG GAA TCA GAA TCA GC‐3′ and reverse primer, 5′‐ACG GTG GCA GGT TAG GAA AT‐3′.
Results
Ifnar1 −/– MEF undergo spontaneous transformation after long‐term cell culture. Primary MEF were prepared from 12–15‐day‐old embryos of wild‐type and Ifnar1−/– mice, and were subsequently maintained in culture in 10‐cm dishes. After 6–8‐week culture, transformed foci appeared from Ifnar1−/– MEF at a frequency of 3 × 10−5 (Fig. 1a,b). On the other hand, wild‐type MEF did not form any transformed foci (Fig. 1a,b). This finding suggests that Ifnar1−/– MEF are predisposed to undergo spontaneous transformation, albeit at a low frequency. We next examined whether the Ifnar1−/– MEF derived from the transformed foci exhibit tumorigenicity in nude mice. After six independent clones from each focus were s.c. injected into nude mice at 1.0 × 106 cells/site, tumors developed within 4 weeks (Fig. 1c), although they did not show any signs of invasion and metastasis. Tumor formation from all of the six clones tested (F1–F6) was observed at 100% frequency (tumors were developed in all four infection sites). These results indicate that the deficiency of the IFNAR1 receptor subunit predisposes MEF to tumorigenic transformation, suggesting that IFNAR1‐mediated signaling or the IFNAR1 subunit itself may have a tumor suppressive function.
Figure 1.
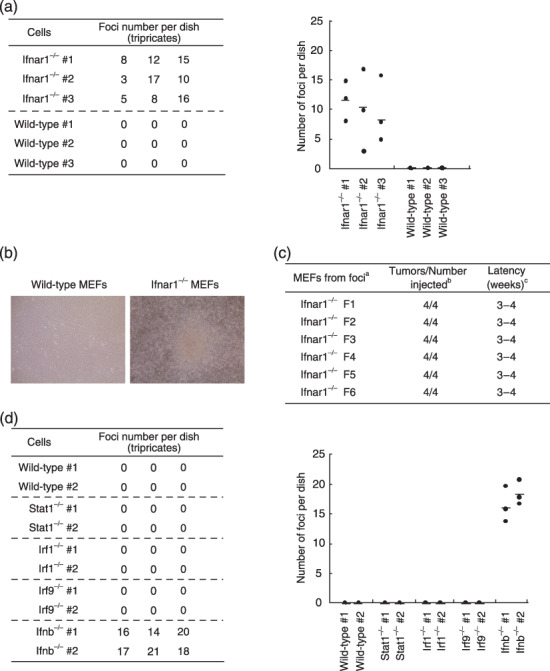
Spontaneous transformation of Ifnar1−/– MEF. (a) Primary wild‐type or Ifnar1−/– MEF (3 × 106 cells) were plated on 10‐cm culture dishes, and after 4‐ to 6‐week cell culture, numbers of foci formed in the dish were counted. Assay was done with three different MEF in three independent dishes. Average of the numbers of foci observed in the three dishes are shown as a horizontal line in the graph (right). (b) Appearance of a cell colony observed after 50‐day culture. (c) Six cell clones (F1–F6) were isolated from separate foci that formed after long‐term cell culture, were inoculated in nude mice for tumorigenicity. aMEF genotypes; bnumber of tumors arising per number of sites injected; cperiod until visible tumor formation. (d) Primary wild‐type or other mutant MEF lacking signaling molecules downstream of the IFNAR were plated on 10‐cm culture dishes, and were examined for the formation of colonies during the long‐term culture, as described in (a). IFNAR, IFN‐α/β receptor; MEF, mouse embryonic fibroblasts.
Tumor suppressive effect of the IFNAR1 subunit is ligand‐dependent. It has been previously shown that IFN‐α/β are constitutively produced by cultured cells at low levels.( 15 , 16 ) On the basis of these findings, one can easily infer that this spontaneous transformation of Ifnar1−/– MEF may result from the disruption of a weak signaling by the constitutively produced IFN‐α/β through the IFNAR. We next examined whether the tumor suppressive effect of the IFNAR1 is ligand‐independent or not. Because it was previously reported that expression of IFN‐α mRNA is barely detectable in MEF in absence of IFN‐β,( 16 ) Ifnb−/– MEF were tested as to whether they form transformed foci in the same experimental setting in which Ifnar1−/– MEF underwent spontaneous transformation as described above. As shown in Figure 1(d), Ifnb−/– MEF developed transformed foci, and the numbers of the transformed foci were more than Ifnar1−/– MEF (Fig. 1a).
