

Abstract
Lung cancers have become the leading cause of cancer deaths in Japan, claiming more than 55 000 lives annually. Unfortunately, substantial improvement in terms of cure rates has not been achieved over the last two decades, although during the same period of time in‐depth basic knowledge of the molecular mechanisms, which underlies carcinogenesis and progression of this deadly group of neoplasms, has accumulated at an amazing pace. It has consequently become evident that they have many shared but also distinct features, when comparisons are made not only with other common epithelial cancers of adults, such as colon cancer, but also within the various histologic types of lung cancers themselves. This review article provides an up‐date on cutting‐edge research into the following three different topics, from which important new insights have been obtained. The first concerns genetic instability, especially chromosome instability, and checkpoint failure in lung cancers. Second, we deal with EGFR mutations, which shows revealing specificities in various aspects. Finally, advances in the expression profiling analysis of both transcriptomes and proteomes of lung cancers are summarized. (Cancer Sci 2005; 96: 63–68)
There are over one million deaths a year worldwide due to lung cancer, making it one of the most prevalent and deadly neoplasms. There is no doubt that smoking of tobacco is the most important causative factor in its development, the disease in fact apparently being rare before the widespread use of tobacco. Japan has been experiencing a steep increase in lung cancer cases, following the footsteps of the USA and other western countries, such as the UK, where increase in tobacco consumption was subsequently followed by an abrupt rise in lung cancer occurrence. Our current problem can be regarded as a ringing alarm bell for other Asian nations, such as China, in which tobacco consumption is now rapidly increasing. Lung cancer currently claims more than 55 000 lives annually in Japan, with a no more than 15% cure rate, meaning that the number of deaths remains unacceptably high. A better understanding of the molecular pathogenesis of this fatal disease is therefore an urgent issue in order to develop better diagnostic approaches and new targeted therapies, as well as effective means for its prevention.
Lung cancers can be classified into two major entities, based on their clinicopathologic characteristics, that is, small cell lung cancer (SCLC) and non‐small cell lung cancer (NSCLC), the latter being further divided into three major histologic types, the adenocarcinoma, squamous cell carcinoma and large cell carcinoma. In the development of squamous carcinoma, oncogenic triggering converts normal bronchial epithelium into hyperplastic, metaplastic and dysplastic lesions, leading to the subsequent emergence of carcinoma in situ (CIS) and then overt squamous cell carcinomas. Adenocarcinomas are also considered to develop, at least in part, through stepwise morphological changes, with atypical adenomatous hyperplasia (AAH) generally thought to be a premalignant precursor lesion. It has been clearly demonstrated that overt cancers carry multiple genetic and epigenetic alterations occurring during initiation of carcinogenesis and subsequent progression. Since cell‐cycle regulation and checkpoint functions are crucial for maintaining genomic integrity, their abrogation is thought to contribute to genomic instability, playing important roles even in the early steps of cancer development. (1) Many of the tumor suppressor genes and oncogenes altered in lung cancer are known to contribute to the regulation of cell cycle progression in either a direct or an indirect manner, and a considerable proportion of the lung cancer‐related gene products are components of vital checkpoint mechanisms. It is notable that exposure to carcinogens in tobacco smoke appears to leave fingerprints of the resulting insult to the genome, for example, as distinct mutational spectra of p53 and KRAS.
In this review article on the molecular pathogenesis of lung cancers, we will confine ourselves to concisely summarizing recent advances in three different topics, rather than attempting a comprehensive coverage of all the related subjects (for this purpose, see other review articles by the authors 2 , 3 , 4 ). The three topics dealt in this article are: (i) chromosome instability and alterations of cell cycle checkpoints; (ii) the clinicopathologic impact of recent identification of frequent EGFR mutations in a specific type of adenocarcinomas; and (iii) accumulating new insights obtained through expression profiling analysis with both genomic and proteomic approaches.
Chromosomal instability and alterations of cell cycle checkpoints
Non‐random chromosomal deletion and loss of heterozygosity (LOH) are hallmarks for the involvement of tumor suppressor genes residing in the affected chromosomal regions, while oncogene amplification can be identified by cytogenetic findings, such as homogenously staining lesions and double minutes. In contrast to certain leukemias, lymphomas and sarcomas, specific balanced translocations are not present in lung cancers, whose karyotypes are frequently very complex. (5) Loss of chromosomal arm 3p is among the first non‐random genomic aberrations identified in lung cancer. In addition to this common loss of 3p, chromosomal losses are also frequent on 4q, 5q, 8p, 10q, 13q and 17p in SCLC, and 8p, 9p, 13q, 17p in NSCLC. Chromosomal gains are frequent on 3q, 5p and 8q in SCLC, and 1q, 3q, 5p and 8q in NSCLC. Allelic losses and amplifications of multiple small chromosomal regions have also been documented through detailed LOH and comparative genomic hybridization (CGH) analyses. It is expected that systematic array CGH analysis will allow further refinement of the affected regions, leading to the identification of candidate genes for the responsible targets.
