

Abstract
Transforming growth factor (TGF)‐β signaling has interesting characteristics in the context of cancer. Although perturbations of TGF‐β signaling are strongly implicated in cancer progression, TGF‐β signaling has both tumor‐suppressive and tumor‐promoting effects. For example, TGF‐β inhibits cancer cell proliferation in some cellular contexts, but promotes it in others. Although several approaches to treating cancer have been considered using TGF‐β‐based therapeutic strategies, the contradictory behaviors of TGF‐β have made these approaches complex. To put them to practical use, either the tumor‐suppressive or tumor‐promoting arm needs to be specifically manipulated. However, there is virtually no method to specifically regulate a certain cell response induced by TGF‐β. In this review, we first consider the basic machinery of TGF‐β signaling, and describe several cell responses induced by TGF‐β stimulation in specific contexts. Mechanisms by which TGF‐β can induce several responses in a cellular context‐dependent fashion are discussed with established paradigms and models. We also address perspectives on the specific control of only a subset of numerous cell responses induced by TGF‐β stimulation. Such methods will aid specific regulation of either the tumor‐suppressive or tumor‐promoting arm of the TGF‐β pathway and in realization of TGF‐β‐based treatment of malignant tumors. (Cancer Sci 2010; 101: 306–312)
Analyses of clinical tumor samples have revealed that transforming growth factor (TGF)‐β signaling is strongly implicated in tumor progression. Components of the TGF‐β signaling pathway, TGF‐β type II receptor (TβRII) and Smad4, are commonly inactivated through mutation or allelic loss of heterozygosity in several types of carcinoma.( 1 ) TβRII‐inactivating mutations are frequently found in colon cancers associated with microsatellite instability.( 2 ) Smad4 is located on chromosome 18q, which is often mutated or completely lost in pancreatic cancer.( 3 ) These results provide evidence that the TGF‐β signaling pathway functions as a tumor suppressor that cancers must bypass for their progression.
However, TGF‐β signaling is also known to act as a tumor promoter. Increased TGF‐β1 expression by tumor cells correlates with progression of colorectal and prostate cancers.( 4 , 5 ) TGF‐β immunostaining also correlates with metastases in breast, prostate, and colorectal cancers.( 5 , 6 , 7 ) Moreover, TGF‐β staining has been shown to be stronger in invading local lymph node metastases than in the primary tumor sites in breast and colorectal cancers.( 8 , 9 ) These findings indicate that excessive TGF‐β stimulation is an indispensable prerequisite for the tumor progression.
Retrospective clinical studies have uncovered “cellular context”‐dependent and complex functions of TGF‐β signaling. Depending on numerous factors, including tumor origin, pathological type, and microenvironment, TGF‐β may act as a tumor‐suppressive or tumor‐promoting factor in cancer progression.( 10 , 11 ) However, the mechanisms of such “Jekyll and Hyde” behaviors of TGF‐β signaling have not been fully elucidated. In this review, we begin by describing basic systems of TGF‐β signaling. We then examine the cell responses induced by TGF‐β signaling, especially in tumor progression. Finally, we discuss probable, proposed, or established mechanisms of cellular context‐dependent diversity of TGF‐β‐induced cell responses and perspectives on their use.
Basics of TGF‐β Signaling Machinery
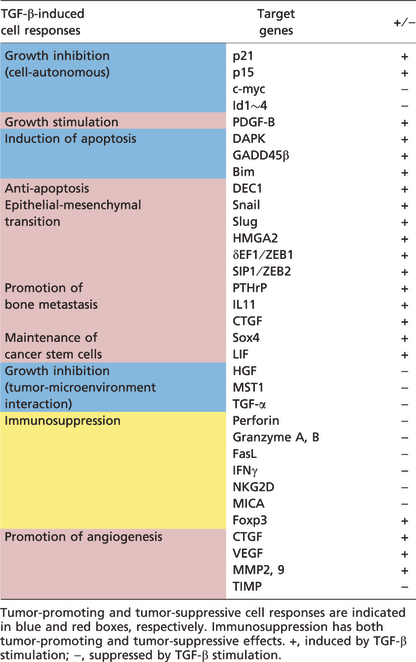
TGF‐β, along with activins, bone morphogenetic proteins, nodal, and growth and differentiation factors, constitute the TGF‐β family.( 12 , 13 , 14 ) Members of the TGF‐β family bind to two distinct receptor types, type II and type I receptors. Five type II receptors and seven type I receptors are present in mammals (Fig. 1). TβRII is the specific receptor for TGF‐β. Among the seven type I receptors, ALK5, also known as TβRI, transduces TGF‐β signaling. Both type II and type I receptors contain serine/threonine kinase domains in their intracellular portions. Binding of the ligand causes formation of heterotetrameric active receptor complexes, resulting in phosphorylation of the type I receptor by the type II receptor in an established receptor complex. Phosphorylation of the type I receptor enables recruitment of receptor‐regulated Smads (R‐Smads). The type I receptor then phosphorylates R‐Smads, allowing them to form heteromeric complexes with the common‐partner Smad (co‐Smad) and translocate into the nucleus. Of the five R‐Smads in mammals, Smad2 and Smad3 transduce TGF‐β signaling. Smad4 is the only known common‐partner Smad in mammals. Smad complexes bind specific DNA sequences in the promoters or enhancers of target genes (Table 1), and the Smad‐binding element (SBE) is defined as the sequence 5′‐AGAC‐3′ or its reverse complement 5′‐GTCT‐3′. Smad complexes recruit co‐activators, co‐repressors, and chromatin remodeling factors to the regulatory regions of target genes, and thereby regulate TGF‐β‐induced cell responses. TGF‐β receptors also transduce signals through non‐Smad pathways, including Erk, p38, and c‐Jun N‐terminal (JNK) MAP kinases, PI3K‐Akt, and small GTPase pathways.( 15 )
Figure 1.