Tumor suppressive effect of the IFNAR1‐mediated constitutive signaling is independent of ISGF3 and AAF. To gain further insights into the mechanism for the spontaneous transformation of Ifnar1−/– MEF, we analyzed the transforming property of several mutant MEF from mice that lack Stat1, IRF9 or IRF1, all of which are key signaling molecules downstream of the IFNAR. However, we did not observe the formation of foci derived from any of these mutant cell cultures (Fig. 1d). These findings indicate that the spontaneous transformation of Ifnar1−/– MEF may be caused by the disruption of an ISGF3 and AAF‐independent, non‐canonical pathway(s) downstream of the IFNAR.
Primary Ifnar1 −/– MEF are susceptible to tumorigenic transformation by expressing the activated c‐Ha‐Ras oncoprotein. To examine whether Ifnar1−/– MEF manifest oncogene susceptibility, we expressed an activated form of c‐Ha‐Ras oncoprotein in primary Ifnar1−/– MEF, and then tested their tumorigenic activity in nude mice. As shown Figure 2, the frequency of tumor formation by the Ras‐expressing Ifnar1−/– MEF was more than 70% (clone #1, six out of six sites injected; clone #2, four out of six sites), whereas Ras‐expressing Trp53−/– or Irf1−/– MEF formed tumors in every site injected. In contrast, no tumors appeared from Ifnar1−/– MEF without the expression of activated Ras (zero out of six sites injected) (Fig. 2). Similarly, primary Ifnb−/– MEF showed tumor development in nude mice only when accompanied with the expression of activated Ras (Fig. 2). According to some previous reports showing that neoplastic transformation of primary cells requires activation of an additional oncogene,( 22 , 24 ) the above results for Ifnar1−/– MEF and Ifnb−/– MEF suggest that, although its activity is not stronger than those of p53 and IRF1, the IFNAR1 may play a putative role as a tumor suppressor, wherein IFNAR1‐mediated constitutive IFN‐α/β signaling is involved. On the other hand, Stat1−/– MEF and Irf9−/– MEF did not exhibit such a transforming property even in the presence of the activated Ras oncoprotein (Fig. 2), which is consistent with the result for the spontaneous transformation (Fig. 1d).
Figure 2.

Oncogenic transformation of primary MEF deficient in IFNAR1, its downstream molecules, or IFN‐β, by the expression of c‐Ha‐Ras oncoprotein. In vivo tumorigenesis of mutant MEF lacking molecules involved in IFN‐α/β‐mediated signaling. Primary MEF, freshly prepared from Ifnar1−/– or ifnb−/– embryos, were infected with pGDV12ras retrovirus, which directs the expression of an activated form of c‐Ha‐Ras oncoprotein carrying an oncogenic G12V mutation.( 21 ) These cells were subjected to tumorigenesis assay in nude mice as described in Fig. 1 (c). IFN‐β, β‐interferon; IFNAR, IFN‐α/β receptor; MEF, mouse embryonic fibroblasts.
IFNAR1 deficiency does not affect the growth properties of MEF. We further examined the growth properties of primary Ifnar1−/– MEF. Ifnar1−/– MEF showed no significant difference in (3H)thymidine uptake during their growth with serum‐containing media (Fig. 3a). In addition, primary Ifnar1−/– MEF were also characterized for their senescence. As shown in Figure 3(b), there is no abnormality in their ability to senescence, as compared with that of wild‐type MEF. Telomerase activity could be detected in primary Ifnar1−/– MEF at a comparable level to that observed in wild‐type MEF (data not shown).
Figure 3.
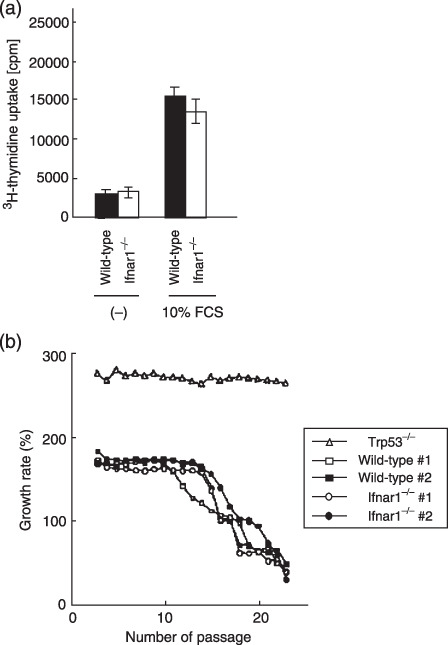
Normal growth properties of Ifnar1−/– MEF. (a) (3H)thymidine incorporation assay using primary wild‐type and Ifnar1−/– MEF. Indicated cells (1.0 × 105) of the indicated cells were plated and grown in media containing 10% FCS for 32 h followed by a (3H)thymidine pulse, and 16 h later, the (3H)thymidine uptake levels were measured by scintillation counting. Values of the average and standard deviation of three measures are shown. (b) Replicative senescence in Ifnar1−/– MEF. Wild‐type, Ifnar1−/– and Trp53−/– MEF were routinely cultured over a period of 23 passages. MEF from two different embryo preparations were used for each genotype (#1 and #2). Cells were counted at subculture to determine the population doublings. Wild‐type and Ifnar1−/– MEF but not Trp53−/– MEF reached replicative senescence after 12–14 passages. cpm, count per minute; FCS, fetal calf serum; MEF, mouse embryonic fibroblasts.