The frequent presence of complex chromosomal changes suggests significant roles in the development of lung cancer, and it is well known that both numerical and structural aberrations may occur. Failure in cell cycle checkpoint control and defects in the DNA double‐strand break repair system are thought to play important roles in the development of chromosome instability (CIN). We have shown that persistent numerical CIN is present in human lung cancer cell lines with a notable association with aneuploid karyotypes, and demonstrated that aneuploidy is not the result of a few past catastrophic processes of missegregations, but rather a reflection of the persistence of an unstable karyotypic state. (6) This feature appears to be common in epithelial cancers in adults, which often show complex aneuploidy. As for the underlying mechanisms, it is notable that mice, deficient in the Mad2 mitotic checkpoint gene, which is important for correct chromosome segregation, have been shown to specifically develop lung tumors characterized by CIN. (7) In human lung cancers, mitotic checkpoint impairment appears to be present at a considerably high frequency, (8) while mutations in the MAD1 and BUB1 mitotic checkpoint genes have been detected in human lung cancer cells, albeit relatively rarely. 9 , 10 , 11 In addition, p53 inactivation appears to play an indirect role in the acquisition of the CIN phenotype. (6)
Double strand breaks (DSB) can be introduced into genome by intracellular stresses, such as oxidative damage, as well as by environmental factors, such as ionizing irradiation, and this highly detrimental form of DNA damage potentially serves as causes of erroneous rejoining of the broken genome. Cell‐cycle checkpoints are built‐in safeguards to prevent damaged cells from starting DNA replication (the G1/S checkpoint), from progressing with replication (the intra S checkpoint), or from going into mitosis (the G2 checkpoint). (12) Lung cancers are known to carry frequent G1/S checkpoint abrogation due to p53 mutations, the most frequent genetic alterations found so far in this common epithelial cancer of adults, (13) and it has been shown that the G2 checkpoint is also frequently impaired in SCLC in a histological type‐specific manner. (14) Failure of G2 checkpoint activation in the presence of DNA damage would generate broken chromosomes, the fates of which include: degradation, healing as truncated forms, and incorrect fusion to generate translocated chromosomes. Fusion between two centromere‐containing chromosomes can trigger the highly unstable breakage‐fusion‐bridge cycle. Multiple components of the DNA damage G2 checkpoint mechanism appear to be involved in the development of lung cancers. CHK1 and CHK2 are preferentially activated by their respective upstream kinases, ATR and ATM, leading to phosphorylation of Cdc25C, a phosphatase that normally activates Cdc2, and resulting in its sequestration and inactivation of Cdc25C by 14‐3‐3 proteins. It is interesting that 14‐3‐3ɛ was recently shown to be involved in a homozygous deletion identified in a SCLC cell line, and that reconstitution of 14‐3‐3ɛ partially restored its G2 checkpoint impairment. (14) Another member of the 14‐3‐3 family, 14‐3‐3σ, which is transactivated by p53 in response to DNA damage, has been shown to contribute to the maintenance of G2 arrest through nuclear exclusion of the Cdc2/cyclin B1. (15) Notably, epigenetic inactivation of 14‐3‐3σ, due to aberrant DNA hypermethylation of its promoter region, has been shown to be frequent in SCLC 16 , 17 as well as in a few other types of human cancers, including, breast, gastric and hepatocellular carcinomas. We have also found that CHK2 is somatically mutated in lung cancers at a low frequency. 18 , 19 It remains to be investigated whether there may be an, as yet, unidentified major target gene responsible for the observed checkpoint defects, or whether there might be a large number of affected genes each playing a role in a small proportion of cases.
In addition, our recent work by Nakagawa et al. provided direct evidence that decatenation at the G2 checkpoint, (20) which ensures sufficient chromatid decatenation by topoisomerase II before entering into mitosis, (21) is impaired in a proportion of human lung cancer cell lines. Therefore, the G2 checkpoint may also be important. This is clinically relevant, with considerable interest in the potential association between decatenation impairment and hypersensitivity to catalytic topoisomerase inhibitors, such as ICRF‐193, (20) since this could ultimately lead to the development of an attractive strategy for lung cancer treatment, that is, selective killing of targeted cancer cells without causing major toxicity in normal cells. The CHFR gene, which has been postulated to play a key role in the prophase checkpoint, is also known to be inactivated by DNA hypermethylation in lung cancers. (22) The findings so far obtained, clearly indicate that multiple cell cycle checkpoints are impaired in lung cancers (Fig. 1), conceivably providing a driving force to acquisition of genetic instability, including CIN, and therefore contributing to the development of lung cancers.
Figure 1.
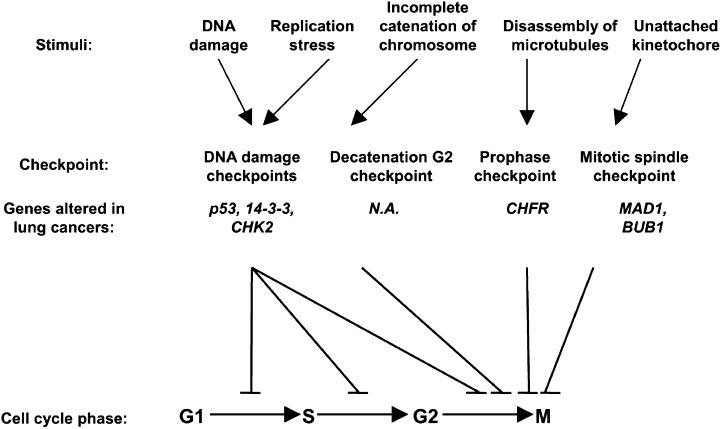
Cell cycle checkpoints perturbed in human lung cancers. N.A., not available.