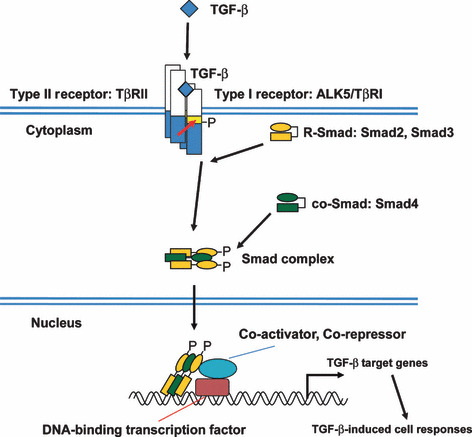
Intracellular transforming growth factor‐β (TGF‐β) signal transduction through Smad proteins. TGF‐β signals are transduced by type II receptor (TβRII) and type I receptor (ALK5), and their downstream Smad proteins. Activated Smad complex interacts with DNA‐binding transcription factors and binds to the promoter regions of TGF‐β target genes. co‐Smad, common‐partner Smad; P, phosphate; R‐Smad, receptor‐regulated Smad.
Table 1.
Major downstream target genes of transforming growth factor‐β (TGF‐β) signaling in each cell response
Diversity of TGF‐β‐Induced Cell Responses
Several in vitro and in vivo studies have uncovered cellular context‐dependent diversity in TGF‐β‐induced cell responses (Fig. 2). Because of such diversity, TGF‐β can be both tumor‐suppressive and pro‐tumorigenic in a cellular context‐dependent fashion.
Figure 2.
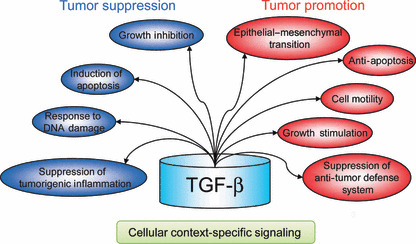
Cell responses induced by transforming growth factor‐β (TGF‐β). TGF‐β suppresses tumorigenesis and tumor progression through some mechanisms (indicated as blue circles), but cancer cells also take advantage of its several pro‐tumorigenic properties (red circles).
TGF‐β inhibits progression of cell cycle phase G1 in multiple types of carcinoma through two sets of events: inhibition of cyclin‐dependent kinase (CDK) functions and elimination of proliferative drivers. In carcinoma cells, TGF‐β induces expression of p15Ink4b, which inhibits cyclin D‐CDK4/6 complexes, and of p21Waf1, which inhibits cyclin E/A‐CDK2 complexes.( 16 , 17 ) TGF‐β also represses expression of c‐myc, a transcription factor that promotes cell proliferation, and Id proteins, nuclear factors that inhibit cell differentiation.( 18 , 19 ) These effects contribute strongly to TGF‐β‐mediated cytostatic responses in some cellular contexts. However, in other cellular contexts, TGF‐β can promote tumor cell proliferation by stimulating production of autocrine mitogenic factors. Some tumor cells, including glioma and osteosarcoma cells, proliferate in response to TGF‐β through induction of platelet‐derived growth factor A or B (PDGF‐A or B), allowing them to take advantage of tumor‐promoting aspects of TGF‐β.( 20 , 21 , 22 )
In addition to regulation of the cell cycle, TGF‐β can limit cancer formation through activation of the apoptotic pathway. Downstream targets for pro‐apoptotic functions of TGF‐β include death‐associated protein kinase (DAPK), growth arrest and DNA‐damage‐inducible β (GADD45β), and Bcl2‐like 11 (Bim).( 23 , 24 , 25 , 26 ) In contrast, TGF‐β also exhibits anti‐apoptotic effects through induction of differentially expressed in chondrocytes 1 (DEC1) in certain conditions.( 27 )
TGF‐β can also enhance migratory and invasive capacities of cancer cells. Migration of epithelial cells requires loss of cell–cell contacts and acquisition of fibroblastic properties. This process is known as epithelial–mesenchymal transition (EMT), and TGF‐β is one of the major inducers of it.( 28 , 29 ) Numerous factors are reported to mediate TGF‐β‐induced EMT, including Snail, Slug, HMGA2, δEF1/ZEB1, and SIP1/ZEB2. These factors are induced by TGF‐β signaling to repress expression of E‐cadherin, a cell–cell adhesion receptor.
In addition to its role in local invasion, TGF‐β also promotes distant metastases. For example, TGF‐β accelerates bone metastases through induction of pro‐osteolytic factors, such as parathyroid hormone‐related protein (PTHrP)( 30 , 31 , 32 ). TGF‐β‐induced PTHrP stimulates production of RANK ligand (RANKL) in osteoblasts, which in turn promotes osteoclast differentiation and bone resorption.( 33 ) TGF‐β signaling also induces other osteolytic genes, including IL‐11 and connective tissue growth factor (CTGF). IL‐11 stimulates expression of osteoclastogenic factors RANKL and granulocyte‐macrophage colony‐stimulating factor in osteoblasts, whereas CTGF promotes not only invasion but also angiogenesis.( 34 )
Recently, rare populations of cells with increased tumor‐initiating capacity have been identified in multiple types of systemic cancer, and are referred to as “cancer stem cells.”( 35 , 36 , 37 ) Such identifications have been followed by demonstration of the novel roles of TGF‐β signaling in tumorigenesis.( 38 ) TGF‐β‐induced EMT results in increased tumorigenicity of CD44+/CD24low breast cancer stem cells.( 39 ) TGF‐β signaling is also required for self‐renewal capacities of glioma stem cells through induction of Sox4 and leukemia inhibitory factor.( 40 , 41 ) TGF‐β‐mediated maintenance of cancer stem cells contributes to progression of primary tumors as well as initiation of macroscopic metastases by disseminated cancer cells.