Irreversible transforming property of Ifnar1 −/– MEF. We investigated whether the transforming property of Ifnar1−/– MEF can be normalized by the reconstitution of IFNAR1 expression. Both spontaneously transformed and ras‐transformed Ifnar1−/– MEF were infected with IFNAR1‐expressing retrovirus, and tested in triplicate for their anchorage‐independent proliferation in methylcellulose gel. As indicated in Figure 4(a), the colony‐forming potential of both transformed cells in gel was not affected by the expression of IFNAR1, although these IFNAR1‐transfected MEF can transmit signals through the reconstituted IFNAR1, in response to IFN‐β treatment (Fig. 4b). These results suggest that Ifnar1−/– MEF may acquire an irreversible genetic alteration(s) during their spontaneous transformation.
Figure 4.
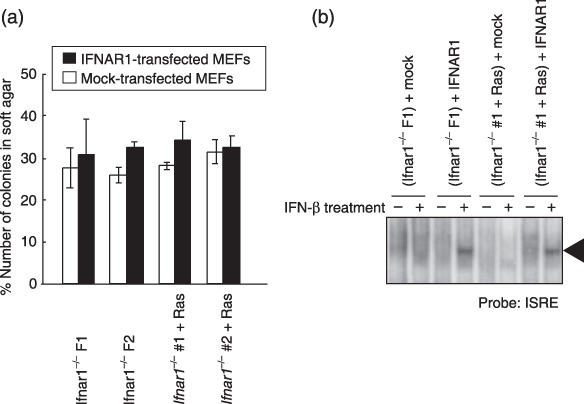
Irreversible transforming property of Ifnar1−/– MEFs. (a) Methylcellulose gel assay. Two randomly selected clones of either spontaneously transformed Ifnar1−/– MEFs (F1 and F2) or Ifnar1−/– MEFs transformed by activated Ras expression (#1 and #2) were infected with a retrovirus that directs the expression of mouse IFNAR1 (solid bars) and the control retrovirus (open bars). In this assay, a total of 5000 viable cells were counted under a microscope, and the number of colonies formed among them was scored. Values are indicated as percentage of total cells examined. (b) DNA binding activity of ISGF3 in transformed Ifnar1−/– MEF reconstituted by the expression of IFNAR1. The transformed Ifnar1−/– MEFs (F1 and #1) retrovirally expressing IFNAR1 or mock are stimulated with IFN‐β (500 U/mL), and subjected to EMSA by using ISRE probe. EMSA, electromobility shift assays; IFN‐β, β‐interferon; IFNAR, IFN‐α/β receptor; ISRE, interferon‐stimulated response element; MEF, mouse embryonic fibroblasts.
IFNAR1 deficiency predisposes mice to skin papilloma formation induced by DMBA treatment. Because the IFNAR1 deficiency intrinsically predisposes cells to transformation as described above (1, 2), it can be presumed that Ifnar1−/– mice may show an increased incidence of chemically induced papillomas, which are well characterized as a skin carcinogenesis model.( 23 ) To test the in vivo role of the IFNAR1‐mediated signaling in tumor development, groups of wild‐type, Ifnar1+/– and Ifnar1−/– mice were initiated with a single treatment with DMBA and promoted twice a week with TPA. Papilloma formation was first observed in Ifnar1−/– mice following 9 weeks of promotion, whereas the appearance of papillomas was delayed in the wild‐type mice (occurring at 17 weeks after promotion) (Fig. 5a). At 19‐week postpromotion, the average number of papillomas observed in the Ifnar1−/– mice was approximately 10‐fold more than that of the wild‐type mice (Fig. 5a). Of note, the rate of appearance of papillomas in Ifnar1+/– mice was almost between Ifnar1−/– and wild‐type mice (Fig. 5a), which is consistent with the observation that the expression level of IFNAR1 protein in the dermal tissues of Ifnar1+/– mice is nearly half that of wild‐type mice (Fig. 5b). These results suggest an Ifnar1 gene dosage effect in developing tumor cells.