Specific Involvement of EGFR and ras Mutations in Lung Adenocarcinomas
In the 1980s, various oncogenes were shown to be altered in lung cancers, and the myc and ras gene families are among the best studied in relation to their pathogenesis. (23) Gene amplification of one of the members of the myc family is detectable in about 25% of SCLC cell lines as well as in 5–15% of primary tumor specimens, while overexpression of one of the members is detectable in virtually all SCLC. Therefore, the vast majority of these lesions appear to have both p53 mutations and MYC overexpression, consistent with the notion that inactivation of the ARF/MDM2/p53 pathway, which is nearly ubiquitous in SCLC, may be required for Myc‐induced transformation. By contrast, K‐ras mutations are detected almost exclusively in adenocarcinomas, usually at codon 12, in association with a poor prognosis of surgically treated cases, (24) whereas H‐ras or N‐ras are only very rarely mutated in any type of lung cancer. K‐ras mutations are predominantly G to T transversions, as is also the case for p53 mutations, implying their creation through DNA adduct formation from tobacco exposure. In addition, mutations in the BRAF gene are present in approximately 3% of adenocarcinomas without K‐ras mutations, 25 , 26 consistent with K‐ras and BRAF ultimately signaling through the same pathway.
The discovery of mutations of the two prototype tumor suppressor genes, namely Rb and p53, in 1989, 27 , 28 shifted much of the attention of investigators in this field, including ourselves, to the involvement of tumor suppressor inactivation, with development of candidate gene approaches and positional and functional cloning efforts. However, recent reports on EGFR mutations by two Boston groups have generated considerable interest because of the highly characteristic and clinically useful nature of this genetic change. 29 , 30 Therefore, the field of oncogene activation is being revisited in relation to lung carcinogenesis. EGFR mutations have been found to be more frequent in cases with an adenocarcinoma histology and female gender, and in Japanese patients (in comparison with American patients), and the presence of EGFR mutations was shown to have a potential predictive value for sensitivity to gefitinib (Iressa, ZD1839), a small molecule tyrosine kinase inhibitor that targets EGFR. Interestingly, Paez et al. observed a marked predominance of EGFR mutations in 26% (15 of 58) of specimens from Japanese patients, compared to only 2% (1 of 61) in a series from American patients. (30) In addition, a group at the Memorial Sloan‐Kettering Cancer Center has recently confirmed the utility of detecting EGFR mutations as a marker for predicting responsiveness to gefitinib. (31) We have extended their findings by analyzing 277 Japanese NSCLC patients, and found EGFR mutations to be present exclusively in adenocarcinomas, with a single exception (an adenosquamous carcinoma case), at a frequency of 59% and 26% in female and male cases, respectively. (32) The vast majority of the mutations proved to be either deletions around codons 746–750 or a missense mutation substituting leucine with arginine at codon 858, generally affecting three functionally important structures (αC helix, activation loop, P‐loop) flanking the ATP binding pocket of the tyrosine kinase domain (Fig. 2). In this connection, it is of interest that EGFR activates several downstream substrates in addition to the RAS‐MAPK pathway, such as the PI3K/AKT and JAK/STAT pathways. PIK3CA, which encodes the p110α catalytic subunit of PI3K, is mutated in lung cancers, (33) though at low frequency, and a recent study by Sordella et al. showed that mutated‐EGFR targets the PI3K/AKT and JAK/STAT pathways rather than the RAS‐MAPK pathway. (34) The findings of a recent immunohistochemical investigation carried out by Cappuzzo et al. are consistent with this notion, that is, gefitinib‐treated patients with P‐Akt‐positive tumors had a better response, disease control, and time to progression than those with P‐Akt‐negative tumors. (35) In addition, we have shown, for the first time, that patients with EGFR mutations survive for a longer period than those without EGFR mutations when treated with gefitinib for recurrent disease after surgery. (36) Although these findings must be confirmed in a prospective clinical trial, screening for EGFR mutations appears to be a promising means to target drugs to specific molecules. This may consequently become a common practice for treating pulmonary adenocarcinoma patients in the near future, making individualized therapy a reality.
Figure 2.
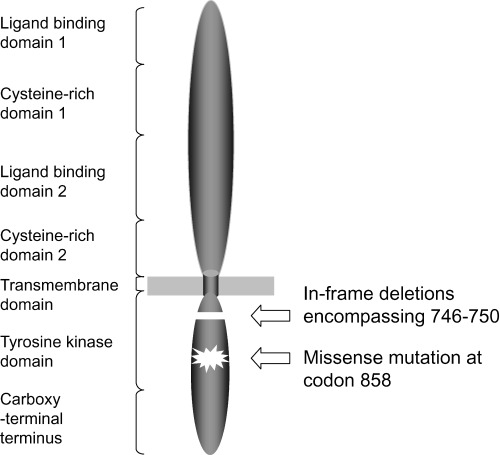
Schematic diagram of the structure of EGFR as well as of the locations of two major types of mutations.