The roles of TGF‐β in tumor progression are not limited to cell‐autonomous signaling. Recent studies have indicated that TGF‐β signal transduction in stromal fibroblasts plays important roles in suppression of tumorigenesis in adjacent epithelium. Although the mechanism of this has not been fully determined, it may include negative regulation of secreted factors such as hepatocyte growth factor, macrophage stimulating 1, and TGF‐α.( 42 , 43 )
In CD8+ cytotoxic T cells, TGF‐β represses the production of cytolytic factors, including the pore‐forming protein perforin, the caspase‐activating secreted factors granzyme A and B, and the pro‐apoptotic cytokines Fas‐ligand and γ‐interferon.( 44 ) TGF‐β can also decrease the expression of the activating immunoreceptor NKG2D in CD8+ T cells and natural killer (NK) cells and of the NKG2D ligand MHC class I polypeptide‐related sequence A in glioma cells.( 45 ) TGF‐β also stimulates the generation of regulatory T cells, which inhibit CD4+ effector T cell functions, through induction of Foxp3. TGF‐β also induces generation of IL‐17‐producing Th17 cells, which regulate NK cells and macrophages.( 46 ) These effects of TGF‐β orchestrate suppression of immune and inflammatory reactions. Integrative suppression of immune reactions can make TGF‐β both tumor‐suppressive and tumor‐promoting, through suppression of tumorigenic inflammation or anti‐tumor defense systems, respectively.
TGF‐β can also induce a pro‐angiogenic environment and stimulate tumor angiogenesis, which is critical for growth and dissemination of tumor cells. TGF‐β signaling stimulates expression of key angiogenic factors such as CTGF and vascular endothelial growth factor.( 47 , 48 ) In addition, TGF‐β can induce the expression, secretion, and activity of matrix metalloproteases 2 and 9, and down‐regulate expression of tissue inhibitor of metalloproteinase in tumor and endothelial cells.( 49 ) These metalloprotease activities result in enhancement of migratory and invasive properties of endothelial cells required for tumor angiogenesis. Conversely, TGF‐β inhibits angiogenesis under certain conditions. In pancreatic cancer and diffuse‐type gastric cancer, TGF‐β induces the production of thrombospondin‐1, a potent angiogenic inhibitor, and perturbations of TGF‐β signaling result in accelerated angiogenesis.( 50 , 51 , 52 )
As discussed above, TGF‐β can act as a tumor‐suppressive factor in some cellular contexts and a tumor‐promoting factor in others. This inherent diversity makes TGF‐β‐based therapeutic strategies complex. Moreover, the reasons for the remarkable plasticity of TGF‐β signaling have been poorly elucidated.
Mechanisms of Cellular Context‐Dependent Diversity of TGF‐β‐Induced Cell Responses
The cells comprising one human body, whether they are normal or abnormal, are all derived from a single cell. However, they behave differently in response to TGF‐β stimulation due to slight but crucial differences. Moreover, even in the same type of cell, the responses induced by TGF‐β differ depending on differences in environmental factors.( 11 ) How, then, does TGF‐β exhibit multiple “colors” in cellular context‐dependent fashion? Here, we discuss the mechanisms responsible for the chaotic diversity of TGF‐β signaling.
Signal cross‐talk. TGF‐β is able to induce certain cell responses in the presence of other types of signaling, but fails to induce such responses in the absence of such signaling. Additional signaling is thus required for some TGF‐β‐induced cell responses (Fig. 3a).
Figure 3.
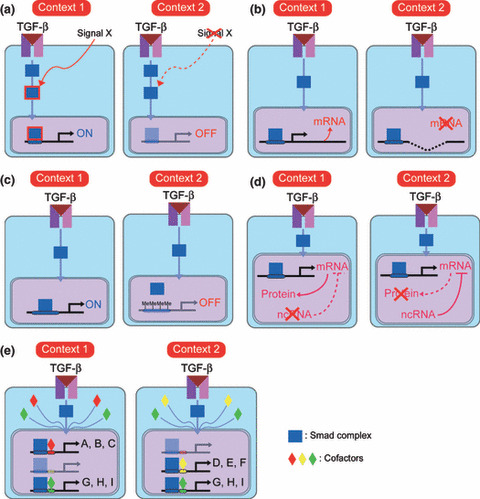
Several mechanisms of cellular context‐specific diversity of transforming growth factor‐β (TGF‐β)‐induced cell responses. (a) “Signal cross‐talk” model. In “context 1”, but not “context 2”, signal X is transduced in cells to modify downstream transducers of TGF‐β signaling and induce a certain “context 1”‐specific cell response. (b) “Genetic alterations” model. In “context 1”, expression of a certain target gene is induced by TGF‐β signaling. In “context 2”, the gene is deleted at the chromosomal level and TGF‐β stimulation fails to induce its expression. (c) “Epigenetics” model. In “context 1”, promoter regions of certain TGF‐β target genes adopt an “open conformation” and are exposed to the Smad complex. Conversely, in “context 2”, promoter regions of the same target genes adopt a “closed conformation” and the Smad complex fails to access the Smad‐binding elements. This difference results in differential responses to TGF‐β stimulation. (d) “Non‐coding RNA” model. In “context 2”, transcribed mRNAs of TGF‐β target genes are negatively regulated by non‐coding RNA (ncRNA). In “context 1”, such ncRNA is not expressed, resulting in translation of the mRNAs. (e) “Cofactors” model. Almost all TGF‐β target genes (A–I) are regulated by Smad proteins. Expression profiles of cofactors of Smad proteins differ between “context 1” and “context 2”, resulting in different responses to TGF‐β stimulation.