Figure 5.
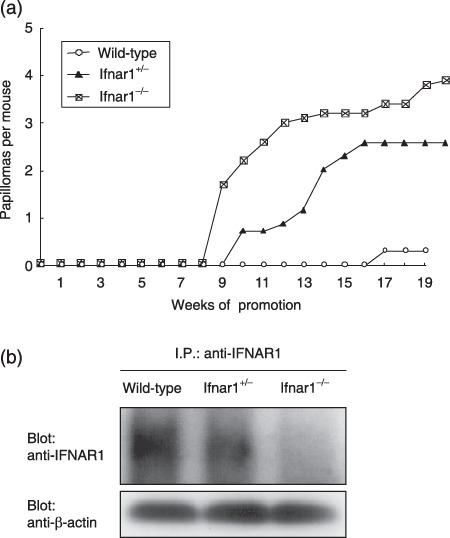
Development of skin papillomas in Ifnar1−/– mice. (a) Groups of wild‐type, Ifnar1+/– and Ifnar1−/– mice (10 mice per group) were initiated by a single treatment with DMBA and promoted twice a week with TPA for 15 weeks. The average number of papillomas (over 2 mm in diameter) per mouse was plotted every week after the start of carcinogen treatment. (b) Constitutive expression levels of IFNAR1 protein in the skin tissues of wild‐type, Ifnar1+/– and Ifnar1−/– mice. Immunoprecipitates with anti‐IFNAR1 were subjected to western blotting analysis with anti‐IFNAR1. The loading amount of each lane was also confirmed by Western blotting with an aliquot of whole cell lysates that were used for immunoprecipitation. DMBA, 7,12‐dimethylbenz(a)anthracene; TPA, 12‐O‐tetradecanoylphorbol 13‐acetate; IFNAR, α/β‐interferon receptor; I.P., Immunoprecipitation.
Decreased expression levels of type I IFN receptor in gastric cancer tissues. Finally, we examined the mRNA expression level of type I IFN receptor subunits IFNAR1 and IFNAR2 in gastric cancer tissue specimens, by quantitative RT‐PCR analysis (Fig. 6). As for the IFNAR1 mRNA expression, a significant decrease in the expression (70% > C/N ratios which represent the ratio derived by division of the expression level of cancerous tissues with those of the corresponding non‐cancerous tissues) was observed in 57% (eight of 14 samples) of the cancerous tissues tested. On the other hand, only a third (four of 11) of the cancerous tissues showed a decreased expression (70% > C/N ratios) of IFNAR2 mRNA (Fig. 6). The rate of gastric cancer patients, who revealed a significantly decreased expression level of either IFNAR1 or IFNAR2, reached 82% (nine of 11) of all the tested tissues. There is no evident relationship between the decreased expression levels of the type I IFN receptor subunits and various clinicopathological factors.
Figure 6.
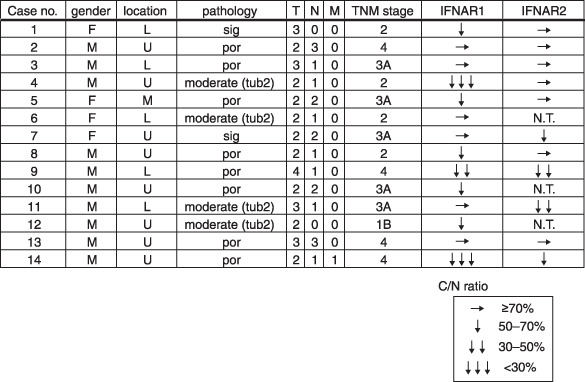
Decreased expression levels of the type I IFN receptor subunits in human gastric cancer tissues. Total RNA were extracted from cancerous and non‐cancerous tissues of 14 patients with various types of gastric cancers, and were analyzed by quantitative RT‐PCR with specific primers. C/N ratios were calculated by comparing the expression level of cancerous tissues with that of non‐cancerous tissue. L, lower region of the stomach; M, middle region of the stomach; N.T., not tested; por, poor differentiated carcinoma; RT‐PCR, reverse transcription polymerase chain reaction; sig, signet ring cell carcinoma; tub2, tubular adenocarcinoma (moderately differentiated type); U, upper region of the stomach.