Notably, our multivariate logistic regression analysis demonstrated a non‐smoking status and adenocarcinoma histology, but not female gender, to independently contribute to the occurrence of EGFR mutations, suggesting that the apparent difference in frequency observed between female and male may result from differences in lifestyle, including smoking habit, rather than involvement of the sex hormone‐related environment, at least in the Japanese population. This is in a striking contrast to other genetic and epigenetic changes previously described in lung cancers. For instance, mutations in the p53 and K‐ras genes are known to be more frequent in smokers than in non‐smokers. 37 , 38 Alterations of the FHIT gene encompassing a fragile site at 3p14.2, a chromosomal region frequently affected in lung cancers, are also more prevalent in smokers than in non‐smokers. (39) The difference in the underlying mechanisms between EGFR mutations and the other genetic and epigenetic alterations reported previously, is also consistent with the occurrence of mutations in EGFR and K‐ras in a mutually exclusive manner, as well as with the fact that K‐ras mutations in adenocarcinomas without EGFR mutations are mostly G to T transversions, which may reflect exposure to carcinogens in tobacco smoke. (32) It is also worthy of mention that Yatabe et al. have found that even within adenocarcinomas, EGFR mutations appear to occur in a distinct subset, that is, terminal respiratory unit‐type adenocarcinomas, which is a specific type of lesion conceivably arising from peripheral lung epithelial cells, including alveolar cells and non‐ciliated bronchiolar epithelium. 40 , 41 It should be stressed that EGFR mutations appear to be extremely specific to lung cancers, mutations being reported to be present in none of 95 primary tumors and 108 cancer‐derived cell lines, of diverse tumor types. (29) At present, it is not clear why the occurrence of EGFR mutations shows such specificity in terms of the smoking status and histology. In our opinion, there may be at least two possible explanations, which are not necessarily mutually exclusive. First, as yet unidentified carcinogens other than those in tobacco smoke might be involved in the development of adenocarcinomas in non‐smoking individuals through the imposition of EGFR mutations in their genome. Cooking oil fumes or HPV 16/18 infection, both of which reportedly have associations with lung cancer occurring in non‐smoking women in Taiwan, might be relevant. Alternatively, EGFR mutations might provide selective advantage only in peripheral lung epithelial cells. In this regard, it is interesting that EGF stimulates anchorage‐independent growth of our HPL1D cell line, the only human peripheral lung epithelial line so far established. (42) These intriguing points await future clarification.
Genomic and Proteomic Profiling of Lung Cancers for Better Understanding and Future Applications
The recent rapid progress in microarray technology has made it possible to analyze gene expression profiles on a genome‐wide basis in order to better understand molecular pathogenesis of human cancers, as well as to search for molecular markers for classification and prediction of outcome (Fig. 3). While lung cancers are known to be very heterogeneous in various aspects, recent expression profiling studies have clearly shown the presence of several distinct subclasses. 43 , 44 , 45 , 46 , 47 , 48 SCLC express a set of genes which are related to neuroendocrine differentiation, including ASH1, a key transcription factor that is indispensable for the development of neuroendocrine cells of the lung. Squamous cell carcinomas show marked elevation of a group of keratin isoforms related to squamous differentiation, as well as of p63, a p53 homolog that is believed to play a role in squamous cell differentiation. Although these results are not unexpected, considering the characteristics evident even on routine pathology examination, it is certainly interesting that expression‐profiling analysis has proven sufficiently powerful to detect the presence of distinct expression profiles, even within a subgroup of tumors with adenocarcinoma histology. Garber et al. identified three subgroups in adenocarcinomas through unsupervised hierarchical clustering analysis, (43) while Bhattacharjee et al. made a subclassification into four subgroups. (44) We have also reported the identification of four distinct subsets of adenocarcinomas based on unsupervised hierarchical clustering analysis. (48) Similarly, Virtanen et al. observed adenocarcinomas to form three distinct clusters. (46) Although the expression signatures identified in these studies do not show complete correspondence with each other, there is a high degree of consistence in the fact that all these studies have identified a subset of adenocarcinomas with high‐level expression of genes related to differentiation of normal peripheral lung epithelial cells, such as TTF‐1 and SP‐C. Notable features of this subclass are a significantly higher proportion in female non‐/light smokers and the tumors are well‐differentiated, suggesting that they may arise from cells committed to becoming peripheral lung epithelial cells, that is, terminal respiratory unit‐type adenocarcinomas. (41) With these lesions, smoking presumably has no or only a weak influence. In fact, we note that these features correspond to those of adenocarcinoma cases with a high frequency of EGFR mutations and hence higher sensitivity to the gefitinib treatment (see above). Indeed, we observed EGFR mutations in 50% of the cases belonging to this cluster (unpublished observation). Therefore, it will be interesting to examine adenocarcinoma cases for expression profiles and the presence of EGFR mutations in a comprehensive manner in order to verify this intriguing possibility.
Figure 3.
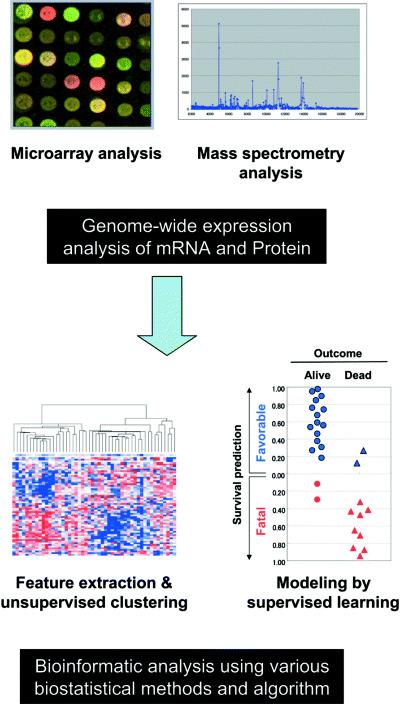
Expression profiling analyzes of both transcriptome and proteome of lung cancers.