It is now generally accepted that TGF‐β is a tumor suppressor in the early phase of tumorigenesis, but can be converted to a tumor promoter during cancer progression.( 10 ) Adorno et al. showed that this switching of TGF‐β from tumor suppressor to promoter is due to additional mutation of p53.( 53 ) In the early stages of tumorigenesis, TGF‐β, acting as a tumor suppressor, inhibits proliferation of tumor cells in concert with wild‐type p53. In contrast, TGF‐β‐activated Smads and mutant p53 cooperatively abrogate the abilities of p63 to down‐regulate sharp‐1 and cyclin G2 expression and to suppress metastasis. As proof of this, gain of mutant p53 expression in non‐invasive tumor cells enhances the pro‐invasive and migratory effects of TGF‐β, whereas loss of mutant p53 expression in aggressive tumors impairs their metastatic potential.
Another key player in tumor progression, Ras, is also involved in the diversity of TGF‐β‐induced cell responses.( 54 ) Induction of Snail, a key regulator of EMT, by TGF‐β is strongly dependent on cooperation with active Ras signals, and silencing of Ras abolishes Snail induction by TGF‐β in pancreatic cancer Panc‐1 cells.( 55 ) In addition, transfection of constitutively active Ras leads to enhanced induction of Snail by TGF‐β, while induction of neither plasminogen activator inhibitor‐1 nor Smad7 is affected by Ras signaling. TGF‐β‐induced Snail is also regulated by glycogen synthase kinase (GSK)‐3β through two phosphorylation motifs within Snail, one for protein degradation and the other for subcellular localization.( 56 ) These findings indicate that the activity of GSK‐3β is one of the major factors determining whether TGF‐β can induce EMT in a certain environment, and that Wnt, PI3K/Akt, and MAP kinase signaling pathways might contribute to environment‐dependent diversity of TGF‐β‐induced cell responses.
In this fashion, TGF‐β‐induced cell responses can be determined by cooperatively acting signaling pathways, which are derived from tumor‐cell‐autonomous and/or tumor‐microenvironment interactions.
Genetic alterations. All cells except immune cells have nearly identical blueprints, or genomes, under physiological conditions. However, cancer cells have several genetic alterations yielding a survival advantage. Deletion or amplification of TGF‐β target genes in a certain cell change its responsiveness to TGF‐β stimulation (Fig. 3B). A subset of glioma cells sustain homozygous deletion of the p15Ink4b locus on chromosome 9p21.( 57 ) Loss of p15Ink4b weakens the tumor‐suppressive arm of the TGF‐β pathway, and cancer cells could benefit from tumor‐ and/or host‐derived TGF‐β ligand. Thus, genetic alterations of downstream genes contribute to cellular context‐specific plasticity of TGF‐β signaling and enhance either the tumor‐suppressive or tumor‐promoting arm of the TGF‐β pathways.
Epigenetics. Gene expression induced by TGF‐β stimulation is regulated by epigenetic mechanisms, including histone modulation and DNA methylation (Fig. 3C).
The precise organization of chromatin is critical for many cellular processes, including transcription, and dynamic changes in chromatin structure are directly influenced by post‐translational modifications of the amino‐terminal tails of the histones. Specific amino acids within these histone tails are targets for a number of post‐translational modifications, including acetylation, methylation, phosphorylation, ADP‐ribosylation, and ubiquitination.( 58 ) These covalent modifications alter the interaction of the histone tails with DNA or with chromatin‐associated proteins that might be required for different downstream transcription profiles and cellular processes. Histone status differs from one type of cell to another, and differences in histone status of promoters and/or enhancers of target genes could lead to different changes in TGF‐β‐mediated transcription profile, resulting in different TGF‐β‐induced cell responses. Switching of TGF‐β between tumor‐suppressive and tumor‐promoting roles might be due, in part, to alteration of histone status. In fact, modification of histones varies drastically during tumorigenesis, and the disruption of many chromatin‐modifying proteins is coincident with the formation of various malignant tumors.( 59 )
TGF‐β‐induced gene expression is also affected by DNA methylation status. TGF‐β induces PDGF‐B expression in glioblastoma U373MG cells, but fails to affect PDGF‐B expression in another glioblastoma cell line, U87MG cells.( 20 ) TGF‐β thus promotes proliferation of U373MG cells but inhibits that of U87MG cells. This phenomenon is explained by DNA methylation of Smad‐binding elements of PDGF‐B promoter. In addition, hypomethylation of PDGF‐B promoter is associated with poor prognosis in glioma patients. DNA methylation status in cells can thus determine whether a certain cell response is induced by TGF‐β or not.
Epigenetic regulation of downstream genes dictates the ability of TGF‐β to induce a certain response in a given cell, and contributes to the cell type‐specific cell responses induced by TGF‐β stimulation.
Non‐coding RNA. Even if TGF‐β stimulation activates promoter regions to the same degree in two different contexts, differences in post‐transcriptional regulation can lead to differences in levels of expression of proteins between the two, resulting in different cell responses to TGF‐β stimulation (Fig. 3d). One of the most investigated factors linked to post‐transcriptional regulation is non‐coding RNA.( 60 , 61 ) Recently, EMT, one of the critical cell responses of the tumor‐promoting arm of TGF‐β signaling, was reported to be regulated by the microRNA‐200 family (miR‐200a, miR‐200b, miR‐200c, miR‐141, and miR‐429).( 62 ) These microRNAs cooperatively regulate expression of the E‐cadherin transcriptional repressors δEF1/ZEB1 and SIP1/ZEB2, which are induced by TGF‐β and implicated in EMT. Manipulation of the miR‐200 family regulates EMT and induces the opposite reaction, mesenchymal–epithelial transition. As levels of expression of the miR‐200 family might differ from cell to cell, they at least in part determine at downstream gene levels whether TGF‐β induces EMT.