Discussion
In this study, we found that IFNAR1 deficiency resulted in spontaneous transformation in MEF. This finding is intriguing in terms of the possibility that the IFNAR1 gene might be a tumor susceptible genes that are involved in the multistage genetic events for tumorigenesis.( 25 ) The cooperative role of c‐Ha‐ras with other oncogenes in tumorigenesis of primary cells has been reported. Primary MEF are usually not transformed by expressing an activated c‐Ha‐ras alone unless accompanied by the activation of a second oncogene (e.g. polyoma large T antigen gene)( 24 ) or the inactivation of a tumor suppressor gene (e.g. Irf1 gene).( 22 ) Our current data (Fig. 2) with Ifnar1−/– MEF showed that a defect in the Ifnar1 alleles rendered the cells to be susceptible to the activated c‐Ha‐ras oncogene, indicating a possible role of IFNAR1 as a tumor suppressor. Another characteristic aspect for tumor suppressors includes spontaneous development of tumors in their mutant mice. In fact, it has been reported that Trp53−/– mice develop various types of tumors,( 26 ) and that mice develop pituitary tumors when one allele of the Rb gene is disrupted.( 27 ) However, we could not observe any spontaneous tumor development in Ifnar1−/– mice aged up to at least 12 months old (data not shown). Therefore, IFNAR1 deficiency may result in enhancement of cellular susceptibility to transformation by oncogenes, as similarly observed in cells deficient in IRF1, which is known to be a tumor susceptible gene.( 22 , 28 ) However, the latency for tumor formation of Ras‐expressing Ifnar1−/– MEF in nude mice is longer (3–6 weeks) than that of Ras‐expressing Irf1−/– MEF (1–3 weeks), and the tumorigenic rates of Ras‐expressing Ifnar1−/– MEFs are less (75%) than those of Irf1−/– MEF (100%) (Fig. 2). These results suggest that IFNAR1‐mediated tumor suppressive effect seems to be distinct from that of IRF1, and not as potent as compared with that of IRF1.
Similar to our observation, there is a report by another group about anti‐oncogenic activity mediated by the IFNAR1 subunit,( 29 ) showing that exogenous expression of IFNAR1 subunit renders a human erythroleukemic cell line, K‐562, to markedly suppress not only in vitro growth properties but also tumorigenicity in nude mice. They concluded that this effect by expression of IFNAR1 is independent of ligands because the IFN‐α/βgenes are completely deleted in K‐562 cells.( 1 ) In contrast, our results showed that the tumor suppressive effect mediated by IFNAR1 is ligand‐dependent (1, 2). In fact, it has been previously shown that low levels of IFN‐α/β are detectable not only in supernatants from cultures of MEF but also in some tissues from normal healthy subjects, although in the absence of viral infection.( 10 , 12 , 15 , 16 ) Recently, we have demonstrated the biological importance of this constitutive IFN‐α/β signaling in antiviral defense.( 8 ) In this context, the present data indicates another aspect of a weak signal by constitutively produced IFN‐α/β in terms of its preventive role against cellular transformation.
We have also provided some mechanistic insights into the tumor suppressive effect of the constitutive IFN‐α/β signaling. A series of evidences regarding antitumor activities of type I IFN has been so far reported.( 30 , 31 ) Some of IFN‐inducible genes were shown to encode proteins that mediate tumor suppressor activities through a direct action on tumor cells: for example, IRF1, PKR, and OAS, TRAIL and caspase‐4/8,( 28 , 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 ) all of which are induced via activation of transcriptional complexes including ISGF3 and AAF. In this regard, no transforming properties were observed in MEF that lack any of the receptor downstream molecules, such as Stat1, IRF9 and IRF1 (1, 2). These results suggest a possible involvement of an unconventional, Stat1‐independent signaling pathway in the tumor suppressive effect of the constitutive IFN‐α/β signaling.
Because the above results suggest that the constitutive IFN‐α/β signaling may contribute to tumor suppression via a Stat1‐independent pathway(s), target gene(s) responsible for this AAF and ISGF3‐independent pathway(s) were investigated. As shown in our previous DNA chip analysis performed by Stat1−/– MEF treated with IFN‐β,( 40 ) Usp18/UBP43, which was the mRNA with the most marked induction belongs to the family of ubiquitin isopeptidases and specifically cleaves ISG15, a ubiquitin‐like molecule.( 41 ) Interestingly, F‐box proteins are known to function as substrate recognition factors for E3 ubiquitin‐ligase complexes, and the related gene, Fbxo39, as well as ISG15 gene were also induced in a Stat1‐independent manner. One hypothesis might be speculated that the cellular transformation of Ifnar1−/– MEF is possibly related to dysregulation of the protein modification by ubiquitin‐like molecules including ISG15, because this protein modification system is considered as a critical regulatory process for the control of cell cycle or stress responses.( 42 , 43 ) It will be required to characterize their detailed involvement.