In addition, the advantages of expression profiling analysis have been shown through the identification of previously undefined subgroups in other types of lung cancers. The existence of two clinically relevant subsets of squamous cell carcinomas has been suggested, with distinct gene expression signatures and markedly different postoperative survival, 1 , 48 while two prognostically significant subtypes of high‐grade neuroendocrine tumors, which appear to be independent of the currently employed pathologic subclassification, have been identified. (49) Based on the hypothesis that genes specifically expressed in various types of NSCLC may be associated with developmentally regulated genes and pathways, Borczuk et al. conducted an interesting exercise. (50) They found that the gene set specific for adenocarcinoma histology corresponded to those expressed in the late stage (terminal sac and alveolar stages) of murine lung development, whereas the large‐cell carcinoma set was associated with genes expressed in the earlier pseudoglandular and canalicular stages. It is also interesting to note that a metastatic signature, obtained by comparing expression profiles of adenocarcinoma metastases of multiple tumor types to unmatched primary adenocarcinomas, was shown to be associated with a poor prognosis, (51) as to be expected from the fact that deaths from lung adenocarcinomas are in most cases attributable to metastasis.
Expression profiling analysis, aimed at individualized patient outcome prediction, is another important and much needed application, since survival or death is a matter of all or nothing, and currently available information regarding what percentage of those at a certain disease stage are likely to survive after a certain period of time is insufficient in many respects. In this regard, there are two good examples of obvious benefit. Beer et al. identified prognosis‐related genes using the t‐test, and constructed a prognosis prediction classifier of adenocarcinomas by defining a risk index as a linear combination of gene‐expression values for selected genes weighted by their estimated regression coefficient in the preceding COX regression analysis. (45) We have also succeeded in constructing individualized outcome classifiers of NSCLC, using gene expression profiling data‐ and a weighted‐voting algorithm‐based approach with genes selected by the signal‐to‐noise metric. (48) Reasoning from potential differences in outcome‐related expression signatures in two major histologic types of NSCLC, histologic type‐specific outcome classifiers were further constructed, showing an accuracy of more than 90% for the prediction of 5‐year survival or death. A study by Endoh et al., in which they selected 44 candidate genes from those previously identified through exploratory expression profiling analysis, which was validated by real‐time reverse transcriptase‐polymerase chain reaction using a completely separate large cohort, points to a sensible direction for the next step aimed at translation into the clinical setting. (52) It can be envisaged that the establishment of new disease classifications and highly accurate, individualized outcome classifiers for identifying those who are at high risk of future failure, and therefore most eligible for intensive adjuvant therapy with the intention of eradicating undetectable micrometastases, sources of future recurrence, would be a realistic immediate goal. In addition, expression profiling employing sophisticated bioinformatic analyzes should soon transform current strategies for clinical evaluation of drug combinations for cancer treatment, making it possible to provide more sound treatment decisions than in current practice.
In addition to analyzing global changes in the transcriptome, recent advances in biomedical technology have enabled new proteomic approaches to be developed. Yanagisawa et al. used matrix‐assisted laser desorption/ionization mass spectrometry to identify more than 1 600 protein peaks from 79 cases of lung tumors. (53) Biostatistical selection of differentially expressed peaks allowed discrimination between normal and malignant tissue, subclassification of primary tumors, identification of patients with nodal involvement, and classification according to their prognosis based on 15 distinct peaks on mass spectrometry. Protein profiling of serum or plasma using high throughput mass spectroscopy has also been reported for lung cancers, distinguishing cases and controls based on key protein patterns with 50–70% detection rates and a 10% false positive rate. (54) Although anonymous peaks or protein profile spectra might be useful for classification, identification and functional investigation of these proteins are essential for understanding the underlying molecular biology. In this regard, we should mention that candidate molecular markers have been identified, 53 , 55 and far more examples will be found with recently developed sophisticated technologies for proteomic analysis, such as multidimensional liquid chromatography combined with tandem mass spectrometry. This should make it possible to further extend appropriate means of generating and exploiting new biological insights and clinical applications for the benefit of patients.
Conclusions
Accumulating evidence clearly indicates that perturbation of the integrity of the genome leads to the genesis and progression of lung cancers. It is becoming evident that the sequential accumulation of various genetic and epigenetic alterations confers various capabilities on lung cancer cells, including escape from growth inhibitory signals as well as from excess shortening of telomeres, resistance to apoptosis, sensitivity to stimuli for proliferation and angiogenesis, and invasive and metastatic characteristics. In the coming decades, taking advantage of the ample information on the human genome sequence as well as emerging new technologies including sophisticated informatics, we should be able to acquire a complete picture of lung cancer biology and revolutionize the prevention, diagnosis and treatment strategies for this presently fatal disease.
Acknowledgment
We are grateful to all members of our lung cancer research group at Aichi Cancer Center, not only for helpful discussion and comments on the manuscript, but also for continuing fruitful collaborations for many years.