Another group of microRNAs, miR‐106b‐25 and miR‐17‐92 clusters, have been reported to affect the TGF‐β‐tumor suppressor pathway.( 63 , 64 ) The miR‐106b‐25 cluster is composed of the highly conserved miR‐106b, miR‐93, and miR‐25, which accumulate in different types of cancer, such as gastric cancer, neuroblastoma, and multiple myeloma. The miR‐17‐92 cluster contains miR‐17, miR‐18a, miR‐19a, miR‐20a, miR‐19b‐1, and miR‐92a‐1. Tumorigenic roles of the miR‐17‐92 cluster are suggested by its frequent amplification and overexpression in diffuse large B‐cell lymphoma and small cell lung carcinoma. Recent studies have uncovered functional implications of miR‐106b‐25 and miR‐17‐92 clusters in TGF‐β‐induced cell cycle arrest and apoptosis.( 63 , 64 ) In fact, these miRNAs silence two main downstream effectors playing central roles in these cell responses, the CDK inhibitor p21Waf1 and the pro‐apoptotic gene Bim. Furthermore, overexpression of miR‐106b and miR‐93 interferes with TGF‐β‐induced cell cycle arrest, and overexpression of miR‐25 prevents TGF‐β‐mediated apoptosis. These reports indicate that profiles of expression of miR‐106b‐25 and miR‐17‐92 clusters affect whether TGF‐β signaling can have tumor‐suppressive effects.
The profiles of expression of non‐coding RNA in cells vary depending on cellular contexts,( 65 ) as a result of which TGF‐β stimulation produces cellular context‐specific cell responses.
Cofactors. In the nucleus, activated Smad complexes cooperate with other DNA‐binding transcription factors to elicit specific transcriptional regulation, as the affinity of the activated Smad complex for the SBE is insufficient to support association with endogenous promoters of target genes except those with multiple SBE clusters. Numerous DNA‐binding transcription factors have been shown to interact with Smads.( 66 ) Various families of transcription factors, such as the forkhead, homeobox, zinc‐finger, AP1, Ets, and basic helix–loop–helix (HLH) families, serve as Smad partners.( 67 , 68 ) Each Smad–cofactor combination targets a particular set of genes, determined by the presence of cognate binding sequence element combinations in the regulatory regions of target genes. TGF‐β‐induced gene responses are thus classified by groups of genes that are jointly controlled by a given Smad–cofactor combination.( 14 ) Through this combinatorial interaction with different transcription factors, a common TGF‐β stimulus can activate or repress hundreds of target genes at once. A group of genes that are simultaneously regulated by a common Smad–cofactor complex is termed a “synexpression group.” Cells of different types or exposed to different conditions express different repertoires of Smad transcriptional partners, thus linking their TGF‐β responses to their cellular context (Fig. 3e). Such gene responses orchestrate the successful maintenance of homeostasis, and aberrant regulation of such responses might lead to various diseases, including malignant tumor.
Recently, it was shown that a novel negative regulator of TGF‐β signaling, human homolog of maternal Id‐like molecule (HHM), suppresses TGF‐β signaling in cell response‐specific fashion.( 69 ) HHM has a HLH domain, but lacks a basic DNA‐binding domain, making it structurally similar to Id transcriptional repressors.( 70 ) Among the several TGF‐β‐induced cell responses, cell cycle arrest is suppressed by HHM, but EMT is not. HHM interacts with DNA‐binding oligodendrocyte transcription factor 1 (Olig1), a newly identified Smad‐binding cofactor, and interferes with the interaction between Olig1 and Smad proteins. Olig1 and R‐Smads interact with each other on chromosomes and yield synergistic upregulation of expression of TGF‐β target genes whose promoter regions include Olig1‐binding sequence(s) and Smad‐binding sequence(s) in close vicinity. HHM abrogates the interaction between Olig1 and the activated Smad complex, and as a consequence HHM inhibits the transcription induced by the Olig1–Smad complex at the transcriptional level. As HHM interacts with some but not all Smad‐binding transcription factors, HHM abrogates only a subset of Smad–cofactor complexes, including the Olig1–Smad complex. HHM thus inhibits cell responses that are regulated by Smad–cofactor synexpression groups targeted by HHM, but fails to affect cell responses that are controlled by Smad–cofactor synexpression groups not targeted by HHM. We noted that the targets of HHM are not limited to the Olig1–Smad synexpression group, as there were some TGF‐β target genes regulated by HHM but not included in the Olig1–Smad synexpression group on microarray analysis.( 69 )
As HHM targets multiple synexpression groups, it is still inappropriate for “specific” regulation of a given cell response. However, the paradigmatic mechanism of inhibition of HHM, inhibition of “restricted” Smad–cofactor synexpression groups through interaction with Smad‐binding transcription factors, can be used. A low molecular weight compound that interacts with only one Smad‐binding transcription factor would abrogate activity of only one Smad–cofactor synexpression group, resulting in specific inhibition of a certain cell response (Fig. 4).
Figure 4.
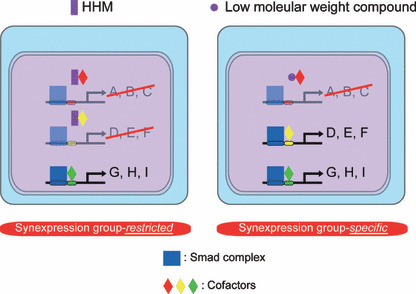
Development of a synexpression‐specific inhibitor of transforming growth factor‐β (TGF‐β) signaling. One of many TGF‐β‐induced cellular responses, human homolog of maternal Id‐like molecule (HHM) inhibits a subset of synexpression groups. An HHM‐like low molecular weight compound, which abrogates only one type of Smad–cofactor complex, inhibits only one TGF‐β‐induced cellular response.