It has been previously reported that fibroblasts lacking Ku70, a DNA repair factor, undergo spontaneous transformation similar to Ifnar1−/– MEF. However, the transformation frequency of Ifnar1−/– MEF (Fig. 1) is much lower (~3 × 10−5), as compared with that (~4 × 10−2) of Ku70‐deficient fibroblasts.( 44 ) This suggests that the underlying mechanism may not be a direct effect of the weak IFN‐α/β signaling on the DNA repair system. Recently, we found a new linkage between IFN‐α/β signaling and the tumor suppressor p53, indicating that IFN‐induced p53 contributes to IFN‐α/β‐mediated antitumor action.( 9 ) In this context, we also examined the constitutive expression levels of p53 protein in wild‐type and Ifnar1−/– MEF. However, no significant difference was detected between these MEF (data not shown). In addition, because the data shown in Figure 4 suggest that the transformation of Ifnar1−/– MEF is irreversible, we further investigated whether these transformed Ifnar1−/– MEF acquire any genetic abnormalities. However, no obvious abnormality could be found in the result by comparative genomic hybridization analysis (data not shown). Further analyses will be required to identify a genetic alteration(s) involved in this transformation process.
Deficiency of IFNAR1 predisposes mice to skin papilloma formation induced by DMBA/TPA treatment (Fig. 5). However, the current data cannot clarify whether this effect is direct or indirect. In this respect, indirect activities of the constitutive IFN‐α/β signaling, such as by activating the immune system, ought to be considered when evaluating the effects of type I IFN on tumor development in vivo (Fig. 5). In relation to this, Schreiber's group recently reported that type I IFN are important components for the cancer immunoediting process wherein protective antitumor responses are developed through their effect on the immune systems.( 45 ) In this regard, we previously found an intriguing aspect of the constitutive IFN‐α/β signaling, which is critical for the enhancement of CD8+ T‐cell activation.( 20 ) Together with this observation, the tumor suppressive effect of the constitutive IFN‐α/β signaling may also be linked to the activation of the immune system as well as its direct action. Notably, we found significantly decreased mRNA levels of both IFNAR1 and IFNAR2 in human gastric cancer tissues (Fig. 6). In addition, we investigated genetic polymorphisms in the human IFNAR1 gene, and have identified five known and two novel SNP and one novel dinucleotide repeat polymorphism. However, we could not find any association for these polymorphisms between approximately 400 gastric cancer patients and approximately 300 control individuals (unpublished observation, Tadatsugu Taniguchi and Ken Yamamoto, 2005). These findings might suggest that the impaired expression of IFNAR1 in gastric tissues may occur during tumor development and contribute to the development or progression of gastric cancers, although some microenvironments that contribute to the impairment of IFN signaling may also be involved. Further analyses will be required to clarify the mechanism of action by constitutive type I IFN signaling.
Collectively, our findings revealed a novel significance of the Stat1‐independent signaling by constitutively produced IFN‐α/β in prevention of cellular transformation. Furthermore, it can be speculated that this constitutive IFN‐α/β signaling may be involved in maintaining the constitutive expression levels of target molecule(s) which regulates the cell‐intrinsic protective system against oncogenesis. The current study may further provide an interesting aspect of the weak IFN‐α/β signaling in terms of a therapeutic application with low doses of IFN‐α/β for cancer prevention.
Acknowledgments
We thank Johji Inazawa for comparative genomic hybridization analysis, Ken Yamamoto for sharing the SNP data, Toray for murine IFN‐β, David Savitsky for critical reading of the manuscript, and Masashi Shishido and Rie Takeda for technical assistance. This work was supported by Kakenhi (Grant‐in‐Aid for Scientific Research) on Priority Areas ‘Integrative Research Toward the Conquest of Cancer’ from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and grants from the Ichiro Kanehara Foundation, Sumitomo Foundation, Senri Life Science Foundation, and the Naito Foundation.