References
- 1. Hartwell L. Defects in a cell cycle checkpoint may be responsible for the genomic instability of cancer cells. Cell 1992; 71: 543–6. [DOI] [PubMed] [Google Scholar]
- 2. Osada H, Takahashi T. Genetic alterations of multiple tumor suppressors and oncogenes in the carcinogenesis and progression of lung cancer. Oncogene 2002; 21: 7421–34. [DOI] [PubMed] [Google Scholar]
- 3. Takahashi T. Lung cancer: an ever increasing store of in‐depth basic knowledge and the beginning of its clinical application. Oncogene 2002; 21: 6868–9. [DOI] [PubMed] [Google Scholar]
- 4. Takahashi T, Sidransky D. Biology of lung cancer. In: Murray JF, Nadel JA, Mason RJ, Broaddus VC, Eds. Textbook of Respiratory Medicine, 4th edn. Philadelphia: Elsevier Science, (in press). [Google Scholar]
- 5. Balsara BR, Testa JR. Chromosomal imbalances in human lung cancer. Oncogene 2002; 21: 6877–83. [DOI] [PubMed] [Google Scholar]
- 6. Haruki N, Harano T, Masuda A, Kiyono T, Takahashi T, Tatematsu Y, Shimizu S, Mitsudomi T, Konishi H, Osada H, Fujii Y. Persistent increase in chromosome instability in lung cancer: possible indirect involvement of p53 inactivation. Am J Pathol 2001; 159: 1345–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Michel LS, Liberal V, Chatterjee A, Kirchwegger R, Pasche B, Gerald W, Dobles M, Sorger PK, Murty VV, Benezra R. MAD2 haplo‐insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature 2001; 409: 355–9. [DOI] [PubMed] [Google Scholar]
- 8. Takahashi T, Haruki N, Nomoto S, Masuda A, Saji S, Osada H. Identification of frequent impairment of the mitotic checkpoint and molecular analysis of the mitotic checkpoint genes, hsMAD2 and p55CDC, in human lung cancers. Oncogene 1999; 18: 4295–300. [DOI] [PubMed] [Google Scholar]
- 9. Nomoto S, Haruki N, Takahashi T, Masuda A, Koshikawa T, Fujii Y, Osada H. Search for in vivo somatic mutations in the mitotic checkpoint gene, hMAD1, in human lung cancers. Oncogene 1999; 18: 7180–3. [DOI] [PubMed] [Google Scholar]
- 10. Gemma A, Seike M, Seike Y, Uematsu K, Hibino S, Kurimoto F, Yoshimura A, Shibuya M, Harris CC, Kudoh S. Somatic mutation of the hBUB1 mitotic checkpoint gene in primary lung cancer. Genes Chromosomes Cancer 2000; 29: 213–8. [DOI] [PubMed] [Google Scholar]
- 11. Sato M, Sekido Y, Horio Y, Takahashi M, Saito H, Minna JD, Shimokata K, Hasegawa Y. Infrequent mutation of the hBUB1 and hBUBR1 genes in human lung cancer. Jpn J Cancer Res 2000; 91: 504–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Elledge SJ. Cell cycle checkpoints: preventing an identity crisis. Science 1996; 274: 1664–72. [DOI] [PubMed] [Google Scholar]
- 13. Robles AI, Linke SP, Harris CC. The p53 network in lung carcinogenesis. Oncogene 2002; 21: 6898–907. [DOI] [PubMed] [Google Scholar]
- 14. Konishi H, Nakagawa T, Harano T, Mizuno K, Saito H, Masuda A, Matsuda H, Osada H, Takahashi T. Identification of frequent G (2) checkpoint impairment and a homozygous deletion of 14–3−3epsilon at 17p13.3 in small cell lung cancers. Cancer Res 2002; 62: 271–6. [PubMed] [Google Scholar]
- 15. Hermeking H, Lengauer C, Polyak K, He TC, Zhang L, Thiagalingam S, Kinzler KW, Vogelstein B. 14–3−3 sigma is a p53‐regulated inhibitor of G2/M progression. Mol Cell 1997; 1: 3–11. [DOI] [PubMed] [Google Scholar]
- 16. Osada H, Tatematsu Y, Yatabe Y, Nakagawa T, Konishi H, Harano T, Tezel E, Takada M, Takahashi T. Frequent and histological type‐specific inactivation of 14–3−3sigma in human lung cancers. Oncogene 2002; 21: 2418–24. [DOI] [PubMed] [Google Scholar]
- 17. Yatabe Y, Osada H, Tatematsu Y, Mitsudomi T, Takahashi T. Decreased expression of 14–3−3sigma in neuroendocrine tumors is independent of origin and malignant potential. Oncogene 2002; 21: 8310–9. [DOI] [PubMed] [Google Scholar]
- 18. Haruki N, Saito H, Tatematsu Y, Konishi H, Harano T, Masuda A, Osada H, Fujii Y, Takahashi T. Histological type‐selective, tumor‐predominant expression of a novel CHK1 isoform and infrequent in vivo somatic CHK2 mutation in small cell lung cancer. Cancer Res 2000; 60: 4689–92. [PubMed] [Google Scholar]
- 19. Matsuoka S, Nakagawa T, Masuda A, Haruki N, Elledge SJ, Takahashi T. Reduced expression and impaired kinase activity of a Chk2 mutant identified in human lung cancer. Cancer Res 2001; 61: 5362–5. [PubMed] [Google Scholar]
- 20. Nakagawa T, Hayashita Y, Maeno K, Masuda A, Sugito N, Osada H, Yanagisawa K, Ebi H, Shimokata K, Takahashi T. Identification of decatenation G2 checkpoint impairment independently of DNA damage G2 checkpoint in human lung cancer cell lines. Cancer Res 2004; 64: 4826–32. [DOI] [PubMed] [Google Scholar]