Conclusion
Numerous mechanisms play roles in the cellular context‐dependent diversity of TGF‐β signaling, so we cannot thoroughly explain the overall design of multiple “colors” of TGF‐β signaling based only on the mechanisms discussed here. Moreover, several “gearwheels” synchronously regulate any given cell response induced by TGF‐β stimulation. However, these mechanisms will provide methods for specifically regulating only one (or some) of the cell responses induced by TGF‐β signaling. They will give us an answer to the inherent complexity of TGF‐β‐based therapeutic strategies and open the way to specific control of the tumor‐suppressive or tumor‐promoting arm of the TGF‐β pathways. For this purpose, we should keep trying to build the rationale for regulating certain cell responses and to develop methods for manipulating specific modules of TGF‐β signaling.
Acknowledgments
This work was supported by KAKENHI (Grant‐in‐Aid for Scientific Research) on Priority Areas “New strategies for cancer therapy based on advancement of basic research” and the Global COE Program (Integrative Life Science Based on the Study of Biosignaling Mechanisms) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan. H.I. has been supported by a Tetsumon scholarship for the Ph.D.‐M.D. program of the University of Tokyo and by a research fellowship of the Japan Society for the Promotion of Science for Young Scientists.
References
- 1. Levy K, Hill CS. Alteration in components of the TGF‐β superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev 2006; 17: 41–58. [DOI] [PubMed] [Google Scholar]
- 2. Markowitz S, Wang J, Myeroff L et al. Inactivation of the type II TGF‐β receptor in colon cancer cells with microsatellite instability. Science 1995; 268: 1336–8. [DOI] [PubMed] [Google Scholar]
- 3. Hahn SA, Seymour AB, Hoque AT et al. Allelotype of pancreatic adenocarcinoma using xenograft enrichment. Cancer Res 1995; 55: 4670–5. [PubMed] [Google Scholar]
- 4. Tsushima H, Kawata S, Tamura S et al. High levels of transforming growth factor β1 in patients with colorectal cancer: association with disease progression. Gastroenterology 1996; 110: 375–82. [DOI] [PubMed] [Google Scholar]
- 5. Wikström P, Stattin P, Franck‐Lissbrant I, Damber JE, Bergh A. Transforming growth factor β1 is associated with angiogenesis, metastasis, and poor clinical outcome in prostate cancer. Prostate 1998; 37: 19–29. [DOI] [PubMed] [Google Scholar]
- 6. Walker RA, Dearing SJ. Transforming growth factor β1 in ductal carcinoma in situ and invasive carcinomas of the breast. Eur J Cancer 1992; 28: 641–4. [DOI] [PubMed] [Google Scholar]
- 7. Friedman E, Gold LI, Klimstra D, Zeng ZS, Winawer S, Cohen A. High levels of transforming growth factor β1 correlate with disease progression in human colon cancer. Cancer Epidemiol Biomarkers Prev 1995; 4: 549–54. [PubMed] [Google Scholar]
- 8. Dalal BI, Keown PA, Greenberg AH. Immunocytochemical localization of secreted transforming growth factor‐β1 to the advancing edges of primary tumors and to lymph node metastases of human mammary carcinoma. Am J Pathol 1993; 143: 381–9. [PMC free article] [PubMed] [Google Scholar]
- 9. Picon A, Gold LI, Wang J, Cohen A, Friedman E. A subset of metastatic human colon cancers expresses elevated levels of transforming growth factor β1. Cancer Epidemiol Biomarkers Prev 1998; 7: 497–504. [PubMed] [Google Scholar]
- 10. Roberts AB, Wakefield LM. The two faces of transforming growth factor β in carcinogenesis. Proc Natl Acad Sci USA 2003; 100: 8621–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bierie B, Moses HL. Tumour microenvironment: TGFβ: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer 2006; 6: 506–20. [DOI] [PubMed] [Google Scholar]
- 12. Miyazawa K, Shinozaki M, Hara T, Furuya T, Miyazono K. Two major Smad pathways in TGF‐β superfamily signalling. Genes Cells 2002; 7: 1191–204. [DOI] [PubMed] [Google Scholar]
- 13. Feng XH, Derynck R. Specificity and versatility in TGF‐β signaling through Smads. Annu Rev Cell Dev Biol 2005; 21: 659–93. [DOI] [PubMed] [Google Scholar]
- 14. Massagué J. TGFβ in cancer. Cell 2008; 134: 215–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Derynck R, Zhang YE. Smad‐dependent and Smad‐independent pathways in TGF‐β family signalling. Nature 2003; 425: 577–584. [DOI] [PubMed] [Google Scholar]
- 16. Datto MB, Li Y, Panus JF, Howe DJ, Xiong Y, Wang XF. Transforming growth factor β induces the cyclin‐dependent kinase inhibitor p21 through p53‐independent mechanism. Proc Natl Acad Sci USA 1995; 92: 5545–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hannon GJ, Beach D. p15INK4B is a potential effector of TGF‐β‐induced cell cycle arrest. Nature 1994; 371: 257–61. [DOI] [PubMed] [Google Scholar]
- 18. Yagi K, Furuhashi M, Aoki H et al. c‐myc is a downstream target of the Smad pathway. J Biol Chem 2002; 277: 854–61. [DOI] [PubMed] [Google Scholar]
- 19. Miyazono K, Miyazawa K. Id: a target of BMP signaling. Science’s STKE 2002; E40. [DOI] [PubMed] [Google Scholar]
- 20. Bruna A, Darken RS, Rojo F et al. High TGFβ‐Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF‐B gene. Cancer Cell 2007; 11: 147–60. [DOI] [PubMed] [Google Scholar]