References
- 1. Novick D, Cohen B, Rubinstein M. The human interferon alpha/beta receptor: characterization and molecular cloning. Cell 1994; 77: 391–400. [DOI] [PubMed] [Google Scholar]
- 2. Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem 1998; 67: 227–64. [DOI] [PubMed] [Google Scholar]
- 3. Uze G, Lutfalla G, Gresser I. Genetic transfer of a functional human interferon alpha receptor into mouse cells: cloning and expression of its cDNA. Cell 1990; 60: 225–34. [DOI] [PubMed] [Google Scholar]
- 4. Decker T, Lew DJ, Darnell JE Jr. Two distinct α‐interferon‐dependent signal transduction pathways may contribute to activation of transcription of the guanylate‐binding protein gene. Mol Cell Biol 1991; 11: 5147–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bluyssen AR, Durbin JE, Levy DE. ISGF3 gamma p48, a specificity switch for interferon activated transcription factors. Cytokine Growth Factor Rev 1996; 7: 11–7. [DOI] [PubMed] [Google Scholar]
- 6. Darnell JE Jr, Kerr IM, Stark GR. Jak‐STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994; 264: 1415–21. [DOI] [PubMed] [Google Scholar]
- 7. Haque SJ, Williams BR. Identification and characterization of an interferon (IFN)‐stimulated response element‐IFN‐stimulated gene factor 3‐independent signaling pathway for IFN‐α. J Biol Chem 1994; 269: 19523–9. [PubMed] [Google Scholar]
- 8. Taniguchi T, Takaoka A. A weak signal for strong responses: interferon‐a/b revisited. Nat Rev Mol Cell Biol 2001; 2: 378–86. [DOI] [PubMed] [Google Scholar]
- 9. Takaoka A, Hayakawa S, Yanai H et al . Integration of interferon‐α/β signalling to p53 responses in tumour suppression and antiviral defence. Nature 2003; 424: 516–23. [DOI] [PubMed] [Google Scholar]
- 10. Bocci V. The physiological interferon response. Immunol Today 1985; 6: 7–9. [DOI] [PubMed] [Google Scholar]
- 11. De Maeyer E, De Maeyer‐Guignard J. Interferons and Other Regulatory Cytokines. New York: John Wiley and Sons, 1988. [Google Scholar]
- 12. Gresser I. Biologic effects of interferons. J Invest Dermatol 1990; 95: 66S–71S. [DOI] [PubMed] [Google Scholar]
- 13. Tovey MG, Streuli M, Gresser I et al . Interferon messenger RNA is produced constitutively in the organs of normal individuals. Proc Natl Acad Sci USA 1987; 84: 5038–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hata N, Sato M, Takaoka A, Asagiri M, Tanaka N, Taniguchi T. Constitutive IFN‐α/β signal for efficient IFN‐α/β gene induction by virus. Biochem Biophys Res Commun 2001; 285: 518–25. [DOI] [PubMed] [Google Scholar]
- 15. Hida S, Ogasawara K, Sato K et al . CD8 (+) T cell‐mediated skin disease in mice lacking IRF‐2, the transcriptional attenuator of interferon‐α/β signaling. Immunity 2000; 13: 643–55. [DOI] [PubMed] [Google Scholar]
- 16. Takaoka A, Mitani Y, Suemori H et al . Cross talk between interferon‐γ and ‐α/β signaling components in caveolar membrane domains. Science 2000; 288: 2357–60. [DOI] [PubMed] [Google Scholar]
- 17. Mitani Y, Takaoka A, Kim SH et al . Cross talk of the interferon‐α/β signalling complex with gp130 for effective interleukin‐6 signalling. Genes Cells 2001; 6: 631–40. [DOI] [PubMed] [Google Scholar]
- 18. Honda K, Yanai H, Negishi H et al . IRF‐7 is the master regulator of type‐I interferon‐dependent immune responses. Nature 2005; 434: 772–7. [DOI] [PubMed] [Google Scholar]
- 19. Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol 2008; 26: 535–84. [DOI] [PubMed] [Google Scholar]
- 20. Ogasawara K, Hida S, Weng Y et al . Requirement of the IFN‐alpha/beta‐induced CXCR3 chemokine signalling for CD8+ T cell activation. Genes Cells 2002; 7: 309–20. [DOI] [PubMed] [Google Scholar]
- 21. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997; 88: 593–602. [DOI] [PubMed] [Google Scholar]
- 22. Tanaka N, Ishihara M, Kitagawa M et al . Cellular commitment to oncogene‐induced transformation or apoptosis is dependent on the transcription factor IRF‐1. Cell 1994; 77: 829–39. [DOI] [PubMed] [Google Scholar]