- 21. Deming PB, Cistulli CA, Zhao H, Graves PR, Piwnica‐Worms H, Paules RS, Downes CS, Kaufmann WK. The human decatenation checkpoint. Proc Natl Acad Sci USA 2001; 98: 12044–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mizuno K, Osada H, Konishi H, Tatematsu Y, Yatabe Y, Mitsudomi T, Fujii Y, Takahashi T. Aberrant hypermethylation of the CHFR prophase checkpoint gene in human lung cancers. Oncogene 2002; 21: 2328–33. [DOI] [PubMed] [Google Scholar]
- 23. Minna JD, Battey JF, Birrer M, Brooks BJ, Cuttitta F, DeGreve J, Gazdar AF, Johnson BE, Nau MM, Sausville EA. Chromosomal deletion, gene amplification, alternative processing, and autocrine growth factor production in the pathogenesis of human lung cancer. Princess Takamatsu Symp 1986; 17: 109–22. [PubMed] [Google Scholar]
- 24. Rodenhuis S, Van De Wetering ML, Mooi WJ, Evers SG, Van Zandwijk N, Bos JL. Mutational activation of the K‐ras oncogene. A possible pathogenetic factor in adenocarcinoma of the lung. N Engl J Med 1987; 317: 929–35. [DOI] [PubMed] [Google Scholar]
- 25. Naoki K, Chen TH, Richards WG, Sugarbaker DJ, Meyerson M. Missense mutations of the BRAF gene in human lung adenocarcinoma. Cancer Res 2002; 62: 7001–3. [PubMed] [Google Scholar]
- 26. Brose MS, Volpe P, Feldman M, Kumar M, Rishi I, Gerrero R, Einhorn E, Herlyn M, Minna J, Nicholson A, Roth JA, Albelda SM, Davies H, Cox C, Brignell G, Stephens P, Futreal PA, Wooster R, Stratton MR, Weber BL. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res 2002; 62: 6997–7000. [PubMed] [Google Scholar]
- 27. Harbour JW, Lai SL, Whang‐Peng J, Gazdar AF, Minna JD, Kaye FJ. Abnormalities in structure and expression of the human retinoblastoma gene in SCLC. Science 1988; 241: 353–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Takahashi T, Nau MM, Chiba I, Birrer MJ, Rosenberg RK, Vinocour M, Levitt M, Pass H, Gazdar AF, Minna JD. p53: a frequent target for genetic abnormalities in lung cancer. Science 1989; 246: 491–4. [DOI] [PubMed] [Google Scholar]
- 29. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non‐small‐cell lung cancer to gefitinib. N Engl J Med 2004; 350: 2129–39. [DOI] [PubMed] [Google Scholar]
- 30. Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304: 1497–500. [DOI] [PubMed] [Google Scholar]
- 31. Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, Mardis E, Kupfer D, Wilson R, Kris M, Varmus H. EGF receptor gene mutations are common in lung cancers from ‘never smokers’ and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA 2004; 101: 13306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kosaka T, Yatabe Y, Endoh H, Takahashi T, Mitsudomi T. Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer Res 2004; 64: 8919–23. [DOI] [PubMed] [Google Scholar]
- 33. Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004; 304: 554. [DOI] [PubMed] [Google Scholar]
- 34. Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib‐sensitizing EGFR mutations in lung cancer activate anti‐apoptotic pathways. Science 2004; 305: 1163–7. [DOI] [PubMed] [Google Scholar]
- 35. Cappuzzo F, Magrini E, Ceresoli GL, Bartolini S, Rossi E, Ludovini V, Gregorc V, Ligorio C, Cancellieri A, Damiani S, Spreafico A, Paties CT, Lombardo L, Calandri C, Bellezza G, Tonato M, Crino L. Akt phosphorylation and gefitinib efficacy in patients with advanced non‐small‐cell lung cancer. J Natl Cancer Inst 2004; 96: 1133–41. [DOI] [PubMed] [Google Scholar]
- 36. Mitsudomi T, Kosaka T, Endoh H, Horio Y, Hida T, Mori S, Hatooka S, Shinoda M, Takahashi T and Yatabe Y. Mutations of the epidermal growth factor receptor gene predict prolonged survival after gefitinib treatment in patients with non‐small cell lung cancer with postoperative recurrence. J Clin Oncol (in press). [DOI] [PubMed] [Google Scholar]
- 37. Rodenhuis S, Slebos RJ, Boot AJ, Evers SG, Mooi WJ, Wagenaar SS, Van Bodegom PC, Bos JL. Incidence and possible clinical significance of K‐ras oncogene activation in adenocarcinoma of the human lung. Cancer Res 1988; 48: 5738–41. [PubMed] [Google Scholar]
- 38. Suzuki H, Takahashi T, Kuroishi T, Suyama M, Ariyoshi Y, Ueda R. p53 mutations in non‐small cell lung cancer in Japan: association between mutations and smoking. Cancer Res 1992; 52: 734–6. [PubMed] [Google Scholar]
- 39. Sozzi G, Sard L, De Gregorio L, Marchetti A, Musso K, Buttitta F, Tornielli S, Pellegrini S, Veronese ML, Manenti G, Incarbone M, Chella A, Angeletti CA, Pastorino U, Huebner K, Bevilaqua G, Pilotti S, Croce CM, Pierotti MA. Association between cigarette smoking and FHIT gene alterations in lung cancer. Cancer Res 1997; 57: 2121–3. [PubMed] [Google Scholar]