- 21. Jennings MT, Pietenpol JA. The role of transforming growth factor β in glioma progression. J Neurooncol 1998; 36: 123–40. [DOI] [PubMed] [Google Scholar]
- 22. Matsuyama S, Iwadate M, Kondo M et al. SB‐431542 and Gleevec inhibit transforming growth factor‐β‐induced proliferation of human osteosarcoma cells. Cancer Res 2003; 63: 7791–8. [PubMed] [Google Scholar]
- 23. Jang CW, Chen CH, Chen CC, Chen JY, Su YH, Chen RH. TGF‐β induces apoptosis through Smad‐mediated expression of DAP‐kinase. Nat Cell Biol 2002; 4: 51–8. [DOI] [PubMed] [Google Scholar]
- 24. Takekawa M, Tatebayashi K, Itoh F, Adachi M, Imai K, Saito H. Smad‐dependent GADD45β expression mediates delayed activation of p38 MAP kinase by TGF‐β. EMBO J 2002; 21: 6473–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yoo J, Ghiassi M, Jirmanova L et al. Transforming growth factor‐β‐induced apoptosis is mediated by Smad‐dependent expression of GADD45β through p38 activation. J Biol Chem 2003; 278: 43001–7. [DOI] [PubMed] [Google Scholar]
- 26. Ohgushi M, Kuroki S, Fukamachi H et al. Transforming growth factor β‐dependent sequential activation of Smad, Bim, and caspase‐9 mediates physiological apoptosis in gastric epithelial cells. Mol Cell Biol 2005; 25: 10017–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ehata S, Hanyu A, Hayashi M et al. Transforming growth factor‐β promotes survival of mammary carcinoma cells through induction of antiapoptotic transcription factor DEC1. Cancer Res 2007; 67: 9694–703. [DOI] [PubMed] [Google Scholar]
- 28. Moustakas A, Heldin CH. Signaling networks guiding epithelial‐mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci 2007; 98: 1512–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xu J, Lamouille S, Derynck R. TGF‐β‐induced epithelial to mesenchymal transition. Cell Res 2009; 19: 156–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ehata S, Hanyu A, Fujime M et al. Ki26894, a novel transforming growth factor‐β type I receptor kinase inhibitor, inhibits in vitro invasion and in vivo bone metastasis of a human breast cancer cell line. Cancer Sci 2007; 98: 127–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guise TA, Yin JJ, Taylor SD et al. Evidence for a causal role of parathyroid hormone‐related protein in the pathogenesis of human breast cancer‐mediated osteolysis. J Clin Invest 1996; 98: 1544–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yin JJ, Selander K, Chirgwin JM et al. TGF‐β signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest 1999; 103: 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kondo H, Guo J, Bringhurst FR. Cyclic adenosine monophosphate/protein kinase A mediates parathyroid hormone/parathyroid hormone‐related protein receptor regulation of osteoclastogenesis and expression of RANKL and osteoprotegerin mRNAs by marrow stromal cells. J Bone Miner Res 2002; 17: 1667–79. [DOI] [PubMed] [Google Scholar]
- 34. Kingsley LA, Fournier PG, Chirgwin JM, Guise TA. Molecular biology of bone metastasis. Mol Cancer Ther 2007; 6: 2609–17. [DOI] [PubMed] [Google Scholar]
- 35. Sims AH, Howell A, Howell SJ, Clarke RB. Origins of breast cancer subtypes and therapeutic implications. Nat Clin Pract Oncol 2007; 4: 516–25. [DOI] [PubMed] [Google Scholar]
- 36. Dietrich J, Imitola J, Kesari S. Mechanisms of disease: the role of stem cells in the biology and treatment of gliomas. Nat Clin Pract Oncol 2008; 5: 393–404. [DOI] [PubMed] [Google Scholar]
- 37. Iwasaki H, Suda T. Cancer stem cells and their niche. Cancer Sci 2009; 100: 1166–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Watabe T, Miyazono K. Roles of TGF‐β family signaling in stem cell renewal and differentiation. Cell Res 2009; 19: 103–15. [DOI] [PubMed] [Google Scholar]
- 39. Mani SA, Guo W, Liao MJ et al. The epithelial‐mesenchymal transition generates cells with properties of stem cells. Cell 2008; 133: 704–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ikushima H, Todo T, Ino Y, Takahashi M, Miyazawa K, Miyazono K. Autocrine TGF‐β signaling maintains tumorigenicity of glioma‐initiating cells through Sry‐related HMG‐box factors. Cell Stem Cell 2009; 5: 504–14. [DOI] [PubMed] [Google Scholar]
- 41. Peñuelas S, Anido J, Prieto‐Sánchez RM et al. TGF‐β increases glioma‐initiating cell self‐renewal through the induction of LIF in human glioblastoma. Cancer Cell 2009; 15: 315–27. [DOI] [PubMed] [Google Scholar]
- 42. Bhowmick NA, Chytil A, Plieth D et al. TGF‐β signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 2004; 303: 848–51. [DOI] [PubMed] [Google Scholar]
- 43. Cheng N, Bhowmick NA, Chytil A et al. Loss of TGF‐β type II receptor in fibroblasts promotes mammary carcinoma growth and invasion through upregulation of TGF‐α‐, MSP‐ and HGF‐mediated signaling networks. Oncogene 2005; 24: 5053–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Thomas DA, Massagué J. TGF‐β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005; 8: 369–80. [DOI] [PubMed] [Google Scholar]