- 23. Kemp CJ, Donehower LA, Bradley A, Balmain A. Reduction of p53 gene dosage does not increase initiation or promotion but enhances malignant progression of chemically induced skin tumors. Cell 1993; 74: 813–22. [DOI] [PubMed] [Google Scholar]
- 24. Weinberg DH, Kelly TJ. Requirement for two DNA polymerases in the replication of simian virus 40 DNA in vitro. Proc Natl Acad Sci USA 1989; 86: 9742–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med 2004; 10: 789–99. [DOI] [PubMed] [Google Scholar]
- 26. Donehower LA, Harvey M, Slagle BL et al . Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992; 356: 215–21. [DOI] [PubMed] [Google Scholar]
- 27. Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature 1992; 359: 295–300. [DOI] [PubMed] [Google Scholar]
- 28. Nozawa H, Oda E, Nakao K et al . Loss of transcription factor IRF‐1 affects tumor susceptibility in mice carrying the Ha‐ras transgene or nullizygosity for p53. Genes Dev 1999; 13: 1240–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Colamonici OR, Porterfield B, Domanski P et al . Ligand‐independent anti‐oncogenic activity of the alpha subunit of the type I interferon receptor. J Biol Chem 1994; 269: 27275–9. [PubMed] [Google Scholar]
- 30. Belardelli F, Ferrantini M, Proietti E, Kirkwood JM. Interferon‐alpha in tumor immunity and immunotherapy. Cytokine Growth Factor Rev 2002; 13: 119–34. [DOI] [PubMed] [Google Scholar]
- 31. Kirkwood J. Cancer immunotherapy: the interferon‐α experience. Semin Oncol 2002; 29: 18–26. [DOI] [PubMed] [Google Scholar]
- 32. Clemens MJ. Interferons and apoptosis. J Interferon Cytokine Res 2003; 23: 277–92. [DOI] [PubMed] [Google Scholar]
- 33. Harada H, Kitagawa M, Tanaka N et al . Anti‐oncogenic and oncogenic potentials of interferon regulatory factors‐1 and ‐ 2. Science 1993; 259: 971–4. [DOI] [PubMed] [Google Scholar]
- 34. Harada H, Kondo T, Ogawa S et al . Accelerated exon skipping of IRF‐1 mRNA in human myelodysplasia/leukemia; a possible mechanism of tumor suppressor inactivation. Oncogene 1994; 9: 3313–20. [PubMed] [Google Scholar]
- 35. Kimchi A, Shure H, Revel M. Anti‐mitogenic function of interferon‐induced (2′‐5′) oligo (adenylate) and growth‐related variations in enzymes that synthesize and degrade this oligonucleotide. Eur J Biochem 1981; 114: 5–10. [DOI] [PubMed] [Google Scholar]
- 36. Koromilas AE, Li S, Matlashewski G. Control of interferon signaling in human papillomavirus infection. Cytokine Growth Factor Rev 2001; 12: 157–70. [DOI] [PubMed] [Google Scholar]
- 37. Meurs EF, Galabru J, Barber GN, Katze MG, Hovanessian AG. Tumor suppressor function of the interferon‐induced double‐stranded RNA‐activated protein kinase. Proc Natl Acad Sci USA 1993; 90: 232–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tanaka N, Ishihara M, Lamphier MS et al . Cooperation of the tumor suppressors IRF‐1 and p53 in response to DNA damage. Nature 1996; 382: 816–8. [DOI] [PubMed] [Google Scholar]
- 39. Zhou A, Paranjape J, Brown TL et al . Interferon action and apoptosis are defective in mice devoid of 2′,5′‐oligoadenylate‐dependent RNase L. EMBO J 1997; 16: 6355–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Takaoka A, Wang Z, Choi MK et al . DAI (DLM‐1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007; 448: 501–5. [DOI] [PubMed] [Google Scholar]
- 41. Malakhov MP, Malakhova OA, Kim KI, Ritchie KJ, Zhang DE. UBP43 (USP18) specifically removes ISG15 from conjugated proteins. J Biol Chem 2002; 277: 9976–81. [DOI] [PubMed] [Google Scholar]
- 42. Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem 1998; 67: 425–79. [DOI] [PubMed] [Google Scholar]
- 43. Hochstrasser M. Evolution and function of ubiquitin‐like protein‐conjugation systems. Nat Cell Biol 2000; 2: E153–7. [DOI] [PubMed] [Google Scholar]
- 44. Li GC, Ouyang H, Li X et al . Ku70: a candidate tumor suppressor gene for murine T cell lymphoma. Mol Cell 1998; 2: 1–8. [DOI] [PubMed] [Google Scholar]
- 45. Dunn GP, Bruce AT, Sheehan KC et al . A critical function for type I interferons in cancer immunoediting. Nat Immunol 2005; 6: 722–9. [DOI] [PubMed] [Google Scholar]