- 40. Yatabe Y, Kosaka T, Takahashi T and Mitsudomi T. EGFR mutation is specific for terminal respiratory unit type adenocarcinoma. Am J Surg Pathol (in press). [DOI] [PubMed] [Google Scholar]
- 41. Yatabe Y, Mitsudomi T, Takahashi T. TTF‐1 expression in pulmonary adenocarcinomas. Am J Surg Pathol 2002; 26: 767–73. [DOI] [PubMed] [Google Scholar]
- 42. Masuda A, Kondo M, Saito T, Yatabe Y, Kobayashi T, Okamoto M, Suyama M, Takahashi T. Establishment of human peripheral lung epithelial cell lines (HPL1) retaining differentiated characteristics and responsiveness to epidermal growth factor, hepatocyte growth factor, and transforming growth factor beta1. Cancer Res 1997; 57: 4898–904. [PubMed] [Google Scholar]
- 43. Garber ME, Troyanskaya OG, Schluens K, Petersen S, Thaesler Z, Pacyna‐Gengelbach M, Van De Rijn M, Rosen GD, Perou CM, Whyte RI, Altman RB, Brown PO, Botstein D, Petersen I. Diversity of gene expression in adenocarcinoma of the lung. Proc Natl Acad Sci USA 2001; 98: 13784–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bhattacharjee A, Richards WG, Staunton J, Li C, Monti S, Vasa P, Ladd C, Beheshti J, Bueno R, Gillette M, Loda M, Weber G, Mark EJ, Lander ES, Wong W, Johnson BE, Golub TR, Sugarbaker DJ, Meyerson M. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci USA 2001; 98: 13790–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Beer DG, Kardia SL, Huang CC, Giordano TJ, Levin AM, Misek DE, Lin L, Chen G, Gharib TG, Thomas DG, Lizyness ML, Kuick R, Hayasaka S, Taylor JM, Iannettoni MD, Orringer MB, Hanash S. Gene‐expression profiles predict survival of patients with lung adenocarcinoma. Nat Med 2002; 8: 816–24. [DOI] [PubMed] [Google Scholar]
- 46. Virtanen C, Ishikawa Y, Honjoh D, Kimura M, Shimane M, Miyoshi T, Nomura H, Jones MH. Integrated classification of lung tumors and cell lines by expression profiling. Proc Natl Acad Sci USA 2002; 99: 12357–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kikuchi T, Daigo Y, Katagiri T, Tsunoda T, Okada K, Kakiuchi S, Zembutsu H, Furukawa Y, Kawamura M, Kobayashi K, Imai K, Nakamura Y. Expression profiles of non‐small cell lung cancers on cDNA microarrays: identification of genes for prediction of lymph‐node metastasis and sensitivity to anti‐cancer drugs. Oncogene 2003; 22: 2192–205. [DOI] [PubMed] [Google Scholar]
- 48. Tomida S, Koshikawa K, Yatabe Y, Harano T, Ogura N, Mitsudomi T, Some M, Yanagisawa K, Takahashi T, Osada H. Gene expression‐based, individualized outcome prediction for surgically treated lung cancer patients. Oncogene 2004; 23: 5360–70. [DOI] [PubMed] [Google Scholar]
- 49. Jones MH, Virtanen C, Honjoh D, Miyoshi T, Satoh Y, Okumura S, Nakagawa K, Nomura H, Ishikawa Y. Two prognostically significant subtypes of high‐grade lung neuroendocrine tumours independent of small‐cell and large‐cell neuroendocrine carcinomas identified by gene expression profiles. Lancet 2004; 363: 775–81. [DOI] [PubMed] [Google Scholar]
- 50. Borczuk AC, Gorenstein L, Walter KL, Assaad AA, Wang L, Powell CA. Non‐small‐cell lung cancer molecular signatures recapitulate lung developmental pathways. Am J Pathol 2003; 163: 1949–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet 2003; 33: 49–54. [DOI] [PubMed] [Google Scholar]
- 52. Endoh H, Tomida S, Yatabe Y, Konishi H, Osada H, Tajima K, Kuwano H, Takahashi T, Mitsudomi T. Prognostic model of pulmonary adenocarcinoma by expression profiling of eight genes as determined by quantitative real‐time reverse transcriptase polymerase chain reaction. J Clin Oncol 2004; 22: 811–9. [DOI] [PubMed] [Google Scholar]
- 53. Yanagisawa K, Shyr Y, Xu BJ, Massion PP, Larsen PH, White BC, Roberts JR, Edgerton M, Gonzalez A, Nadaf S, Moore JH, Caprioli RM, Carbone DP. Proteomic patterns of tumour subsets in non‐small‐cell lung cancer. Lancet 2003; 362: 433–9. [DOI] [PubMed] [Google Scholar]
- 54. Sidransky D, Irizarry R, Califano JA, Li X, Ren H, Benoit N, Mao L. Serum protein MALDI profiling to distinguish upper aerodigestive tract cancer patients from control subjects. J Natl Cancer Inst 2003; 95: 1711–7. [DOI] [PubMed] [Google Scholar]
- 55. Campa MJ, Wang MZ, Howard B, Fitzgerald MC, Patz EF Jr. Protein expression profiling identifies macrophage migration inhibitory factor and cyclophilin a as potential molecular targets in non‐small cell lung cancer. Cancer Res 2003; 63: 1652–6. [PubMed] [Google Scholar]