- 45. Friese MA, Wischhusen J, Wick W et al. RNA interference targeting transforming growth factor‐β enhances NKG2D‐mediated antiglioma immune response, inhibits glioma cell migration and invasiveness, and abrogates tumorigenicity in vivo . Cancer Res 2004; 64: 7596–603. [DOI] [PubMed] [Google Scholar]
- 46. Li MO, Flavell RA. TGF‐β: a master of all T cell trades. Cell 2008; 134: 392–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kang Y, Siegel PM, Shu W et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003; 3: 537–49. [DOI] [PubMed] [Google Scholar]
- 48. Sánchez‐Elsner T, Botella LM, Velasco B, Corbí A, Attisano L, Bernabéu C. Synergistic cooperation between hypoxia and transforming growth factor‐β pathways on human vascular endothelial growth factor gene expression. J Biol Chem 2001; 276: 38527–35. [DOI] [PubMed] [Google Scholar]
- 49. Derynck R, Akhurst RJ, Balmain A. TGF‐β signaling in tumor suppression and cancer progression. Nat Genet 2001; 29: 117–29. [DOI] [PubMed] [Google Scholar]
- 50. Schwarte‐Waldhoff I, Volpert OV, Bouck NP et al. Smad4/DPC4‐mediated tumor suppression through suppression of angiogenesis. Proc Natl Acad Sci USA 2000; 97: 9624–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Komuro A, Yashiro M, Iwata C et al. Diffuse‐type gastric carcinoma: progression, angiogenesis, and transforming growth factor β signaling. J Natl Cancer Inst 2009; 101: 592–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kiyono K, Suzuki HI, Morishita Y et al. c‐Ski overexpression promotes tumor growth and angiogenesis through inhibition of transforming growth factor‐β signaling in diffuse‐type gastric carcinoma. Cancer Sci 2009; 100: 1809–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Adorno M, Cordenonsi M, Montagner M et al. A mutant‐p53/Smad complex opposes p63 to empower TGFβ‐induced metastasis. Cell 2009; 137: 87–98. [DOI] [PubMed] [Google Scholar]
- 54. Oft M, Peli J, Rudaz C, Schwarz H, Beug H, Reichmann E. TGF‐β1 and Ha‐Ras collaborate in modulating the phenotypic plasticity and invasiveness of epithelial tumor cells. Genes Dev 1996; 10: 2462–77. [DOI] [PubMed] [Google Scholar]
- 55. Horiguchi K, Shirakihara T, Nakano A, Imamura T, Miyazono K, Saitoh M. Role of Ras signaling in the induction of snail by transforming growth factor‐β. J Biol Chem 2009; 284: 245–53. [DOI] [PubMed] [Google Scholar]
- 56. Zhou BP, Deng J, Xia W et al. Dual regulation of Snail by GSK‐3β‐mediated phosphorylation in control of epithelial‐mesenchymal transition. Nat Cell Biol 2004; 6: 931–40. [DOI] [PubMed] [Google Scholar]
- 57. Jen J, Harper JW, Bigner SH et al. Deletion of p16 and p15 genes in brain tumors. Cancer Res 1994; 54: 6353–8. [PubMed] [Google Scholar]
- 58. Rice JC, Allis CD. Histone methylation versus histone acetylation: new insights into epigenetic regulation. Curr Opin Cell Biol 2001; 13: 263–73. [DOI] [PubMed] [Google Scholar]
- 59. Esteller M. Cancer epigenomics: DNA methylomes and histone‐modification maps. Nat Rev Genet 2007; 8: 286–98. [DOI] [PubMed] [Google Scholar]
- 60. Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post‐transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet 2008; 9: 102–14. [DOI] [PubMed] [Google Scholar]
- 61. Winter J, Jung S, Keller S, Gregory RI, Diederichs S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol 2009; 11: 228–34. [DOI] [PubMed] [Google Scholar]
- 62. Gregory PA, Bert AG, Paterson EL et al. The miR‐200 family and miR‐205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 2008; 10: 593–601. [DOI] [PubMed] [Google Scholar]
- 63. Petrocca F, Visone R, Onelli MR et al. E2F1‐regulated microRNAs impair TGFβ‐dependent cell‐cycle arrest and apoptosis in gastric cancer. Cancer Cell 2008; 13: 272–86. [DOI] [PubMed] [Google Scholar]
- 64. Ventura A, Young AG, Winslow MM et al. Targeted deletion reveals essential and overlapping functions of the miR‐17 through 92 family of miRNA clusters. Cell 2008; 132: 875–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lu J, Getz G, Miska EA et al. MicroRNA expression profiles classify human cancers. Nature 2005; 435: 834–38. [DOI] [PubMed] [Google Scholar]
- 66. Lin X, Chen YG, Feng XH. The TGF‐β Family. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, 2008; Chapter 10. [Google Scholar]
- 67. Koinuma D, Tsutsumi S, Kamimura N et al. Chromatin immunoprecipitation on microarray analysis of Smad2/3 binding sites reveals roles of ETS1 and TFAP2A in transforming growth factor β signaling. Mol Cell Biol 2009; 29: 172–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Koinuma D, Tsutsumi S, Kamimura N, Imamura T, Aburatani H, Miyazono K. A promoter‐wide analysis of Smad4 binding sites in human epithelial cells. Cancer Sci 2009; 100: 2133–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ikushima H, Komuro A, Isogaya K et al. An Id‐like molecule, HHM, is a synexpression group‐restricted regulator of TGF‐β signalling. EMBO J 2008; 27: 2955–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Seto A, Ikushima H, Suzuki T et al. Crystallization and preliminary X‐ray diffraction analysis of GCIP/HHM transcriptional regulator. Acta Crystallogr Sect F Struct Biol Cryst Commun 2009; 65: 21–4. [DOI] [PMC free article] [PubMed] [Google Scholar]